

Abstract
Human monoclonal antibodies based on IgG and IgA have shown promise as topical microbicide candidates to protect women from HIV infection. Their application has been limited, however, by the inability of gel and conventional intravaginal ring designs, the predominant topical vaginal product formulations, to effectively deliver biomolecules in a coitally-independent fashion with retention of bioactivity. We have developed intravaginal rings (IVRs) using a novel pod-IVR platform that delivers ovine immunoglobulin G (ov-IgG) as a model for IgG and IgA human monoclonal antibodies. In vitro release of ov-IgG from the pod-IVRs was sustained for 14 days. Facile control of release rate was achieved by changing the size of delivery channels in the ring structure, and the feasibility of ov-IgG delivery in the range 0.5 to 30 mg day-1 from a ten-pod IVR was demonstrated. The activity of ov-IgG in pod-IVR formulations was maintained as confirmed by ELISA binding assay. Pod-IVRs delivering ov-IgG show promise for the effective sustained topical delivery of antibody-based microbicides. This significantly broadens the range of microbicides that can be delivered in a sustained fashion from IVRs and enables a new arsenal of topical biologic microbicide candidates beyond small molecule antiretrovirals.
Introduction
The global estimated human immunodeficiency virus (HIV) incidence is more than 34 million, and over than 2.5 million HIV-1 infections are still acquired annually despite significant efforts in the development of broad-spectrum microbicides and an effective vaccine.1 Both tenofovir gel2,3 and oral tenofovir and emtricitabine combination4 microbicides have shown promise in preventing sexual HIV transmission in clinical trials, but trial failures of other microbicide candidates5-9 indicate that new effective and safe microbicide candidates are needed urgently. The first candidates studied for topical HIV prevention were broadly-acting, non-specific microbicides such as nonoxynol-9,5,6 λ- and κ-carrageenan (Carraguard),8 or naphthalene sulfonate polymer (PRO 2000 gel).7 More recently microbicide efforts have focused on antiretroviral drugs such as tenofovir,2-4 dapivirine,10 or MIV-150.11 Antiretrovirals target specific stages of the virus lifecycle such as viral entry (CCR5 agonists), viral DNA replication (reverse transcriptase inhibitors), or viral genome insertion (integrase inhibitors). The high concentrations of these compounds in gel formulations have the potential for adverse safety effects by damaging the highly sensitive cervico-vaginal mucosal tissues, and CCR5 agonists are not active against ×4 and dual tropic viruses.12
As an alternative, antibody-based microbicides applied topically to the vagina may play an important role in protecting women from HIV infection, from both efficacy and safety perspectives.12 The broadly neutralizing human monoclonal antibodies (bNAbs) b12,13,14 2G12,15 2F5,16 and 4E1016 have demonstrated efficacy against SHIV infection in macaque models. These bNAbs neutralize a diverse range of primary HIV-1 isolates,12,17 and more potent bNabs against a wider range of HIV-1 isolates have subsequently been identified including PG9, PG16, VRC01, and multiple PTG bNAbs.18-21 The bNAb VRC01 protected against HIV-1 vaginal transmission in a mouse model and is the first in vivo demonstration of bNAb efficacy in human target cells.12 The target of a bNAb microbicide is not limited to HIV: passive immunization against herpes simplex virus-2 (HSV-2) by FcRN-transported IgG delivered to the female genital tract was obtained in a mouse model.22
The practical application of bNABs as a topical microbicide has thus far been limited by the inability of gels and conventional intravaginal ring designs,23 the predominant topical vaginal product formulations, to effectively deliver biomolecules in a coitally-independent fashion with retention of antibody bioactivity. Morrow et al developed an insert vaginal ring for delivery of hydrophilic and macromolecular drugs and demonstrated release of the antibody 2F5, but the delivery was only sustained over a maximum of 5 days with limited control of release rate.24 The pod-IVR,25 a novel modular ring design consisting of polymer coated solid drug cores (“pods”) incorporated into a silicone IVR, was specifically designed for simultaneous delivery of multiple drugs, and in particular, relatively hydrophilic antiviral agents that are difficult to release from traditional matrix and reservoir IVRs. In pod-IVRs, the release rate for each drug pod is controlled independently, determined by the size of one or more delivery channels that are mechanically formed in the elastomer backbone during fabrication, as well as the pod's biocompatible polymer coating and the total number of pods per IVR. The pod-IVR platform has been applied to delivery of tenofovir (TFV),26,27 tenofovir disoproxil fumarate (TDF),27 and acyclovir (ACV)28 individually and in combination.29 Pod-IVRs have the capability for simultaneous delivery of drug combinations spanning a wide-range of physicochemical properties. A five-drug multipurpose protection (MPT) pod-IVR that simultaneously delivers, with independently controlled release rates, three antiretroviral drugs against HIV (tenofovir, nevirapine, and sequinavir) and a progestin-estrogen contraceptive (etonogestrel and estradiol) has been developed and its pharmacokinetics investigated in a sheep model.30 Described here is the application of the pod-IVR platform to delivery of monoclonal antibodies and other highly water-soluble biological molecules using the ovine Immunoglobulin G (ov-IgG) antibody as a model for IgG and IgA human monoclonal antibodies.
Materials and Methods
Preparation of IgG antibody solid formulation
Protein G purified sheep immunoglobulin G (ov-IgG, > 95% pure) was obtained from Innovative Research (Novi, MI, USA) as a 4.7 mg mL-1 solution in pH 7.4 phosphate buffer solution (PBS, 0.02M sodium phosphate, 0.15M NaCl). A 12 mL aliquot of ov-IgG solution was freeze dried to obtain ∼200 mg of a white, voluminous powder containing approximately 56 mg ov-IgG (29%), 105 mg NaCl (54%), and 21 mg Na2HPO4 (11%) and 11 mg Na2HPO4 (6%). The freeze-drying cycle consisted of initial flash freezing of the solution by immersing the lyophilizer flask in liquid nitrogen followed by drying on a lyophilizer (Virtis Freezemobile 12SL, SP Industries, Warminster, PA) at 0.1 torr for 16 h.
Manufacture of silicone pod-intravaginal rings
Intravaginal rings of the pod-IVR design containing ov-IgG were prepared using methods previously reported.25 Briefly, cylindrical cores of 40 mg dry ov-IgG solid formulation (3.2 mm diam. × 2 mm ht.) were formed using compaction with a pellet press (Globe Pharma MTCM-I, North Brunswick, NJ). The compressed ov-IgG cores were coated with two layers of poly(D,L-lactide) (PLA, 10-18 kDa, ester-terminated) (Resomer R 202 S, Evonik Industries AG, Essen, Germany) from a 5% PLA solution in 2:1 dichloromethane:ethyl acetate to form ov-IgG pods. A 6 μL aliquot of PLA solution was dropped on one flat end of the cylindrical core using an automatic pipette and allowed to dry. The core was inverted and a second 6 μL aliquot applied to the opposite flat end. After drying for ∼4h, a second PLA layer was applied using the same technique. The PLA-coated ov-IgG pods were embedded in silicone ring segments with one delivery channel per pod (channel diameter 0.75 – 2.0 mm) as described previously.25 Delivery channel diameters reported here are the punch size used to form the channel. The measured channel diameter has been shown previously to agree closely with the punch diameter. 25
In vitro studies
Studies to measure the in vitro release of ov-IgG into a simplified vaginal fluid simulant (VFS) were carried out on IVRs containing one ov-IgG pod. The VFS was adapted from Owen and Katz31 and consisted of 25 mM acetate buffer (pH 4.2) with NaCl added to yield a 200 mOs solution. For all in vitro release studies, the IVRs were placed in glass vials containing 10 mL VFS at 25 ± 2°C and with shaking at 60 rpm on an orbital shaker. Aliquots of the release medium were removed at specified time intervals and analyzed by UV absorption spectroscopy at 280 nm (OD280) using a Spectramax Plus384 96-well plate reader (Molecular Devices, Sunnyvale, CA, USA). The concentration of ov-IgG in the release solution was calculated using the Beer-Lambert law and the absorption coefficient 1.36 mg-1 cm-1 mL provided by the manufacturer on the product datasheet. Data fitting was carried out by non-linear least squares minimization using Excel (Microsoft, Redmond, WA) software.
Fluorescence imaging of PLA polymer coatings
The thickness and uniformity of the PLA coating was evaluated by adding a Rhodamine 6G fluorescent dye to the coating solution and imaging sections of pods using fluorescence microscopy. Rhodamine 6G perchlorate (Acros, Geel, Belgium) was dissolved in dichloromethane and an aliquot added to the 5% PLA solution to obtain a dye concentration of 20 mM. Pods were coated with the fluorescent PLA solution as described above. The cylindrical pods were embedded in silicone and cut on two different cross-sections parallel and perpendicular to the round cylindrical pod faces for imaging. Low magnification (6×) light images were acquired using a 6×-50× stereo zoom microscope (Edmund Optics, Barrington, NJ) with a Nikon Coolpix 995 digital camera. Fluorescence images were acquired with an EVOS fl fluorescence microscope (AMG, Mill Creek, WA) using a filter set that overlaps the Rhodamine 6G absorption (λmax = 530 nm) and emission (λmax = 552 nm) bands [EVOS RFP light cube: λex=530 nm, 40 nm bandwidth; λem = 593 nm, 40 nm bandwidth]. Thickness of the coating was measured using the ImageJ software package (National Institutes of Health, Bethesda, MD).32,33 The number of pixels per micrometer for each image was determined using the Set Scale function of ImageJ and the calibrated scale bar provided by the EVOS fl software. The thickness of the fluorescent PLA film in each image was determined using the Measure function in ImageJ at ten points equally spaced across the pod surface shown in each image. Thickness data is reported as mean ± standard deviation (SD) for the set of ten measurements.
ELISA measurements
The binding activity of ov-IgG was analyzed by Enzyme-Linked ImmunoSorbant Assay (ELISA). Rabbit F(ab')2 Anti-sheep IgG (coating antigen) and Rabbit Anti-sheep IgG(H+L)-HRP (detection antibody) were obtained from SouthernBiotech (Birmingham, AL, USA). MaxiSorp 96-well flat-bottom immune plates, wash buffer (1× PBS), Tween 20, TMB substrate solution, and SEA BLOCK blocking buffer were purchased from Thermo Scientific (Rockford, IL, USA). ELISA plates were prepared by sequentially incubating overnight at 4°C with 100 μL per well of coating antigen (2 ng μL-1 in 1× PBS), 1 h at room temperature with 200 μL per well of SEA BLOCK blocking buffer, 1 h at room temperature with 100 μL of standard or sample, and finally 100 μL of detection antibody diluted 1:5000 in wash buffer. Plates were washed three times with wash buffer between each incubation step. Plates were developed by adding 100 μL TMB Substrate solution to each well, followed by 50 μL 4N H2SO4 once color development was observed in low concentration standards. The absorption at 450 nm (OD450) was measured using a SpectraMAX Plus384 micro plate reader. All ov-IgG samples were adjusted to a concentration of 0.41 mg mL-1 as determined by OD280 then further diluted 1:200 prior to ELISA measurement. For each ELISA experiment, a single plate was used for all ov-IgG standards and samples. Samples and standards were measured in triplicate. Standards at eight concentrations from 0 to 1000 ng/mL (as determined by OD280) were prepared from a new batch of ov-IgG purchased within 1 week of the ELISA measurement. Plots of standard OD450 from ELISA versus concentration from OD280 measurement were fit using non-linear least-squares regression to a second-order polynomial with GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA). The OD450 value for each sample was converted to concentration of ov-IgG exhibiting antigen binding activity [IgG] using the calibration curve obtained for a set of standards on the same ELISA plate. The fraction of initial antigen binding activity was calculated as,
where [IgG] is the concentration of active antibody in the sample and [IgG]o is the concentration of active antibody in the original ov-IgG solution used to prepare the IVRs and stored continuously at -80°C.
Results
A photograph of a pod-IVR with four ov-IgG pods and a cross-sectional drawing showing the delivery channel and pod configuration is shown in Figure 1. In vitro release of ov-IgG from single-pod IVRs into VFS followed pseudo-zero order kinetics for up to 14 days. The release rate for ov-IgG from the pod-IVR is determined by the size and number of the delivery channels, the number of pods per ring, and the pod coating material and thickness. Figure 2 shows the cumulative release of ov-IgG from single pod IVRs with a with delivery channels of four different diameters between 0.75 mm and 2.0 mm. Figure 3 and Table 1 show the daily release rate calculated from the slope of the cumulative release plots in Figure 2 as a function of delivery channel cross-sectional area. A release rate range of 0.53-3.0 mg day-1 is obtained by varying the delivery channel size. For a pod-IVR with a maximum of ten pods per ring, the maximum number of pods in a human-sized IVR, this will allow incremental variation of the release over a 50-fold range simply by changing the delivery channel size and number of pods in the ring. Release of ov-IgG from pod-IVRs is described by a passive diffusion model illustrated in Figure 4 and described below. Non-linear least squares fits of the in vitro release data to the model are shown in Figure 2. The linear portion of each release curve was fit using the zero-order equations and the non-linear portion to the first-order equations. The resulting model parameters from the fit are shown in Table 1.
Figure 1.
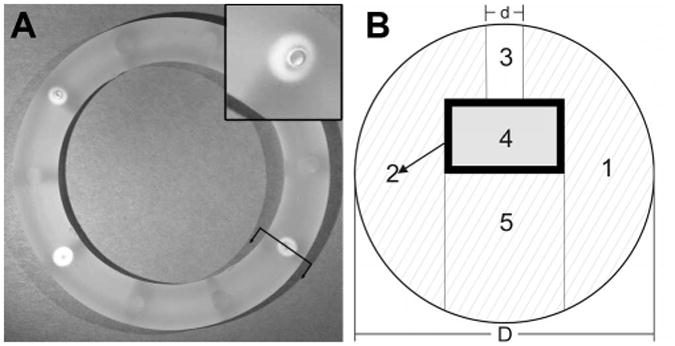
(A) Human and sheep-sized pod-IVR with four ov-IgG pods. The inset shows a close-up of a delivery channel and pod. The ring outer diameter is 56 mm. (B) Cross-sectional drawing [orientation indicated by arrows in (A)] of IVR through center of drug pod showing silicone ring, 1, containing embedded ov-IgG core, 4, with polylactic acid coating, 2, and delivery channel, 3, sealed in place with silicone backfill, 5. The ring cross-sectional diameter, D, is 8 mm. The delivery channel diameter, d, is 0.75 – 2.0 mm depending on desired ov-IgG release rate.
Figure 2.

Average cumulative release of ov-IgG into VFS as a function of delivery channel size (N=3). Pod-IVRs contained one ∼50 mg pod per ring. Solid lines are fits of the data to a passive diffusion model as described in Figure 4 and in the text. The daily release rate was obtained from the slope of the linear portion of each release profile curve.
Figure 3.

Average daily release rate obtained from release profiles in Figure 2 as a function of delivery window area for ov-IgG release from a pod-IVR. Delivery channel area is the cross sectional area of the delivery channel. [Area = π • (diameter/2)2].
Table 1. Delivery channel configuration, release rate data, and model fit parameters.
| Delivery Channel | Zero-order Release rate (mg day-1) | Model Fit Parameters | |||
|---|---|---|---|---|---|
|
|
|
||||
| Diameter (mm) | Area (mm2) | C0 (mg cm-3) | P (cm d-1) | V (cm3) | |
| 0.75 | 0.44 | 0.53 | 9.5 | 12.5 | n/a |
| 1.0 | 0.79 | 0.70 | 7.5 | 11.9 | n/a |
| 1.5 | 1.8 | 1.7 | 7.5 | 11.5 | 0.68 |
| 2.0 | 3.1 | 3.0 | 10.7 | 14.1 | 0.72 |
Figure 4.
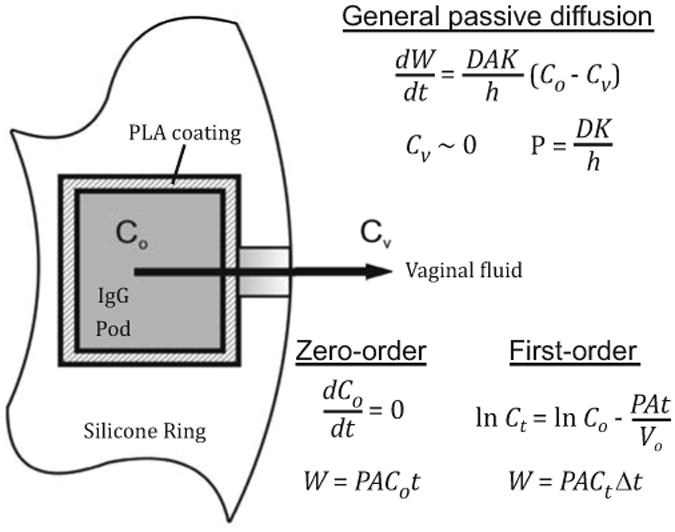
Passive diffusion model of ov-IgG release from a pod-IVR. Co is the concentration of dissolved ov-IgG in the pod (inside the PVA membrane) and Cv is the concentration in the bulk VFS release medium. The shading of the pod interior and delivery channel represents the gradient established between Co and Cv (dark to light represents high concentration to low). Release of ov-IgG from the ring is described by the general passive diffusion equations (Refs. 41, 42) solved for zero-order and first-order conditions. Model variables and constants: cumulative amount of ov-IgG released, W (mg); ov-IgG diffusivity (D, cm s-1); membrane area, A (cm2); partition coefficient, K; diffusion length (incorporating membrane and delivery channel), h (cm); permeability, P (cm s-1), time, t (d). For calculations under first-order conditions, Δt is the time interval used for the calculation of concentration of ov-IgG in the pod at time t, Ct.
Figure 5 shows an image of a PLA coated pod sectioned along a plane parallel to the circular end face. The PLA layer containing red Rhodamine 6G dye is clearly visible and uniformly distributed around the circumference of the pod. Figure 6 shows fluorescence images of the Rhodamine 6G-PLA film on a pod sectioned parallel (Figure 6a) and perpendicular (Figure 6b) to the cylindrical end face. For the pod side surface (Figure 6a), the mean PLA film thickness is 136 ± 8 μm (mean ± SD, n = 10 measurements). For the pod end surface (Figure 6b), the face of the pod that abuts the delivery channel, the mean PLA film thickness is 112 ± 7 μm (mean ± SD, n = 10 measurements).
Figure 5.

Light stereo-microscope image (6×) of PLA-coated ov-IgG pod sliced along a cross-section parallel to the circular end face of the cylindrical pod. The PLA coating solution contained 20 mM Rhodamine 6G dye. The uniform, thin PLA film appears as a red band around the circumference of the pod.
Figure 6.
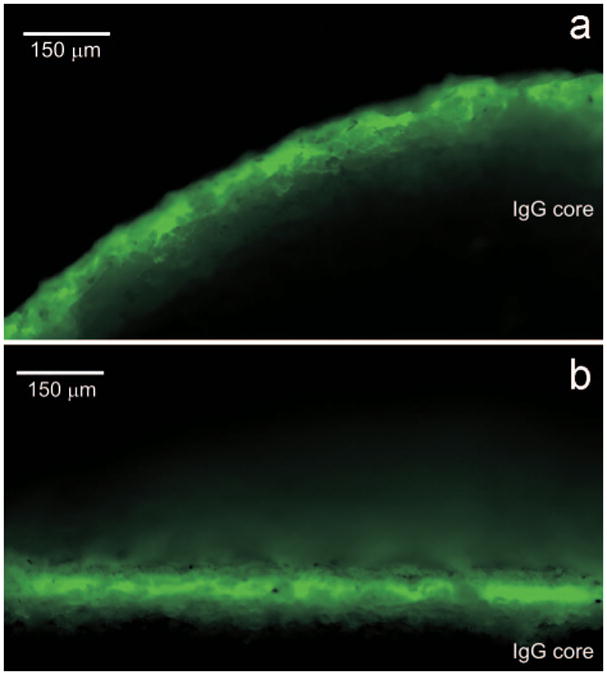
False color fluorescence images at 10× magnification of the ov-IgG pod from Figure 5 showing the uniformly applied polymer film in green. Two different cross sections were obtained by slicing the cylindrical pod parallel (a) and perpendicular (b) to the circular end face that abuts the delivery channel.
The specific binding activity of ov-IgG following formulation in a pod-IVR and in subsequent in vitro release studies was assessed using ELISA with a Rabbit F(ab')2 Anti-sheep IgG coating antigen and Rabbit Anti-sheep IgG(H+L)-HRP detection antibody. Figure 7 shows the percent binding activity observed during the manufacture and in vitro testing of ov-IgG pod IVRs and after 6.5 months of storage of an ov-IgG IVR at 4°C. Because the magnitude of the ELISA response is dependent on a number of factors including efficiency of antigen binding to the 96-well plate and the enzyme activity in the detection antibody, the OD450 values from different ELISA experiments cannot be compared directly. Consequently, the fraction of active ov-IgG in each sample is calculated relative to a sample of the original ov-IgG prior to lyophilization. The initial lyophilization step to obtain dry ov-IgG results in a decrease of fractional binding activity to 0.61±0.03 (40% loss). The pod-IVR fabrication process does not lead to additional loss of activity, ELISA showing 0.58±0.08 activity for pod-IVRs stored 9 days at 4°C following manufacture. The fractional activity of ov-IgG in the pod-IVRs is unchanged (0.62±0.08) following storage for 196 days (6.5 months) at 4°C, indicating that the solid-state pod-IVR formulation stabilizes the ov-IgG toward loss of activity during long-term storage. The acidic aqueous VFS used in the in vitro dissolution experiments leads to a dramatic loss of ov-IgG binding activity, but the pod-IVR formulation significantly stabilizes the antibody relative to ov-IgG dissolved in VFS. Analysis of an IVR following 14 days of dissolution testing in VFS showed that the fraction of antigen binding activity in the residual ov-IgG in the wetted pod (not released) was 0.32±0.02, a 48% decrease compared to the lyophilized ov-IgG solid formulation. The fractional binding activity of ov-IgG in the dissolution medium following 14 days of cumulative release was 0.12±0.03 (80% decrease).
Figure 7.

Fraction of antigen binding activity compared to ov-IgG prior to drying and pod-IVR formulation as measured by ELISA: A. ov-IgG stored 14 days in PBS at -80°C (as received); B. lyophilized ov-IgG before IVR formulation; C. ov-IgG excised from pod-IVR following 9 days storage at 4°C; D. residual ov-IgG in pod-IVR following 14 day in vitro release into in VFS; E. ov-IgG stored 196 days in PBS at -80°C (as received); F. ov-IgG excised from pod-IVR following 196 days storage at 4°C. Gray bars indicate one ELISA experiment conducted following the in vitro release; hatched bars indicate a second ELISA measurement conducted following the 6.5 month stability study.
Discussion
Pod-IVR design and in vitro release of ov-IgG
The pod-IVR described above demonstrates sustained in vitro release of an ov-IgG antibody with the ability to control release rate by simple modification of the elastomer ring structure, providing a novel, generalized platform for delivery of monoclonal antibodies to the female genital tract. Conventional IVR formulations are typically based on two designs: (1) a matrix IVR with the drug either dissolved in or homogeneously dispersed as amorphous or crystalline solids throughout a polymeric matrix, or (2) a reservoir IVR with a drug-loaded polymer core similar to a matrix ring covered by an outer non-medicated polymer layer to control drug release.23 Both designs require that drug molecules diffuse through the structural ring polymer for release into vaginal fluids and have typically been limited to delivery of hydrophobic (logP > 3) small-molecule drugs.23,34,35 The use of a hydrophilic polyurethane in place of silicone or ethylene-co-vinyl acetate (EVA) in matrix and reservoir IVRs has allowed delivery of the more hydrophilic tenofovir (logP = -2.3)36 and tenofovir disoproxil fumarate37 (logP = 1.25)38. Traditional matrix and reservoir IVR approaches, however, are unsuitable for large biomolecules such as antibodies because they do not diffuse through the elastomeric ring structure, and a novel approach to IVR design is required. The only report of an IVR delivering an antibody was by Malcolm et al who prepared rod-insert IVRs (RiRs) consisting of an elastomeric ring body with inserts containing drug in compressed solid or gel form.24,39 The RiRs released bovine serum albumin (BSA) and the antibody 2F5 in vitro, but control of release rate was limited and 2F5 delivery was sustained for less than one week.
The pod-IVR platform can deliver large, highly soluble biomolecules because drug release is not dependent on diffusion through the bulk ring elastomer, but only on passive diffusion across a thin, polymeric membrane and through large physical delivery channels in the ring elastomer material. The biocompatible PLA membranes coating the ov-IgG cores are of uniform thickness across the cylindrical pod face and sides as shown in Figures 5 and 6. Diffusion occurs primarily across the circular end surface that abuts the delivery channel, with a mean PLA thickness of 124 ± 7 μm observed. Modification of the release rate on a per-pod basis is achieved with a single polymer composition and thickness through variation of the delivery channel size for each pod and the number of pods in the IVR. Rather than modifying the polymer composition and thickness to modulate drug release rate, a polymer composition and thickness is chosen to provide a range of accessible release rates that are subsequently tuned using the delivery channel size and number, both easily changed as the delivery channels are mechanically punched during the ring assembly. In the case of technologies relying on the diffusion through a polymer as the primary determinant of active pharmaceutical agent (API) release, changing the release rate target requires a much more substantial effort.
The use of one polymer material and thickness for all release rates has important implications for manufacturability and regulatory approval. Delivery channels are formed during the ring assembly step, and manufacturing of pods is identical regardless of the target release rate, allowing a single coating method to be developed and optimized for application to numerous different IVR configurations and release targets. This is particularly important for antibodies and other APIs with stability issues as the conditions during manufacturing can lead to API degradation (i.e. loss of antibody activity), and the pod fabrication process can be developed once in an optimized fashion. Additionally, changing the polymer composition to obtain a different release rate requires that the new polymer demonstrate safety and API compatibility. This means that changing the IVR release rate potentially requires significant additional safety and compatibility studies. For the pod-IVR, release rate changes can be accomplished with mechanical means described above that do not involve a change in polymers.
Sustained release of ov-IgG from single-pod pod-IVRs was maintained for up to 14 days in vitro. The release rate was controlled from 0.5 to 3 mg day-1 through modifications to the size of the delivery channels as described previously for pod-IVRs delivering small molecule antiviral and hormonal agents.25,30 Because pod-IVRs in the human/sheep size can contain up to ten pods,25 an ov-IgG dosing range of 0.5-30 mg per day may be obtained by varying the number of pods per ring. Multiple delivery channels per pod could be used to further increase the daily release rate, or to select rates within those obtained with single channels as has been demonstrated in pod-IVRs releasing small-molecule antiretroviral agents.25,30 For all dissolution studies in VFS, pseudo-zero order release kinetics were obtained for release of 80-85% of the total ov-IgG mass, with release continuing at a decreasing rate until >95% of the total mass was released. All of the pods used in this work contain ∼50 mg solid material (29% ov-IgG, ∼14 mg). Because all pods release simultaneously, the amount of time that release is sustained (the number of days the IVR may be used) is dependent on the amount of ov-IgG in each pod and the release rate per pod. For the configurations evaluated here, a range of 4-20 days is required for release of 83% of the total 14 mg ov-IgG per pod. Although pods in the work presented here are 3.2 mm diameter and contain ∼50 mg solid mass, a human-sized pod-IVR may accommodate pods as large as 4.8 mm diameter and 220 mg total solid (62 mg ov-IgG). This indicates a maximum possible deliverable ov-IgG loading of ∼500 mg released over 17-86 days at the daily release rates obtained here. These calculations assume a 29% IgG formulation as obtained by simple lyophilization of the ov-IgG as used here and release of 80% of the total IgG mass. A solid antibody formulation designed specifically for pod-IVR delivery could have a significantly higher IgG fraction, allowing a larger dosing range to be obtained.
Delivery mechanism
Delivery of ov-IgG from a pod-IVR follows the mechanism of a polymer membrane controlled-permeation delivery system.40 Release of ov-IgG involves passive diffusion across the PLA membrane and through the delivery channel to the vaginal fluids, and can be modeled using Fick's first and second laws of diffusion.41,42 Figure 4 shows the application of this model to the pod-IVR system. Following pod-IVR insertion, vaginal fluid (simulant in the case of in vitro release studies) fills the delivery channel, diffuses across the PLA membrane into the pod core, and dissolves ov-IgG until a saturation concentration, Co, is reached inside the pod membrane. Under sink conditions, the concentration of ov-IgG in vaginal fluid (Cv) is such that Co≫Cv, and a steady state condition with a constant concentration gradient between the inside of the pod and the vaginal fluid is established. Release of ov-IgG under this steady-state condition follows the zero-order equations in Figure 4. Following release of ∼80% of the IgG in the pod, the steady-state condition is no longer maintained, and the effective IgG concentration inside the pod decreases with time, resulting in first-order release kinetics. Figure 2 shows the cumulative daily release of ov-IgG from four pod-IVRs with differing delivery channel size overlaid with fits of the data to the model in Figure 4. Table 1 lists the parameters used in the model fits. For zero-order release kinetics, values are obtained from the model for ov-IgG concentration in the pod (Co) and ov-IgG permeability (P), a constant that incorporates the diffusion coefficient in the membrane, the delivery channel length (h), and a coefficient (K) related to partitioning of ov-IgG into and out of the PLA membrane. The first-order release model incorporates a volume term, Vo. This is not an actual pod volume; rather, it is an effective volume related to the volume of solution on the high-concentration side of the membrane required to obtain identical parameter values in a two-compartment passive diffusion model. The values of these parameters obtained from the model fits are consistent across the 0.75 - 2.0 mm delivery channel range: Co = 8.8 ± 1.6 mg cm-3, P = 12.5 ± 1.1, and Vo = 0.70 ± 0.03 (mean ± SD). Additional studies are required for a complete understanding of the partitioning and diffusion of the large antibodies through the PLA membrane. Unlike small-molecule drugs that diffuse through intact PLA membranes, diffusion of the larger antibodies may be dominated by penetration of cracks or channels in the PLA film. For pod-IVRs with 0.75 and 1.0 mm diameter delivery channels, a small jump in concentration and slight change in release rate is observed around day 7-8. This may indicate a mechanistic change or a complex combination of other factors such as the formation of and release through cracks in the polymer. It is clear from the model, however, that antibody release is membrane-controlled to be zero-order for release of the first 80% of ov-IgG, with modulation of the zero-order rate determined by the delivery channel size.
Formulation stability
A primary technical challenge with antibody-based delivery systems is stabilizing the protein structure during formulation and storage to retain the desired antigen binding activity. The stability of monoclonal antibodies and other proteins during manufacture, formulation, and storage has been reviewed extensively,43-46 and is affected by physical (denaturation, covalent and non-covalent aggregation, adsorption, and precipitation) and chemical (deamination, oxidation, and glycation) processes. Of particular importance to solid antibody formulation stability are aggregation43,47 and the effect of moisture.48,49 The drying methods employed and the stabilizing excipients used in solid formulations are complex factors that are critical to the safety and efficacy of pharmaceutical antibodies.44,50-56 Although antibodies exhibit a high degree of structural similarity, antigen binding specificity is determined by surface-exposed amino acid sequences, and each specific antibody requires a unique solid-state formulation to maintain binding activity.46
For this proof-of-principal study, simple lyophilization of ov-IgG together with its buffer components was used to prepare the solid formulation incorporated into pod-IVRs. This non-optimized preparation method leads to a loss of approximately 40% of antigen binding activity compared to ov-IgG as received in solution. The methods and formulations required to obtain solid antibody formulations that maintain antibody activity and exhibit long-term stability are highly antibody-specific.46,47 Because ov-IgG is used as a model monoclonal antibody, the development of a solid ov-IgG formulation optimized with respect to maintaining antigen-binding activity would require significant effort and serve no purpose. As demonstrated by the ELISA assay data in Figure 7, the 0.61 fractional antigen binding activity of the lyophilized solid ov-IgG material is maintained during the core compression, PLA coating, and IVR assembly steps during manufacturing, and the pod-IVR formulation exhibits no loss of initial lyophilized ov-IgG binding activity for more than 6 months when IVRs are stored at 4°C. Consequently, the pod-IVR platform described here can be applied to any antibody solid formulation. Optimized drying methods,50,51 including spray-drying and lyophilization; the incorporation of stabilizing excipients such as buffers, anions, polymers, carbohydrates, and surfactants in the core formulation45,57; and dry, sealed IVR packaging may lead to stability improvements that remove the cold-chain requirement for storage altogether.
In vitro release
The in vitro method described in this work is not intended to precisely mimic physiological conditions. Rather, the goal is to measure an in vitro release rate that can be subsequently correlated with an in vivo release rate in a sheep or other animal model to obtain an in vivo-in vitro correlation (IVIVC) that may serve as a predictive tool for iterative development of pod-IVRs. Based on prior work with pod-IVRs releasing TFV,26,27 TDF,27 and ACV,28 the in vitro method used here involved measurement of cumulative release from single-pod IVRs in vials containing pH 4.2 VFS at 25°C with continuous agitation. The VFS leads to additional loss of ov-IgG activity as measured by ELISA. After 14 days of cumulative release, the residual ov-IgG wetted by VFS but remaining in the pod-IVR has a fractional activity of 0.32, a loss of approximately 50% of the activity observed for ov-IgG in pod-IVRs stored under dry conditions at 4°C. Because release of ov-IgG from the pod-IVR is a zero order process, the loss of 50% of the IgG activity for the antibody in the wetted pod over 14 days in pH 4.2 VFS during an in vitro study is equivalent to the daily release rate decreasing by 50% over 14 days. This level of activity loss is significantly less than the four-fold decrease over 14 days of the in vitro daily release rate observed for silicone matrix IVRs delivering the small-molecule antiretroviral dapivirine that are currently in Phase III clinical trial.58 The fractional activity of ov-IgG released from the ring is 0.12 after 14 days in aqueous solution at pH 4.2, a loss of 80% compared to the ov-IgG in the lyophilized powder. The loss of binding activity over 14 days is 2.6 times less in the VFS-wetted pod than in VFS solution, demonstrating a protective function of the concentrated solid core pod-IVR formulation for biological agents that are unstable in solution. The amount of antibody activity loss that could be accommodated by increasing the daily release rate to maintain levels above the therapeutic index will be dependent on the monoclonal antibody and stabilizing excipients in the dry solid formulation.
This in vitro system most likely overestimates ov-IgG instability relative to an in vivo system. Acidic conditions are known to accelerate IgG aggregation and denaturation,47 and no stabilizing proteins or carbohydrates were present to reduce aggregation and denaturation. In vitro binding activity was measured following 14 days of cumulative release into VSF. For topical delivery to the vagina, antibodies will be continuously released with rapid uptake into the vaginal mucosa.59,60 The binding activity of ov-IgG following release measured here is not predictive of in vivo behavior, and only serves characterize the in vitro system. Antibody instability is increased in dilute solution and in absorptive containers, and without added detergent or carrier proteins such as BSA, IgG adsorbs to glass and plastic, rapidly denaturing with concomitant loss of activity.46 In vitro concentration measurements were obtained using OD280 and are a measure of total protein in solution, not intact, active antibody. Adsorptive losses may reduce the magnitude of IgG concentration measured in these experiments, but the linear relationship between delivery channel size and ov-IgG release rate indicates that this simple in vitro system should be predictive of in vivo behavior through an IVIVC as has been demonstrated for pod-IVRs delivering antiretroviral drugs.26,29
Implications for multiple drug delivery
Pod-IVRs delivering ov-IgG show promise for the effective sustained topical delivery of antibody-based microbicides. The unique design of the pod-IVR allows these highly-soluble antibodies to be delivered in combination with small-molecule microbicides or contraceptive hormones, molecules with radically different release characteristics compared to IgG. A pod-IVR has been reported that simultaneously delivers three small-molecule antiretroviral drugs (tenofovir, saquinavir, and nevirapine) in combination with a progestin-estrogen contraceptive (etonogestrel and estradiol) at levels required for putative efficacy at preventing sexual HIV infection and pregnancy.30 Each pod in the IVR acts as an independent drug delivery device, and up to ten pods of different composition and number and size(s) of delivery channels may be combined in a single ring. Because release of an active agent from one pod is not dependent on the other pods in the IVR, any combination of pods may be incorporated in a single ring to target one or more indications. The ability to deliver multiple active agents is important both in terms of efficacy of prophylaxis of HIV alone and in combination with other sexually transmitted infections and in reducing the emergence of drug resistance.
Conclusion
The novel pod-IVR design presented here provides a platform for the intravaginal delivery of multiple antiviral compounds including antibodies. In vitro release studies demonstrate the sustained delivery ov-IgG as a model for human-derived monoclonal antibodies against HIV and HSV. Delivery at pre-determined rates may be controlled over more than one order of magnitude. Controlled, sustained release of ov-IgG from a pod-IVR was demonstrated in vitro with control of the release rate achieved through modification of the pod-IVR delivery channel size.
Acknowledgments
This work was supported by internal funding from Auritec Pharmaceuticals and the Oak Crest Institute of Science. Research reported in this publication also was supported, in part, by the National Institute of Allergy And Infectious Diseases of the National Institutes of Health under Award Number U19AI096398. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors gratefully acknowledge their support.
References
- 1.Global HIV/AIDS response: epidemic update and health sector progress towards universal access: progress report 2011. Geneva: World Health Organization; 2011. [accessed Jul. 21, 2014]. http://whqlibdoc.who.int/publications/2011/9789241502986_eng.pdf. [Google Scholar]
- 2.Karim QA, Karim SSA, Frohlich JA, Grobler AC, Baxter C, Mansoor LE, Kharsany ABM, Sibeko S, Mlisana KP, Omar Z, Gengiah TN, Maarschalk S, Arulappan N, Mlotshwa M, Morris L, Taylor D. Effectiveness and Safety of Tenofovir Gel, an Antiretroviral Microbicide, for the Prevention of HIV Infection in Women. Science. 2010;329(5996):1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karim S, Kashuba A, Werner L, Karim Q. Drug Concentrations After Topical and Oral Antiretroviral Pre-exposure Prophylaxis: Implications for HIV Prevention in Women. Lancet. 2011;378(9787):279–281. doi: 10.1016/S0140-6736(11)60878-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapia M, Guanira-Carranza JV, Ramirez-Cardich ME, Montoya-Herrera O, Fernandez T, Veloso VG, Buchbinder SP, Chariyalertsak S, Schechter M, Bekker LG, Mayer KH, Kallas EG, Amico KR, Mulligan K, Bushman LR, Hance RJ, Ganoza C, Defechereux P, Postle B, Wang FR, McConnell JJ, Zheng JH, Lee J, Rooney JF, Jaffe HS, Martinez AI, Burns DN, Glidden DV. Preexposure Chemoprophylaxis for HIV Prevention in Men Who Have Sex with Men. N Engl J Med. 2010;363(27):2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Damme L, Ramjee G, Alary M, Vuylsteke B, Chandeying V, Rees H, Sirivongrangson P, Tshibaka LM, Ettiègne-Traoré V, Uaheowitchai C, Karim SSA, Mâsse B, Perriëns J, Laga M. Effectiveness of COL-1492, a nonoxynol-9 vaginal gel, on HIV-1 transmission in female sex workers: a randomised controlled trial. Lancet. 2002;360(9338):971–977. doi: 10.1016/s0140-6736(02)11079-8. [DOI] [PubMed] [Google Scholar]
- 6.Roddy RE, Zekeng L, Ryan KA, Tamoufé U, Weir SS, Wong EL. A Controlled Trial of Nonoxynol 9 Film to Reduce Male-to-Female Transmission of Sexually Transmitted Diseases. N Engl J Med. 1998;339(8):504–510. doi: 10.1056/NEJM199808203390803. [DOI] [PubMed] [Google Scholar]
- 7.McCormack S, Ramjee G, Kamali A, Rees H, Crook AM, Gafos M, Jentsch U, Pool R, Chisembele M, Kapiga S, Mutemwa R, Vallely A, Palanee T, Sookrajh Y, Lacey CJ, Darbyshire J, Grosskurth H, Profy A, Nunn A, Hayes R, Weber J. PRO2000 vaginal gel for prevention of HIV-1 infection (Microbicides Development Programme 301): a phase 3, randomised, double-blind, parallel-group trial. Lancet. 2010;376(9749):1329–1337. doi: 10.1016/S0140-6736(10)61086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skoler-Karpoff S, Ramjee G, Ahmed K, Altini L, Plagianos MG, Friedland B, Govender S, De Kock A, Cassim N, Palanee T, Dozier G, Maguire R, Lahteenmaki P. Efficacy of Carraguard for prevention of HIV infection in women in South Africa: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9654):1977–1987. doi: 10.1016/S0140-6736(08)61842-5. [DOI] [PubMed] [Google Scholar]
- 9.Marrazzo J, Ramjee G, Nair G, Palanee T, Mkhize B, Nakabiito, Taljaard M, Piper J, Gomez Feliciano K, Chirenje M. Pre-exposure prophylaxis for HIV in women: daily oral tenofovir, oral tenofovir/emtricitabine or vaginal tenofovir gel in the VOICE study (MTN 003). Presented March 4, 2013 at the 20th Conference on Retroviruses and Opportunistic Infections; Atlanta, GA. Oral Abstract 26LB. [Google Scholar]
- 10.Nel A, Smythe S, Young K, Malcolm K, McCoy C, Rosenberg Z, Romano J. Safety and pharmacokinetics of dapivirine delivery from matrix and reservoir intravaginal rings to HIV-negative women. J Acquir Immun Defic Syndr. 2009;51:416. doi: 10.1097/qai.0b013e3181acb536. [DOI] [PubMed] [Google Scholar]
- 11.Singer R, Mawson P, Derby N, Rodriguez A, Kizima L, Menon R, Goldman D, Kenney J, Aravantinou M, Seidor S, Gettie A, Blanchard J, Piatak M, Lifson JD, Fernandez-Romero JA, Robbiani M, Zydowsky TM. An Intravaginal Ring That Releases the NNRTI MIV-150 Reduces SHIV Transmission in Macaques. Sci Trans Med. 2012;4(150) doi: 10.1126/scitranslmed.3003936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veselinovic M, Preston Neff C, Mulder LR, Akkina R. Topical gel formulation of broadly neutralizing anti-HIV-1 monoclonal antibody VRC01 confers protection against HIV-1 vaginal challenge in a humanized mouse model. Virology. 2012;432(2):505–510. doi: 10.1016/j.virol.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veazey RS, Shattock RJ, Pope M, Kirijan JC, Jones J, Hu QX, Ketas T, Marx PA, Klasse PJ, Burton DR, Moore JP. Prevention of virus transmission to macaque monkeys by a vaginally applied monoclonal antibody to HIV-1 gp120. Nature Med. 2003;9(3):343–346. doi: 10.1038/nm833. [DOI] [PubMed] [Google Scholar]
- 14.Parren P, Marx PA, Hessell AJ, Luckay A, Harouse J, Cheng-Mayer C, Moore JP, Burton DR. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J Virology. 2001;75(17):8340–8347. doi: 10.1128/JVI.75.17.8340-8347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hessell AJ, Rakasz EG, Poignard P, Hangartner L, Landucci G, Forthal DN, Koff WC, Watkins DI, Burton DR. Broadly Neutralizing Human Anti-HIV Antibody 2G12 Is Effective in Protection against Mucosal SHIV Challenge Even at Low Serum Neutralizing Titers. Plos Pathogens. 2009;5(5) doi: 10.1371/journal.ppat.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hessell AJ, Rakasz EG, Tehrani DM, Huber M, Weisgrau KL, Landucci G, Forthal DN, Koff WC, Poignard P, Watkins DI, Burton DR. Broadly Neutralizing Monoclonal Antibodies 2F5 and 4E10 Directed against the Human Immunodeficiency Virus Type 1 gp41 Membrane-Proximal External Region Protect against Mucosal Challenge by Simian-Human Immunodeficiency Virus SHIVBa-L. J Virology. 2010;84(3):1302–1313. doi: 10.1128/JVI.01272-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mascola JR, Montefiori DC. The Role of Antibodies in HIV Vaccines. In Paul WE, Littman DR, Yokoyama WM, editors. Ann Rev Immun. 2010;28:413–444. doi: 10.1146/annurev-immunol-030409-101256. ed. [DOI] [PubMed] [Google Scholar]
- 18.Corti D, Langedijk JPM, Hinz A, Seaman MS, Vanzetta F, Fernandez-Rodriguez BM, Silacci C, Pinna D, Jarrossay D, Balla-Jhagjhoorsingh S, Willems B, Zekveld MJ, Dreja H, O'Sullivan E, Pade C, Orkin C, Jeffs SA, Montefiori DC, Davis D, Weissenhorn W, McKnight A, Heeney JL, Sallusto F, Sattentau QJ, Weiss RA, Lanzavecchia A. Analysis of Memory B Cell Responses and Isolation of Novel Monoclonal Antibodies with Neutralizing Breadth from HIV-1-Infected Individuals. Plos One. 2010;5(1) doi: 10.1371/journal.pone.0008805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Koff WC, Wilson IA, Burton DR, Poignard P, Protocol GPI. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477(7365):466–U117. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu XL, Yang ZY, Li YX, Hogerkorp CM, Schief WR, Seaman MS, Zhou TQ, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. Rational Design of Envelope Identifies Broadly Neutralizing Human Monoclonal Antibodies to HIV-1. Science. 2010;329(5993):856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Euler Z, Bunnik EM, Burger JA, Boeser-Nunnink BDM, Grijsen ML, Prins JM, Schuitemaker H. Activity of Broadly Neutralizing Antibodies, Including PG9, PG16, and VRC01, against Recently Transmitted Subtype B HIV-1 Variants from Early and Late in the Epidemic. J Virology. 2011;85(14):7236–7245. doi: 10.1128/JVI.00196-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li ZL, Palaniyandi S, Zeng RY, Tuo WB, Roopenian DC, Zhu XP. Transfer of IgG in the female genital tract by MHC class I-related neonatal Fc receptor (FcRn) confers protective immunity to vaginal infection. Proc Nat Acad Sci. 2011;108(11):4388–4393. doi: 10.1073/pnas.1012861108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malcolm R, Edwards K, Kiser P, Romano J, Smith T. Advances in microbicide vaginal rings. Antiviral Res. 2010;88(Suppl. 1):S30–39. doi: 10.1016/j.antiviral.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Morrow RJ, Woolfson AD, Donnelly L, Curran R, Andrews G, Katinger D, Malcolm RK. Sustained release of proteins from a modified vaginal ring device. Eur J Pharmaceut Biopharmaceut. 2011;77(1):3–10. doi: 10.1016/j.ejpb.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baum MM, Butkyavichene I, Gilman J, Kennedy S, Kopin E, Malone AM, Nguyen C, Smith TJ, Friend DR, Clark MR, Moss JA. An intravaginal ring for the simultaneous delivery of multiple drugs. J Pharm Sci. 2012;101(8):2833–2843. doi: 10.1002/jps.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moss JA, Malone AM, Smith TJ, Butkyavichene I, Cortez C, Gilman J, Kennedy S, Kopin E, Nguyen C, Sinha P, Hendry M, Guenthner P, Holder A, Martin A, McNicholl J, Mitchell J, Pau CP, Srinivasan P, Smith JM, Baum MM. Safety and pharmacokinetics of intravaginal rings delivering tenofovir in pig-tailed macaques. Antimicrob Agents Chemother. 2012;56(11):5952–5960. doi: 10.1128/AAC.01198-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moss JA, Baum MM, Malone AM, Kennedy S, Kopin E, Nguyen C, Gilman J, Butkyavichene I, Willis RA, Vincent KL, Motamedi M, Smith TJ. Tenofovir and tenofovir disoproxil fumarate pharmacokinetics from intravaginal rings. AIDS. 2012;26(6):707–710. doi: 10.1097/QAD.0b013e3283509abb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller M, Malone A, Carpenter C, Lo Y, Huang M, Corey L, Willis R, Nguyen C, Kennedy S, Gunawardana M, Guerrero D, Moss J, Baum M, Smith T, Herold B. Safety and Pharmacokinetics of Acyclovir in Women Following Release from a Silicone Elastomer Vaginal Ring. J Antimicrob Chemother. 2012;67(8):2005–2012. doi: 10.1093/jac/dks151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moss J, Malone A, Smith T, Kennedy S, Kopin E, Nguyen C, Gilman J, Butkyavichene I, Vincent K, Motamedi M, Friend D, Clark M, Baum M. Simultaneous Delivery of Tenofovir and Acyclovir via an Intravaginal Ring. Antimicrob Agents Chemother. 2012;56(2):875–882. doi: 10.1128/AAC.05662-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moss JA, Malone AM, Smith TJ, Kennedy S, Nguyen C, Vincent KL, Motamedi M, Baum MM. Pharmacokinetics of a mutipurpose pod-intravaginal ring simultaneously delivering five drugs in an ovine model. Antimicrob Agents Chemother. 2013;57(8):3994–3997. doi: 10.1128/AAC.00547-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owen DH, Katz DF. A Vaginal Fluid Simulant. Contraception. 1999;59:91–95. doi: 10.1016/s0010-7824(99)00010-4. [DOI] [PubMed] [Google Scholar]
- 32.Abramoff MD, Magalhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics Int. 2004;11(7):36–42. [Google Scholar]
- 33.Rasband WS. ImageJ [computer program] Bethesda, Maryland: U. S. National Institutes of Health; 1997-2011. [accessed Jul. 21, 2014]. http://imagej.nih.gov/ij/ [Google Scholar]
- 34.Malcolm RK, Fetherston SM, McCoy CF, Boyd P, Major I. Vaginal rings for delivery of HIV microbicides. Int J Womens Health. 2012;4:595. doi: 10.2147/IJWH.S36282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiser PF, Johnson TJ, Clark JT. State of the Art in Intravaginal Ring Technology for Topical Prophylaxis of HIV Infection. AIDS Rev. 2012;14(1):62–77. [PubMed] [Google Scholar]
- 36.Johnson TJ, Gupta KM, Fabian J, Albright TH, Kiser PF. Segmented polyurethane intravaginal rings for the sustained combined delivery of antiretroviral agents dapivirine and tenofovir. Eur J Pharmaceut Sci. 2010;39(4):203–212. doi: 10.1016/j.ejps.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 37.Mesquita PMM, Rastogi R, Segarra TJ, Teller RS, Torres NM, Huber AM, Kiser PF, Herold BC. Intravaginal ring delivery of tenofovir disoproxil fumarate for prevention of HIV and herpes simplex virus infection. J Antimicrob Chemother. 2012;67(7):1730–1738. doi: 10.1093/jac/dks097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Product Monograph - Viread. Gilead Sciences, Inc.; Foster City, CA: 2012. [accessed Jul. 21, 2014]. www.gilead.ca/pdf/ca/viread_pm_english.pdf. [Google Scholar]
- 39.Pattani A, Lowry D, Curran RM, McGrath S, Kett VL, Andrews GP, Malcolm RK. Characterisation of protein stability in rod-insert vaginal rings. Int J Pharmaceut. 2012;430(1-2):89–97. doi: 10.1016/j.ijpharm.2012.03.036. [DOI] [PubMed] [Google Scholar]
- 40.Perrie Y, Rades T. Pharmaceutics: Drug Delivery and Targeting. 2nd. London: Pharmaceutical Press; 2012. pp. 122–123. [Google Scholar]
- 41.Ma JKH, Hadzija B. Basic Physical Pharmacy. Burlington, MA: Jones & Bartlett Publishers; 2013. pp. 193–198. [Google Scholar]
- 42.Perrie Y, Rades T. Pharmaceutics: Drug Delivery and Targeting. 2nd. London: Pharmaceutical Press; 2012. pp. 101–106. [Google Scholar]
- 43.Chang LQ, Pikal MJ. Mechanisms of Protein Stabilization in the Solid State. J Pharm Sci. 2009;98(9):2886–2908. doi: 10.1002/jps.21825. [DOI] [PubMed] [Google Scholar]
- 44.Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci. 2007;96(1):1–26. doi: 10.1002/jps.20727. [DOI] [PubMed] [Google Scholar]
- 45.Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Stability of Protein Pharmaceuticals: An Update. Pharm Res. 2010;27(4):544–575. doi: 10.1007/s11095-009-0045-6. [DOI] [PubMed] [Google Scholar]
- 46.Daugherty AL, Mrsny RJ. Formulation and delivery issues for monoclonal antibody therapeutics. Adv Drug Deliv Rev. 2006;58(5-6):686–706. doi: 10.1016/j.addr.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 47.Vazquez-Rey M, Lang DA. Aggregates in Monoclonal Antibody Manufacturing Processes. Biotechnol Bioeng. 2011;108(7):1494–1508. doi: 10.1002/bit.23155. [DOI] [PubMed] [Google Scholar]
- 48.Breen ED, Curley JG, Overcashier DE, Hsu CC, Shire SJ. Effect of moisture on the stability of a lyophilized humanized monoclonal antibody formulation. Pharm Res. 2001;18(9):1345–1353. doi: 10.1023/a:1013054431517. [DOI] [PubMed] [Google Scholar]
- 49.Chang LQ, Shepherd D, Sun J, Tang XL, Pikal MJ. Effect of sorbitol and residual moisture on the stability of lyophilized antibodies: Implications for the mechanism of protein stabilization in the solid state. J Pharm Sci. 2005;94(7):1445–1455. doi: 10.1002/jps.20363. [DOI] [PubMed] [Google Scholar]
- 50.Abdul-Fattah AM, Kalcinia DS, Pikal MI. The challenge of drying method selection for protein pharmaceuticals: Product quality implications. J Pharm Sci. 2007;96(8):1886–1916. doi: 10.1002/jps.20842. [DOI] [PubMed] [Google Scholar]
- 51.Abdul-Fattah AM, Truong-Le V, Yee L, Nguyen L, Kalonia DS, Cicerone MT, Pikal MJ. Drying-induced variations in physico-chemical properties of amorphous pharmaceuticals and their impact on stability (I): Stability of a monoclonal antibody. J Pharm Sci. 2007;96(8):1983–2008. doi: 10.1002/jps.20859. [DOI] [PubMed] [Google Scholar]
- 52.Andya JD, Maa YF, Costantino HR, Nguyen PA, Dasovich N, Sweeney TD, Hsu CC, Shire SJ. The effect of formulation excipients on protein stability and aerosol performance of spray-dried powders of a recombinant humanized anti-IgE monoclonal antibody. Pharm Res. 1999;16(3):350–358. doi: 10.1023/a:1018805232453. [DOI] [PubMed] [Google Scholar]
- 53.Heljo VP, Filipe V, Romeijn S, Jiskoot W, Juppo AM. Stability of rituximab in freeze-dried formulations containing trehalose or melibiose under different relative humidity atmospheres. J Pharm Sci. 2013;102(2):401–414. doi: 10.1002/jps.23392. [DOI] [PubMed] [Google Scholar]
- 54.Remmele RL, Krishnan S, Callahan WJ. Development of Stable Lyophilized Protein Drug Products. Curr Pharmaceut Biotechnol. 2012;13(3):471–496. doi: 10.2174/138920112799361990. [DOI] [PubMed] [Google Scholar]
- 55.Schule S, Schulz-Fademrecht T, Garidel P, Bechtold-Peters K, Friess W. Stabilization of IgG1 in spray-dried powders for inhalation. Eur J Pharmaceut Biopharmaceut. 2008;69(3):793–807. doi: 10.1016/j.ejpb.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 56.Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci. 2004;93(6):1390–1402. doi: 10.1002/jps.20079. [DOI] [PubMed] [Google Scholar]
- 57.Warne NW. Development of high concentration protein biopharmaceuticals: The use of platform approaches in formulation development. Eur J Pharmaceutics Biopharmaceutics. 2011;78:208–212. doi: 10.1016/j.ejpb.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 58.Malcolm K, Woolfson D, Russell J, Tallon P, McAuley L, Craig D. Influence of silicone elastomer solubility and diffusivity on the in vitro release of drugs from intravaginal rings. J Controlled Release. 2003;90:217–225. doi: 10.1016/s0168-3659(03)00178-0. [DOI] [PubMed] [Google Scholar]
- 59.Woof JM, Mestecky J. Mucosal immunoglobulins. Immunol Rev. 2005;206:64–82. doi: 10.1111/j.0105-2896.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- 60.Phalipon A, Cardona A, Kraehenbuhl JP, Edelman L, Sansonett PJ, Corthesy B. Secretory component: a new role in secretory IgA-mediated immune exclusion. Immunity. 2002;17:107–115. doi: 10.1016/s1074-7613(02)00341-2. [DOI] [PubMed] [Google Scholar]