

Abstract
Peroxisomes are degraded by a selective type of autophagy known as pexophagy. Several different types of pexophagy have been reported in mammalian cells. However, the mechanisms underlying how peroxisomes are recognized by autophagy-related machinery remain elusive. PEX3 is a peroxisomal membrane protein (PMP) that functions in the import of PMPs into the peroxisomal membrane and has been shown to interact with pexophagic receptor proteins during pexophagy in yeast. Thus, PEX3 is important not only for peroxisome biogenesis, but also for peroxisome degradation. However, whether PEX3 is involved in the degradation of peroxisomes in mammalian cells is unclear. Here, we report that high levels of PEX3 expression induce pexophagy. In PEX3-loaded cells, peroxisomes are ubiquitinated, clustered, and degraded in lysosomes. Peroxisome targeting of PEX3 is essential for the initial step of this degradation pathway. The degradation of peroxisomes is inhibited by treatment with autophagy inhibitors or siRNA against NBR1, which encodes an autophagic receptor protein. These results indicate that ubiquitin- and NBR1-mediated pexophagy is induced by increased expression of PEX3 in mammalian cells. In addition, another autophagic receptor protein, SQSTM1/p62, is required only for the clustering of peroxisomes. Expression of a PEX3 mutant with substitution of all lysine and cysteine residues by arginine and alanine, respectively, also induces peroxisome ubiquitination and degradation, hence suggesting that ubiquitination of PEX3 is dispensable for pexophagy and an endogenous, unidentified peroxisomal protein is ubiquitinated on the peroxisomal membrane.
Keywords: peroxisome, autophagy, pexophagy, PEX3, peroxisomal membrane protein, ubiquitin, NBR1, SQSTM1/p62
Introduction
Peroxisomes are single-membrane-bound organelles that mediate numerous metabolic activities, such as β-oxidation of very long chain fatty acids, detoxification of hydrogen peroxides, and the synthesis of ether phospholipids and bile acids in mammalian cells.1-3 Impaired peroxisome biogenesis results in the development of peroxisome biogenesis disorders, including Zellweger syndrome, neonatal adrenoleukodystrophy, infantile Refsum disease, and rhizomelic chondrodysplasia punctata.4,5 Each cell contains several hundred peroxisomes, and peroxisome number is controlled by a balance between peroxisomal biogenesis and degradation. Peroxisomes are degraded by several different pathways, including the Lon protease system, ALOX15 (arachidonate 15-lipoxygenase)-mediated autolysis, and autophagy in mammalian cells.6
Pexophagy, a type of autophagy specific for peroxisomes, is the major degradation pathway active in peroxisome homeostasis. In mouse livers, peroxisome numbers are increased by feeding with peroxisome proliferators, such as phthalate esters, and excess peroxisomes are degraded specifically upon the withdrawal of peroxisome proliferators. This peroxisome degradation is impaired in autophagy-related protein 7 (atg7) knockout mouse livers.7,8 To analyze the molecular mechanisms of mammalian pexophagy, an experimental system for the induction and analysis of pexophagy needs to be established in cultured cells.
We have reported that pexophagy can be induced by manipulating nutritional availability in cell culture conditions.9 Upon replenishment from short starvation, the autophagy-related protein LC3-II, the form of LC3 conjugated to phosphatidylethanolamine,10,11 interacts with PEX14. PEX14 functions in peroxisome matrix protein import as a component of the docking complex on the peroxisomal membrane and directly interacts with PEX5, a cytosolic receptor for matrix proteins.12-17 LC3-II interacts with PEX14 in a competitive manner with PEX5, and the affinity of LC3-II for PEX14 increases upon cell starvation.
Kim et al.18 report that pexophagy is induced by the expression of ubiquitin (Ub)-fused peroxisomal membrane proteins (PMPs) with a topology in which Ub faces the cytosol. SQSTM1 (sequestosome 1), an autophagic receptor protein, is required for this pathway. SQSTM1 mediates the selective autophagy of Ub-positive protein aggregates by binding to both ubiquitinated substrates and LC3.19-21 As occurs in Ub-mediated selective autophagy,22,23 peroxisomes are recognized by SQSTM1 and degraded, suggesting that the presentation of Ub on the cytosolic surface of the peroxisomal membrane is sufficient to induce SQSTM1-dependent pexophagy. However, it remains unknown whether Ub is exposed on the peroxisome surface under physiological conditions.
More recently, Deosaran et al.24 have reported that pexophagy is also induced by overexpression of NBR1 (neighbor of BRCA1 gene 1). NBR1 is similar to SQSTM1 in that it binds to both Ub and LC3 and is required for Ub-mediated selective autophagy of cytosolic protein aggregates.25,26 Deosaran et al. also show that overexpression of NBR1 induces peroxisome clustering around a NBR1-positive punctate structure, and that these clustered peroxisomes are degraded by an autophagic pathway. It is noteworthy that a membrane-binding region of NBR1 is required for its colocalization with peroxisomes and the induction of peroxisome clustering, and the Ub-binding domain plays a regulatory role in the colocalization. The coiled-coil domain of NBR1 also plays a role in the clustering.24 However, it is unclear what is the target for NBR1 on peroxisomes.
As described above, 3 different types of pexophagies, PEX14-mediated, artificial Ub-mediated, and NBR1-induced, have been reported in mammalian cells. Because these reports suggest that mutually distinct key factors are essential for each type of pexophagy, it is difficult to understand the relationship, if one exists, between these different pexophagies. Therefore, a more comprehensive understanding of the regulation of pexophagy is required. To obtain deeper insight into mammalian pexophagy, additional pexophagy-inducing methods need to be discovered.
In yeast pexophagy, Pex3, a PMP that functions in PMP import into the peroxisomal membrane,27-31 is suggested to mediate the recognition step in pexophagy. Pichia pastoris Atg3032 and Saccharomyces cerevisiae Atg3633 have been identified as pexophagy-specific receptors that link peroxisomes to the autophagosome formation site by binding to both Pex3 and other autophagic machinery components. Overexpression of these proteins induces pexophagy,32,33 similar to the effect of NBR1 overexpression in mammalian cells. Thus, we hypothesized that PEX3 functions not only in peroxisomal membrane biogenesis, but also in pexophagy in mammalian cells.
In the present study, we investigated whether ectopic expression of PEX3 induces pexophagy in mammalian cells. An expression of PEX3 induced the ubiquitination of peroxisomal proteins, thereby leading to the translocation of NBR1 to the peroxisomal membrane for degradation. Under these conditions, peroxisomes were clustered in a SQSTM1-dependent manner, although SQSTM1 was not required for peroxisome degradation. Thus, the exogenous expression of PEX3 likely leads to activation of the endogenous Ub conjugation system required for peroxisome degradation.
Results
PEX3 overexpression induces pexophagy
To monitor the induction of pexophagy in mammalian cells, we focused on PEX3 as a target for pexophagy-related receptor proteins, as observed in yeast,32,33 and investigated whether PEX3 interacts with pexophagy-specific machinery, subsequently leading to peroxisomal degradation. To do this, we expressed PEX3 in Chinese hamster ovary (CHO)-K1 cells, HeLa cells, and mouse embryonic fibroblasts (MEFs). Peroxisomes were significantly decreased in cells expressing high levels of PEX3 (Fig. 1A, a and b). By contrast, such degradation was not discernible in cells expressing PEX14 (Fig. 1A, e) or those transfected with the empty vector (Fig. 1A, i and j). Since mitochondrial depolarization and endoplasmic reticulum stress were not induced and the levels of these organelles were not decreased, it appeared that peroxisomes were eliminated preferentially by PEX3 overexpression (Fig. S1). Figure 1B shows the percentages of cells with fewer than 20 peroxisomes that were calculated from the cells exogenously expressing PEX3 or PEX14 shown in Figure 1A, a and e, respectively. The drastic decrease in the number of peroxisomes was observed in almost half the cells expressing PEX3 (Fig. 1B).

Figure 1. PEX3 overexpression induces pexophagy. (A) CHO-K1 cells were transfected with PEX3-HA2 (a–d), PEX3-HA2 (e–h) and empty vector (i and j), as indicated. After 24 h, the cells were fixed and immunostained with antibodies against ABCD3/PMP70 (a, e, and i), PEX14 (b, f, and j), and HA (c, g, and k). Merged views are shown (d, h, and l). (B and C) The percentage of cells showing fewer than 20 peroxisomes was calculated from 50 cells transfected with PEX3-HA2, PEX14-HA2 or empty vector alone as shown in (A, a, e, and i) (B) and those transfected with PEX3-HA2 in the presence of autophagy inhibitors (C). Data are presented as the mean ± SD of 3 replicates. 3-MA: 3-methyladenine (10 mM); BafA1: bafilomycinA1 (100 nM). (D) HeLa cells were transfected with PEX3-HA2 and PEX14-HA2 alone in the absence (-) or presence of autophagy inhibitors. After 24 h, the cells were lysed with SDS-PAGE sample buffer and analyzed by SDS-PAGE and immunoblotting with antibodies against ACOX1 (acyl-CoA oxidase), a peroxisomal matrix protein, and TUBA/α-tubulin for a loading control. (E) WT MEF (a–f) and atg5 KO MEFs (g–l) were transfected with PEX3-HA2 and immunostained with antibodies against PEX14 (a, d, g, and j) and HA (b, e, h, and k). (F) The percentage of cells showing less than 20 peroxisomes was calculated as in (B and C). Scale bars: 10 μm.
To assess whether peroxisomes are eliminated by autophagy following PEX3 overexpression, the percentages of cells showing peroxisome elimination were also determined in the presence of the autophagy inhibitors 3-methyladenine and bafilomycinA1. Under these conditions, the percentages of cells exhibiting peroxisome elimination were significantly decreased (Fig. 1C). We also analyzed the abundance of peroxisomes by immunoblotting of ACOX1 (acyl-CoA oxidase1), a peroxisomal matrix protein. The protein level of ACOX1 was decreased by overexpression of PEX3-HA2, but not PEX14-HA2 (Fig. 1D, left panels). Furthermore, this was abrogated in the presence of autophagy inhibitors (Fig. 1D, right panels). We likewise examined peroxisome elimination in MEF cells deficient in ATG5, an essential factor for lipidation of LC3. As expected, marked change in the number of peroxisomes was not observed in atg5-knockout (KO) MEFs (Fig. 1E, j) following PEX3 overexpression, while peroxisomes were drastically decreased in normal MEFs (Fig. 1E, d). No apparent changes in peroxisome number were detected in empty vector transfected MEFs (Fig. 1E, a and g). These morphological results were confirmed by quantifying the numbers of peroxisomes in both types of MEFs (Fig. 1F). These results suggest that peroxisomes are degraded by autophagy, and therefore pexophagy is likely induced by PEX3 overexpression.
Next, we investigated the morphological effects of PEX3-induced pexophagy. In the course of macropexophagy, peroxisomes are enclosed by autophagosomes,9 which are then recruited into lysosomes for degradation. To investigate the processes of such recruitment, we expressed PEX3-HA2 in HeLa cells and analyzed by immunofluorescence microscopy its colocalization with LC3B and LAMP1, which are marker proteins for autophagosomes and lysosomes, respectively. LC3B colocalized with a peroxisome matrix protein, CAT (catalase), in PEX3-expressing cells (Fig. 2A, f; Fig. 2B), but not in the vector-transfected cells (Fig. 2A, a; Fig. 2B). The level of LC3-II, a lipidated form of LC3-I required for autophagy, was elevated by overexpression of PEX3 (Fig. 2C). Peroxisomes were primarily associated with lysosomes following PEX3 overexpression in the presence of chloroquine, an inhibitor of lysosomal acidification (Fig. 2D, p). Figure 2E shows percentages of LAMP1-positive peroxisomes in Figure 2D, in which peroxisome signals can be observed in lysosomes only in the presence of chloroquine. We interpreted these morphological data as indicative of macropexophagy.

Figure 2. Peroxisomes are recruited into lysosomes upon PEX3 overexpression. (A) HeLa cells were transfected with empty vector (a–e) and PEX3-HA2 (f–j). After 12 h, the cells were fixed and immunostained with antibodies against LC3B (b and g), CAT (catalase) (c and h), and HA (d and i). Merged views of LC3B and CAT are shown in (a and f). (B) The percentage of peroxisome signals colocalized with LC3B was calculated by Metamorph version 7.6 software from 10 cells. (C) CHO-K1 cells transfected with empty vector and PEX3-HA2 were lysed and analyzed by SDS-PAGE and immunoblotting with antibodies against LC3 (upper panel), HA (middle panel), and TUBA (bottom panel). (D) HeLa cells were transfected with empty vector (a–j) and PEX3-HA2 (k–t) in the presence of 20 μM chloroquine, a lysosome inhibitor (f–j and p–t). After 24 h, the cells were fixed and immunostained with antibodies against LAMP1 (b, g, l, and q), CAT (c, h, m, and r) and HA (d, i, n, and s). Merged views of immunostained peroxisomes and LAMP1 are shown in (a, f, k, and p). (E) The percentage of peroxisome signals colocalized with LAMP1 was calculated using Metamorph version 7.6 software from 10 cells. Scale bars: 10 μm.
Peroxisomal membrane targeting of PEX3-HA2 is essential for the induction of pexophagy
In the course of the pexophagy induced by PEX3 overexpression, the expressed PEX3-HA2 localized to the mitochondria in addition to the peroxisomes (Fig. 3B, f–j). This mitochondrial mislocalization of PEX3-HA2 was apparently an artifact of overexpression. To verify that pexophagy is not dependent on the mitochondrial localization of PEX3-HA2, we analyzed the effect of expression of the PEX3-HA2 variants shown in Figure 3A. PEX3 has a peroxisomal targeting signal sequence and a transmembrane domain at the N-terminal region, and an N-terminal deletion mutant of PEX3 (Cyto-PEX3) mislocalizes to the cytosol.29 To investigate the effect of mitochondrial mistargeting of PEX3-HA2, we generated Mito-PEX3-HA2, a construct in which the N-terminal region of TOMM20, a mitochondrial outer membrane protein was fused to Cyto-PEX3-HA2. As expected, Mito-PEX3-HA2 and Cyto-PEX3-HA2 localized to the mitochondria (Fig. 3B, k–o) and the cytosol (Fig. 3B, p–t), respectively. The percentage of cells exhibiting pexophagy was not increased by expression of Mito-PEX3-HA2 or Cyto-PEX3-HA2 (Fig. 3C). These results suggest that peroxisomal targeting of PEX3 is essential for pexophagy, but the mitochondrial mislocalization is dispensable.

Figure 3. Peroxisomal membrane targeting of Pex3 is essential for the induction of pexophagy. (A) Schematic representation and alignment of N-terminal amino acid sequence of the PEX3-HA2 variants, including Mito-PEX3-HA2 and Cyto-PEX3-HA2. The 39 N-terminal amino acid peroxisome-targeting sequences were replaced with the 69 N-terminal amino acids of TOMM20 (Mito-PEX3) or deleted (Cyto-PEX3). (B) CHO-K1 cells were transfected with empty vector or PEX3-HA2 variants. After 12 h, the cells were fixed and immunostained with antibodies against HA (b, g, l, and q), TOMM20 (c, h, m, and r) and PEX14 (d, i, n, and s). Merged views of the PEX3-HA2 variants and TOMM20 are shown in (f, k, and p). (C) The percentage of cells exhibiting pexophagy was calculated as in Figure 1B. Data are presented as the mean ± SD of 3 replicates.
Autophagic receptor proteins translocate to peroxisomes upon PEX3 overexpression
In yeast, Pex3 interacts with pexophagic receptor proteins, including Atg30 and Atg36.32,33 Therefore, we studied next whether an autophagic receptor protein is involved in PEX3-induced pexophagy in mammalian cells. The autophagic receptor proteins, SQSTM1 and NBR1, have been suggested to be involved in pexophagy in mammalian cells.18,24 Therefore, we assessed the interaction between these autophagic receptors and PEX3 by immunoprecipitation (IP) and yeast 2-hybrid (Y2H) assay. FLAG-SQSTM1, FLAG-NBR1, and FLAG-PEX19 were coexpressed separately with PEX3-HA2 in CHO-K1 cells. PEX19, a cytoplasmic chaperone-like protein, functions in PMP import into the peroxisomal membrane via interaction with PEX3.34-36 Therefore, we used PEX19 as a positive control for IP analysis and Y2H assay. PEX3-HA2 did not coimmunoprecipitate with FLAG-SQSTM1 or FLAG-NBR1 (Fig. 4A, upper panel, lanes 7, 8), while an interaction between PEX3-HA2 and FLAG-PEX19 was detected (Fig. 4A, upper panel, lane 5). Likewise, in Y2H assay, the direct interactions of PEX3 with receptor proteins were not detected, while positive controls were clearly detectable (PEX3 and PEX19 or LC3B and receptors) (Fig. 4B). Intracellular localization of these receptors was examined by immunofluorescence microscopy with anti-SQSTM1 and anti-NBR1 antibodies. In contrast to the results of IP analysis and Y2H assay, these receptors colocalized with peroxisomes upon PEX3 overexpression (Fig. 4C, f; Fig. 4D, f). We also performed subcellular fractionation analysis of these receptors. Upon overexpression of PEX3, the relative level of NBR1 was increased in the peroxisome-enriched fraction, while that of SQSTM1 appeared not to be altered (Fig. 4E, lane 8). These results suggest that these autophagic receptors translocate to peroxisomes and more readily effective transition of NBR1, but that this localization is not directly mediated by PEX3. Furthermore, PEX14-positive peroxisomes were found in larger foci in PEX3-overexpressed cells (Fig. 4C, h; Fig. 4D, h). It is conceivable that peroxisomes are clustered by PEX3 overexpression in a manner similar to that of damaged mitochondria in parkin-mediated mitophagy.37,38

Figure 4. Autophagic receptor proteins are colocalized with peroxisomes upon PEX3 overexpression. (A) CHO-K1 cells were cotransfected with PEX3-HA2 and FLAG-PEX19, empty vector, and FLAG-SQSTM1 or FLAG-NBR1. After 12 h, the cells were lysed with buffer containing 1% Triton X-100. The cell lysates were subjected to immunoprecipitation with anti-FLAG M2 affinity beads. Samples eluted from the affinity beads were analyzed by SDS-PAGE and immunoblot analysis with antibodies against HA (upper panel) and FLAG (lower panel). (B) Interaction of PEX3 with autophagic receptors was assessed by yeast 2-hybrid assay. Host strain MaV203 was transformed with the Gal4 DNA binding domain (BD) DNA sequence fused to PEX3 or LC3B indicated at the top of panels and Gal4 DNA activation-domain (AD) fused to PEX19, SQSTM1 or NBR1 indicated on the left of panels. Transformants were assayed for growth on synthetic complete medium (SC) without Leu, Trp (left panel) and His in the presence of 25 mM 3-aminotriazole (3-AT) (middle panel) and β-galactosidase activity (right panel). (C and D) CHO-K1 cells were transfected with empty vector (a–e) or PEX3-HA2 (f–j). After 12 h, the cells were fixed and immunostained with antibodies against SQSTM1 (C, b and g) and NBR1 (D, b and g), PEX14 (C and D, c and h) or HA (C and D, d and i). Merged views of receptor proteins and peroxisomes are shown in (C and D, a and f). Quantification of the colocalization of receptor proteins with peroxisomes are shown in the right bar graphs of (C and D). Data are presented as the mean ± SD from 10 cells. Scale bars: 10 μm. (E) Subcellular fractionation of HeLa cells transfected with empty vector (right panels) and PEX3-HA2 (left panels) as in (C and D).
NBR1 is essential for pexophagy upon PEX3 overexpression
To investigate whether these receptors are essential for pexophagy, we used siRNA to knock down (KD) endogenous SQSTM1 and NBR1 expression in HeLa cells (Fig. 5A, top and middle panels). Under these KD conditions, PEX3-mediated pexophagy was observed by immunostaining with an anti-PEX14 antibody. These siRNA treatments did not affect the peroxisome number (Fig. 5B). Peroxisomes were detected in cells that had been treated with siRNA against NBR1 alone (Fig. 5C, g) and in cells treated with siRNA against both SQSTM1 and NBR1 (Fig. 5C, j), while peroxisomes were degraded in cells that had been treated with siRNA against luciferase (as a control; Fig. 5C, a) or SQSTM1 alone (Fig. 5C, d). The degree of peroxisome degradation was quantified as in Figure 1B, confirming the decrease in PEX3-mediated pexophagy in NBR1 siRNA-treated cells (Fig. 5D). These results suggest that NBR1, but not SQSTM1, is required for the induction of pexophagy upon PEX3 overexpression.

Figure 5. NBR1, but not SQSTM1, is required for pexophagy upon PEX3 overexpression. (A) HeLa cells were transfected with siRNA against luciferase (Control), SQSTM1, NBR1, or both SQSTM1 and NBR1. Two days later, the cells were lysed with SDS-PAGE sample buffer and analyzed by SDS-PAGE and immunoblotting with antibodies against SQSTM1 (upper panel), NBR1 (middle panel) and TUBA (bottom panel). (B and C) The cells treated with siRNAs were transfected with empty vector (B) or PEX3-HA2 (C). After 24 h, the cells were fixed and immunostained with antibodies against PEX14 (a, d, g, and j) and HA (b, e, h, and k). Scale bars, 10 μm. (D) The percentage of cells exhibiting pexophagy upon PEX3-HA2 overexpression was calculated as in Figure 1C. Data are presented as the mean ± SD of 3 replicates.
UBA domain of NBR1 is required for specific translocation of NBR1 to peroxisomes upon PEX3 overexpression
NBR1 contains multiple domains, including phox/Bem1 (PB1), coiled-coil (CC), LC3-interacting region (LIR), membrane-interacting amphipathic α-helix (Juxta-UBA, J-UBA), and UBA (ubiquitin-associated) domains (Fig. 6A, black boxes).39 To determine which domain is required for the colocalization of NBR1 with peroxisomes, EGFP-NBR1 variants harboring respective domain-deletion mutations were coexpressed with PEX3-HA2 in HeLa cells pretreated with NBR1 siRNA for 2 d as shown in Figure 5A. The NBR1 variants included a PB1-defective mutant with a D50R substitution and an LIR mutant with a LIR2 deletion and Y750A substitution as well as a series of domain-deletion mutants (Fig. 6B).25 The NBR1 variants, except for D50R, were coimmunoprecipitated with FLAG-SQSTM1, hence indicating that the N-terminal regions of the variants are functional (Fig. 6C). The full-length, D50R, CCΔ, LIR mut, J-UBAΔ, and UBAΔ EGFP-NBR1 variants colocalized with peroxisomes (Fig. 6D, a, f, k, p, u, and z). However, the CCΔ and UBAΔ variants showed a significantly lower level of colocalization ratio as compared with the other variants, and the ratio of UBAΔ was lower than that of CCΔ (Fig. 6E). These results suggest that the UBA domain of NBR1 contributes to the specificity of the translocation of NBR1 and that the CC domain assists the NBR1 translocation. Furthermore, NBR1 CΔ harboring both J-UBAΔ and UBAΔ was completely diffused in the cytosol (Fig. 6D, e’; Fig. 6E), suggesting that the J-UBA domain partially contributes to the translocation to peroxisomes of NBR1 via its nonspecific membrane-binding.

Figure 6. The LC3 interaction region and UBA domain of NBR1 are required for pexophagy. (A) Schematic representation of NBR1. NBR1 contains a phox and Bem1 (PB1) domain (amino acid alignment positions at residue 1–86), coiled-coil (CC) domain (289–330), LC3-interacting region (LIR) (541–637 and 746–755), juxta-UBA domain (894–933) and a UBA domain (934–988) as indicated by black shading. (B) HeLa cells with NBR1 knockdown, as shown in Figure 5A, were cotransfected with PEX3-HA2 and siRNA-resistant EGFP-NBR1 domain-deletion mutants as indicated at the top of each lane. After 12 h, the cells were lysed with SDS-PAGE sample buffer and analyzed by SDS-PAGE followed by immunoblotting with antibodies against GFP (upper panels), HA (middle panel) and TUBA (bottom panel). (C) To verify whether siRNA-resistant EGFP-NBR1 variants are functional, IP analysis was performed with FLAG-SQSTM1. HeLa cells were cotransfected with FLAG-SQSTM1 and EGFP-NBR1s. After 24 h, the cells were lysed in IP buffer containing 1% Triton X-100. The cell lysates were subjected to IP analysis with anti-GFP antibody. IP samples were analyzed by SDS-PAGE and immunoblotting with antibodies against GFP (upper panels) and FLAG (lower panel). (D) To analyze the colocalization between peroxisomes and EGFP-NBR1 domain-deletion mutants, the cells were fixed and immunostained with antibodies against PEX14 (c, h, m, r, w, b’, and g’) and HA (d, i, n, s, x, c’, and h’). Merged views of EGFP-NBR1s and PEX14 are shown in (a, f, k, p, u, z, and e’). Scale bars: 10 μm. (E) Percentages of NBR1 signals colocalized with peroxisomes were calculated with Metamorph version 7.6 software from 10 cells. (F) The percentage of cells exhibiting pexophagy was calculated as in Figure 1B. Data are presented as the mean ± SD of 3 replicates. Asterisks indicate significant differences from full-length (FL). *P < 0.05, **P < 0.01.
The LIR and UBA domains of NBR1 are required for pexophagy upon PEX3 overexpression
Next, we determined which domain of NBR1 is required for the degradation of peroxisomes. Figure 6F shows a statistical analysis of peroxisome degradation in cells expressing EGFP (control) or the EGFP-NBR1 mutants shown in Figure 6B. Peroxisome degradation was not complemented by expression of the EGFP-NBR1 LIR mutant, UBAΔ, or CΔ in the NBR1 KD background. By contrast, normal pexophagy was observed in cells expressing the EGFP-NBR1 full-length and J-UBAΔ (Fig. 6F). These results suggest that the LIRs of NBR1 are required for pexophagy and contribute to this process by mediating the connection between peroxisomes and autophagosomes. The C-terminal region, including the UBA domain, is also required for peroxisome degradation. This finding is consistent with the results of localization analysis (Fig. 6D) and suggests that transport of NBR1 to peroxisomes is a prerequisite for pexophagy. The J-UBA domain has been shown to be required for transport of NBR1 to peroxisomes and pexophagy induced by NBR1 overexpression.24 However, pexophagy is mediated in J-UBA-independent manner upon PEX3 overexpression (Fig. 6F). This difference in the J-UBA domain dependency suggests that targeting of NBR1 to peroxisomes occurs via mutually distinct mechanisms. Peroxisome degradation was mildly decreased in EGFP-NBR1D50R- or CCΔ-expressing cells (Fig. 6F), implying that the interaction between SQSTM1 and NBR1 and self-oligomerization of NBR1 increases the efficiency of pexophagy.
SQSTM1 is required for peroxisome clustering
In mammalian cells, SQSTM1 mediates the clustering of damaged mitochondria in parkin-mediated mitophagy, although it is dispensable for mitochondrial degradation.37,38 Similarly, as described above, SQSTM1 colocalized with larger peroxisomal foci (Fig. 4C, f), although it was not essential for peroxisome degradation (Fig. 5D). Therefore, we next investigated whether SQSTM1 is involved in the clustering of peroxisomes. To compare the size of peroxisomal foci, PEX3-HA2 was expressed in HeLa cells treated with SQSTM1 siRNA, and the cells were fixed and immunostained before the peroxisomal foci were degraded. Larger peroxisomal foci were not discernible in either SQSTM1- or SQSTM1- and NBR1-KD cells (Fig. 7A, d and j), while the larger foci were detectable in NBR1-KD cells, similar to control (Fig. 7A, a and g). The percentage of cells with the large foci was significantly decreased in SQSTM1-deficient cells (Fig. 7B), strongly suggesting that SQSTM1 functions in peroxisome clustering.
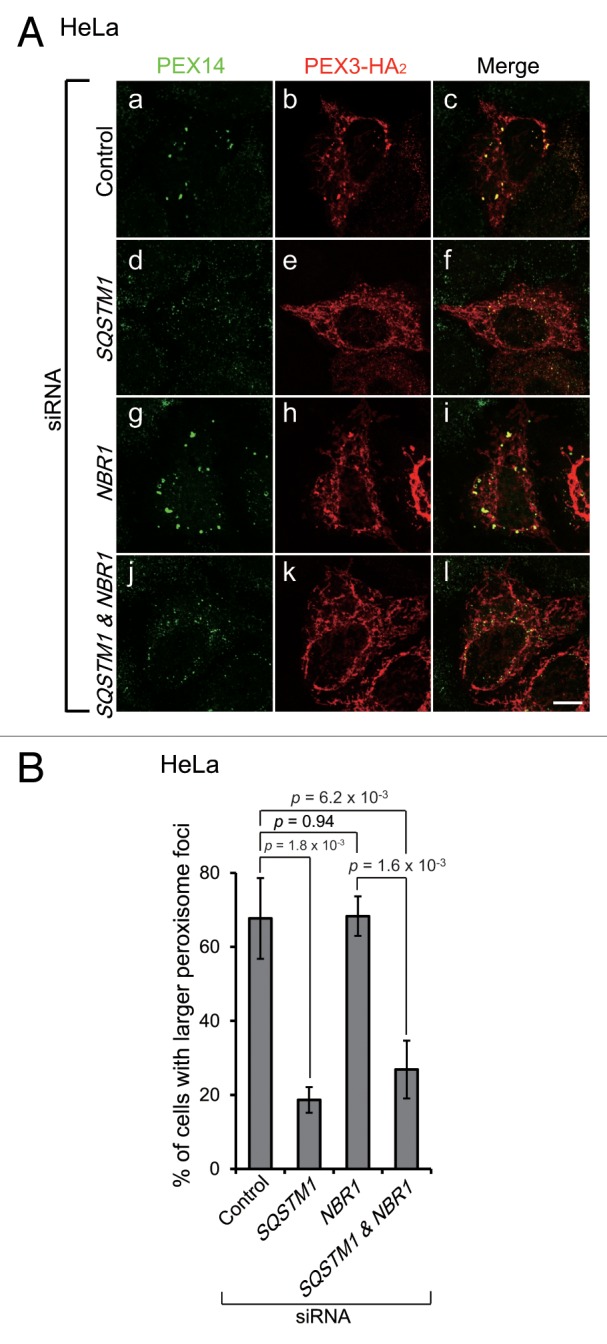
Figure 7. SQSTM1 is required for peroxisome clustering. (A) HeLa cells treated with siRNA against autophagic receptor proteins as indicated were transfected with PEX3-HA2. After 12 h, the cells were fixed and immunostained with antibodies against PEX14 (a, d, g, and j) and HA (b, e, h, and k). Scale bars: 10 μm. (B) The percentage of cells with larger peroxisome-foci over 0.3 μm2 as average area was calculated as in Figure 1B. Data are presented as the mean ± SD of 3 replicates.
Peroxisomal proteins are ubiquitinated upon PEX3 overexpression prior to pexophagy
Based on the finding that NBR1 is transported to peroxisomes in a UBA domain-dependent manner in PEX3-induced pexophagy (Fig. 6D), we hypothesized that a ubiquitin signal is present on the peroxisomal membrane. Indeed, immunofluorescence microscopy with anti-ubiquitin antibody showed ubiquitin-positive signals at PEX3-loaded peroxisomes (Fig. 8A, f; Fig. 8B), while ubiquitin-positive peroxisomes were not discernible in vector transfected cells (Fig. 8A, a; Fig. 8B). Next, we assessed whether the ubiquitin-positive signal represents an ubiquitinated form of PEX3 using a PEX3 K-less mutant in which all lysine residues were substituted with arginine. The PEX3-HA2 K-less mutant targeted normally to peroxisomes and restored peroxisomal membrane biogenesis when expressed in the Pex3-deficient pex3 CHO mutant, ZPG208 (data not shown). Ectopically expressed PEX3-HA2 WT and K-less mutant were then immunoprecipitated with anti-HA antibody and analyzed by SDS-PAGE and immunoblotting with anti-ubiquitin antibody. PEX3-HA2 WT was detected with anti-ubiquitin antibody (Fig. 8C, lane 5), and Mito-PEX3-HA2 and Cyto-PEX3-HA2 were also slightly ubiquitinated (data not shown), although no ubiquitin-positive signals were observed at peroxisomes following expression of Mito-PEX3-HA2 or Cyto-PEX3-HA2 (Fig. 8A, p and u; Fig. 8B). Interestingly, immunofluorescence signals of ubiquitin were observed in the K-less mutant-loaded peroxisomes (Fig. 8A, k; Fig. 8B) similar to WT, even though ubiquitination of the K-less mutant was not detected by immunoblot analysis (Fig. 8C, lane 6). Next, we investigated whether cysteine residues of PEX3 are ubiquitinated by IP analysis under non-reducing condition. Ubiquitination of PEX3 K-less mutant was detected at a low level under the nonreducing condition, hence suggesting that cysteine residues may be ubiquitinated (Fig. 8C, lane 12). Moreover, we verified whether the peroxisomal ubiquitination is induced with PEX3-CA mutant in which all cysteine residues are substituted with alanine and K-less PEX3-CA. The peroxisomal ubiquitination was induced by overexpression of any of the PEX3 mutants, thereby indicating that the ubiquitination of cysteine residues of PEX3 is not responsible for the peroxisomal ubiquitination (Fig. S2). Furthermore, peroxisome degradation was induced even in PEX3-HA2 K-less, CA, K-less-CA mutant-expressing cells (Fig. 8D). Taken together, these results strongly suggest that a currently unidentified peroxisomal membrane protein is ubiquitinated in the PEX3-mediated pexophagy and functions in NBR1 recruitment as a target for aberrant peroxisomes.

Figure 8. Peroxisomes are ubiquitinated upon PEX3 overexpression. (A) CHO-K1 cells transiently expressing WT PEX3-HA2, the K-less PEX3-HA2 mutant, Mito-PEX3-HA2 or Cyto-PEX3-HA2 for 12 h were fixed and immunostained with antibodies against ubiquitin (b, g, l, q, and v), PEX14 (c, h, m, r, and w) and HA (d, i, n, s, and x). Merged views of ubiquitin and PEX14 are shown in (a, f, k, p, and u). Scale bar: 10 μm. (B) Percentages of peroxisome signals colocalized with ubiquitin were calculated with Metamorph version 7.6 software from 10 cells. (C) CHO-K1 cells were transiently transfected with empty vector, PEX3-HA2 or the K-less mutant. After 12 h, the cells were lysed and denatured by TSD or TSN (without DTT, for nonreducing condition) buffers, and then diluted with TNN buffer for IP with an anti-HA antibody as described in Materials and Methods. Each IP sample, indicated at the top of each lane, was subjected to immunoblot analysis with antibodies against ubiquitin (upper panel) and HA (lower panel). (D) The percentage of cells exhibiting pexophagy was calculated as in Figure 1B. Data are presented as the mean ± SD of 3 replicates.
Discussion
Peroxisome number is maintained by a balance between peroxisome biogenesis and degradation. In mammalian cells, up to 80% of peroxisomes are degraded by pexophagy as evidenced by degradation analysis of atg7 conditional knockout mice, demonstrating that pexophagy functions as a major component of peroxisome homeostasis.7,8,40 To elucidate the molecular mechanism of mammalian pexophagy, several different conditions that induce pexophagy have been reported in mammalian cultured cells.9,18,24 In the present study, we discovered a novel condition for PEX3-mediated pexophagy in mammalian cells, analogous to the yeast pexophagy system.32,33 Unexpectedly, although the interaction between PEX3 and the mammalian autophagic receptors SQSTM1 and NBR1 was not detected, peroxisomes were degraded by NBR1-mediated pexophagy following ectopic PEX3 expression. Furthermore, an unidentified endogenous peroxisomal protein was ubiquitinated following increased PEX3 expression. This condition is the first that has been shown to induce endogenous ubiquitination on the peroxisomal membrane.
Generally, the poly-Ub chain linked to substrate proteins is used as a signal for targeting to the proteasome for degradation, as well as several other cellular events, including selective autophagy.22,23 In Ub-mediated selective autophagy, Ub-binding proteins function as a receptor at the recognition step of ubiquitinated substrates. These autophagic receptors have several similar domains, including a UBA domain and LIR, through which they bind to both Ub and LC3, respectively, to link the substrates to an autophagosome. Ub-positive aggregates, cell-invading bacteria, and damaged mitochondria have been reported as substrates for Ub-mediated selective autophagy.20,21,37,38,41,42 In addition, artificial Ub-presenting peroxisomes have been suggested to be substrates for Ub-mediated pexophagy,18 implying that Ub-presentation on the cytosolic surface of the target organelle can induce Ub-mediated selective organelle autophagy. Thus, in the course of Ub-mediated organelle autophagy, the most important step might be the ubiquitination of target proteins on the organelle membrane.
In the preset study, Ub signals were observed on peroxisomes following high-level PEX3 expression, as shown by immunofluorescence staining with an anti-Ub antibody (Fig. 8A), and PEX3 was found to be ubiquitinated under these conditions (Fig. 8C). Furthermore, pexophagy was also induced with concomitant peroxisome ubiquitination upon ectopic expression of the PEX3 K-less, CA, and K-less CA mutants (Fig. 8D). Consequently, ubiquitination on the membrane of peroxisomes occurs not only on PEX3, but also on an endogenous substrate, and this endogenous ubiquitination is sufficient to induce pexophagy. No Ub signals were observed at peroxisomes upon expression of Mito-PEX3 or Cyto-PEX3 (Fig. 8A), suggesting that peroxisome targeting of PEX3 is crucial for the ubiquitination of the peroxisomal membrane. Although a more detailed mechanism of how PEX3 induces peroxisomal ubiquitination remains to be elucidated, the pexophagy induction system established here will be a highly useful tool for identifying this substrate and its Ub-ligase.
Although PEX3-induced pexophagy is similar to the NBR1-induced pexophagy,24 some differences exist, particularly with respect to the role of the C-terminal region of NBR1. In NBR1-induced pexophagy, peroxisomes are clustered around NBR1-positive punctate structures and then degraded by pexophagy. The J-UBA domain is required in this system for both clustering and the degradation of peroxisomes, while the UBA domain is required only for degradation. Thus, interaction between Ub and the UBA domain is dispensable for clustering, but indispensable for subsequent degradation. Because ubiquitination of peroxisomes is rarely observed under normal conditions (Fig. 8A), nonubiquitinated peroxisomes are likely to cluster artificially along with cytosolic NBR1-positive puncta comprised of ubiquitinated proteins. However, it is unclear how NBR1 enriches peroxisome clustering around NBR1-positive punctate structures via the J-UBA domain. In contrast to NBR1-induced pexophagy, the UBA domain, not the J-UBA domain, is required for peroxisome degradation, while both domains contribute to the translocation of NBR1 to the ubiquitinated peroxisomes upon PEX3 overexpression (Fig. 6). Given that significant Ub signals are observed only upon PEX3 overexpression (Fig. 8B), we suspect that the differences in NBR1 dependency are derived from the role of the Ub signal in each pathway.
SQSTM1 is required for ubiquitin-mediated pexophagy18 and enhances the efficiency of degradation in NBR1-induced pexophagy.24 In the present study, SQSTM1 likewise enhances the efficiency of degradation via interaction with NBR1, as evidenced in the pexophagy analysis using NBR1D50R mutant (Fig. 6F). Furthermore, SQSTM1 is required for peroxisome clustering following PEX3 overexpression (Fig. 7). The translocation of SQSTM1 to peroxisomes also depends on its UBA domain, and overexpression of SQSTM1 increases the peroxisomal cluster size in a PB1 domain-dependent manner (data not shown). Based on these data, we suggest that SQSTM1 functions in the clustering of aberrant ubiquitinated peroxisomes and may increase the efficiency of peroxisome degradation by enhancing this clustering.
Based on the finding that an unidentified endogenous peroxisomal protein is ubiquitinated by an endogenous ubiquitin conjugation system upon PEX3 overexpression, we believe that peroxisomes possess an inherent ubiquitin conjugation system for peroxisomal proteins in response to a situation that is mimicked by PEX3 overexpression. Physiological relevance of the elevated PEX3 level remains elusive. However, we find that endogenous PEX3 is constitutively degraded by the proteasome system (Fig. S3). Such degradation implies the physiological importance of regulation of PEX3 level on peroxisomal membrane and several situations of elevated PEX3 level could be a trigger for pexophagy. Significant amounts of Ub signal are not observed following starvation or NBR1 overexpression, suggesting that endogenous Ub-mediated pexophagy induced by PEX3 overexpression is only one type of the pexophagy system. A physiological role for this type of pexophagy could be defined by making use of the Ub-mediated pexophagy model that we have established here.
Materials and Methods
Cell culture, DNA transfection, and RNAi-mediated knockdown (KD)
CHO-K1 cells were cultured with Ham F-12 medium (Invitrogen, 21700-018), and HeLa and MEF cells were cultured with DMEM medium (Invitrogen, 12800-082); both media types were supplemented with 10% fetal bovine serum and cells were maintained at 37 °C under 5% CO2. DNA transfection of CHO-K1 cells was performed using Lipofectamine reagent (Invitrogen, 18324-012) and HeLa and MEF cells were transfected using Lipofectamine 2000 (Invitrogen, 11668-019) according to the manufacturer’s instructions. siRNA-mediated KD of SQSTM1 and NBR1 in HeLa cells was performed with Stealth RNAi siRNAs (Invitrogen). siRNAs were transfected into HeLa cells using Lipofectamine 2000 according to the manufacturer’s instructions. For NBR1 complementation analysis, siRNA-resistant NBR1 variants were cotransfected with PEX3-HA2 48 h after siRNA treatment. The sequences of the siRNAs are listed in Table S1.
Chemicals
3-methyladenine (Sigma-Aldrich, M9281) was directly dissolved in culture media. Bafilomycin A1 (Sigma-Aldrich, B1793) and chloroquine (Nacalai Tesque, 08660-62) were prepared from stocks in DMSO and water, respectively. 3-aminotriazole was from Sigma-Aldrich (A8056).
Plasmids
cDNA encoding SQSTM1, NBR1, and LC3B were amplified by PCR. For N-terminal fusion constructs, a fragment of EGFP and FLAG tag were ligated into the BamHI-EcoRI site of pcDNATM3.1/Zeo(+) vector (Invitrogen, V86020), and then PCR fragments of SQSTM1, NBR1, and LC3B were cloned into these vectors via EcoRI-XhoI or NotI-XbaI sites. Domain-deletions and siRNA-resistant silent mutations of EGFP-NBR1 were introduced by an inverse PCR-mediated strategy.
To generate PEX3-HA2 constructs, PEX3, Cyto-PEX3, and Mito-PEX3 fragments were ligated with the C-terminal tandem HA fusion vector, pcDNAZeo/C-HA2.43 BamHI-XhoI fragments of PEX3 and PEX3 N39Δ from C-terminal EGFP fusion constructs44 were inserted into the same sites of pcDNAZeo/C-HA2. The Mito-PEX3 fragment from pcDNAZeo/Mito-PEX3-EGFP45 was likewise ligated by the same strategy. To produce the K-less mutant of PEX3-HA2, 21 lysine residues were substituted with arginine at the DNA level by point mutation with the respective primers.
The PEX14 fragment was amplified by PCR from His-PEX1446 and inserted into pcDNAZeo/C-HA2 at the BamHI-XhoI site. All primers used in this study are listed in Table S2.
Immunoprecipitation (IP)
FLAG-tagged PEX19, SQSTM1, and NBR1 were coexpressed separately for 12 h with PEX3-HA2 in CHO-K1 cells. The cells were lysed with IP buffer [1.0% TritonX-100 (Nacarai tesque, 35501-15), 50 mM TRIS-HCl, pH 7.5, 150 mM NaCl, 1.0 mM EDTA, 1.0 mM PMSF, and a cocktail of protease inhibitors: 10 μg/ml of each pepstatin A, leupeptin, E64, antipain, and chymostatin (Peptide institute, 4397, 4041, 4096, 4062, and 4063, respectively) and 5 μg/ml of Aprotinin (Sigma-Aldrich, A1153)], and then the FLAG-tagged protein complexes were immunoprecipitated using anti-FLAG M2 agarose beads (Sigma-Aldrich, A2220). After washing the beads 3 times with IP buffer, the immunoprecipitated proteins were eluted and analyzed by SDS-PAGE and immunoblotting with an anti-HA antibody. An IP assay of ubiquitinated PEX3-HA2 was performed under denaturing conditions. PEX3-HA2 variants were expressed for 12 h in CHO-K1 cells, which were then lysed and denatured with TSD buffer (50 mM TRIS-HCl, pH 7.5, 1.0% SDS, 5.0 mM DTT) or TSN buffer [50 mM TRIS-HCl, pH 7.5, 1.0% SDS, and 5.0 mM N-ethylmaleimide (Nacarai tesque, 15512-82)] for nonreducing condition by sonication and heating at 95 °C for 15 min. The lysates were diluted with TNN buffer [50 mM TRIS-HCl, pH 7.5, 250 mM NaCl, 1.0% NP-40 (Nacarai tesque, 25223-04), and a cocktail of protease inhibitors, as described above]. PEX3-HA2 variants were immunoprecipitated with an anti-HA rabbit polyclonal antibody14 and protein A-Sepharose beads (GE Healthcare, 17-0780-01). After washing 3 times with TNN buffer, the immunoprecipitated PEX3-HA2 variants were analyzed by SDS-PAGE and immunoblotting with anti-Ub antibody.
Subcellular fractionation
The cells were homogenized with a potter-elvehjem homogenizer and the postnuclear supernatant (PNS) fraction was prepared by a centrifugation at 800 × g. PNSs were further fractionated by sequential centrifugation at 2,000 × g (2K), 20,000 × g (20 K), and 100,000 × g (100 K). These fractions were analyzed by SDS-PAGE and immunoblotting.
Antibodies
Rabbit polyclonal antibodies against ACOX1/acyl-CoA oxidase1,47 PEX14,17 ABCD3/PMP70,47,48 and HA (raised against a synthetic peptide containing the HA epitope, CYPYDVPDYASLRS),14 and guinea pig polyclonal antibodies against PEX1449 and CAT/catalase (raised against full-length human catalase) were described previously. Rabbit polyclonal antibodies against MAP1LC3B (PM036), SQSTM1 (PM045), and GFP (598), and a mouse monoclonal antibody against ubiquitin (1B3: MK-11-3, FK2: D058-3), were purchased from Medical and Biological Laboratories. Mouse monoclonal antibodies against GFP (B-2; sc-9996) and TOMM20 (F-10; sc-17764) were purchased from Santa Cruz Biotechnology. Mouse monoclonal antibodies against HA (16B12; Covance, MMS-101P), NBR1 (M05; Abnova, H00004077-M05), TUBA/α-tubulin (DM1A; Abcam, ab7291), FLAG (M2; Sigma-Aldrich, F1804), and a rabbit polyclonal antibodies against LAMP1 (Abcam, ab24170) and FLAG (Sigma, F7425) were purchased.
Morphological analysis
Cells were fixed in 4.0% paraformaldehyde for 15 min and then permeabilized with 0.1% TritonX-100 for 30 min. Immunofluorescence staining was performed with the indicated primary antibodies and Alexa Fluor 488-, 568- and 647-conjugated secondary antibodies (Invitrogen, A11029, A11036, and A21450, respectively) in 1% BSA (Nacarai tesque, 01281-97) in PBS (0.2 g of KCl, 0.2 g of KH2PO4, 2.896 g of Na2HPO4·H2O, 8 g of NaCl per liter). Cells were monitored with a confocal laser scanning microscope (LSM710; Carl Zeiss, Carl Zeiss Promenade 10, 07745 Jena, Germany). The number of peroxisomes was quantified in at least 50 cells using ImageJ software (National Institute of Health). The confocal images were converted to threshold images, and the number of peroxisomes was calculated with the Analyze Particles program of ImageJ. To determine the colocalization ratio, threshold images were analyzed with the Measure Colocalization program of Metamorph version 7.6 software (Molecular Devices). All data are presented as the mean ± SD of 3 independent experiments.
Yeast 2-hybrid assay
Construction of plasmids each encoding GAL4 DNA-binding domain (BD) or transcription activation domain (AD), the transformation of the host MaV203 of S. cerevisiae and β-galactosidase assay were performed using a ProQuestTM Two-Hybrid System (Invitrogen), according to the manufacturer’s instructions. cDNAs including PEX3, LC3B, PEX19, SQSTM1, and NBR1 were amplified by PCR. NBR1 cDNA variants each encoding N-terminal amino acid residues at 1–480 and C-terminal residues at 481–988 were also amplified. PCR fragments of PEX3 and LC3B were ligated into SalI-NotI site of pDBLeu coding for GAL4 BD. PEX19, SQSTM1, and NBR1 were ligated into SalI-NotI site of pPC86 GAL4 AD. All pairs of BD containing plasmid and AD containing plasmid were cotransformed into S. cerevisiae Mav203.
Statistical analysis
Statistical analyses were performed using the unpaired Student t test. P < 0.05 was considered statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank N Mizushima for providing atg5 KO MEFs. We also thank K Shimizu for preparing the figures and the other members of our laboratory for discussions. This work was supported in part by Grants-in-Aid for Scientific Research (numbers 19058011, 20370039, 24247038, 25112518, and 26116007 to YF) and The Global COE Program and Grants for Excellent Graduate Schools from the Ministry of Education, Culture, Sports, Science and Technology of Japan and CREST grant (to YF) from the Science and Technology Agency of Japan, and grants (to YF) from Takeda Science Foundation and Japan Foundation for Applied Enzymology. SY was a Research Fellow of the Japan Society for the Promotion of Science.
Glossary
Abbreviations:
- CC
coiled-coil
- CHO
Chinese hamster ovary
- EGFP
enhanced green fluorescence protein
- HA
hemagglutinin antigen
- IP
immunoprecipitation
- J-UBA
juxta-UBA
- KD
knockdown
- LIR
LC3-interacting region
- MAP1LC3 (LC3)
microtubule-associated protein 1 light chain 3
- MEF
mouse embryonic fibroblast
- NBR1
neighbor of BRCA1 gene 1
- PB1
phox/Bem1
- PMP
peroxisomal membrane protein
- SQSTM1
sequestosome 1
- Ub
ubiquitin
- UBA
ubiquitin-associated
- Y2H
yeast 2-hybrid
References
- 1.De Duve C, Baudhuin P. Peroxisomes (microbodies and related particles) Physiol Rev. 1966;46:323–57. doi: 10.1152/physrev.1966.46.2.323. [DOI] [PubMed] [Google Scholar]
- 2.van den Bosch H, Schutgens RBH, Wanders RJA, Tager JM. Biochemistry of peroxisomes. Annu Rev Biochem. 1992;61:157–97. doi: 10.1146/annurev.bi.61.070192.001105. [DOI] [PubMed] [Google Scholar]
- 3.Wanders RJ. Peroxisomes, lipid metabolism, and peroxisomal disorders. Mol Genet Metab. 2004;83:16–27. doi: 10.1016/j.ymgme.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 4.Fujiki Y. Peroxisome biogenesis and peroxisome biogenesis disorders. FEBS Lett. 2000;476:42–6. doi: 10.1016/S0014-5793(00)01667-7. [DOI] [PubMed] [Google Scholar]
- 5.Gould SJ, Valle D. Peroxisome biogenesis disorders: genetics and cell biology. Trends Genet. 2000;16:340–5. doi: 10.1016/S0168-9525(00)02056-4. [DOI] [PubMed] [Google Scholar]
- 6.Yokota S, Dariush Fahimi H. Degradation of excess peroxisomes in mammalian liver cells by autophagy and other mechanisms. Histochem Cell Biol. 2009;131:455–8. doi: 10.1007/s00418-009-0564-6. [DOI] [PubMed] [Google Scholar]
- 7.Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, Chiba T, Tanaka K, Kominami E. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem. 2006;281:4035–41. doi: 10.1074/jbc.M512283200. [DOI] [PubMed] [Google Scholar]
- 8.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hara-Kuge S, Fujiki Y. The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp Cell Res. 2008;314:3531–41. doi: 10.1016/j.yexcr.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–12. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 12.Fransen M, Terlecky SR, Subramani S. Identification of a human PTS1 receptor docking protein directly required for peroxisomal protein import. Proc Natl Acad Sci U S A. 1998;95:8087–92. doi: 10.1073/pnas.95.14.8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otera H, Fujiki Y. Pex5p imports folded tetrameric catalase by interaction with Pex13p. Traffic. 2012;13:1364–77. doi: 10.1111/j.1600-0854.2012.01391.x. [DOI] [PubMed] [Google Scholar]
- 14.Otera H, Harano T, Honsho M, Ghaedi K, Mukai S, Tanaka A, Kawai A, Shimizu N, Fujiki Y. The mammalian peroxin Pex5pL, the longer isoform of the mobile peroxisome targeting signal (PTS) type 1 transporter, translocates the Pex7p.PTS2 protein complex into peroxisomes via its initial docking site, Pex14p. J Biol Chem. 2000;275:21703–14. doi: 10.1074/jbc.M000720200. [DOI] [PubMed] [Google Scholar]
- 15.Otera H, Okumoto K, Tateishi K, Ikoma Y, Matsuda E, Nishimura M, Tsukamoto T, Osumi T, Ohashi K, Higuchi O, et al. Peroxisome targeting signal type 1 (PTS1) receptor is involved in import of both PTS1 and PTS2: studies with PEX5-defective CHO cell mutants. Mol Cell Biol. 1998;18:388–99. doi: 10.1128/mcb.18.1.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otera H, Setoguchi K, Hamasaki M, Kumashiro T, Shimizu N, Fujiki Y. Peroxisomal targeting signal receptor Pex5p interacts with cargoes and import machinery components in a spatiotemporally differentiated manner: conserved Pex5p WXXXF/Y motifs are critical for matrix protein import. Mol Cell Biol. 2002;22:1639–55. doi: 10.1128/MCB.22.6.1639-1655.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimizu N, Itoh R, Hirono Y, Otera H, Ghaedi K, Tateishi K, Tamura S, Okumoto K, Harano T, Mukai S, et al. The peroxin Pex14p. cDNA cloning by functional complementation on a Chinese hamster ovary cell mutant, characterization, and functional analysis. J Biol Chem. 1999;274:12593–604. doi: 10.1074/jbc.274.18.12593. [DOI] [PubMed] [Google Scholar]
- 18.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105:20567–74. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8:1986–90. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- 21.Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–96. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–69. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 24.Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott-Schwartz J, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci. 2013;126:939–52. doi: 10.1242/jcs.114819. [DOI] [PubMed] [Google Scholar]
- 25.Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–16. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 26.Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy. 2009;5:732–3. doi: 10.4161/auto.5.5.8566. [DOI] [PubMed] [Google Scholar]
- 27.Fang Y, Morrell JC, Jones JM, Gould SJ. PEX3 functions as a PEX19 docking factor in the import of class I peroxisomal membrane proteins. J Cell Biol. 2004;164:863–75. doi: 10.1083/jcb.200311131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujiki Y, Matsuzono Y, Matsuzaki T, Fransen M. Import of peroxisomal membrane proteins: the interplay of Pex3p- and Pex19p-mediated interactions. Biochim Biophys Acta. 2006;1763:1639–46. doi: 10.1016/j.bbamcr.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 29.Matsuzaki T, Fujiki Y. The peroxisomal membrane protein import receptor Pex3p is directly transported to peroxisomes by a novel Pex19p- and Pex16p-dependent pathway. J Cell Biol. 2008;183:1275–86. doi: 10.1083/jcb.200806062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato Y, Shibata H, Nakano H, Matsuzono Y, Kashiwayama Y, Kobayashi Y, Fujiki Y, Imanaka T, Kato H. Characterization of the interaction between recombinant human peroxin Pex3p and Pex19p: identification of TRP-104 IN Pex3p as a critical residue for the interaction. J Biol Chem. 2008;283:6136–44. doi: 10.1074/jbc.M706139200. [DOI] [PubMed] [Google Scholar]
- 31.Sato Y, Shibata H, Nakatsu T, Nakano H, Kashiwayama Y, Imanaka T, Kato H. Structural basis for docking of peroxisomal membrane protein carrier Pex19p onto its receptor Pex3p. EMBO J. 2010;29:4083–93. doi: 10.1038/emboj.2010.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farré JC, Manjithaya R, Mathewson RD, Subramani S. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev Cell. 2008;14:365–76. doi: 10.1016/j.devcel.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Motley AM, Nuttall JM, Hettema EH. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012;31:2852–68. doi: 10.1038/emboj.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuzono Y, Matsuzaki T, Fujiki Y. Functional domain mapping of peroxin Pex19p: interaction with Pex3p is essential for function and translocation. J Cell Sci. 2006;119:3539–50. doi: 10.1242/jcs.03100. [DOI] [PubMed] [Google Scholar]
- 35.Matsuzono Y, Kinoshita N, Tamura S, Shimozawa N, Hamasaki M, Ghaedi K, Wanders RJA, Suzuki Y, Kondo N, Fujiki Y. Human PEX19: cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane assembly. Proc Natl Acad Sci U S A. 1999;96:2116–21. doi: 10.1073/pnas.96.5.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuzono Y, Fujiki Y. In vitro transport of membrane proteins to peroxisomes by shuttling receptor Pex19p. J Biol Chem. 2006;281:36–42. doi: 10.1074/jbc.M509819200. [DOI] [PubMed] [Google Scholar]
- 37.Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–80. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mardakheh FK, Auciello G, Dafforn TR, Rappoport JZ, Heath JK. Nbr1 is a novel inhibitor of ligand-mediated receptor tyrosine kinase degradation. Mol Cell Biol. 2010;30:5672–85. doi: 10.1128/MCB.00878-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Till A, Lakhani R, Burnett SF, Subramani S. Pexophagy: the selective degradation of peroxisomes. Int J Cell Biol. 2012;2012:512721. doi: 10.1155/2012/512721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita N, Yoshimori T. Ubiquitination-mediated autophagy against invading bacteria. Curr Opin Cell Biol. 2011;23:492–7. doi: 10.1016/j.ceb.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–8. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okumoto K, Kametani Y, Fujiki Y. Two proteases, trypsin domain-containing 1 (Tysnd1) and peroxisomal lon protease (PsLon), cooperatively regulate fatty acid β-oxidation in peroxisomal matrix. J Biol Chem. 2011;286:44367–79. doi: 10.1074/jbc.M111.285197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghaedi K, Tamura S, Okumoto K, Matsuzono Y, Fujiki Y. The peroxin pex3p initiates membrane assembly in peroxisome biogenesis. Mol Biol Cell. 2000;11:2085–102. doi: 10.1091/mbc.11.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yagita Y, Hiromasa T, Fujiki Y. Tail-anchored PEX26 targets peroxisomes via a PEX19-dependent and TRC40-independent class I pathway. J Cell Biol. 2013;200:651–66. doi: 10.1083/jcb.201211077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Itoh R, Fujiki Y. Functional domains and dynamic assembly of the peroxin Pex14p, the entry site of matrix proteins. J Biol Chem. 2006;281:10196–205. doi: 10.1074/jbc.M600158200. [DOI] [PubMed] [Google Scholar]
- 47.Tsukamoto T, Yokota S, Fujiki Y. Isolation and characterization of Chinese hamster ovary cell mutants defective in assembly of peroxisomes. J Cell Biol. 1990;110:651–60. doi: 10.1083/jcb.110.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Honsho M, Tamura S, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y. Mutation in PEX16 is causal in the peroxisome-deficient Zellweger syndrome of complementation group D. Am J Hum Genet. 1998;63:1622–30. doi: 10.1086/302161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mukai S, Ghaedi K, Fujiki Y. Intracellular localization, function, and dysfunction of the peroxisome-targeting signal type 2 receptor, Pex7p, in mammalian cells. J Biol Chem. 2002;277:9548–61. doi: 10.1074/jbc.M108635200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.