

Abstract
The identification of EGFR mutations and ALK rearrangements in nonsmall cell lung cancer (NSCLC) has led to the rapid development of targeted therapies and significant changes in the treatment paradigm. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) and crizotinib are now standard therapies for patients with the appropriate molecular alteration. Current investigations are determining the mechanisms of resistance to targeted therapies and developing novel agents to combat resistance. For patients with KRAS mutant NSCLC, a phase III trial of the MEK inhibitor, selumetinib, has been initiated. For patients without a defined mutation or a mutation without a known targeted therapy, immunotherapy, ganetespib, nintedanib and MET inhibitors in combination with EGFR TKIs are in development. Preliminary results of phase III trials raise doubts about the future development of dacomitinib as a second-line agent.
Keywords: anaplastic lymphoma kinase, dacomitinib, epidermal growth factor receptor, ganetespib, nintedanib, onartuzumab, targeted therapy, tivantinib
Introduction
Lung cancer remains the leading cause of cancer-related mortality in the United States and in the world [Jemal et al. 2011; Siegel et al. 2013]. The majority of the patients with lung cancer have the nonsmall cell (NSCLC) subtype and the majority of patients have advanced disease, defined as stage IIIB or IV, at the time of diagnosis [Govindan et al. 2006]. Under the previous staging system, American Joint Committee on Cancer (AJCC) TNM 6th edition, patients with malignant pleural and pericardial effusions were considered stage IIIB, often referred to as ‘wet IIIB,’ and were included in advanced stage trials [Greene et al. 2002]. Under the current staging system, AJCC TNM 7th edition, patients with malignant pleural or pericardial effusions are considered metastatic lesions (M1a) and patients with these conditions are considered as stage IV disease [Goldstraw et al. 2007]. In first-line cooperative group trials in the United States, the most common histology was adenocarcinoma (approximately 45–55% of the cases), followed by squamous histology (approximately 20–30% of the cases) and large cell histology (approximately 10–15% of cases) [Wakelee et al. 2006; Kelly et al. 2013]. Squamous histology is closely associated with tobacco use and the prevalence of squamous histology may vary depending on the prevalence of tobacco use [Kenfield et al. 2008].
The goals of treatment for patients with advanced stage disease are to improve overall survival (OS) and health-related quality of life (HRQOL), and to reduce disease-related symptoms. Historically, patients with advanced NSCLC were treated with a platinum-based doublet therapy without regard to histology. However, in a phase II trial of bevacizumab, a monoclonal antibody against the vascular endothelial growth factor (VEGF) A, a prohibitive rate of severe pulmonary hemorrhage was observed in patients with squamous histology [Johnson et al. 2004]. Consequently, patients with squamous histology were excluded from subsequent trials of bevacizumab. After the approval by the US Food and Drug Administration (FDA) of pemetrexed, analyses from phase III trials revealed the activity of pemetrexed is limited to patients with nonsquamous histology [Scagliotti et al. 2009]. Thus, patients with NSCLC are frequently divided into squamous and nonsquamous cohorts for treatment selection and drug development. An overview of the commonly used treatments for patients with nonsquamous and squamous stage IV disease with a good performance status is presented in Figures 1 and 2.
Figure 1.

Commonly used treatment paradigms for advanced stage non-small cell lung cancer for non-squamous histology.
A: Crizotinib is approved by the US Food and Drug Administration without regard to line of therapy.
B: Bevacizumab is a treatment option for patients without contraindication (e.g. hemoptysis, uncontrolled hypertension).
ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; TKI: tyrosine kinase inhibitor.
Figure 2.
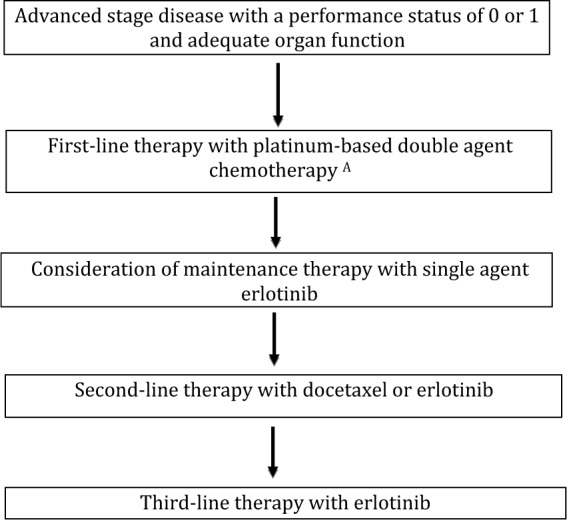
Commonly used therapies for advanced non-small cell lung cancer with squamous histology.
A: Pemetrexed and bevacizumab are not approved by the US Food and Drug Administration for use in patients with squamous histology non=small cell lung cancer.
The identification of EGFR mutations and ALK rearrangements in NSCLC has further subdivided patients with advanced NSCLC [Lynch et al. 2004; Paez et al. 2004; Soda et al. 2007]. In the United States, patients with a known EGFR mutation can be treated with an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) in the first-line setting, and crizotinib is approved by the US FDA for patients with an ALK rearrangement without regard to the line of therapy. It is estimated that 10–15% of all NSCLC harbor an EGFR mutation and that 3–5% harbor an ALK rearrangement [Soda et al. 2007; Sequist et al. 2008].
A frequent clinical question is which NSCLC tumors should be tested for these uncommon but clinically important molecular alterations. These alterations are more prevalent in younger patients, patients with adenocarcinoma histology, or a history of never or light smoking [Rosell et al. 2009; Shaw et al. 2009] In NSCLC with adenocarcinoma histology it is estimated that 5-10% of tumors have an ALK rearrangement and 10–20% have an EGFR mutation [Kris et al. 2011]. EGFR mutations have been detected in tumors from patients with a significant history of tobacco use, suggesting that the history of tobacco use is not sufficient to exclude patients from molecular testing [D′Angelo et al. 2011; Lindeman et al. 2013]. The current diagnostic standard is to test for EGFR and ALK molecular alterations in all nonsquamous tumors regardless of clinical characteristics [Lindeman et al. 2013].
The need for routine testing for EGFR mutations and ALK rearrangements for patients with squamous histology is debated, in part due to the low prevalence of these molecular alterations. The rate of EGFR mutations in patients with squamous histology is reported to be 1–15% [Chou et al. 2005; Kim et al. 2005; Pallis et al. 2007; Park et al. 2009; Miyamae et al. 2011]. One issue with basing the decision to perform molecular testing on histology is that there can be significant interobserver variability among pathologists in the classification of squamous and nonsquamous histology when hematoxylin–eosin slides are used [Grilley-Olson et al. 2013]. Given the clinical implications of the classification between squamous and nonsquamous histology, pathologists frequently use immunohistochemistry (IHC) to assist in the classification. A single IHC test is not sufficient to distinguish between squamous and nonsquamous histology. The expression of p63 and the N-terminal truncated p40 (ΔNp63) and the absence of thyroid transcription factor (TTF-1) expression are consistent with squamous histology [Bishop et al. 2012; Pelosi et al. 2012]. Poorly differentiated adenocarcinomas may express p63 and rarely squamous histology may have focal expression of TTF-1. Thus, the presence of p63 or the absence of TTF-1 is not sufficient to determine histology. The use of IHC improves the classification of NSCLC subtypes [Steinfort et al. 2012].
A retrospective study found the rate of EGFR and KRAS mutations in squamous cell carcinoma when IHC classification was used to identify histology was 0% [95% confidence interval (CI): 0–3.8%] [Rekhtman et al. 2012]. A review of previous cases of EGFR and KRAS mutations reported in patients with squamous histology revealed that the histology in the vast majority of the cases would be reclassified as poorly differentiated adenocarcinoma or adenosquamous with the addition of IHC staining to morphological assessment histology [Rekhtman et al. 2012]. These data suggest that, with better histological classification, the rate of EGFR and KRAS mutations observed in squamous histology tumors will be lower than the rate observed in previous series. A reasonable approach is to test patients with squamous histology for EGFR mutations and ALK rearrangements if the histological diagnosis is uncertain, if the histology is adenosquamous, or in a patient with squamous histology with a limited history of tobacco use.
Unfortunately, acquired resistance to targeted therapy generally develops within 1 to 2 years. In the general NSCLC patient population, patients who receive first-line chemotherapy experience disease progression within 6 months and patients receiving second-line therapy generally experience disease progression within 3 or 4 months. Thus, there is a significant need for novel agents, and increasingly novel agents are being developed in molecularly or histologically defined patient populations.
EGFR mutant NSCLC
For patients with a known EGFR activating mutation (exon 19 deletion and exon 21 L858R point mutation) treatment with an EGFR TKI (e.g. erlotinib, gefitinib or afatinib) is a standard first-line therapy [Keedy et al. 2011]. Multiple phase III trials have compared EGFR TKI therapy with platinum-based doublet chemotherapy as first-line therapy for patients with EGFR mutant NSCLC. These trials have consistently demonstrated an improvement in objective response rate (ORR), progression-free survival (PFS) and HRQOL with EGFR TKI compared with chemotherapy [Mok et al. 2009; Maemondo et al. 2010; Mitsudomi et al. 2010; Zhou et al. 2011; Rosell et al. 2012; Sequist et al. 2013; Yang et al. 2013a]. The ORR and PFS observed in EGFR TKI arms were approximately 60–80% and 10–15 months, respectively. A median OS of approximately 20–30 months was observed in both treatment arms. A high crossover rate from chemotherapy to EGFR TKI therapy is thought to be responsible for the similar OS. Once a patient experiences disease progression on EGFR TKI, the available treatment options are local radiation if the patient has an isolated site of progression and continuation of the EGFR TKI, continuation of the EGFR TKI in combination with chemotherapy, discontinuation of EGFR TKI and initiation of chemotherapy alone [Weickhardt et al. 2012; Goldberg et al. 2013; Ohashi et al. 2013; Yu et al. 2013a]. The optimal treatment paradigm is unclear and there are limited data available to base treatment decisions. Most physicians select treatment based on the individual patient since there is no defined standard therapy.
In several studies, a biopsy at the time of disease progression has been performed to characterize the molecular mechanisms of acquired resistance [Sequist et al. 2011b; Yu et al. 2013a]. One study of 37 paired pre- and post-EGFR TKI samples revealed that five patients (14%) had transformed to small cell lung cancer (SCLC), and the most commonly identified mechanisms of resistance were an acquired EGFR exon 20 T790M mutation in 18 patients (49%), MET amplification in 2 patients (5%), and PIK3CA mutations in 2 patients (5%) [Sequist et al. 2011b]. A larger study of 155 patients with acquired resistance on EGFR TKI therapy revealed that 98 patients had an acquired EGFR exon 20 T790M (63%), 4 had SCLC (3%), and MET amplification (5%); HER2 amplification was observed in 3 of 24 patients (13%) [Yu et al. 2013]. The development of BRAF mutations as a mechanism of acquired resistance has been observed as well [Ohashi et al. 2012]. Patients with the EGFR exon 20 T790M are resistant to EGFR TKI, but appear to have a more favorable prognosis and indolent disease course [Oxnard et al. 2011]. Collectively, these data suggest that multiple mechanisms are responsible for EGFR TKI resistance and there is value in performing a repeat biopsy at the time of disease progression, especially if conversion to SCLC is suspected.
In a retrospective analysis of patients receiving afatinib in 3 clinical trials, 14 patients were identified as having a de novo T790M mutation alone (n = 3) or combination with other mutations (n = 11) [Yang et al. 2013b]. The ORR, median PFS and median OS observed with afatinib in this patient population were 14.3% (n = 2), 2.9 months (95% CI: 0.3–13.8) and 14.9 months (95% CI: 1.5–30.5). These data suggest the single-agent activity of afatinib in NSCLC with a T790M mutation is low. A single-arm phase Ib trial investigated afatinib and cetuximab, a monoclonal antibody against the extracellular domain of EGFR, in patients who had experienced disease progression on erlotinib or gefitinib [Janjigian et al. 2012]. Patients were required to have an EGFR mutation or a response or stable disease for ≥6 months on prior EGFR TKI. Of the 100 patients enrolled, an EGFR T790M was detected in 53 tumor samples and not detected in 42 tumor samples. The primary grade ≥3 toxicities were rash (18%), diarrhea (7%) and fatigue (9%). The ORR was 40%, 94% of patients experienced disease control (defined as response or stable disease), and the median PFS was 4.7 months. The ORR in the patients with a T790M and without a T790M was 38% and 47%, respectively. A similar phase I/II trial of erlotinib and cetuximab did not demonstrate efficacy [Janjigian et al. 2011]. The combination of afatinib and cetuximab appears to have greater activity than single-agent afatinib in patients who develop progressive disease after an EGFR TKI therapy and warrants further investigation.
Another method of combating acquired resistance is the development of an EGFR TKI that is active against both the T790M mutation as well as the baseline activating EGFR mutations (Table 1). CO-1686 is active against the activating EGFR mutations and the T790M mutation, and has limited inhibition of EGFR wildtype which may reduce the rate of rash and diarrhea. CO-1686 is being investigated in an ongoing phase I/II trial in patients with EGFR mutant NSCLC who have experienced disease progression on an EGFR TKI [ClinicalTrials.gov identifier: NCT01526928]. The phase I portion of the trial revealed six of nine patients with an acquired T790M mutation experienced an objective response [Soria et al. 2013]. Patients have not experienced rash, and the most common toxicities (all grades) observed in 56 patients were nausea (20%), diarrhea (20%), fatigue (20%), vomiting (15%), and decreased appetite (10%). This trial is ongoing to determine the recommended dose for phase II trials and to determine the optimal formulation of the agent for further investigation. AZD9291 is an irreversible EGFR TKI with activity against activating EGFR mutations and the T790M mutation. AZD9291 is being investigated in a phase I trial [ClinicalTrials.gov identifier: NCT01802632]. Initial efficacy data are available from the dose escalation cohort and the expansion cohort for patients T790M mutations [Ranson et al. 2013]. The ORR in all patients was 46% and the response rate in patients T790M mutation-positive NSCLC was 58%. Only grade 1 or 2 rash and diarrhea have been observed in the multiple dose cohorts. Dose escalation continues to further define toxicity and to determine the recommended dose for phase II trials. Both these agents require further investigation but have demonstrated promise for patients who have acquired resistance to EGFR TKI.
Table 1.
Select ongoing phase II or III trials for patients with EGFR mutant NSCLC or ALK rearranged NSCLC.44
| Agent | Patient population | Phase | NCT number | Primary endpoint (s) |
|---|---|---|---|---|
| CO-1686 | EGFR mutation positive previously treated with EGFR TKI | I/II | 01526928 | Grade 3 or 4 adverse events, ORR |
| AZD9291 | EGFR mutation positive previously treated with EGFR TKI | I with expansion cohort | 01802632 | Safety and tolerability |
| LDK378 | ALK positive, crizotinib naïve | II | 01685138 | ORR |
| LDK378 | ALK positive, previously untreated | III (LDK378 versus platinum/pemetrexed) |
01828099 | PFS |
| CH5424802 | ALK positive, previously treated with crizotinib | II | 01871805 | ORR |
| Ganetespib | ALK positive, and have failed up to three therapies | II | 01562015 | ORR |
ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; NCT, National Clinical Trial; NSCLC, non-small cell lung cancer; ORR, objective response rate; PFS, progression-free survival; TKI, tyrosine kinase inhibitor.
NSCLC with an ALK rearrangement
Crizotinib is currently approved by the US FDA for patients whose tumors demonstrate an ALK rearrangement without regard to line of therapy. A phase III trial compared crizotinib with standard second-line chemotherapy (pemetrexed or docetaxel) in patients with NSCLC with an ALK rearrangement. Patients assigned to the crizotinib compared with the chemotherapy arm experienced a higher ORR (65% versus 20%, p < 0.001), longer PFS [hazard ratio (HR): 0.49, 95% CI: 0.37–0.64; p = 0.001; median 7.7 and 3.0 months, respectively), and a better HRQOL [Shaw et al. 2013]. The OS was similar in the crizotinib and chemotherapy arms (HR: 1.02, 95% CI: 0.68–1.54; p=0.54; median 20.3 and 22.8 months, respectively). The ORR and median PFS in the pemetrexed and docetaxel treated patients were 29% (95% CI: 21–39%), and 7% (95% CI: 2–16%), respectively, and 4.2 and 2.6 months, respectively. These data suggest a higher ORR in NSCLC with ALK rearrangements with pemetrexed than in unselected nonsquamous NSCLC. A phase III trial comparing crizotinib with carboplatin or cisplatin and pemetrexed in patients with NSCLC with an ALK rearrangement and nonsquamous histology has completed accrual [ClinicalTrials.gov identifier: NCT01154140]. This trial will provide data on the efficacy of crizotinib compared with standard double agent platinum-based therapy in the first-line setting. For patients who have isolated progression in the brain or oligometastatic progression, local therapy with radiation and continuing crizotinib may be a treatment option, and patients who have more indolent and asymptomatic disease progression continuing crizotinib beyond disease progression are reasonable options [Weickhardt et al. 2012; Ou et al. 2014].
In a case series of 18 patients who underwent biopsy after experiencing disease progression on crizotinib, 4 patients had a mutation within the ALK tyrosine kinase domain and an additional patient had amplification to ALK fusion gene [Katayama et al. 2012]. Other mechanisms of resistance identified include amplification of KIT and increased autophosphorylation of the EGFR, suggesting activation of the EGFR pathway as a mechanism of resistance. A similar case series of 11 patients identified secondary mutations in the tyrosine kinase domain in 4 patients, ALK copy number gain in 2 patients (1 patient also demonstrated an ALK resistance mutation), KRAS mutation in 2 patients (1 without the evidence of the original ALK rearrangement), 1 patient developed EGFR mutant NSCLC without evidence of a persistent ALK rearrangement, and 1 patient developed an ALK rearrangement negative NSCLC [Doebele et al. 2012]. The limited data available suggest multiple mechanisms are responsible for crizotinib resistance including ‘gate keeper mutations,’ copy number gain, and gain or loss of oncogenic driver mutations.
One concern with crizotinib is a relatively low penetration of the blood–brain barrier. In one study, 5 hours after taking crizotinib the crizotinib concentration in the cerebrospinal fluid (CSF) was 0.0014 µmol, and the CSF to plasma ratio was 0.0026 [Costa et al. 2011]. The CSF levels were significantly below the 50% growth inhibition of ALK rearranged cell lines. The pharmacokinetic data and clinical data raise concerns that the brain may be a ‘pharmacokinetic sanctuary’ and patients may experience intracranial disease progression while having extracranial disease control [Weickhardt et al. 2012; Camidge, 2013]. This pattern of disease progression is relevant as novel ALK inhibitors enter clinical trials.
Several ‘second generation’ ALK inhibitors are currently in development that may have potential therapeutic advantages compared with crizotinib (Table 1). LDK378 (now known as ceritinib) is a more potent and selective ALK inhibitor than crizotinib and was investigated in phase I trial of patients with malignancies that harbored ALK rearrangement. A total of 26 patients with ALK rearranged NSCLC who had experienced disease progression on crizotinib were treated with LDK378 at a dose of ≥400 mg daily and 21 patients responded (86%) including brain metastases responses [Mehra et al. 2012]. A second ALK inhibitor, CH5424802 (now known as alectinib), has activity against ALK rearranged cell lines with the gatekeeper mutation L1196M in cell lines and xenograft models [Sakamoto et al. 2011]. This agent was investigated in phase I/II trial and patients enrolled could not have received previous therapy with an ALK inhibitor. In the phase II portion, 43 of 46 patients (93.5%, 95% CI: 82.1–98.6%) experienced an ORR; the PFS was unavailable at the time of analysis [Seto et al. 2013]. In addition, AP26113 is a dual ALK/EGFR inhibitor with activity in cell lines with ALK rearrangement with gate keeper mutations [Camidge et al. 2013]. In a phase I study among patients with ALK rearranged NSCLC with previous therapy with crizotinib, 12 of the 16 (75%) patients responded and responses were observed in 4 of 5 (80%) patients with untreated or progressing brain metastases. All of these agents are early in development, but the preliminary evidence indicates activity in patients who have progressed on crizotinib, increased ALK selectivity and greater potency, and the potential for intracranial disease responses. Several agents that inhibit the heat-shock protein (HSP) 90 have also revealed single activity in patients with ALK rearrangement. Trials are ongoing with these agents (Table 1) [Sequist et al. 2010; Socinski et al. 2013].
KRAS mutant NSCLC
KRAS mutations are the most common mutation detected in NSCLC and are associated with a history of tobacco use and adenocarcinoma histology. The rate of KRAS mutations observed in patients with adenocarcinoma and squamous histology reported in a recent analysis were 34% and 6%, respectively [Shepherd et al. 2013]. The rate of KRAS mutations observed among former/current smokers and never smokers in a recent meta-analysis were 25% and 6%, respectively [Mao et al. 2010]. Unfortunately, a targeted therapy is not available for this patient population and the utility of routine clinical testing is debated [Roberts and Stinchcombe, 2013]. MEK1/MEK2 are two downstream kinases in the RAS-RAF-MEK-ERK pathway and inhibition of MEK is one strategy to block signaling [Janne et al. 2013]. Selumetinib is a MEK1/MEK2 inhibitor, and preclinical evidence revealed activity in KRAS mutant xenograft models and synergy with docetaxel. A randomized phase II trial investigated docetaxel (75 mg/m2 every 3 weeks) with and without selumetinib in patients with KRAS mutant NSCLC who had progressed after first-line therapy; the primary end-point was OS. Patients assigned to the docetaxel and selumetinib (n = 44) compared with the docetaxel arm (n = 43) experienced a statistically significant improvement in ORR (37% versus 0%, p < 0.0001) and PFS (HR: 0.58, 80% CI: 0.42–0.79; p = 0.014), and a numerically superior OS (HR: 0.80, 80% CI: 0.56–1.14; p = 0.21). The rate of grade 3 or 4 neutropenia observed in the docetaxel and selumetinib and docetaxel arms was 82% and 67%, respectively, and the rate of febrile neutropenia was 18% and 0%, respectively. The rate of any adverse event leading to hospitalization in the docetaxel and selumetinib and docetaxel alone arms was 48% and 19%, respectively. While the efficacy results of this phase II study are promising, the toxicity observed in the combination arm is concerning. A phase III trial of docetaxel with selumetinib or placebo as second-line therapy for patients with KRAS mutant NSCLC has been initiated [ClinicalTrials.gov identifier: NCT01933932]. The primary endpoint is PFS and patients in both treatment arms receive prophylactic pegylated granulocyte colony stimulating factor.
Immunotherapy
Historically NSCLC was not thought to be susceptible to immunotherapy, but several recent trials have challenged this perception. In order for tumors to develop, grow and metastasize, the malignant cells must evade the immune system. Malignant cells are able to avoid immune detection and destruction by modifying several immune ‘check points.’ Programmed death 1 (PD-1) is an immune check point receptor which is expressed on activated T cells and is part of the process of immunosuppression [Topalian et al. 2012]. PD-ligand-1 (PD-L1) and PD ligand-2 (PD-L2) are ligands which bind to the PD-1 receptor, and are expressed on some tumor cells and stromal cells [Topalian et al. 2012]. Blockade of the PD-1 receptor and PD-L1 ligand interaction leads to T-cell stimulation and can overcome tumor immune resistance.
A monoclonal antibody against PD-1, BMS-936558 (now known as nivolumab), was investigated in a phase I trial in patients with advanced cancer and multiple tumor types. The ORR observed in patients with advanced NSCLC was 18% (14 of 76 patients). To assess the role of intratumoral PD-L1 expression, IHC analysis was performed on pretreatment specimens from 42 patients. Among the 17 patients with PD-L1 negative tumors, no responses were observed; in contrast among the 25 patients with PD-L1 positive tumors, nine responses were observed (p = 0.006). Long-term follow up of the phase I trial revealed an ORR of 16% and a median OS of 9.6 months (95% CI: 7.4–13.7) [Brahmer et al. 2013]. Among patients with nonsquamous histology the ORR and median OS were 15% and 9.6 months (95% CI: 5.3–13.7), respectively, and among patients with squamous histology the ORR and median OS were 19% and 9.2 months (95% CI: 7.6 to not reached), respectively. The initial impression was that nivolumab was more active in squamous NSCLC, but with longer follow up and larger numbers it appears to have similar activity in both squamous and nonsquamous NSCLC. On long-term follow up, the most common grade 3 or 4 adverse events were fatigue, pneumonitis and elevated aspartate aminotransferase (2% each); two-drug related death due to pneumonitis were observed.
A similar trial of an anti-PD-L1 monoclonal antibody, BMS-936559, enrolled patients with advanced cancer including 75 patients with NSCLC [Brahmer et al. 2012]. Of the 49 patients with NSCLC evaluable for response, 5 patients experienced an objective response and 6 additional patients experienced stable disease for at least 24 weeks [Brahmer et al. 2012]. MPDL3280A, a monoclonal antibody that binds PD-L1, was investigated in a phase I study advanced NSCLC [Spigel et al. 2013]. At the time of analysis, 53 patients were enrolled and 37 patients were evaluable for response. The ORR was 24% (9 of 37 patients) and responses were observed in both squamous and nonsquamous histology. A preliminary analysis revealed that the ORR among patients with PD-L1 expression was 100% (4 of 4 patients) and the ORR among patients without PD-L1 expression was 15% (4 of 26 patients).
At this time, numerous trials investigating a variety of immunotherapy agents are ongoing in advanced NSCLC. The role of PD-L1 expression as a biomarker and the optimal method of testing for PD-L1 expression are areas of investigation. The data from available clinical trials are too limited and immature to determine if targeting PD-1 or PD-L1 will have greater efficacy and/or a lower toxicity.
Multi-targeted tyrosine kinases
Several phase II and III trials have investigated agents that target multiple tyrosine kinases; the majority of these agents inhibit angiogenesis through inhibition of the VEGF receptors [Scagliotti et al. 2010, 2012a, 2012b]. Nintedanib (BIBF1120) is a multi-targeted tyrosine kinase that inhibits VEGF receptors 1–3, fibroblast growth factor receptor 1–3 and platelet derived growth factor receptor. A phase III trial investigated docetaxel with nintedanib or placebo in patients who have experienced disease progression after first-line chemotherapy [Reck et al. 2013]. The primary endpoint was improvement in PFS by independent radiological review (IRC) and OS was the secondary endpoint. Patients assigned to the nintedanib arm compared with the placebo arm experienced a statistically significant improvement in PFS (HR: 0.79, 95% CI: 0.68–0.92; p=0.0019; median PFS of 3.4 and 2.7 months, respectively). The absolute difference in median PFS was modest. In the intent-to-treat patient (ITT) population, a significant difference in OS was not observed (HR: 0.94, 95% CI: 0.83–1.05; p = 0.2720). In a subset analysis of patients with adenocarcinoma histology (n = 568), patients assigned to the nintedanib compared with the placebo arm experienced a significantly longer OS (HR: 0.83, 95% CI: 0.70–0.99; p = 0.0359). In the squamous histology subset (n = 555), a statistically significant difference in OS was not observed (HR: 1.01, 95% CI: 0.85–1.21; p = 0.8907). The rate of drug-related grade ≥3 adverse events was higher in the nintedanib than the placebo arm (50.8% and 42%, respectively); the most common adverse events that were observed at a higher rate in the nintedanib arm were diarrhea, nausea and elevated liver function tests. The rate of grade ≥3 hemorrhage and hypertension were similar in the two treatment arms.
HSP 90 inhibitor
HSPs are part of a protein complex that forms a chaperone complex which regulates protein folding, stability and function [Shimamura and Shapiro, 2008]. Many of the client proteins are involved oncogenesis and inhibiting the HSP complex may successfully inhibit multiple oncogenic pathways [Socinski et al. 2013]. This class of agents has demonstrated activity in patients with NSCLC with an ALK rearrangement as discussed previously. Ganetespib, a HSP-90 inhibitor, has demonstrated single-agent activity in NSCLC, and preclinical data indicates synergy between chemotherapy and ganetespib [Socinski et al. 2013]. This agent was investigated in a randomized phase IIb trial in patients who had experienced disease progression after first-line therapy. The coprimary endpoints were PFS in patients with KRAS mutant NSCLC and patients with an elevated lactate dehydrogenase (LDH), and secondary endpoints were PFS and OS in patients with adenocarcinoma. After the trial was initiated, enrollment was restricted to adenocarcinoma histology due to concerns about pulmonary hemorrhage and a lack of efficacy in the nonadenocarcinoma histology cohort. In patients with adenocarcinoma histology (n = 252), patients assigned to the ganetespib containing compared with the docetaxel alone arm experienced a nonstatistically significant difference in PFS (HR: 0.83, 90% CI: 0.64–1.06; p = 0.108) and a statistically significant difference in OS (HR: 0.73, 90% CI: 0.55–0.98; p = 0.041). Patients assigned to the ganetespib compared with docetaxel alone experienced a numerically higher rate of diarrhea (48% versus 16%), fatigue (34% versus 24%), and grade 3 or 4 febrile neutropenia (11% versus 2%). When time since diagnosis of advanced disease was analyzed (>6 months versus ≤6 months from time of diagnosis), patients with a diagnosis of advanced disease >6 months appeared to benefit from ganetespib and patients with a diagnosis of advanced disease ≤6 months appeared to benefit from standard therapy (p-value for interaction = 0.0064). It is unclear if this observation is due to the multiple comparisons, a difference in the biology of NSCLC, or a difference in treatment effect; therefore, it should be interpreted with caution. A phase III trial is enrolling patients with adenocarcinoma with >6 months from diagnosis is comparing docetaxel/ganetespib with docetaxel [ClinicalTrials.gov identifier: NCT10798485].
Dacomitinib
Dacomitinib is an irreversible inhibitor of EGFR, HER2 and HER4. It was compared to erlotinib in a randomized phase II trial in patients with advanced NSCLC who had progressed on one or two lines of chemotherapy (n = 188) [Ramalingam et al. 2012]. Patients were not selected based on EGFR mutation status and 30 patients (16%) enrolled had EGFR mutant NSCLC. The primary endpoint was PFS and patients assigned to the dacomitinib compared with the erlotinib arm experienced a statistically significant improvement in PFS in the ITT population (HR: 0.66, 95% CI: 0.47–0.91; p = 0.012). Several subset analyses were performed for PFS based on EGFR and KRAS mutational status. Among patients with KRAS wildtype tumors/EGFR wildtype (n = 100), a statistically significant improvement in PFS was observed with dacomitinib compared with erlotinib (HR: 0.61, 95% CI: 0.37–0.99; p = 0.043) and among patients with KRAS wildtype tumors (HR: 0.55, 95% CI: 0.35–0.85; p = 0.006). Among patients with EGFR mutant NSCLC, a numerically longer PFS was observed in the patients assigned to dacomitinib compared with erlotinib (HR: 0.46, 95% CI: 0.18–1.18; p = 0.098). A phase III trial compared dacomitinib with erlotinib in the second-line setting; the coprimary endpoints were PFS by IRC in the ITT patients and in the KRAS wildtype patient populations [ClinicalTrials.gov identifier: NCT01360554] [Pfizer, 2012]. A phase III trial, BR.26, compared dacomitinib with placebo in patients who have progressed after first-line chemotherapy and erlotinib or gefitinib; the primary endpoint was OS [ClinicalTrials.gov identifier: NCT01000025]. Neither of these trials met the primary endpoints based on the preliminary press release and additional data be will presented in the future [Pfizer, 2014]. An ongoing phase III trial is comparing dacomitinib with gefitinib as first-line therapy for patients with EGFR mutant NSCLC; the primary endpoint is PFS by IRC [ClinicalTrials.gov identifier: NCT01774721].
MET inhibitors in combination with EGFR TKI
MET is a tyrosine kinase receptor that is directly involved in cell proliferation, survival and invasion, and is commonly dysregulated in malignant cells [Trusolino et al. 2010]. MET is activated by binding of hepatocyte growth factor (HGF) and onartuzumab is a monoclonal antibody that binds to the extracellular domain of MET and prevents HGF binding [Spigel et al. 2013a]. A randomized phase II trial investigated erlotinib with onartuzumab or placebo in patients who had progressed after one or two standard therapies (n = 137). A preplanned subset analysis was performed to assess the efficacy of onartuzumab in patients with tumors that demonstrated MET overexpression as assessed by IHC. In the MET-negative patients squamous histology was more common (42% versus 15%) and never-smokers were less common (5% versus 20%). Patients were not selected based on EGFR mutational status, and 13 patients with EGFR mutations were enrolled in the trial; 6 patients in the placebo arm and 7 patients in the onartuzumab arm
In the ITT patient population, there was no significant difference in PFS and OS. In the subset analysis patients with MET-positive tumors by IHC (n = 66), a statistically significant longer PFS (HR: 0.53, 95% CI: 0.283–0.99; p = 0.04) and OS (HR: 0.37, 95% CI: 0.19–0.72; p = 0.002) was observed with the addition of onartuzumab. Conversely among patients MET-negative tumors (n = 62) patients, a worse PFS (HR: 1.82, 95% CI: 0.99–3.32; p = 0.05) and OS (HR: 1.78, 95% CI: 0.79–3.99; p = 0.16) was observed with the addition of onartuzumab. The rates of rash, diarrhea, fatigue and nausea were similar in the two treatment arms. The rate of peripheral edema (all grades) was numerically higher in the onartuzumab than the placebo arm (23.2% and 7.5%, respectively). The ongoing phase III trial is comparing erlotinib with onartuzumab or placebo in MET-positive patients by IHC who have progressed on one or two lines of therapy [ClinicalTrials.gov identifier: NCT01456325].
Tivantinib is an oral MET inhibitor and a randomized phase II trial revealed an improvement in OS among patients with nonsquamous who were assigned to erlotinib and tivantinib compared with erlotinib and placebo [Sequist et al. 2011a]. Patients were not selected based on EGFR mutation status for the phase II trial and the majority of patients enrolled were EGFR wildtype (85%). A phase III trial of erlotinib and tivantinib or placebo in patients with nonsquamous histology was initiated. The trial was stopped after a planned interim analysis revealed that the trial would not meet the primary endpoint of improvement in OS (HR: 0.98, 95%CI: 0.84–1.15; p = 0.81) [Scagliotti et al. 2013]. The rates of rash, diarrhea and asthenia/fatigue were similar in the two treatment arms. The rate of grade 3 or 4 neutropenia was numerically higher in the erlotinib and tivantinib than the erlotinib and placebo arm (10% and 1.0%, respectively). Patients were not selected based on EGFR mutation status for the phase III trial and 109 patients with EGFR mutant NSCLC were enrolled. The OS in the EGFR mutant subgroup in the erlotinib and tivantinib and erlotinib and placebo arms was similar (HR: 0.72, 95% CI: 0.35–1.48).
A retrospective subset analysis of patients with high MET expression by IHC (n = 211) revealed patients assigned to the erlotinib and tivantinib compared with the erlotinib and placebo experienced statistically significant improvement in OS (HR: 0.70; p = 0.03). Patients with low MET expression by IHC (n = 234) assigned to the erlotinib and tivantinib and erlotinib placebo experienced similar OS (HR: 0.90; p = 0.53). The clinical characteristics (e.g. gender, histology, performance status, smoking history, rate of brain metastases) were similar of the high and low MET expression subgroups. Subsequent to the completion of the trial, cell-line data indicated that the cytotoxic activity of tivantinib was not based on MET inhibition alone but inhibition of microtubule assembly as well [Basilico et al. 2013; Katayama et al. 2013]. The future development of tivantinib in combination with EGFR TKI in NSCLC is unclear, and any potential development will most likely require selection of patients by MET expression.
Conclusion
The identification of EGFR mutations and ALK rearrangements has led to the rapid development of targeted therapies and changes in the treatment paradigms for these patient populations. Current investigations are focused on determining the mechanisms of resistance to targeted therapy and developing novel agents to combat mechanisms of resistance. KRAS mutations are the most common mutations in NSCLC, and a phase III trial investigating selumetinib, a MEK1/MEK2 inhibitor, in patients with KRAS mutant NSCLC has been initiated. For patients without a defined mutation or mutation without a known targeted therapy immunotherapy, ganetespib, nintedanib and MET inhibitors in combination with EGFR TKI therapy are second-line agents in development. The future development of dacomitinib in the second-line setting is in doubt based on the preliminary results of phase III trials.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare no conflict of interest in preparing this article.
References
- Basilico C., Pennacchietti S., Vigna E., Chiriaco C., Arena S., Bardelli A., et al. (2013) Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin Cancer Res 19: 2381–2392 [DOI] [PubMed] [Google Scholar]
- Bishop J., Teruya-Feldstein J., Westra W., Pelosi G., Travis W., Rekhtman N. (2012) P40 (Deltanp63) is superior to P63 for the diagnosis of pulmonary squamous cell carcinoma. Mod Pathol 25: 405–415 [DOI] [PubMed] [Google Scholar]
- Brahmer J., Horn L., Antonia S., Spigel D., Gandhi L., Sequist L., et al. (2013) Survival and long-term follow-up of the phase I trial of nivolumab (anti-PD-1; BMS-936558; ONO-4538) in patients (pts) with previously treated advanced non-small cell lung cancer (NSCLC). J Clin Oncol 31: abstract 8030. [Google Scholar]
- Brahmer J., Tykodi S., Chow L., Hwu W., Topalian S., Hwu P., et al. (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366: 2455–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camidge D. (2013) Taking aim at ALK across the blood-brain barrier. J Thorac Oncol 8: 389–390 [DOI] [PubMed] [Google Scholar]
- Camidge D., Bazhenova L., Salgia R., Weiss G., Langer C., Shaw A., et al. (2013) First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: updated results. J Clin Oncol 31: abstract 8031. [Google Scholar]
- Chou T., Chiu C., Li L., Hsiao C., Tzen C., Chang K., et al. (2005) Mutation in the tyrosine kinase domain of epidermal growth factor receptor is a predictive and prognostic factor for gefitinib treatment in patients with non-small cell lung cancer. Clin Cancer Res 11: 3750–3757 [DOI] [PubMed] [Google Scholar]
- Costa D., Kobayashi S., Pandya S., Yeo W., Shen Z., Tan W., et al. (2011) Csf Concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 29: e443–445 [DOI] [PubMed] [Google Scholar]
- D′Angelo S., Pietanza M., Johnson M., Riely G., Miller V., Sima C., et al. (2011) Incidence of EGFR exon 19 deletions and l858r in tumor specimens from men and cigarette smokers with lung adenocarcinomas. J Clin Oncol 29: 2066–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebele R., Pilling A., Aisner D., Kutateladze T., Le A., Weickhardt A., et al. (2012) Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 18: 1472–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg S., Oxnard G., Digumarthy S., Muzikansky A., Jackman D., Lennes I., et al. (2013) Chemotherapy with erlotinib or chemotherapy alone in advanced non-small cell lung cancer with acquired resistance to EGFR tyrosine kinase inhibitors. Oncologist 18: 1214–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstraw P., Crowley J., Chansky K., Giroux D., Groome P., Rami-Porta R., et al. (2007) The IASLC lung cancer staging project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM classification of malignant tumours. J Thorac Oncol 2: 706–714 [DOI] [PubMed] [Google Scholar]
- Govindan R., Page N., Morgensztern D., Read W., Tierney R., Vlahiotis A., et al. (2006) Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol 24: 4539–4544 [DOI] [PubMed] [Google Scholar]
- Greene F., Page D., Fleming I., Fritz A., Balch C., Haller D., et al. (2002) American Joint Committee on Cancer (AJCC) Cancer Staging Handbook. Springer: New York [Google Scholar]
- Grilley-Olson J., Hayes D., Moore D., Leslie K., Wilkerson M., Qaqish B., et al. (2013) Validation of interobserver agreement in lung cancer assessment: hematoxylin-eosin diagnostic reproducibility for non-small cell lung cancer: the 2004 World Health Organization classification and therapeutically relevant subsets. Arch Pathol Lab Med 137: 32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjigian Y., Azzoli C., Krug L., Pereira L., Rizvi N., Pietanza M., et al. (2011) Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin Cancer Res 17: 2521–2527 [DOI] [PubMed] [Google Scholar]
- Janjigian Y., Smit E., Horn L., Groen H., Camidge R., Gettinger S., et al. (2012) Activity of afatinib/cetuximab in patients (pts) with EGFR mutant non-small cell lung cancer (NSCLC) and acquired resistance (AR) to EGFR inhibitors. In: Proeedings of 37th European Society of Medical Oncology (ESMO) Congress, Vienna Austria September 28-October 2, 2012: abstract#1227O. [Google Scholar]
- Janne P., Shaw A., Pereira J., Jeannin G., Vansteenkiste J., Barrios C., et al. (2013) Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol 14: 38–47 [DOI] [PubMed] [Google Scholar]
- Jemal A., Bray F., Center M., Ferlay J., Ward E., Forman D. (2011) Global cancer statistics. CA Cancer J Clin 61: 69–90 [DOI] [PubMed] [Google Scholar]
- Johnson D., Fehrenbacher L., Novotny W., Herbst R., Nemunaitis J., Jablons D., et al. (2004) Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol 22: 2184–2191 [DOI] [PubMed] [Google Scholar]
- Katayama R., Aoyama A., Yamori T., Qi J., Oh-Hara T., Song Y., et al. (2013) Cytotoxic activity of tivantinib (ARQ 197) is not due solely to C-MET inhibition. Cancer Res 73: 3087–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama R., Shaw A., Khan T., Mino-Kenudson M., Solomon B., Halmos B., et al. (2012) Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med 4: 120ra117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy V., Temin S., Somerfield M., Beasley M., Johnson D., Mcshane L., et al. (2011) American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol 29: 2121–2127 [DOI] [PubMed] [Google Scholar]
- Kelly K., Chansky K., Mack P., Lara P., Jr., Hirsch F., Franklin W., et al. (2013) Chemotherapy outcomes by histologic subtypes of non-small-cell lung cancer: analysis of the southwest oncology group database for antimicrotubule-platinum therapy. Clin Lung Cancer 14: 627–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenfield S., Wei E., Stampfer M., Rosner B., Colditz G. (2008) Comparison of aspects of smoking among the four histological types of lung cancer. Tob Control 17: 198–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K., Jeong J., Kim Y., Na K., Kim Y., Ahn S., et al. (2005) Predictors of the response to gefitinib in refractory non-small cell lung cancer. Clin Cancer Res 11: 2244–2251 [DOI] [PubMed] [Google Scholar]
- Kris M., Johnson B., Kwiatkowski D., Iafrate A., Wistuba I., Aronson S., et al. (2011) Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: the NCI’s Lung Cancer Mutation Consortium (LCMC). J Clin Oncol 29(Suppl.): abstract CRA7506. [Google Scholar]
- Lindeman N., Cagle P., Beasley M., Chitale D., Dacic S., Giaccone G., et al. (2013) Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study Of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol 8: 823–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch T., Bell D., Sordella R., Gurubhagavatula S., Okimoto R., Brannigan B., et al. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350: 2129–2139 [DOI] [PubMed] [Google Scholar]
- Maemondo M., Inoue A., Kobayashi K., Sugawara S., Oizumi S., Isobe H., et al. (2010) Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 362: 2380–2388 [DOI] [PubMed] [Google Scholar]
- Mao C., Qiu L., Liao R., Du F., Ding H., Yang W., et al. (2010) KRAS mutations and resistance to EGFR-TKIS treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer 69: 272–278 [DOI] [PubMed] [Google Scholar]
- Mehra R., Camidge D., Sharma S., Felip E., Tan D., Vansteenkiste J., et al. (2012) First-in-human phase I study of the alk inhibitor LDK378 in advanced solid tumors. J Clin Oncol 30(Suppl.): abstract 3007. [Google Scholar]
- Mitsudomi T., Morita S., Yatabe Y., Negoro S., Okamoto I., Tsurutani J., et al. (2010) Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 11: 121–128 [DOI] [PubMed] [Google Scholar]
- Miyamae Y., Shimizu K., Hirato J., Araki T., Tanaka K., Ogawa H., et al. (2011) Significance of epidermal growth factor receptor gene mutations in squamous cell lung carcinoma. Oncol Rep 25: 921–928 [DOI] [PubMed] [Google Scholar]
- Mok T., Wu Y., Thongprasert S., Yang C., Chu D., Saijo N., et al. (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361: 947–957 [DOI] [PubMed] [Google Scholar]
- Ohashi K., Maruvka Y., Michor F., Pao W. (2013) Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol 31: 1070–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K., Sequist L., Arcila M., Moran T., Chmielecki J., Lin Y., et al. (2012) Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A 109: E2127–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou S., Janne P., Bartlett C., Tang Y., Kim D., Otterson G., et al. (2014) Clinical benefit of continuing ALK inhibition with crizotinib beyond initial disease progression in patients with advanced ALK-positive NSCLC. Ann Oncol 25: 415–422 [DOI] [PubMed] [Google Scholar]
- Oxnard G., Arcila M., Sima C., Riely G., Chmielecki J., Kris M., et al. (2011) Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 17: 1616–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez J., Janne P., Lee J., Tracy S., Greulich H., Gabriel S., et al. (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304: 1497–1500 [DOI] [PubMed] [Google Scholar]
- Pallis A., Voutsina A., Kalikaki A., Souglakos J., Briasoulis E., Murray S., et al. (2007) ‘Classical’ but not ‘other’ mutations of EGFR kinase domain are associated with clinical outcome in gefitinib-treated patients with non-small cell lung cancer. Br J Cancer 97: 1560–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S., Ha S., Lee J., Lee H., Sim H., Kim Y., et al. (2009) Epidermal growth factor receptor mutations and the clinical outcome in male smokers with squamous cell carcinoma of lung. J Korean Med Sci 24: 448–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelosi G., Fabbri A., Bianchi F., Maisonneuve P., Rossi G., Barbareschi M., et al. (2012) Deltanp63 (P40) and thyroid transcription factor-1 immunoreactivity on small biopsies or cellblocks for typing non-small cell lung cancer: a novel two-hit, sparing-material approach. J Thorac Oncol 7: 281–290 [DOI] [PubMed] [Google Scholar]
- Pfizer (2012) ARCHER 1009 Study (Advanced Research for Cancer targeted pan-HER therapy): Dacomitinib (PF-00299804), a pan-HER inhibitor, vs erlotinib in second- or third-line therapy for advanced non- small cell lung cancer (NSCLC). New York: Pfizer, Inc. Available at: http://www.pfizer.com/Files/News/Asco/Dacomitinib_Archer_1009_Backgrounder.Pdf (accessed 21 January 2014). [Google Scholar]
- Pfizer (2014) Pfizer announces top line results from two phase 3 trials of dacomitinib in patients with refractory advanced non small cell lung cancer. New York: Pfizer, Inc. Available at: http://www.pfizer.com/news/press-release/press-release-detail/pfizer_announces_top_line_results_from_two_phase_3_trials_of_dacomitinib_in_patients_with_refractory_advanced_non_small_cell_lung_cancer (accessed 27 January 2014). [Google Scholar]
- Ramalingam S., Blackhall F., Krzakowski M., Barrios C., Park K., Bover I., et al. (2012) Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 30: 3337–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson M., Pao W., Kim D., Kim S., Ohe Y., Felip E., et al. (2013) Preliminary results from a phase I study with AZD9291: an irreversible inhibitor of epidermal growth factor receptor (EGFR) activating and resistance mutations in non-small-cell lung cancer (NSCLC). Presidential Sessions in the European Journal of Cancer 49: abstract 33. [Google Scholar]
- Reck M., Kaiser R., Mellemgaard A., Douillard J., Orlov S., Krzakowski M., et al. (2013) Nintedanib (BIBF 1120) plus docetaxel in NSCLC patients progressing after first-line chemotherapy: LUME lung 1, a randomized, double-blind phase III trial. J Clin Oncol 21: abstract LBA8011. [Google Scholar]
- Rekhtman N., Paik P., Arcila M., Tafe L., Oxnard G., Moreira A., et al. (2012) Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations. Clin Cancer Res 18: 1167–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts P., Stinchcombe T. (2013) KRAS mutation: should we test for it, and does it matter? J Clin Oncol 31: 1112–1121 [DOI] [PubMed] [Google Scholar]
- Rosell R., Carcereny E., Gervais R., Vergnenegre A., Massuti B., Felip E., et al. (2012) Erlotinib versus standard chemotherapy as first-line treatment for european patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13: 239–246 [DOI] [PubMed] [Google Scholar]
- Rosell R., Moran T., Queralt C., Porta R., Cardenal F., Camps C., et al. (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361: 958–967 [DOI] [PubMed] [Google Scholar]
- Sakamoto H., Tsukaguchi T., Hiroshima S., Kodama T., Kobayashi T., Fukami T., et al. (2011) CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 19: 679–690 [DOI] [PubMed] [Google Scholar]
- Scagliotti G., Hanna N., Fossella F., Sugarman K., Blatter J., Peterson P., et al. (2009) The differential efficacy of pemetrexed according to NSCLC histology: a review of two phase III studies. Oncologist 14: 253-263 [DOI] [PubMed] [Google Scholar]
- Scagliotti G., Krzakowski M., Szczesna A., Strausz J., Makhson A., Reck M., et al. (2012a) Sunitinib plus erlotinib versus placebo plus erlotinib in patients with previously treated advanced non-small-cell lung cancer: a phase III trial. J Clin Oncol 30: 2070–2078 [DOI] [PubMed] [Google Scholar]
- Scagliotti G., Novello S., Ramlau R., Favaretto A., Barlesi F., Akerley W., et al. (2013) Marquee: a randomized, double-blind, placebo-controlled, phase 3 trial of tivantinib (ARQ 197) plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, non-squamous, non-small-cell lung cancer (NSCLC). In: Proceedings of the European Cancer Congress September 27-October 1, 2013: abstract 3410. [Google Scholar]
- Scagliotti G., Novello S., Von Pawel J., Reck M., Pereira J., Thomas M., et al. (2010) Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non-small-cell lung cancer. J Clin Oncol 28: 1835–1842 [DOI] [PubMed] [Google Scholar]
- Scagliotti G., Vynnychenko I., Park K., Ichinose Y., Kubota K., Blackhall F., et al. (2012b) International, randomized, placebo-controlled, double-blind phase III study of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous non-small-cell lung cancer: MONET1. J Clin Oncol 30: 2829–2836 [DOI] [PubMed] [Google Scholar]
- Sequist L., Gettinger S., Senzer N., Martins R., Janne P., Lilenbaum R., et al. (2010) Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol 28: 4953–4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist L., Martins R., Spigel D., Grunberg S., Spira A., Janne P., et al. (2008) First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol 26: 2442–2449 [DOI] [PubMed] [Google Scholar]
- Sequist L., Von Pawel J., Garmey E., Akerley W., Brugger W., Ferrari D., et al. (2011a) Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol 29: 3307–3315 [DOI] [PubMed] [Google Scholar]
- Sequist L., Waltman B., Dias-Santagata D., Digumarthy S., Turke A., Fidias P., et al. (2011b) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3: 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist L., Yang J., Yamamoto N., O′Byrne K., Hirsh V., Mok T., et al. (2013) Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 31: 3327–3334 [DOI] [PubMed] [Google Scholar]
- Seto T., Kiura K., Nishio M., Nakagawa K., Maemondo M., Inoue A., et al. (2013) CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. Lancet Oncol 14: 590–598 [DOI] [PubMed] [Google Scholar]
- Shaw A., Kim D., Nakagawa K., Seto T., Crino L., Ahn M., et al. (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 368: 2385–2394 [DOI] [PubMed] [Google Scholar]
- Shaw A., Yeap B., Mino-Kenudson M., Digumarthy S., Costa D., Heist R., et al. (2009) Clinical features and outcome of patients with non-small-cell lung cancer who harbor EMl4-ALK. J Clin Oncol 27: 4247–4253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd F., Domerg C., Hainaut P., Janne P., Pignon J., Graziano S., et al. (2013) Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol 31: 2173–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T., Shapiro G. (2008) Heat shock protein 90 inhibition in lung cancer. J Thorac Oncol 3: S152–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R., Naishadham D., Jemal A. (2013) Cancer statistics, 2013. CA Cancer J Clin 63: 11–30 [DOI] [PubMed] [Google Scholar]
- Socinski M., Goldman J., El-Hariry I., Koczywas M., Vukovic V., Horn L., et al. (2013) A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res 19: 3068–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda M., Choi Y., Enomoto M., Takada S., Yamashita Y., Ishikawa S., et al. (2007) Identification of the transforming eml4-alk fusion gene in non-small-cell lung cancer. Nature 448: 561–566 [DOI] [PubMed] [Google Scholar]
- Soria J., Sequist L., Gadgeel S., Goldman J., Wakelee H., Varga A., et al. (2013) First-in-human evaluation of CO-1686, an irreversible, highly selective tyrosine kinase inhibitor of mutations of EGFR (activating and T790m). In: Proceedings of ISALC:15th World Conference on Lung Cancer, October 27-30, 2013: abstract 1354. [Google Scholar]
- Spigel D., Ervin T., Ramlau R., Daniel D., Goldschmidt J., Jr., Blumenschein G., Jr., et al. (2013a) Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 31: 4105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spigel D., Gettinger S., Horn L., Herbst R., Gandhi L., Gordon M., et al. (2013b) Clinical activity, safety, and biomarkers of mpdl3280A, an engineered PD-l1 antibody in patients with locally advanced or metastatic non-small cell lung cancer (Nsclc). J Clin Oncol 31: abstract 8008. [Google Scholar]
- Steinfort D., Russell P., Tsui A., White G., Wright G., Irving L. (2012) Interobserver agreement in determining non-small cell lung cancer subtype in specimens acquired by EBUS-TBNA. Eur Respir J 40: 699–705 [DOI] [PubMed] [Google Scholar]
- Topalian S., Hodi F., Brahmer J., Gettinger S., Smith D., Mcdermott D., et al. (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366: 2443–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusolino L., Bertotti A., Comoglio P. (2010) MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 11: 834–848 [DOI] [PubMed] [Google Scholar]
- Wakelee H., Bernardo P., Johnson D., Schiller J. (2006) Changes in the natural history of nonsmall cell lung cancer (NSCLC) –-comparison of outcomes and characteristics in patients with advanced NSCLC entered in Eastern Cooperative Oncology Group Trials before and after 1990. Cancer 106: 2208–2217 [DOI] [PubMed] [Google Scholar]
- Weickhardt A., Scheier B., Burke J., Gan G., Lu X., Bunn P., Jr., et al. (2012) Local ablative therapy of oligoprogressive disease prolongs disease control by tyrosine kinase inhibitors in oncogene-addicted non-small-cell lung cancer. J Thorac Oncol 7: 1807–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Hirsh V., Schuler M., Yamamoto N., O′Byrne K., Mok T., et al. (2013a) Symptom control and quality of life in LUX-lung 3: a phase III study of afatinib or cisplatin/pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations. J Clin Oncol 31: 3342–3350 [DOI] [PubMed] [Google Scholar]
- Yang J., Sequist L., Geater S., Tsai C., Mok T., Schuler M., et al. (2013b) Activity of afatinib in uncommon epidermal growth factor receptor (EGFR) mutations: findings from three prospective trials of afatinibin EGFR mutation-positive lung cancer. Abstract number O03.05. In: Proceedings of ISALC: 15th World Conference on Lung Cancer October 27-30: [Google Scholar]
- Yu H., Arcila M., Rekhtman N., Sima C., Zakowski M., Pao W., et al. (2013a) Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 19: 2240–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Sima C., Huang J., Solomon S., Rimner A., Paik P., et al. (2013a) Local therapy with continued EGFR tyrosine kinase inhibitor therapy as a treatment strategy in EGFR-mutant advanced lung cancers that have developed acquired resistance to EGFR tyrosine kinase inhibitors. J Thorac Oncol 8: 346–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Wu Y., Chen G., Feng J., Liu X., Wang C., et al. (2011) Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (optimal, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 12: 735–742 [DOI] [PubMed] [Google Scholar]