

Abstract
Autophagy, a highly conserved process conferring cytoprotection against stress, contributes to the progression of cerebral ischemia. β-arrestins are multifunctional proteins that mediate receptor desensitization and serve as important signaling scaffolds involved in numerous physiopathological processes. Here, we show that both ARRB1 (arrestin, β 1) and ARRB2 (arrestin, β 2) were upregulated by cerebral ischemic stress. Knockout of Arrb1, but not Arrb2, aggravated the mortality, brain infarction, and neurological deficit in a mouse model of cerebral ischemia. Accordingly, Arrb1-deficient neurons exhibited enhanced cell injury upon oxygen-glucose deprivation (OGD), an in vitro model of ischemia. Deletion of Arrb1 did not affect the cerebral ischemia-induced inflammation, oxidative stress, and nicotinamide phosphoribosyltransferase upregulation, but markedly suppressed autophagy and induced neuronal apoptosis/necrosis in vivo and in vitro. Additionally, we found that ARRB1 interacted with BECN1/Beclin 1 and PIK3C3/Vps34, 2 major components of the BECN1 autophagic core complex, under the OGD condition but not normal conditions in neurons. Finally, deletion of Arrb1 impaired the interaction between BECN1 and PIK3C3, which is a critical event for autophagosome formation upon ischemic stress, and markedly reduced the kinase activity of PIK3C3. These findings reveal a neuroprotective role for ARRB1, in the context of cerebral ischemia, centered on the regulation of BECN1-dependent autophagosome formation.
Keywords: ARRB1, autophagy, cerebral ischemia, BECN1, neuron
Introduction
Stroke is the second most common cause of death and major cause of disability worldwide.1 Approximately, 80% of the cases are ischemic. Cerebral ischemia results in severe intracellular energy stress leading to cell death. Meanwhile, the defense to energy exhaustion and metabolic stress contributes to the survival of neurons under cerebral ischemia.2 Autophagy is a novel cytoprotective mechanism, whereby cells self-digest intracellular organelles as a salvage mechanism for survival during starvation.3,4 Understanding the molecular regulatory mechanisms of autophagy and the relevance of autophagy in various diseases, such as neurodegeneration and ischemic injury, have a potential therapeutic significance.3,4 Recent studies have demonstrated that autophagy represents a critical neuroprotective response against neuronal injury.5,6 As a highly conserved process, autophagy is elegantly controlled by key intracellular signaling pathways and multiple autophagy-related (ATG) proteins.3,4 We previously showed that autophagy contributes to the neuroprotective mechanism of NAMPT (nicotinamide phosphoribosyltransferase) in cerebral ischemic injury.7,8
β-arrestins are a family of cytoplasmic adaptor proteins consisting of 2 members: ARRB1 and ARRB2. As their names imply, they were initially identified as the proteins that function to desensitize G protein-coupled receptors (GPCRs).9,10 Later research shows that β-arrestins also serve a second function in GPCRs internalization.9,10 Moreover, it has recently been realized that β-arrestins also act as scaffold proteins to interact with other proteins, and influence intracellular signaling pathways and manipulate cell biological functions dependent on or independent of GPCRs.9,10 Accumulating evidence has confirmed their importance in human health and disease,11 including our previous work on their critical regulation of insulin resistance and autoimmunity.12,13 In brain, ARRB2 regulates Aβ generation and γ-secretase activity in Alzheimer disease,14 suggesting the importance of β-arrestins in central nervous system (CNS) disease. However, the role of β-arrestins in cerebral ischemia is essentially uncharacterized. Here we demonstrate that ARRB1, but not ARRB2, protects against cerebral ischemic injury via a previously unknown mechanism involving BECN1-dependent autophagosome formation.
Results
Cerebral ischemia stimulates protein expression of β-arrestins in brain
We investigated the protein expression of ARRB1 and ARRB2 in cardio–cerebro–vascular tissues. As shown in Figure 1A, both ARRB1 and ARRB2 were expressed in mouse brain tissue. In a well-established mouse model of cerebral ischemia produced by middle cerebral artery occlusion (MCAO), both ARRB1 and ARRB2 were upregulated in the peri-infarct penumbra tissue (Fig. 1B). We also determined ARRB1 and ARRB2 expression using immunohistochemistry and observed similar results (Fig. 1C; Fig. S1). ARRB1 and ARRB2 were upregulated at 2 h after cerebral ischemia and lasted until 36 h after ischemia (Fig. S2). These results suggest that these 2 proteins might contribute to the pathophysiological process of cerebral ischemia.

Figure 1. Cerebral ischemia stimulates protein expression of β-arrestins in brain. (A) Protein expression of ARRB1 and ARRB2 in cardio–cerebro–vascular tissues. (B) ARRB1 and ARRB2 were upregulated in the penumbra of ischemic hemisphere from the mice subjected to middle cerebral artery occlusion (MCAO). *P < 0.05, determined by Mann-Whitney Test. n = 4. (C) Frozen brain section (20 μM) was stained by antibodies against ARRB1 and ARRB2 respectively and observed under confocal microscopy. Relative fluorescence density was calculated.
Deletion of Arrb1, but not Arrb2, aggravates neuronal injury in cerebral ischemia in vivo and in vitro
To evaluate the exact functions of β-arrestins in cerebral ischemia, we subjected Arrb1 and Arrb2 knockout mice (arrb1−/− and arrb2−/− mice, Fig. S3A and S3B) to MCAO as described previously.8,15 The cerebral blood flow was reduced by ~80% after MCAO, comparable between arrb1−/− and arrb2−/− mice (Fig. S4). Physiological parameters, including pH, pCO2, pO2, and blood pressure, were also comparable between arrb1−/− and arrb2−/− mice (Table S1). At 24 h after MCAO, the mortality (Fig. 2A) and infarct size (Fig. 2C) were higher in arrb1−/− mice compared with wild-type (WT) controls, and the neurological deficit was aggravated in arrb1−/− mice (Fig. 2E). Surprisingly, such phenotypes were not observed in arrb2−/− mice (Fig. 2B, D, and F). In the ischemic penumbra areas of both arrb1−/− and arrb2−/− mice, hematoxylin and eosin staining demonstrated a large number of shrunken, scalloped neurons with pyknotic nuclei (yellow arrow), which indicated the dead neurons (Fig. 2G). Notably, there were still some intact neurons (blue arrow) in WT mice, but not in arrb1−/− mice (Fig. 2G). We did not observe any abnormality of neuron death or survival in 8- to 10-wk-old arrb1−/− mice in normal conditions (data not shown). We also detected apoptosis in the ischemic penumbra areas using TUNEL analysis. As shown in Figure 2H, there were more apoptotic cells (TUNEL-positive) in arrb1−/− mouse brain compared with WT mice. These results indicate that deletion of Arrb1, but not Arrb2, aggravates neuronal injury induced by cerebral ischemia in vivo.

Figure 2. ARRB1, but not ARRB2, is neuroprotective in cerebral ischemia in vivo. (A and B) Mortality at 24 h after MCAO was increased in arrb1−/− mice (A), but not in arrb2−/− mice (B). *P < 0.05, determined by the Chi-Square Test. n = 15 to 32. NS, no significance. (C and D) Infarct size in arrb1−/− (C) and arrb2−/− mice (D) was measured by 2, 3, 5-triphenyltetrazolium chloride (TTC) staining (white). *P < 0.05, determined by ANOVA. n = 6 to 9. NS, no significance. (E and F) Neurological deficit score in arrb1−/− (E) and arrb2−/− mice (F). *P < 0.05, determined by Kruskal-Wallis test. n = 6 to 9. NS, no significance. (G) Hematoxylin and eosin staining showing the morphological characteristics of WT and arrb1−/− mouse brains upon MCAO. Shrunken neurons with pyknotic nuclei are indicated with yellow arrows while intact neurons are indicated with blue arrows. (H) TUNEL analysis showing the apoptotic cells in penumbra of WT and arrb1−/− mouse brain upon MCAO. TUBB3 was stained to show axons of neurons. n = 7. *P < 0.05, determined by t test.
We then isolated neurons from WT and arrb1−/− mice and cultured them in vitro. Oxygen-glucose deprivation (OGD) treatment is a well-established in vitro model of cerebral ischemia.8,15 Upon 2 h OGD treatment, there was an obvious reduction in cell viability (Fig. 3A) in WT neurons. The lactate dehydrogenase (LDH) in the culture supernatant fraction was also increased in WT neurons (Fig. 3B). These changes were further deteriorated in arrb1−/− neurons (Fig. 3A and B). Furthermore, using immunofluorescence staining of TUBB3, a marker of living neuron, we found that the cell number decreased significantly by OGD treatment in WT neurons and arrb1−/− neurons, while the arrb1−/− neurons displayed extensive severe injury under OGD (Fig. 3C).
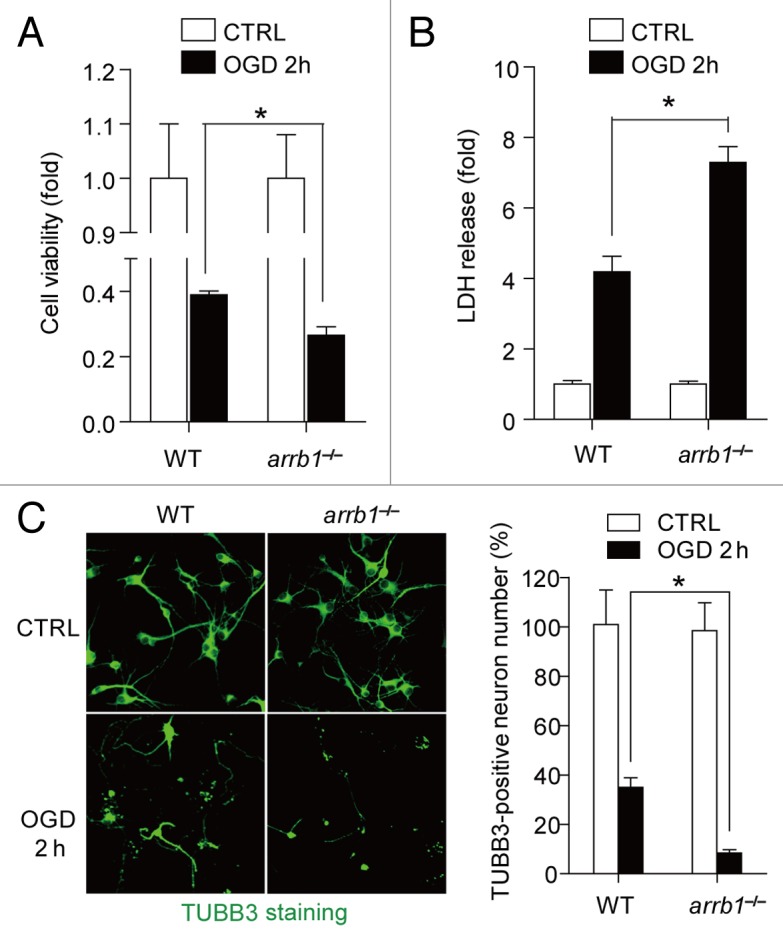
Figure 3. ARRB1 is neuroprotective in neuronal ischemia in vitro. (A) Cell viability in cultured arrb1−/− neurons under control (CTRL) and oxygen glucose deprivation (OGD) conditions were determined by the CCK-8 assay. (B) Cell injury in cultured arrb1−/− neurons under control and OGD conditions were determined by LDH release assay. (C) Number of living neurons was determined by immunofluorescence staining assay with an antibody against the neuron marker TUBB3. *P < 0.05, determined by t test. n = 8.
Deletion of Arrb1 does not influence inflammation, oxidative stress, and NAD+ metabolism in cerebral ischemia
Since post-ischemic inflammation and oxidative stress were tightly associated with post-ischemic brain damage,16,17 we studied the levels of inflammatory factors and oxidative stress. First, the levels of 3 important inflammatory factors, including TNF (tumor necrosis factor), IL6 (interleukin 6 [interferon, β 2]) and CCL2 (chemokine [C-C motif] ligand 2), were measured. We found that ischemic stress stimulated the levels of them in the penumbra area (Fig. 4A–C). However, these inductions were comparable between WT and arrb1−/− mice (Fig. 4A–C). We also found similar phenotypes in levels of malondialdehyde (MDA) and superoxide, 2 markers of oxidative stress (Fig. 4D and E). In addition, we examined the influence of arrb1 knockout on NAMPT, the rate-limiting enzyme for NAD+ biosynthesis.18 NAMPT has been reported to be a determinant of cell death upon nutrition deprivation recently.19 Cerebral ischemia induced NAMPT upregulation in the peri-infarct penumbra, comparable between arrb1−/− and WT mice (Fig. 4F). All above factors appeared unimportant to the neuroprotection of ARRB1 against ischemic brain injury, and we then investigated the possibility of the autophagic defense system.

Figure 4. Knockout of Arrb1 does not alter inflammation, oxidative stress, and NAD+ metabolism during cerebral ischemia. (A–C) Levels of TNF, IL6, and CCL2 in penumbra and contralateral brain tissues at 24 h after cerebral ischemia in WT and arrb1−/− mice were determined by ELISA assays. n = 6 to 9. NS, no significance. (D and E) Levels of MDA and superoxide in penumbra and contralateral brain tissues at 24 h after cerebral ischemia in WT and arrb1−/− mice were determined. n = 6 to 9. MDA, malondialdehyde; NS, no significance. (F) Expression of NAMPT protein, which is the rate-limiting enzyme for NAD+ biosynthesis, in unaffected contralateral and peri-infarct penumbra brain tissues in WT and arrb1−/− mice. n = 4 in each group. *P < 0.05 vs contralateral; NS, no significance.
Deletion of Arrb1 strongly suppresses autophagosome formation upon cerebral ischemic stress
To assess the potential involvement of ARRB1 in autophagy process during cerebral ischemia, we pretreated the WT and arrb1−/− mice with autophagy inhibitor 3-methyladenine (3-MA) at 30 min before MCAO. In WT mice, 3-MA treatment significantly aggravated neuronal cell death (Fig. 5A) and increased cleaved CASP3/caspase-3 expression (Fig. 5B) in the penumbra area, suggesting the neuroprotection of autophagy in cerebral ischemia. This observation was in line with previous reports.5,6,20,21 On the contrary, 3-MA treatment failed to induce any effects in arrb1−/− mice (Fig. 5A and B). And, the neuronal cell death and cleaved CASP3 expression in arrb1−/− mice without 3-MA treatment were similar to those in WT mice with 3-MA treatment (Fig. 5A and B). These results implicate that the neuroprotection of ARRB1 might involve autophagy. To further explore this hypothesis, we injected rapamycin, an inducer of autophagy, into arrb1−/− mice. Injection of rapamycin partly antagonized arrb1 knockout-induced aggravation of brain injury (Fig. 5C).

Figure 5. Deletion of Arrb1 markedly limits the autophagic process and promotes apoptosis-necrosis in the mouse cerebral ischemia model. (A) TUNEL assay showing the brain cell apoptosis in penumbra tissues at 24 h after cerebral ischemia in WT and arrb1−/− mice. **P < 0.01 compared to the WT without 3-MA treatment. n = 8. NS, no significance. (B) Western blotting assay showing the cleaved CASP3 expression in penumbra brain tissues at 24 h after cerebral ischemia in WT and arrb1−/− mice. **P < 0.01 compared to the WT without 3-MA treatment. n = 8. MCAO, middle cerebral artery occlusion; CTRL, control (without MCAO). NS, no significance. (C) Autophagy inducer rapamycin antagonized the deletion of Arrb1-induced aggravation of neuronal injury by MCAO. n = 8. (D) Ultrastructural features in brain tissues of penumbra from arrb1−/− and WT mice at 30 min after MCAO (yellow asterisk indicating autophagosomes). n = 8. N, nuclei. (E) Western blotting images depicting LC3-I, LC3-II, and SQSTM1 and quantification of LC3-II and SQSTM1 levels in arrb1−/− and WT brains. *P < 0.05, determined by the Mann-Whitney Test. n = 5. *P < 0.05, determined by the Mann-Whitney Test. n = 5. (F) Immunohistochemistry staining of LC3 in brain tissues of penumbra from arrb1−/− and WT mice at 30 min after MCAO. *P < 0.05, determined by the Mann-Whitney Test. n = 8.
Next, we directly analyzed autophagy at early stage of cerebral ischemia (30 min after MCAO [in vivo] and 30 min after OGD [in vitro]). Ultrastructural analysis demonstrated the typical autophagosomes in penumbra area of WT mice after MCAO but scarce in arrb1−/− mice (Fig. 5D). LC3-II, which is recruited to phagophore membranes, is an indicator of autophagosome formation.6,22 We tested the LC3-II/ACTB (actin, β) ratio in penumbra tissue of WT and arrb1−/− mice upon MCAO. The LC3-II/ACTB ratio was significantly increased by ischemic insult in WT mice, but to a much lesser extent in arrb1−/− mice (Fig. 5E), indicating that knockout of arrb1 markedly inhibits the conversion from LC3-I to LC3-II. SQSTM1 is an autophagic substrate whose abundance correlates inversely with autophagic activity.22 SQSTM1 protein expression was decreased by ischemic insult in WT mice but, surprisingly, increased in arrb1−/− mice (Fig. 5E). However, similar changes of Sqstm1 mRNA expression were also observed (Fig. S5A). Immunofluorescent analysis showed less LC3 puncta in arrb1−/− mice upon MCAO compared with WT mice (Fig. 5F).
We further studied the essential role of ARRB1 in autophagy in cultured neurons. At 30 min after OGD, LC3 formed puncta-like structure (yellow arrows) in WT neurons (Fig. 6A). However, it still uniformly distributed in the arrb1−/− neurons (Fig. 6A). Notably, at this time point, the signal observed for cleaved CASP3 was stronger in arrb1−/− neurons than in WT neurons (Fig. 6A). We also transfected WT and arrb1−/− neurons with lentivirus expressing GFP-LC3. GFP-LC3 puncta was induced by short-term OGD treatment in WT neurons, but to a lesser extent in arrb1−/− neurons (Fig. 6B). Treatment with bafilomycin A1, a specific inhibitor of the vacuolar ATPase-dependent proton pump that prevents the acidification of lysosomes and disrupts autophagic degradation, further increased GFP-LC3 puncta in WT neurons, and also to a lesser extent in arrb1−/− neurons (Fig. 6B). Immunoblotting assay showed that LC3-II/ACTB ratio was significantly increased in WT neurons upon OGD, which was not observed in arrb1−/− neurons (Fig. 6C). Decrease in autophagosomes could result from enhancement of autophagic degradation or block of autophagic flux. To distinguish between these, we treated WT and arrb1−/− neurons with bafilomycin A1 and subjected them to OGD stress. Bafilomycin A1 markedly increased LC3-II levels in WT neurons but to a lesser extent in arrb1−/− neurons (Fig. 6D). We also investigated SQSTM1 protein and mRNA levels in control and OGD neurons. SQSTM1 protein was downregulated by OGD in WT neurons, but accumulated in arrb1−/− neurons upon OGD stimuli (~3-fold, Fig. 6E). Bafilomycin A1 challenge further increased SQSTM1 protein levels in WT neurons but not in arrb1−/− neurons (Fig. 6F). Sqstm1 mRNA levels were increased in arrb1−/− neurons upon ischemic stress (~1.5-fold, Fig. S5B), suggesting that the increase of SQSTM1 protein in arrb1−/− neurons may not be, or at least not only be, due to an impairment of protein degradation.

Figure 6. Deletion of Arrb1 suppresses autophagosome formation in neuron OGD models. (A) Triple immunohistochemistry staining of TUBB3 (neuron marker), LC3, and cleaved CASP3 in arrb1−/− and WT neurons under control (CTRL) and OGD conditions (see Materials and Methods for details). DAPI was used to stain nuclei. (B) Cultured arrb1−/− and WT neurons were transfected with GFP-LC3 lentivirus and subjected to OGD treatment with or without bafilomycin A1 (Baf A1, 100 nM). *P < 0.05 vs WT, n = 8. (C and D) Western blotting images depicting LC3-I and LC3-II in arrb1−/− and WT neurons with or without bafilomycin A1 (Baf A1). *P < 0.05, determined by the Mann-Whitney Test. n = 5. (E and F) Western blotting images depicting SQSTM1 in arrb1−/− and WT neurons with or without bafilomycin A1 (Baf A1). *P < 0.05, determined by the Mann-Whitney Test. n = 5.
ARRB1 is recruited to the BECN1 core complex upon ischemic stress in neurons
We next asked how ARRB1 regulates neuronal autophagy during cerebral ischemia. Since ARRB1 functions as a scaffold for intracellular signaling transductions,9 we investigated the distribution of ARRB1 protein in primary neurons. In normal conditions, ARRB1 was expressed in both cytoplasm and nucleus (Fig. 7A). Intriguingly, in OGD-treated neurons, some puncta-like structures formed and these structures were ARRB1-positive (Fig. 7A). In view of these results, we speculated that ARRB1 might directly participate in autophagosome formation as a scaffold protein via interacting with some critical autophagic protein complexes. The ULK1 (homolog of yeast Atg1) kinase complex leads to autophagy induction,23,24 while the BECN1 core complex is recruited to the phagophore to initialize the nucleation and elongation of the autophagosome.25,26 We did not detect an interaction between ARRB1 and ULK1 or ULK2 under normal or OGD conditions (Fig. S6). Instead, we found that ARRB1 interacted with BECN1 in the OGD condition, but not in normal conditions (Fig. 7B). To confirm this observation, we studied whether the phosphatidylinositol 3-kinase (PtdIns3K) class III catalytic subunit (PIK3C3/Vps34), an important component of the BECN1 core complex,25 interacted with ARRB1. As expected, the interaction between ARRB1 and BECN1-PIK3C3 was detected under the OGD condition but not normal conditions in WT neurons (Fig. 7B). In arrb1−/− neurons, the interaction between ARRB1 and BECN1-PIK3C3 was not observed (Fig. 7B). Immunofluorescence staining further demonstrated the colocalization of ARRB1 and BECN1 in OGD-treated neurons (Fig. 7C). ARRB1 deletion did not alter the protein levels of BECN1 and PIK3C3 under both normal and ischemic conditions (Fig. S7) and ARRB1 did not participate in the interaction between BECN1 and PIK3C3 under normal conditions (Fig. S8). These interesting results suggest that ARRB1 does not interact with the BECN1 core complex under normal conditions, but is recruited to BECN1 core complex when autophagy is induced upon ischemic stress.

Figure 7. ARRB1 interacts with BECN1 and PIK3C3 upon ischemic stress in neurons. (A) Distribution pattern of ARRB1 protein in WT primary neurons under control and OGD conditions. (B) Interaction between ARRB1 and BECN1-PIK3C3 detected by immunoprecipitation analysis in WT and arrb1−/− neurons under control and OGD conditions. IP, immunoprecipitation; WB, western blotting. (C) Colocalization of ARRB1 and BECN1 in WT primary neurons under OGD stress.
Deletion of Arrb1 impairs the formation of BECN1 core complex upon ischemia stress in neurons
We further asked whether the BECN1 core complex, which is required for autophagosome formation, would be affected by knockout of Arrb1. Immunoprecipitation assay showed the interaction between BECN1 and PIK3C3 upon OGD stress in WT neurons (Fig. 8A and B). However, this interaction was absent in arrb1−/− neurons (Fig. 8A and B). These results indicate that deletion of Arrb1 disrupts the formation of the BECN1 core complex upon ischemic stress. Using the PIK3C3 kinase assay, we showed that deletion of Arrb1 markedly reduced the kinase activity of PIK3C3 (Fig. 8C). We also assessed the influence of ARRB1 on the interaction of BECN1 and BCL2. Arrb1 deletion did not affect the interaction or disassociation between BECN1 and BCL2 under normal and ischemic conditions (Fig. S9).
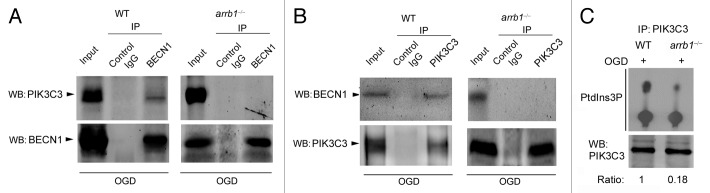
Figure 8. Knockout of Arrb1 abrogates the interaction between BECN1 and PIK3C3 in neurons upon ischemic stress. (A) Cell lysates of WT and arrb1−/− neurons upon OGD were immunoprecipitated with anti-BECN1 antibody and then detected by anti-PIK3C3 antibody. (B) Cell lysates of WT and arrb1−/− neurons upon OGD were immunoprecipitated with anti- PIK3C3 antibody and then detected by anti-BECN1 antibody. WB, western blotting. (C) Lysates from the WT and arrb1−/− neurons were immunoprecipitated with anti-PIK3C3 and assayed for PIK3C3 kinase activity with a thin layer chromatography assay.
Discussion
This is the first report showing ARRB1 is neuroprotective in cerebral ischemia through the coordination of autophagy. In this study, both ARRB1 and ARRB2 were upregulated by ischemic stimuli in mouse MCAO model. However, only knockout of Arrb1 aggravated the neuronal injury induced by experimental ischemia. arrb1−/− mice exhibited more severe infarct size, neuronal deficit, and mortality compared with WT mice in the MCAO model. Deletion of Arrb1 also induced deleterious influences on cultured neurons in the OGD model. Knockout of Arrb1 did not change the inflammation, oxidative stress, and NAD+ metabolism, but greatly inhibited autophagosome formation in neurons upon ischemic stress, which was evidenced by autophagy inhibitor 3-MA treatment, electron microscope assay, immunofluorescence staining, and immunoblotting analysis. Thus, the arrb1 knockout-induced loss in functional autophagy promoted neuron death. Mechanically, it seemed that ARRB1 did not participate in autophagy under normal conditions, but was recruited to BECN1 core complex when autophagy was induced upon ischemic stress. Because ARRB1 interacted with BECN1 and PIK3C3 in ischemic setting, knockout of Arrb1 blocked the formation of the BECN1 core complex and extensively inhibited autophagosome formation and markedly reduced the kinase activity of PIK3C3, the catalytic subunit of PtdIns3K.
It is well described that β-arrestins are regulators for receptors of structurally diverse classes, including 7 membrane-spanning receptors, the nicotinic cholinergic receptors, receptor tyrosine kinases and cytokine receptors.9,10 β-arrestins “arrest” the GPCRs signaling capability through desensitization and internalization. This negative feedback regulation of GPCRs is important for GRCRs signaling termination.9,10 In recent years, β-arrestins have emerged as multifunctional adaptor and scaffold proteins that dynamically assemble a wide range of multiprotein complexes in various critical intracellular signal transduction pathways such as those modulated by WNT-CTNNB1/β-catenin,27 EP300/p-300,28 mitogen-activated protein kinases/extracellular signal-regulated kinases,29 and insulin signaling.12 Thus, β-arrestins participate in many critical physiopathological processes, including schizophrenia,30 depression,31 cardioprotection of β-blocker,32 and insulin resistance.12 However, their function in ischemia-induced cell injury is unknown. A previous work reported that ARRB1 was upregulated in a rat cerebral hypoxia-ischemia model, yet the significance of this upregulation was not studied.33 In our study, using Arrb1 and Arrb2 knockout mice, we provided the first evidence that genetic deletion of Arrb1, but not Arrb2, aggravated the cerebral ischemia-induced neuronal damage. These results indicate that ARRB1 has neuroprotective effects in cerebral ischemia, which may provide new insight into its biological functions.
In addition, we found that deletion of Arrb1 abolished the autophagic process, and prompted apoptosis and/or necrosis at the early stage of cerebral ischemia, pointing out that the arrb1 knockout disrupts the balance between autophagy and apoptosis or necrosis in neurons upon ischemic stress. Generally, autophagy is thought to be a adaptive response with cytoprotective effect in ischemic injury, e.g., myocardial infarct34,35 and liver ischemia,36,37 although the role of autophagy in cerebral ischemia is to some extent controversial, with some studies showing that activation of autophagy is detrimental38-41 while others supporting that autophagy is neuroprotective.5,6,20,21,42 A series of studies demonstrate that inhibition of autophagy, including the use of the pharmacological inhibitor 3-MA,39 or genetic inactivation of Atg740 or Becn1,41 protects against cerebral hypoxia-ischemia-induced neuronal death. In contrast, other studies show that inhibition of autophagy leads to aggravated neuronal injury using similar investigative tools.5,6,21,42 We consider that the differences in research tools, administration route, drug dose, ischemia duration, observation time, and evaluation index may critically affect the ultimate conclusion. The most important issue may be the research tools. Among these studies, Uchiyama's group40 and Zeng's group41 have used genetic tools, while other studies5,6,21,38,39,42 use 3-MA to test the in vivo function of autophagy in cerebral ischemic damage. As 3-MA inhibits all classes of PtdIns3K and the following signaling cascades, the specificity of 3-MA, especially in vivo, should be carefully considered. The side effects of 3-MA may lead to inappropriate conclusions. Besides the research tools, observation time also affects the results. In our previous study,8 we found that autophagy was increased only at 2 h post-cerebral ischemia in NAMPT-overexpressing brain tissue and neurons, which was not observed at 8 and 24 h post-cerebral ischemia. To clarify the exact role of autophagy in cerebral ischemia, more detailed works are warranted.
Our results further demonstrate that deletion of Arrb1 remarkably inhibited autophagosome formation upon ischemic stress. We also found that ARRB1 interacts with BECN1 and PIK3C3 in OGD model but not in normal conditions. Coimmunoprecipitation assays showed that ARRB1 is able to interact with BECN1 and PIK3C3 in neurons. Colocalization staining assays confirmed this phenotype. Additionally, deletion of Arrb1 reduced the kinase activity of PIK3C3. Therefore, we postulated that ARRB1 acts as an essential coordinator of autophagy interacting with BECN1-PIK3C3 to regulate the BECN1 core complex development. The experiments in arrb1−/− neurons (Fig. 8) confirmed this speculation. BECN1 interacts with several cofactors, including ATG14,43,44 PIK3R4/VPS15/p150,43 UVRAG,43 HMGB1,45 and KIAA0226 (1700021K19Rik in mice and RGD1305422 in rats)/Rubicon,44 to regulate the lipid kinase PIK3C3 and promote formation of BECN1 core complexes, thereby inducing autophagy.26 ARRB1 is a scaffold and adaptor protein and plays a role in desensitization, i.e., alterations in receptor sequestration and receptor signaling.9,10 ARRB1 also interacts with other proteins, such as the γ-secretase complex.46 The amino acid residues 241 to 360 of ARRB1 are important for the interaction between the ARRB1 and γ-secretase complex.46 So how ARRB1 exactly interacts with the BECN1 core complex remains an unanswered and intriguing question of fundamental importance. This issue is a limitation of our study and we will explore the interaction domain of ARRB1 and BECN1-PIK3C3 in future investigations. Interestingly, deletion of Arrb1 does not totally abolish autophagy. In fact, in arrb1−/− mice or neurons, mild autophagy was still observed upon ischemic stress (Figs. 5 and 6). This observation suggests that noncanonical BECN1-independent autophagic pathways47,48 might also take part in pathophysiological changes after cerebral ischemia.
SQSTM1 is a stress-inducible intracellular protein known to regulate various signal transduction pathways involved in cell survival and cell death.49 Growing lines of evidence suggest that SQSTM1, as a autophagy substrate, is degraded by autophagy for clearance of protein aggregates and organelles.49 Sqstm1 mRNA level has been reported to be regulated by different factors. For example, Jain et al. report that overexpression of NFE2L2/NRF2 (nuclear factor [erythroid-derived 2]-like 2) increases Sqstm1 mRNA levels.50 If there are simultaneous increases of SQSTM1 protein and mRNA expression, the accumulation of SQSTM1 protein may be not caused by disruption of autophagy.50 In our study, SQSTM1 protein was degraded in WT mice brain tissue or WT neurons after ischemic stress, which was delayed by deletion of Arrb1 (increased about ~3-fold). However, the Sqstm1 mRNA levels were also increased about 3.5-fold in arrb1−/− mice penumbra and 1.5-fold in arrb1−/− neurons respectively. The difference between the in vivo and in vitro Sqstm1 mRNA levels (1.5-fold vs 3.5-fold) may due to the influence of activated glia or infiltrated immune cells such as T cells on Sqstm1 mRNA levels upon ischemia.51,52 Thus, we think the data obtained from cell experiment may be more accurate for reflecting the Sqstm1 mRNA level changes in arrb1−/− neurons. Since the SQSTM1 protein was increased about ~3-fold whereas Sqstm1 mRNA was increased ~1.5-fold in arrb1−/− neurons under ischemic conditions, we considered that the accumulation of SQSTM1 protein in arrb1−/− neurons under ischemic conditions is not, or at least not only, due to an impairment of protein degradation. Because both ARRB1 and SQSTM1 are intracellular adaptor proteins, we propose that there may be some undiscovered molecular links between them. This question needs further investigation.
We have noted that there were some reports on roles of ARRB1 and ARRB2 in neuronal cell death in Alzheimer disease. Thathiah et al. demonstrate that genetic deletion of Arrb2 leads to a reduction in the production of Aβ(40) and Aβ(42) in transgenic AD mice.14 Liu et al. show that genetic ablation of Arrb1 diminishes Aβ pathology and behavioral deficits in transgenic AD mice.46 Although there are great differences between the pathophysiology of cerebral ischemia and Alzheimer disease, we can safely conclude that β-arrestins play pivotal role in neuronal survival.
Collectively, we demonstrate that ARRB1 mediates neuroprotection in cerebral ischemia through coordinating BECN1-dependent autophagy. This action of ARRB1 provides a novel insight into the molecular mechanisms underlying the autophagy-induced neuroprotection in cerebral ischemia, and may be of particular importance in autophagy biology. Furthermore, because autophagy has been found to be associated with not only cerebral ischemia, but also other neurological diseases, such as neurodegeneration, brain microbial infection, and aging, thus our results of the regulation of ARRB1 on autophagy may ultimately help to improve our understandings of the molecular mechanisms and offer new clinical therapeutic directions for these diseases.
Materials and Methods
Mice
Male arrb1−/− and arrb2−/− mice (2- to 4- mo old, body weight > 20 g) were used and described previously.12,13 All experiments were performed in adherence with the National Institutes of Health guidelines on the laboratory animals and were approved by the Scientific Investigation Board of our university.
Materials and reagents
Antibodies against ARRB1 (SAB4501798) and ACTB/β-actin #A1978) were obtained from Sigma-Aldrich Co. LLC. Antibodies against ARRB2 #3857), cleaved CASP3 (9661), BECN1 (3495) and PIK3C3 (4263) were obtained from Cell Signaling Technology, Inc.. NAMPT antibody (sc-67020) was purchased from Santa Cruz Biotechnology, Inc. LC3 antibody (ab58610) was purchased from Abcam. TUBB3/Tuj-1 antibody (ab14545) was from Abcam. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit (G3250) was purchased from Promega. TNF/tumor necrosis factor-α (SEA133Mu, IL6 (interleukin 6, SEA079Mu) and CCL2 (SEA087Mu) ELISA kits were purchased from USCN Life Science. Malondialdehyde (MDA) assay kit (STA-332) was obtained from Cell Biolabs, Inc. Superoxide anion chemiluminescent detection kit (574590) was obtained from Millipore.
Primary neuron culture
Primary mouse neuronal cells were prepared from the cerebral cortex of neonatal animals within 6 h after birth and genotyping, as described previously.8,15 One day after isolation, the cultures were replenished with Neurobasal medium (Life Technologies, 21103-049) supplemented with 2% B27 (Life Technologies, 17504044). Glial growth was suppressed by addition of 5-fluoro-2-deoxyuridine and uridine (10 μM), yielding cultured cells with > 90% neurons, confirmed by RBFOX3/NeuN (neuron marker; Millipore, MAB377) and GFAP (astrocyte marker; Cell Signaling Technology, Inc., 3670) staining. After 5 d in vitro (DIV), the neurons were used for experiments.
In vivo and in vitro cerebral ischemia model
MCAO model (in vivo) was prepared in mice as described previously.8,15 The core temperature (rectum) was maintained by use of a homeothermic heating pad. Cerebral focal ischemia was produced by intraluminal occlusion of the left middle cerebral artery using a silicone rubber-coated nylon monofilament. Cortical blood flow was measured with a laser Doppler flowmeter (Moor Instruments, moorVMS-LDF, Axminster, Devon, UK). The physiological parameters, including arterial blood pressure, arterial pH, arterial pCO2, and arterial pO2, were monitored in mice before and after MCAO using a blood pressure instrument and a blood gas analyzer. Mice were examined for neurological deficits using Bederson method.53 Infarct size was determined by staining with 2,3,5-triphenyltetrazolium chloride (TTC, Amresco LLC., 298-96-4) and was analyzed with Image J software.
OGD model (in vitro) was prepared in cultured neurons as described previously.8,15 Primary mouse cortical neurons were isolated from the cerebral cortex of neonatal mice within 6 h after birth and genotyping, described previously.15 To establish OGD conditions, the cultured neurons (d 7 to 10) were washed 3 times and incubated with glucose-free Earle’s balanced salt solution and placed for different times within a hypoxic chamber (Forma Scientific) that was continuously flushed with 95% N2 and 5% CO2 at 37 °C to obtain < 0.5% O2. Control neuron cultures were placed in EBSS (14155063, Gibco, St. Lawrence, MA, USA) containing glucose (25 mmol/L) and incubated under normal culture conditions for the same period.
Administration of 3-MA
Autophagy inhibitor 3-MA was purchased from Sigma (M9281). Intracerebroventricular (-1.0 mm anteroposterior, 1.4 mm lateral, -4.0 mm dorsoventral solution prepared in saline. Mice were injected at 30 min before the MCAO under anesthesia. relative to bregma) injections were given in the right lateral ventricle with 3 μl of a 20 mg/ml The dosage of 3-MA was selected according to several published studies5,41,54,55 and our previous study.8 According to previous results, this dosage of 3-MA did not affect physiological parameters including pH, pCO2, pO2, and serum glucose level.56
Administration of rapamycin and bafilomycin A1
Rapamycin (200 μg/kg, Cell Signaling Technology, 9904) or vehicle (saline) was injected intraperitoneally. The dose of rapamycin was selected according to the previous reports.57,58 Bafilomycin A1 (100 nM, Santa Cruz Biotechnology, Inc., sc-201550) was added into cultured cells to prevent the acidification of lysosomes and disrupt autophagic degradation.
Tissue sampling
In this study, the ischemic penumbra was used as illustrated in Figure S10. Mice were euthanized and brains were harvested and sectioned into 1.6-mm coronal sections (5 sections) and stored at −80 °C. The middle section (3rd section) was stained with 1.2% TTC for 20 min (37 °C) to determine the infarct area. The infarct area became white and the uninfarct area was red, so the ischemic penumbra in this section was determined. Therefore, we could approximately decide the ischemic penumbra in the adjacent sections (2nd and 4th sections). And the experiments such as TUNEL staining, immunohistochemistry and immunoblotting were performed on the ischemic penumbra of these 2 sections.
Transmission electron microscopy
Tissues were fixed with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.4), followed by post-fixation for 8 h in 1.5% osmium tetraoxide. After dehydration with graded alcohols, the samples were dehydrated in a graded ethanol series and embedded in epoxy resin. Samples were sectioned (80 nm), counterstained with uranylacetate and lead citrate and observed with a transmission electron microscope (Hitachi, H-800, Tokyo, Japan). Images were acquired digitally from a randomly selected pool of 10 to 15 fields under each condition.
Western blotting
Tissues and cells were lysed in RIPA buffer with protease inhibitor cocktail (Pierce, 78430) and used for western blotting as described previously.15,59 Blots were incubated with specific primary antibodies and IRDye800CW-conjugated secondary antibodies (926-32211 and 926-32212, Li-Cor Bioscience, Lincoln, NE, USA).60,61 The image was captured by the Odyssey infrared imaging system (Li-Cor Bioscience, Lincoln, NE, USA). All western blotting experiments were repeated at least 3 times.
Coimmunoprecipitation
Immunoprecipitation was performed as described previously.15,59 Primary neurons were lysed in RIPA buffer with protease inhibitor cocktail. The crude lysates were cleared of insoluble debris by centrifugation at 12,000 g. Immunoprecipitating antibody (3 μg) or normal IgG (negative control, Santa-cruz, sc-2762) were added and incubated on a rotator at 4 °C overnight. The 20 μl G/A agarose beads (Santa-cruz, sc-2003) were added into the 200 μl homogenates or lysates, and incubated for 2 h with gentle agitation. The beads were washed 3 times with the lysis buffer and boiled with 10 μl 2 × sample buffer (Fermentas, R1011). The beads were removed by centrifugation (5 min at 14,000 rpm). The supernatant fraction was collected and used for western blotting.62
Immunochemistry and immunofluorescence
Immunochemistry and immunofluorescence were performed as described previously.63,64 Frozen 20-μm thick brain sections or cultured neurons placed on Confocal dish (Corning, CLS-DL-CC-014) were fixed in 4% paraformaldehyde, blocked by 8% normal donkey serum (Santa Cruz Biotechnology, sc-2044), and incubated in specific primary antibodies as follows: goat anti-LC3 (1:500), mouse anti-TUBB3 (1:1000), rabbit anti-cleaved CASP3 (1:200), mouse anti-ARRB1 (1:200) and rabbit anti-BECN1 (1:500). After being washed 3 times by PBST (0.1% Tween 20 in PBS [70011-044, Gibco]), the sections and cells were incubated with corresponding Alexa 488-conjugated donkey anti-mouse (Molecular Probes, A-21202), Alexa 555-conjugated donkey anti-rabbit (Molecular Probes, A-21432) and Alexa 647-conjugated donkey anti-goat (Molecular Probes, A11055) secondary antibodies. DAPI (Molecular Probes, D1306) was used to stain nuclei.65 The immunofluorescence TUNEL assay was performed according to the instructions of the manufacturer.8,55 Images were obtained by confocal microscopy (Olympus, Fluoview FV1000, Tokyo, Japan).
Quantification of LC3 puncta or GFP-LC3 puncta
For quantitative confocal microscopy of endogenous LC3 puncta, LC3 immunohistochemistry was performed in frozen brain tissues. The 20-μm-thick brain sections were incubated with LC3 primary antibody and then Alexa-555 conjugated secondary antibody. Under confocal microscopy (10 × magnification), the puncta-like structures per field were calculated. For GFP-LC3 puncta quantification, lentivirus expressing GFP-LC366 (Millipore Corporation, #17-10193) was added into the medium of cultured neurons. Three d later, the neurons were subjected to OGD treatment with or without bafilomycin A1 (100 nM).67,68
Inflammatory factors and oxidative stress assays
Mouse non-ischemic and ischemic brain tissues were homogenized in PBS with a protease inhibitor cocktail. After centrifuging at 14,000 rpm for 10 min, the supernatant fraction was collected. TNF, IL6, CCL2 and MDA levels were determined with ELISA kits according to the manufacturer’s instructions. Reaction product was detected by spectrophotometry using microtiter plate reader. Superoxide Anion levels were determined with a chemiluminescent kit (Calbiochem, 574590) and the luminescence was recorded by microtiter plate reader. The levels of TNF, IL6, CCL2, MDA, and superoxide were calculated according to the standard curve.69
Cell viability and injury assay
Cell viability was evaluated by a nonradioactive cell counting kit (CCK-8, Dojindo, CK04-01) assay. Lactate dehydrogenase release analysis was performed with a colorimetric LDH cytotoxicity assay to assess cell injury. Reaction product was detected by spectrophotometry at 450 nm,70 using a microtiter plate reader (Tecan, Maennedorf, Switzerland).
In vitro PIK3C3 lipid kinase assay
The endogenous PIK3C3 lipid kinase assay was performed according to a previous report.71 Cells were lysed and immunoprecipitaed with anti-PIK3C3. Then, the immune complexes were incubated in a buffer (20 mM HEPES, 1 mM EGTA, 0.4 mM EDTA, 5 mM MgCl2, 0.05 mM DTT, 50 mM ATP, 5 mM MnCl2, and 50 mM DTT, pH 7.4) containing 0.2 mg/ml phosphatidylinositol (Sigma, P5766) and 5 μCi 32P-ATP at 37 °C for 45 min. The kinase reactions were terminated by the addition of 20 μl of 8 M HCl and extracted with 160 μl chloroform:methanol (1:1). This extracted phospholipid products were separated on Silica Gel 60A (Merck, 115111). Plates were dried and followed by visualization with a Typhoon Imager (GE Healthcare Biosciences, Piscataway, NJ).
Statistical analysis
Data are expressed as mean ± SEM P values were calculated with one-way analysis of variance (ANOVA) followed by the Tukey post-hoc test with Prism software (GraphPad) unless otherwise noted. Statistical significance was set at P < 0.05.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the National Basic Research Program of China (2009CB521902), the National Natural Science Foundation of China (81100866, 81373414 and 81130061), the Shanghai Municipal Education Commission (12ZZ078), Shanghai Committee of Science and Technology Fund for “Qimingxing” Project (14QA1404700) and the Shanghai “Shu Guang” Project (10GG19).
Glossary
Abbreviations:
- 3-MA
3-methyladenine
- CCL2
chemokine (C-C motif) ligand 2
- CNS
central nervous system
- GPCRs
G protein-coupled receptors
- IL6
interleukin 6 (interferon, beta 2)
- LDH
lactate dehydrogenase
- MCAO
middle cerebral artery occlusion
- MDA
malondialdehyde
- NAMPT
nicotinamide phosphoribosyltransferase
- OGD
oxygen-glucose deprivation
- PIK3C3
phosphatidylinositol 3-kinase, catalytic subunit type 3
- PtdIns3K
phosphatidylinositol 3-kinase
- TNF/TNFα
tumor necrosis factor
- WT
wild type
References
- 1.Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371:1612–23. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- 2.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 3.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–30. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 5.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32:329–39. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 6.Papadakis M, Hadley G, Xilouri M, Hoyte LC, Nagel S, McMenamin MM, Tsaknakis G, Watt SM, Drakesmith CW, Chen R, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med. 2013;19:351–7. doi: 10.1038/nm.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang P, Tian WW, Song J, Guan YF, Miao CY. Deficiency of NG2+ cells contributes to the susceptibility of stroke-prone spontaneously hypertensive rats. CNS Neurosci Ther. 2011;17:327–32. doi: 10.1111/j.1755-5949.2011.00265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 2012;8:77–87. doi: 10.4161/auto.8.1.18274. [DOI] [PubMed] [Google Scholar]
- 9.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 10.Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol Cell. 2006;24:643–52. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 11.Shenoy SK, Lefkowitz RJ. β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci. 2011;32:521–33. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luan B, Zhao J, Wu H, Duan B, Shu G, Wang X, Li D, Jia W, Kang J, Pei G. Deficiency of a beta-arrestin-2 signal complex contributes to insulin resistance. Nature. 2009;457:1146–9. doi: 10.1038/nature07617. [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, Feng Y, Kang J, Liu C, Li Z, Li D, Cao W, Qiu J, Guo Z, Bi E, et al. Critical regulation of CD4+ T cell survival and autoimmunity by beta-arrestin 1. Nat Immunol. 2007;8:817–24. doi: 10.1038/ni1489. [DOI] [PubMed] [Google Scholar]
- 14.Thathiah A, Horré K, Snellinx A, Vandewyer E, Huang Y, Ciesielska M, De Kloe G, Munck S, De Strooper B. β-arrestin 2 regulates Aβ generation and γ-secretase activity in Alzheimer’s disease. Nat Med. 2013;19:43–9. doi: 10.1038/nm.3023. [DOI] [PubMed] [Google Scholar]
- 15.Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC, Zhai QW, Su DF, Miao CY. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann Neurol. 2011;69:360–74. doi: 10.1002/ana.22236. [DOI] [PubMed] [Google Scholar]
- 16.Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. 2007;20:334–42. doi: 10.1097/WCO.0b013e32813ba151. [DOI] [PubMed] [Google Scholar]
- 17.El Kossi MM, Zakhary MM. Oxidative stress in the context of acute cerebrovascular stroke. Stroke. 2000;31:1889–92. doi: 10.1161/01.STR.31.8.1889. [DOI] [PubMed] [Google Scholar]
- 18.Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, Andris F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32:3225–34. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He S, Wang C, Dong H, Xia F, Zhou H, Jiang X, Pei C, Ren H, Li H, Li R, et al. Immune-related GTPase M (IRGM1) regulates neuronal autophagy in a mouse model of stroke. Autophagy. 2012;8:1621–7. doi: 10.4161/auto.21561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9:1321–33. doi: 10.4161/auto.25132. [DOI] [PubMed] [Google Scholar]
- 22.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 26.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–80. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bryja V, Gradl D, Schambony A, Arenas E, Schulte G. Beta-arrestin is a necessary component of Wnt/beta-catenin signaling in vitro and in vivo. Proc Natl Acad Sci U S A. 2007;104:6690–5. doi: 10.1073/pnas.0611356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M, Zhang M, Bao G, Wang F, Zhang X, et al. A nuclear function of beta-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell. 2005;123:833–47. doi: 10.1016/j.cell.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 29.Zheng H, Loh HH, Law PY. Beta-arrestin-dependent mu-opioid receptor-activated extracellular signal-regulated kinases (ERKs) Translocate to Nucleus in Contrast to G protein-dependent ERK activation. Mol Pharmacol. 2008;73:178–90. doi: 10.1124/mol.107.039842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci U S A. 2008;105:13656–61. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, Wetsel WC, Lefkowitz RJ, Gainetdinov RR, Caron MG. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–36. doi: 10.1016/j.cell.2007.11.041. [DOI] [PubMed] [Google Scholar]
- 32.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci U S A. 2007;104:16657–62. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lombardi MS, van den Tweel E, Kavelaars A, Groenendaal F, van Bel F, Heijnen CJ. Hypoxia/ischemia modulates G protein-coupled receptor kinase 2 and beta-arrestin-1 levels in the neonatal rat brain. Stroke. 2004;35:981–6. doi: 10.1161/01.STR.0000121644.82596.7e. [DOI] [PubMed] [Google Scholar]
- 34.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–57. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 35.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 36.Wang D, Ma Y, Li Z, Kang K, Sun X, Pan S, Wang J, Pan H, Liu L, Liang D, et al. The role of AKT1 and autophagy in the protective effect of hydrogen sulphide against hepatic ischemia/reperfusion injury in mice. Autophagy. 2012;8:954–62. doi: 10.4161/auto.19927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evankovich J, Zhang R, Cardinal JS, Zhang L, Chen J, Huang H, Beer-Stolz D, Billiar TR, Rosengart MR, Tsung A. Calcium/calmodulin-dependent protein kinase IV limits organ damage in hepatic ischemia-reperfusion injury through induction of autophagy. Am J Physiol Gastrointest Liver Physiol. 2012;303:G189–98. doi: 10.1152/ajpgi.00051.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–9. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 39.Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–89. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- 40.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–69. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xing S, Zhang Y, Li J, Zhang J, Li Y, Dang C, Li C, Fan Y, Yu J, Pei Z, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy. 2012;8:63–76. doi: 10.4161/auto.8.1.18217. [DOI] [PubMed] [Google Scholar]
- 42.Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, Zhang XY, Qin ZH. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. 2012;8:310–25. doi: 10.4161/auto.18673. [DOI] [PubMed] [Google Scholar]
- 43.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–72. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–76. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ, 3rd, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–92. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X, Zhao X, Zeng X, Bossers K, Swaab DF, Zhao J, Pei G. β-arrestin1 regulates γ-secretase complex assembly and modulates amyloid-β pathology. Cell Res. 2013;23:351–65. doi: 10.1038/cr.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grishchuk Y, Ginet V, Truttmann AC, Clarke PG, Puyal J. Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy. 2011;7:1115–31. doi: 10.4161/auto.7.10.16608. [DOI] [PubMed] [Google Scholar]
- 48.Scarlatti F, Maffei R, Beau I, Ghidoni R, Codogno P. Non-canonical autophagy: an exception or an underestimated form of autophagy? Autophagy. 2008;4:1083–5. doi: 10.4161/auto.7068. [DOI] [PubMed] [Google Scholar]
- 49.Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res. 2012;66:457–62. doi: 10.1016/j.phrs.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 50.Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–91. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paul S, Schaefer BC. Selective autophagy regulates T cell activation. Autophagy. 2012;8:1690–2. doi: 10.4161/auto.21581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdulrahman BA, Khweek AA, Akhter A, Caution K, Tazi M, Hassan H, Zhang Y, Rowland PD, Malhotra S, Aeffner F, et al. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr ΔF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J Biol Chem. 2013;288:2049–58. doi: 10.1074/jbc.M112.411728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–6. doi: 10.1161/01.STR.17.3.472. [DOI] [PubMed] [Google Scholar]
- 54.Yan W, Zhang H, Bai X, Lu Y, Dong H, Xiong L. Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Res. 2011;1402:109–21. doi: 10.1016/j.brainres.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 55.Guo R, Hu N, Kandadi MR, Ren J. Facilitated ethanol metabolism promotes cardiomyocyte contractile dysfunction through autophagy in murine hearts. Autophagy. 2012;8:593–608. doi: 10.4161/auto.18997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qin AP, Liu CF, Qin YY, Hong LZ, Xu M, Yang L, Liu J, Qin ZH, Zhang HL. Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy. 2010;6:738–53. doi: 10.4161/auto.6.6.12573. [DOI] [PubMed] [Google Scholar]
- 57.Chauhan A, Sharma U, Jagannathan NR, Reeta KH, Gupta YK. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav Brain Res. 2011;225:603–9. doi: 10.1016/j.bbr.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 58.Yin L, Ye S, Chen Z, Zeng Y. Rapamycin preconditioning attenuates transient focal cerebral ischemia/reperfusion injury in mice. Int J Neurosci. 2012;122:748–56. doi: 10.3109/00207454.2012.721827. [DOI] [PubMed] [Google Scholar]
- 59.Wang P, Zhang RY, Song J, Guan YF, Xu TY, Du H, Viollet B, Miao CY. Loss of AMP-activated protein kinase-α2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes. 2012;61:1051–61. doi: 10.2337/db11-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He W, Wang Q, Xu J, Xu X, Padilla MT, Ren G, Gou X, Lin Y. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy. 2012;8:1811–21. doi: 10.4161/auto.22145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, Walz G, Huber TB. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8:826–37. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- 62.Tanida I, Yamasaki M, Komatsu M, Ueno T. The FAP motif within human ATG7, an autophagy-related E1-like enzyme, is essential for the E2-substrate reaction of LC3 lipidation. Autophagy. 2012;8:88–97. doi: 10.4161/auto.8.1.18339. [DOI] [PubMed] [Google Scholar]
- 63.Wang P, Xu TY, Guan YF, Su DF, Fan GR, Miao CY. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res. 2009;81:370–80. doi: 10.1093/cvr/cvn288. [DOI] [PubMed] [Google Scholar]
- 64.Wang P, Yang FJ, Du H, Guan YF, Xu TY, Xu XW, Su DF, Miao CY. Involvement of leptin receptor long isoform (LepRb)-STAT3 signaling pathway in brain fat mass- and obesity-associated (FTO) downregulation during energy restriction. Mol Med. 2011;17:523–32. doi: 10.2119/molmed.2010.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Korkmaz G, le Sage C, Tekirdag KA, Agami R, Gozuacik D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy. 2012;8:165–76. doi: 10.4161/auto.8.2.18351. [DOI] [PubMed] [Google Scholar]
- 66.Kobayashi S, Xu X, Chen K, Liang Q. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy. 2012;8:577–92. doi: 10.4161/auto.18980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yefimova MG, Messaddeq N, Harnois T, Meunier AC, Clarhaut J, Noblanc A, Weickert JL, Cantereau A, Philippe M, Bourmeyster N, et al. A chimerical phagocytosis model reveals the recruitment by Sertoli cells of autophagy for the degradation of ingested illegitimate substrates. Autophagy. 2013;9:653–66. doi: 10.4161/auto.23839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zou Z, Yuan Z, Zhang Q, Long Z, Chen J, Tang Z, Zhu Y, Chen S, Xu J, Yan M, et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy. 2012;8:1798–810. doi: 10.4161/auto.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen ML, Yi L, Jin X, Liang XY, Zhou Y, Zhang T, Xie Q, Zhou X, Chang H, Fu YJ, et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy. 2013;9:2033–45. doi: 10.4161/auto.26336. [DOI] [PubMed] [Google Scholar]
- 70.Ambjørn M, Ejlerskov P, Liu Y, Lees M, Jäättelä M, Issazadeh-Navikas S. IFNB1/interferon-β-induced autophagy in MCF-7 breast cancer cells counteracts its proapoptotic function. Autophagy. 2013;9:287–302. doi: 10.4161/auto.22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.