

SUMMARY
The P7C3 class of neuroprotective aminopropyl carbazoles has been shown to block neuronal cell death in models of neurodegeneration. We now show that P7C3 molecules additionally preserve axonal integrity after injury, before neuronal cell death occurs, in a rodent model of blast-mediated traumatic brain injury (TBI). This protective quality may be linked to the ability of P7C3 molecules to activate nicotinamide phosphoribosyltransferase, the rate-limiting enzyme in nicotinamide adenine dinucleotide salvage. Initiation of daily treatment with our recently reported lead agent, P7C3-S243, one day after blast-mediated TBI blocks axonal degeneration and preserves normal synaptic activity, learning and memory, and motor coordination in mice. We additionally report persistent neurologic deficits and acquisition of an anxiety-like phenotype in untreated animals eight months after blast exposure. Optimized variants of P7C3 thus offer hope for identifying neuroprotective agents for conditions involving axonal damage, neuronal cell death, or both, such as occurs in TBI.
INTRODUCTION
Traumatic brain injury (TBI) has emerged as the signature injury of military conflict, estimated to affect 20% of the 2.3 million servicemen and women deployed since 2001. Blast exposure from explosive devices affects soldiers and civilians around the globe, placing them at increased risk for TBI associated with long-term neurologic complications, including cognitive and motor decline, acquisition of psychiatric symptoms, and neuropathological features similar to Alzheimer’s disease (Hoge et al., 2008; Wolf et al., 2009; Shively et al., 2012; Goldstein et al., 2012). While the mechanisms of injury from exposure to the blast-generated shockwave are incompletely understood, the associated sheer forces are known to lead to widespread, diffuse and progressive axonal injury (Nakagawa et al., 2011; Magnuson et al., 2012). Unfortunately, just as there are currently no pharmacologic agents that arrest neuron cell death in any of the wide spectrum of neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease or amyotrophic lateral sclerosis, there are also currently no treatment options for patients with TBI beyond supportive and rehabilitative care.
The lack of pharmacologic strategies to block neuron cell death relates to the failure of target-directed drug discovery programs to develop neuroprotective therapeutics. Phenotypic screening, by contrast, offers the opportunity for discovery of new compounds with a desired biologic effect without bias concerning mechanism (Pieper et al., 2014). With this in mind, we previously implemented an in vivo phenotypic screen in living mice to identify new small drug-like molecules that safely increased the net magnitude of postnatal hippocampal neurogenesis. We designed the screen to capture agents that would either enhance proliferation or block death of newborn hippocampal neural precursor cells (Pieper et al., 2010), and identified an aminopropyl carbazole, named ‘P7C3,’ that was fortuitously endowed with favorable pharmacokinetic properties (Pieper et al., 2010). It was subsequently demonstrated that P7C3 achieved its proneurogenic effect by virtue of blocking death of neural precursor cells. We later demonstrated additional protective benefit of P7C3 molecules, specifically the highly active analog P7C3-A20, in blocking neuronal cell death and improving neurologic outcomes in a variety of rodent models of neurodegenerative disease and injury, including those involving death of mature neurons in other regions of the central nervous system (MacMillan et al., 2010; Naidoo and De Jesús-Cortés et al., 2013; Walker et al., 2014; De Jesus-Cortes et al., 2012; Tesla et al., 2012; Blaya et al., 2014).
Recently, we reported the synthesis and characterization of an optimized member of the P7C3 series, (−)-P7C3-S243, which exhibits improved polarity and lacks the aniline moiety present in other members of the P7C3 series. This analog is readily available as a single enantiomer with selective neuroprotective activity (Naidoo and De Jesús-Cortés et al., 2014). Because blast exposure in rodents results in a phenotype that recapitulates the consequences of blast-mediated TBI experienced by humans (Goldstein et al., 2012; Mohan et al., 2013), this animal model is suitable for evaluating the efficacy of neuroprotective agents. Here, we describe protective efficacy of P7C3-S243 in the mouse model of blast-induced TBI, which has also led to the discovery of a new protective quality of the P7C3-class – the ability to block axonal degeneration preceding neuronal cell death.
RESULTS
To test the neuroprotective efficacy of P7C3-A20 and P7C3-S243, we employed a model of blast-induced TBI in which the blast wave is propagated following rupture of a mylar membrane (Mohan et al., 2013). Briefly, anesthetized mice are placed in an enclosed blast chamber constructed from an air tank partitioned into two sides. A sealed mylar membrane covers a small port between the two halves of the tank. When the pressure is increased in the side without the mouse, the membrane ruptures at ≈22 kilopascal (kPa), generating a blast wave that travels through the mouse’s head. The head of the mouse is untethered and located in a padded holder, while the body is shielded from the blast by a metal tube. As previously reported (Mohan et al., 2013), the intensity of the blast wave in our system is 149.8 ± 2.09 kPa, and the duration of the total pressure (Blast wave + wind gust) is ≈10–15 milliseconds.
We first evaluated hippocampal-dependent memory in the Barnes maze task after blast-injury, with immediate administration of test compounds. The Barnes maze consists of a circular table with holes equally spaced around the perimeter. One of these holes contains an escape cup, such that the mouse can enter the hole and hide in the cup to avoid exposure on the table. Testing in the Barnes maze was initiated 7 days after blast-injury. To begin, mice underwent 4 days of training, during which time they were allowed to find and enter the escape hole, and rest in the protective cup. By the 4th day of training, mice typically learn how to rapidly locate the escape hole, based on visual cues permanently positioned around the table throughout training. After 4 days of training, the cup is removed and the ability of the mouse to remember where the cup was previously located is assessed in the ‘probe test.’ Memory is reflected by measuring the amount of time the mouse spends in the area around where the cup was previously located. Sham-injured animals that were intraperitoneally administered daily vehicle, P7C3-A20, or P7C3-S243 spent 55–60% of their time in the quadrant that contained the escape hole, known as the ‘escape quadrant,’ during the probe test (Figure S1A), indicating that normal memory is not affected by P7C3 compounds in this task. By contrast, blast-injured animals administered vehicle were notably impaired, spending only ≈25% of their time in the escape quadrant, which would be expected by chance alone (Figure S1A). Daily treatment with P7C3-A20 or P7C3-S243 immediately after injury, however, restored the time spent in the escape quadrant to normal levels seen in uninjured mice (Figure S1A).
Delayed Initiation of Treatment with P7C3-S243 Preserves Learning and Memory after Blast-Mediated TBI
On the basis of the promising results seen with administration of P7C3 molecules immediately after injury, we sought to determine whether P7C3-S243 could offer protective efficacy when treatment was initiated later. Because TBI can disrupt the blood-brain barrier (BBB), we first examined BBB integrity over time by means of the Evans blue assay. Evans blue is an azo dye with tight affinity for albumin, such that in serum virtually all Evans blue is albumin-bound. Normally, serum albumin cannot cross the BBB. However, when the BBB is compromised, albumin and its bound Evans blue can enter the CNS. The magnitude of Evans blue accumulation in the brain can then be spectrophotometrically determined in order to monitor disruption of the BBB (Uyama et al., 1988; Hawkins and Egleton, 2006). With this technique, we observed significant disruption of the BBB 6 hours after blast-injury, with integrity of the BBB recovering to normal 18 hours later (Figure S1B). BBB integrity remained intact 102 hours after injury, and 3 days of daily treatment with P7C3-S243 did not compromise BBB integrity in sham-injured mice.
We next tested whether P7C3-S243 could preserve function when treatment was initiated 24 hours after blast-injury, when the BBB was intact. A schematic of this experimental design is illustrated in Figure S1C, and we have previously reported that P7C3-S243 readily enters the brains of mice with intact BBBs (Naidoo and De Jesús-Cortés et al., 2014). We reasoned that this time point for starting treatment represented an important milestone, as most patients would be expected to access treatment within 24 hours of their injury. Every group consisted of 25 male C57/Bl6 wild type mice, aged 12–14 weeks, and data was collected and analyzed in an automated manner blind to treatment group. The most stringent measure of performance in the Barnes maze probe test is percent time in the escape area, defined as a 5 cm radius surrounding the escape hole. Sham-injured animals treated with vehicle or 10 mg/kg/day P7C3-S243 spent ≈39–43% of their time in the escape area, as opposed to only ≈15% for injured animals treated with vehicle (Figure 1A). Daily treatment with P7C3-S243 initiated 24 hours after injury dose-dependently preserved performance in this measure, with 3, 10 and 30 mg/kg/day doses showing complete protection, and 1 mg/kg/day partial, though not statistically significant, protection (Figure 1A). Administration of the active enantiomer (−)-P7C3-S243 at 3 mg/kg/day showed complete protection, while the same dose of the less active enantiomer, (+)-P7C3-S243, showed no efficacy (Figure 1A).
Figure 1. P7C3-S243 Preserves Memory after Blast-Mediated TBI.

(A) Daily IP administration of P7C3-S243 for 11 days in divided daily doses for the total amount indicated dose-dependently preserved memory in the Barnes maze probe test in blast-injured mice, as measured by the most stringent measure of percent time in escape area (5 cm radius around the escape hole). Treatment with an intermediate dose (3 mg/kg/d) of the active (−)-P7C3-S243 enantiomer preserved normal performance to the level displayed by sham-injured mice. By contrast, mice treated with the same dose of the less active (+)-P7C3-S243 enantiomer showed the same deficit as injured mice that were treated with vehicle. (B) Daily administration of P7C3-S243 was initiated at later time periods after injury to define the window of therapeutic efficacy. Whereas both 3 and 30 mg/kg/d doses preserved normal function when treatment was initiated 24 hours after injury, only the 30 mg/kg/d dose was efficacious when treatment was initiated at 36 hours. When treatment was initiated 48 hours after injury, no protective efficacy was noted at any dose. (C) Oral administration of the highly active (−)-P7C3-S243 enantiomer preserved normal hippocampal dependent memory at 3, 10 and 30 mg/kg/day doses. Every group shown consisted of 25 male C57/Bl6 mice, aged 12–14 weeks, and data was collected and scored in an automated manner blind to treatment group. Data are represented as mean ± SEM. Significance was determined by two way ANOVA with Bonferroni post-hoc analysis. p-value labeled as *<0.05, **<0.01, ***<0.001, and ****<0.0001 compared to blast-injured animals treated with vehicle. See also Figure S1 and S2.
Next, we sought to define the time window of treatment efficacy by initiating daily IP administration of P7C3-S243 at later points after injury. Whereas both 3 and 30 mg/kg/d doses preserved normal function when treatment was initiated 24 hours after injury, only the 30 mg/kg/d dose was partially efficacious when initiated 36 hours after injury (Figure 1B). When initiated 48 hours after injury, no protective efficacy was noted at any dose (Figure 1B). Thus, acute memory deficits after blast-mediated TBI can be effectively mitigated when treatment with P7C3-S243 is initiated within 36 hours. Finally, we tested whether oral administration of the highly active (−)-P7C3-S243 enantiomer could achieve protective effect. Indeed, initiation of daily oral administration of (−)-P7C3-S243 at 24 hours after injury preserved memory at 3, 10 and 30 mg/kg/day doses (Figure 1C).
In addition to measuring time spent in the 5 cm radius around the escape hole in the probe test, we also tested other commonly used measures of memory. Specifically, we determined the percent of nose pokes into the correct hole, defined as ‘target entry’, and the percent of time spent in the quadrant of the maze containing the escape hole, defined as ‘percent quadrant time.’ Monitoring of target entry in the treatment groups showed the same protective effects as with the escape area measure (Figure S2A–C). Similar findings were also obtained with the metric of % quadrant time, with the exception that 3 mg/kg/day of the less active (+)-P7C3-S243 enantiomer achieved some measure of protective efficacy at the margin of statistical significance (Figure S2D–F). This modest effect indicates that low-level activity may reside in (+)-P7C3-S243.
As controls in the Barnes maze, we examined the ability of animals to physically participate in the task, as well as to learn the task over the 4 day training period. Physical participation was defined as speed and distance traveled, and none of the groups differed significantly in these measures (Figure S2G–L). Learning was assessed as the percent latency to escape, defined as the percentage of time the mouse took to enter the escape hole on day 4 out of the time originally required on day 1. All animals were able to learn, though mice subjected to blast-mediated TBI and then treated with vehicle, 0.3 mg/kg/day P7C3-S243, or 3 mg/kg/day of the less active enantiomer (+)-P7C3-S243 learned significantly worse than sham-injury mice (Figure S2M). When treatment with 1, 3, 10 or 30 mg/kg/day P7C3-S243, or 3 mg/kg/day (−)-P7C3-S243, was initiated 24 hours after injury, however, mice learned the task as well as sham-injury mice (Figure S2M). Notably, the 1 mg/kg/day dose failed to improve memory in the probe test (Figure 1A), yet still facilitated learning during the training period.
Initiation of administration of 3 and 30 mg/kg/day doses of P7C3-S243 at 36 hours after injury improved learning (Figure S2N), even though the 3 mg/kg/day dose did not improve memory in the probe test (Figure 1B). Similarly, 30 mg/kg/day P7C3-S243 restored normal learning when treatment was initiated 48 hours after injury (Figure S7C), despite having no effect on memory in the probe test (Figure 1C). Finally, oral administration of 1 mg/kg/day (−)-P7C3-S243, which did not preserve memory in the probe test (Figure 1C), also restored normal learning (Figure S2O). Taken together, it can be concluded that treatment with P7C3-S243 mitigates acute neurocognitive deficits after blast-mediated TBI, and improves learning during the training period of the Barnes maze at doses lower than those required for the more challenging task of preserving memory.
To ensure that the improvement observed in mice treated with P7C3-S243 in this assay of learning and memory did not simply reflect an acceleration of what would otherwise be a normal recovery response after injury, we compared performance of sham- vs blast-injured mice eight months after a single blast injury. As shown in figure S2S-U, equivalent deficits in learning and all three measures of memory in the Barnes maze are noted in untreated animals at this chronic time point. Future experiments in our laboratory will determine whether P7C3-S243 or related agents offer protection from the chronic consequences of blast-mediated TBI as well.
P7C3-S243 Preserves Hippocampal Synaptic Function after Blast-Mediated TBI
To test our hypothesis that preserved performance in the Barnes maze was associated with hippocampal function, we investigated synaptic plasticity in the hippocampus. Specifically, we measured both long-term potentiation (LTP) and paired pulse facilitation (PPF) in brain slices derived from mice exposed to blast-injury as a function of treatment with different doses of P7C3-S243 for 9 days, initiated 24 hours after injury, in animals not subjected to behavioral testing. The stimulating electrode activated CA3 Schaeffer collateral axons, and the recording electrode was placed in the stratum radiatum of the CA1 region, where Schaffer collateral axons synapse with dendrites from CA1 pyramidal cells. As shown in Figure 2, blast-injury induced significant deficits in both measures, and low dose (0.3 mg/kg/day) P7C3-S243 had no effect. This dose also did not preserve memory in the Barnes maze probe test (Figure 1). However, treatment with higher doses (3 and 30 mg/kg/day) of P7C3-S243, which did preserve normal memory after injury (Figure 1), preserved both LTP and PPF in the hippocampus (Figure 2). Thus, dose-dependent protective efficacy of P7C3-S243 in behavioral tasks of learning and memory correlated with hippocampal function.
Figure 2. P7C3-S243 Preserves Hippocampal Synaptic Transmission after Blast-Mediated TBI.

Treatment with P7C3-S243 rescued blast-injury-induced deficits in long-term potentiation (LTP) and paired-pulse facilitation (PPF) in the hippocampal CA1 Schaffer-collateral pathway. (A) LTP induced by 12 theta burst stimulation (TBS) is significantly decreased in animals that sustained blast-induced TBI 14 days prior to testing. (B) This deficit was not rescued by treatment with low dose P7C3-S243 (0.3 mg/kg/d) starting 24 hours after injury, but was rescued by treatment with higher doses of (C) 3 and (D) 30 mg/kg/d P7C3-S243. LTP 1 hour after 12 TBS is summarized by quantification of the initial slope in panel (E). (F) Blast-injury-induced PPF deficit of 50 ms inter-pulse interval was also rescued in animals treated with the two higher doses (3 and 30 mg/kg/d) of P7C3-S243. Data are represented as mean ± SEM. Statistics were determined by one-way ANOVA with Tukey’s post hoc test.
P7C3-S243 Blocks Axonal Degeneration after Blast-Mediated TBI
Histologic examination of brain tissue 12 days after blast-injury revealed prominent axonal degeneration in the absence of cell death or acute inflammation. As shown in Figure 3A, silver staining of degenerating axons was markedly increased in the CA1 stratum radiatum, without loss of cell bodies in the dentate gyrus. Automated optical densitometry of silver-stained tissue was used to quantify the magnitude of staining, such that greater impedance of light through the section reflected greater axonal degeneration (Figure 3B). As this is the first time we have reported this automated and unbiased objective method of quantification, we verified that the results correlated with a previously established method of quantifying silver staining through manually delineating the area of interest and determining optical density of silver staining on the same section through NIH Image J software (Northington et al., 2001). Figure S3A shows that our new automated method correlated perfectly with this previously established method. Our analysis showed that degeneration of axons was blocked by treatment with P7C3-S243 at doses of 3 and 30 mg/kg/day when initiated 24 hours after injury. Treatment with 3 mg/kg/day of (−)-P7C3-S243 also offered significant protection from axonal degeneration, whereas the same dose of the less active enantiomer (+)-P7C3-S243 did not. Similar protective efficacy of 3 and 30 mg/kg/day of P7C3-S243 was also observed outside the hippocampus, including corpus callosum, thalamus, cortex, olfactory bulb, striatum and cerebellum (Figure S3B–D). Again, axonal degeneration in these regions preceded frank neuronal cell death (Figure S3E–F) or death of other cell types (Figure S3G–H). P7C3-S243 is likely acting directly on neurons to block axonal degeneration, as we have previously shown that P7C3 does not affect oligodendrocytes (Pieper et al., 2010).
Figure 3. P7C3-S243 Blocks Axonal Degeneration after Blast-Mediated TBI.
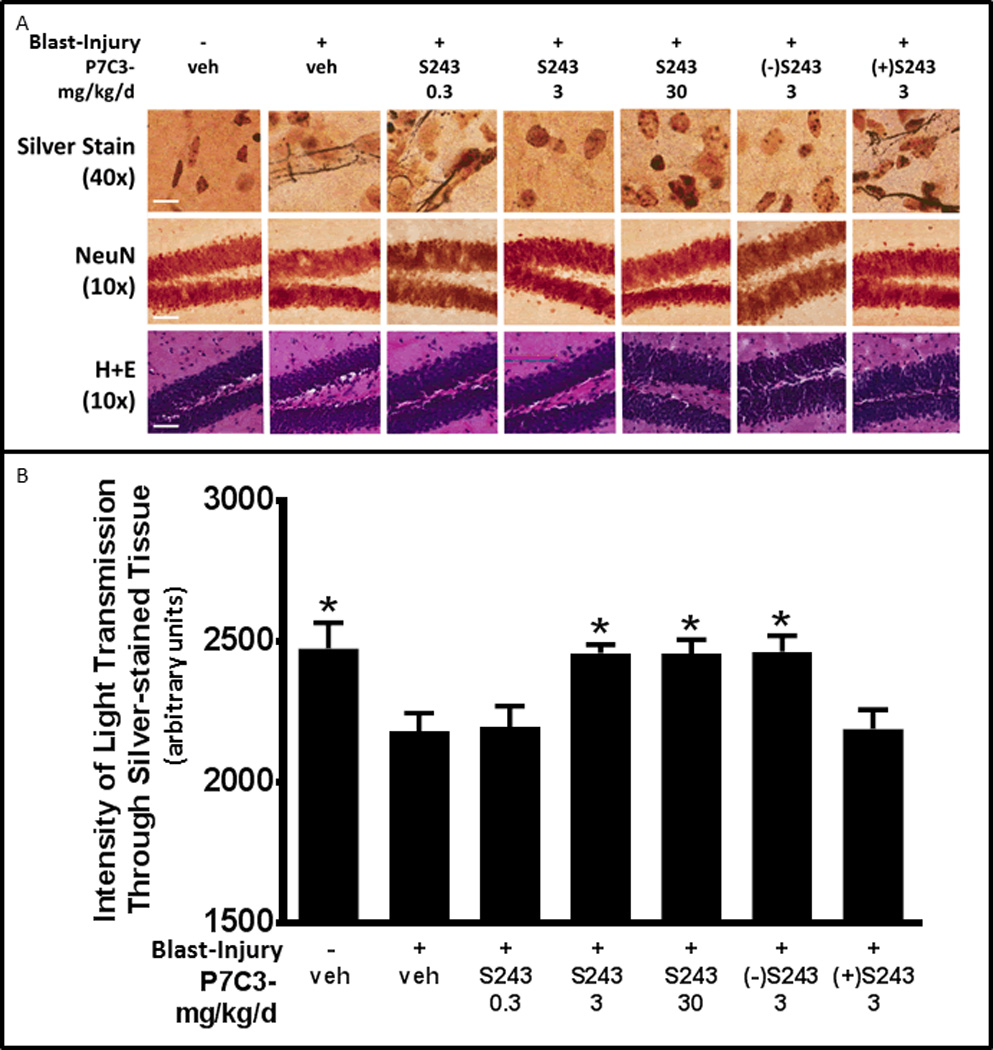
(A) Representative pictures from CA1 stratum radiatum show prominent silver staining of degenerating axons 12 days after blast-injury, in the absence of loss of NeuN of H&E staining. Images shown are representative of typical images from 5 animals in each group, and demonstrate that 3 and 30 mg/kg/day doses of P7C3-S243, initiated 24 hours after blast-injury, block axonal degeneration. Similar protective efficacy is seen in animals treated with 3 mg/kg/day of the highly active enantiomer (−)-P7C3-S243, but not in animals treated with the less active enantiomer (+)-P7C3-S243 (scale bar = 2.5µM). (B) Optical densitometry of light transmitted through silver-stained CA1 stratum radiatum from all animals in each group was used to quantify the protective effect. Signal was quantified for 18 sections for each of the 5 animals, spaced 480 µM apart. Agreater value indicates that more light passed unimpeded through the section by virtue of less silver staining, which reflects less axonal degeneration. Data are represented as mean ± SEM. P-value *<0.05, determined by-two way ANOVA with Bonferroni post-hoc analysis. See also Figure S3.
As neurodegenerative processes can be associated with neuroinflammation, we examined GFAP staining. Somewhat surprisingly, we did not observe elevated GFAP after blast-injury (Figure S3I). As immunohistochemical staining was conducted 12 days after injury, we looked earlier for changes in relative gene expression in the inflammatory IL-1 pathway, which has been suggested to be active in some mouse models of TBI (Lloyd et al., 2008; Clausen et al., 2009). We examined IL-1 pathway expression in the hippocampus 2 and 24 hours after blast-injury, and saw no meaningful changes (Tables S1 and S2). Our data thus show that widespread axonal damage and degeneration, in the absence of acute inflammation or widespread cell death, is the predominant neuropathologic feature in the brains of mice in our model of blast-mediated TBI at these early time points. This finding is reflective of the pathology observed in humans with mild TBI.
To further examine hippocampal structural pathology, we next turned to ultrathin (1 µM) section histology using toluidine blue staining, as well as transmission electron microscopy (TEM) (Figure S4A). As shown in Figure 4, sham-injury mice treated with vehicle displayed densely packed pyramidal neurons in CA1 stratum pyramidale, with abundant dendritic extensions into the stratum radiatum. These parameters did not change in sham-injured mice treated with orally administered (−)-P7C3-S243 (30 mg/kg/d). In blast-injured animals, however, we observed accumulation of chromatolytic and pyknotic neurons in the CA1 stratum pyramidale, accompanied by decreased dendritic extensions in the stratum radiatum. These changes indicate general ongoing pathologic processes associated with neurodegeneration preceding neuronal cell death, and are consistent with our electrophysiologic data in Figure 2. In blast-injured animals treated with low-dose (0.3 mg/kg/day) oral (−)-P7C3-S243, these same pathologic features were present (Figure 4). Initiation of treatment 24 hours after injury with an intermediate dose (3 mg/kg/day) of oral (−)-P7C3-S243, however, appeared to decrease the abundance of pyknotic pathology in the CA1 stratum pyramidale and partially preserve dendritic extension into the stratum radiatum in blast-injured mice. A higher dose (30 mg/kg/d) of oral (−)-P7C3-S243 fully preserved all structural aspects of this region in blast-injured mice.
Figure 4. Toluidine Blue Staining and Transmission Electron Microscopy Visualization of Hippocampal Protection by orally administered (−)-P7C3-S243 after Blast-Injury.

Daily oral administration of the highly active enantiomer (−)-P7C3-S243 for 14 days, starting 24 hours after injury, dose-dependently preserved CA1 morphology, as well as myelin and mitochondrial structures in the hippocampus after blast-injury. Two-weeks after either sham or blast-injury, animals were perfused and processed for ultrastructural pathology. Toluidine blue-stained semithin sections (left panel) of sham-injured mice treated with vehicle or (−)-P7C3-S243 showed normal CA1 histology, with densely packed neurons in the stratum pyramidale (1) and profuse dendritic profiles in the stratus radiatum (2; black arrows). Blast-injured animals treated with vehicle showed accumulation of chromatolytic and pyknotic neurons (white arrow) throughout the stratum pyramidale, as well as fewer dendrites in the stratum radiatum. There is no protection in CA1 morphology at the lowest concentration of blast-injured animals treated with 0.3 mg/kg/d of (−)-P7C3-S243. However, treatment with 3mg/kg/day (−)-P7C3-S243 lowered the abundance of chromatolytic and pyknotic neurons, and resulted in a more densely packed stratum pyramidale. At the highest concentration (30mg/kg/day) of (−)-P7C3-S243, there was complete preservation of CA1 morphology after blast-mediated TBI. Transmission electron micrographs (TEM; right panel) of immediately adjacent ultrathin sections showed normal myelin and axonal mitochondrial structures in the stratum radiatum of sham-injury mice treated with vehicle or (−)-P7C3-S243. Blastinjured mice treated with vehicle or 0.3mg/kg/day of (−)-P7C3-S243 showed degeneration of myelin sheath (red arrows), along with abnormal outer membrane and internal cristae structures within neuronal mitochondria (blue arrows). At 3 and 30mg/kg/day doses, however, both myelin and neuronal mitochondria were preserved. Pictures shown are representative of 4 animals per condition. Scale bars: Toluidine blue: 50uM; TEM: 500nm. See also Figure S4.
The stratum radiatum is where CA3 Schaeffer collateral axons synapse with dendrites of CA1 pyramidal cells. This was the circuit we investigated electrophysiologically (Figure 2), and we next turned to TEM to further characterize blast-injury induced axonal degeneration. As shown in Figure 4, we observed normally myelinated axons in the stratum radiatum of sham-injury mice treated with vehicle or orally administered (−)-P7C3-S243 (30 mg/kg/d). These axons contained intact and healthy-appearing mitochondria. In blast-injured animals treated with vehicle or low dose (0.3 mg/kg/day) oral (−)-P7C3-S243, however, we observed degenerating axons with characteristic unraveling of the myelin sheath. In addition, neuronal mitochondria contained within these degenerating axons showed both swelling and degenerating outer membrane and internal cristae. Initiation of treatment 24 hours after injury with an intermediate dose (3 mg/kg/d) of oral (−)-P7C3-S243 achieved partial resolution of pathological signs in both axons and neuronal mitochondria, and treatment with the higher oral dose (30 mg/kg/d) achieved full protection (Figure 4). To illustrate the robust effect, additional representative figures from each group are shown in Supplemental Figures S4.
P7C3-S243 Preserves Cerebellar Function and Axonal Integrity after Blast-Mediated TBI
Extensive axonal degeneration was also observed in the molecular layer of the cerebellum in blast-injured mice treated with vehicle or low dose (0.3 mg/kg/day) P7C3-S243, again in the absence of cell death (Figure 5A,B). As in other brain regions, axonal degeneration was blocked in the molecular layer by treatment with 3 and 30 mg/kg/day P7C3-S243, as well as 3 mg/kg/day of the highly active enantiomer (−)-P7C3-S243, but not by treatment with the same dose of the less active enantiomer (+)-P7C3-S243 (Figure 5A,B). Because the cerebellum controls coordination, we next assayed balance and coordination, using standard procedures (Luong et al., 2011). Mice were trained to cross an 80 cm long beam over 2 days, and then sham or blast-injured on day 3. Daily oral administration of (−)-P7C3-S243 was initiated 24 hours later, and animals were tested 7 and 28 days after injury. Mice were videotaped during the test, and then analyzed for the number of foot slips by observers blind to treatment group. Seven days after blast-injury, mice showed a trend towards increased number of foot slips, which did not reach statistical significance. By 28 days after injury, however, blast-injured mice displayed a two-fold increase in number of foot slips. This behavioral deficit was normalized by daily oral treatment with (−)-P7C3-S243, initiated 24 hours after injury. Thus, protective efficacy for cerebellar axonal degeneration after injury by (−)-P7C3-S243 correlated with preservation of motor coordination.
Figure 5. P7C3-S243 Blocks Cerebellar Axonal Degeneration and Preserves Balance and Coordination.

(A) Representative pictures from the cerebellar molecular layer show prominent silver staining of degenerating axons 12 days after blast-injury, in the absence of loss of NeuN or H&E staining. Images shown are representative of typical images from 5 animals in each group, and demonstrate that 3 and 30 mg/kg/day doses of P7C3-S243, initiated 24 hours after blast-injury, block axonal degeneration. Similar protective efficacy is seen in animals treated with 3 mg/kg/day of the highly active enantiomer (−)-P7C3-S243, but not in animals treated with the less active enantiomer (+)-P7C3-S243. (scale bar = 2.5µM). (B) Optical densitometry of light transmitted through silver-stained cerebellar molecular layer from all animals in each group was used to quantify the protective effect. The specific tissue area was manually delineated, and signal was quantified for 18 sections for each of the 5 animals, spaced 480 µM apart. A greater value indicates that more light passed unimpeded through the section by virtue of less silver staining, which reflects less axonal degeneration. (C) Seven days after blast-injury, mice show a trend towards impaired balance and coordination with increased foot slips that did not achieve statistical significance. By 28 days, however, blast-injured mice showed a two-fold increase in the number of foot slips relative to sham-injured mice. When daily oral treatment with 6 mg/kg/day of the active enantiomer (−)-P7C3-S243 was initiated 24 hours after blast-injury, however, mice performed normally in this task. Every group shown consisted of 25 male C57/Bl6 mice, aged 12–14 weeks, and data was collected and scored in an automated manner blind to treatment group. Significance was determined by 2 way ANOVA with Bonferroni post-hoc analysis. Data are represented as mean ± SEM. p-value labeled as **<0.01 and ****<0.0001 compared to blast-injured animals treated with vehicle. See also Figure S5, S6.
As with cognitive measures, we verified that the protective effect of treatment with P7C3-S243 did not simply reflect an acceleration of what might otherwise be a normal recovery process after injury. As shown in Figure S5, eight months after a single blast injury, untreated mice continue to show a statistically significant deficit in ability to perform this task of motor coordination.
Acquisition of Anxiety-like Phenotypes Chronically after Blast-Mediated TBI
We additionally analyzed mice at acute and chronic time points in the Laboras system, which allows 24-hour automated collection of normal home cage behavior using a vibration-sensitive plate to monitor motor activity (Xu et al., 2013). Here, we noted acquisition of anxiety-related phenotypes at the chronic time point that were not observed acutely after injury. Specifically, we observed increased grooming (Figure S6A), and increased locomotion and rearing (Figure S6B) in untreated mice eight months after a single blast injury. This phenotype correlates with increased susceptibility of humans to psychiatric symptoms after TBI. Future work will focus on expanding this finding, including determination of whether acute or chronic treatment with P7C3 molecules offer protection from this aspect of TBI as well.
DISCUSSION
Here, we show for the first time that the P7C3 class of neuroprotective chemicals blocks axonal degeneration prior to neuron cell death. Briefly, administration of P7C3-S243, initiated 24 hours after blast-mediated TBI, potently preserves axonal integrity throughout the brain. This axonal rescue is associated with preservation of related measures of synaptic transmission, hippocampal-dependent learning and memory, and motor coordination. Examination of injured mice at chronic time points shows that these behavioral deficits do not resolve on their own in mice that received only vehicle, and also that injured animals additionally attain behavioral phenotypes related to anxiety at this chronic time point after injury. We propose that P7C3-S243 serves as a chemical scaffold upon which new drugs can be designed to treat patients with conditions of axonal degradation, such as occurs in TBI or other neurodegenerative diseases. Such an agent would have broad applicability, as axon degeneration proceeds through unique mechanisms distinct from cell death (Yan et al., 2010), and most forms of neurodegenerative disease involve degradation of synapses and axons preceding loss of neuronal cell bodies (Li et al., 2001; Raff et al., 2002; Coleman and Yao, 2003; Fischer et al., 2004; Gunawardena and Goldstein, 2005; Luo and O’Leary, 2005).
How might P7C3-S243 act to protect axons? In the accompanying manuscript (Wang et al., 2014), we show that active P7C3 variants bind and enhance activity of the enzyme nicotinamide phosphoribosyltransferase (NAMPT). NAMPT synthesizes nicotinamide mononucleotide (NMN) from nicotinamide, the rate-limiting step in nicotinamide adenine dinucleotide (NAD) salvage (Preiss and Handler, 1958), and NAD is known to play a vital role in axon degeneration. For example, the Wallerian degeneration slow (WldS) strain of mice is resistant to axonal degeneration after injury (Lunn et al., 1989) by virtue of a triplicated fusion gene resulting in over-expression of nicotinamide mononucleotide adenylyl-transferase 1 (NMNAT1) (Araki et al., 2004), the enzyme that converts NMN into NAD. Furthermore, it has also been shown that treatment with NAD and NAD precursors, including nicotinamide, nicotinic acid mononucleotide and NMN, or overexpression of NAMPT, achieves axonal protection in vitro (Araki et al., 2004; Sasaki et al., 2006; Wang et al., 2005). Thus, active variants of P7C3 may protect from axonal degeneration by enhancing intracellular production of NAD through enhancing NAMPT activity. In conclusion, it is our hope that the P7C3 family of neuroprotective chemicals will form the basis for a new class of therapeutics applicable to a variety of conditions of nerve cell dysfunction currently lacking treatment options.
EXPERIMENTAL PROCEDURES
Animals
Approval for animal experiments was obtained from the University of Iowa Institutional Animal Care and Use Committee. Mice were singly housed in the University of Iowa vivarium in a temperature-controlled environment with lights on 0600–1800. Mice had ad libitum access to water and standard chow. 8-week old C57BL6 wild-type mice were obtained from Jackson Laboratories.
Blast-Mediated Traumatic Brain Injury
Mice were anesthetized with ketamine/xylazine (1mg/kg and 0.1mg/kg respectively) and placed in an enclosed blast chamber (50 cm long and 33 cm wide) constructed from an air tank partitioned into two sides. One side was pressurized with a 13cm opening between the partitions, and covered with a Mylar membrane. The unpressurized partition contained a restraint 10 cm from the Mylar membrane into which the mouse was placed. The head was freely moving while a metal tube shielded the body. Compressed air was forced into the pressurized partition until the Mylar membrane burst at 22 kPa. The blast wave impacted the test animal inside a foam-lined restraint to reduce blunt impact trauma of the head against the metal tube. The left side of the head was closest the origin of the blast wave. Sham-injured animals were anesthetized in the same way and not subjected to the blast.
P7C3 series of compounds formulation
Compound formulation was conducted using previously described methods (Pieper et al., 2010, Naidoo and De Jesús-Cortés et al. 2014)
Statistics
GraphPad Prism 6 software was used to perform all statistical analyses.
For detailed methodology of blood-brain barrier studies, Barnes maze, immunohistochemistry, electron microscopy, and electrophysiology studies, IL-1 pathway analysis, foot slip assay, and LABORAS assay, see supplemental methods.
Supplementary Material
Acknowledgements
We thank Mike Welsh and Randy Kardon for advice and encouragement, and Noelle Williams for pharmacologic studies. Medical illustration was provided by Teresa A. Ruggle (Department of Internal Medicine Design Center, University of Iowa Hospitals & Clinics) and Victor Powell (Medical Media, Iowa City VA Health Care System). The work was funded by a grant awarded to AAP and SLM from the NIMH (5-RO1-MH087986); grant awards from the Welch Foundation (I-1612) and the Edward N. and Della C. Thome Memorial Foundation to JMM; unrestricted funds provided to SLM by an anonymous donor, NIH grants 1R21MH100086-01 and 5R01NS064159-05 to AGB, the Department of Veterans Affairs, Veterans Health Administration, Rehabilitation Research and Development Service (Center for the Treatment and Prevention of Visual Loss, RR&D Career Development Award (MMH) and Merit Award RX000427), and a National Science Foundation Graduate Research Fellowship to HD J-C).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: T.C.Y., J.K.B and H. De J-C. serve as co-first authors. All three investigators were equally instrumental in design of the study, and conducted experiments and data analysis, and contributed to writing the manuscript and preparation of figures. Specifically, T.C.Y. and J.K.B. led behavioral studies of learning and memory, coordination and anxiety. H. De J.C. and T.C.Y. led ultrastructural electron microscopy hippocampal studies. All three investigators conducted key aspects of histochemical analysis.
REFERENCES
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Blaya MO, Bramlett HM, Naidoo J, Pieper AA, Dietrich WD. Neuroprotective efficacy of a proneurogenic compound after traumatic brain injury. J. Neurotrauma. 2014;31:476–486. doi: 10.1089/neu.2013.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen F, Hanell A, Bjork M, Hillered L, Mir AK, Gram H, Marklund N. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2009;30:385–396. doi: 10.1111/j.1460-9568.2009.06820.x. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer’s disease. Neurobiol. Aging. 2003;24:1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- De Jesús-Cortés H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt JK, Tesla R, Morlock L, Naidoo J, Melito LM, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc. Natl. Acad. Sci. USA. 2012;109:17010–17015. doi: 10.1073/pnas.1213956109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang X-L, Veliske L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Trans. Med. 2012;134:1–16. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS. Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch. Neurol. 2005;62:46–51. doi: 10.1001/archneur.62.1.46. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Egleton RD. Fluorescence imaging of blood-brain barrier disruption. J. Neurosci. Meth. 2006;151:262–267. doi: 10.1016/j.jneumeth.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. soldiers returning from Iraq. N. Engl. J. Med. 2008;385:453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J. Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd E, Somera-Molina K, Van Eldik LJ, Waterson DM, Wainwright MS. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J. Neuroinflammation. 2008;5:5–28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Luong TN, Carlisle HJ, Southwell A, Patterson PH. Assessment of motor balance and coordination in mice using the balance beam. J. Vis. Exp. 2011;10 doi: 10.3791/2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan KS, Naidoo J, Liang J, Melito L, Williams NS, Morlock L, Huntington PJ, Estill SJ, Longgood J, Becker GL, et al. Development of proneurogenic, neuroprotective small molecules. J. Am. Chem. Soc. 2010;133:1428–1437. doi: 10.1021/ja108211m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson J, Leonessa F, Ling GS. Neuropathology of explosive blast traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2012;12:570–579. doi: 10.1007/s11910-012-0303-6. [DOI] [PubMed] [Google Scholar]
- Mohan K, Kecova H, Hernandez-Merino E, Kardon RH, Harper MM. Retina ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury. Invest. Opthalmol. Vis. Sci. 2013;54:3440–3450. doi: 10.1167/iovs.12-11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo J, Bemben CJ, Allwein SR, Liang J, Pieper AA, Ready JM. Development of a scalable synthesis of P7C3-A20, a potent neuroprotective agent. Tett. Lett. 2013;54:4429–4431. [Google Scholar]
- Naidoo J, De Jesús-Cortés H, Huntington P, Estill S, Morlock LK, Starwalt R, Mangano TJ, Williams NS, Pieper AA, Ready JM. Discovery of a neuroprotective chemical, (S)-N-(3,6-dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(−)P7C3-S243], with improved druglike properties. J. Med. Chem. 2014;57:3746–3754. doi: 10.1021/jm401919s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa A, Manley GT, Gean AD, Ohtani K, Armonda R, Tsukamoto A, Yamamoto H, Takayama K, Tominaga T. Mechanisms of primary blast-induced traumatic brain injury: Insights from shock-wave research. J. Neurotrauma. 2011;28:1101–1119. doi: 10.1089/neu.2010.1442. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol. Dis. 2001;8:207–219. doi: 10.1006/nbdi.2000.0371. [DOI] [PubMed] [Google Scholar]
- Pieper AA, Wu X, Han TW, Estill SJ, Dang Q, Wu LC, Reece-Fincanon S, Dudley CA, Richardson JA, Brat DJ, McKnight SL. The neuronal PAS domain protein 3 transcription factor controls FGF-mediated adult hippocampal neurogenesis in mice. Proc.Natl. Acad. Sci. USA. 2005;102:14052–14057. doi: 10.1073/pnas.0506713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen C-H, et al. Discovery of a proneurogenic, neuroprotective chemical. Cell. 2010;142:39–51. doi: 10.1016/j.cell.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper AA, McKnight SL, Ready JM. P7C3 and an unbiased approach to drug discovery for neurodegenerative diseases. Chem. Soc. Rev. 2014 Feb 11; doi: 10.1039/c3cs60448a. [EPub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss J, Handler P. Biosynthesis of diphosphopyridine nucleotide. I. Identification of Intermediates. J. Biol. Chem. 1958;233:488–492. [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Sasacki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J. Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shively SI, Scher AI, Perl DP, Diaz-Arrastia R. Dementia resulting from traumatic brain injury: what is the pathology? Arch. Neurol. 2012;69:1245–1251. doi: 10.1001/archneurol.2011.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, McDaniel L, Knobbe W, Burket A, Tran S, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 2012;109:17016–17021. doi: 10.1073/pnas.1213960109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyama O, Okamura N, Yanase M, Narita M, Kawabata K, Sugita M. Quantitative evaluation of vascular permeability in the gerbil brain after transient ischemia using Evans blue fluorescence. J.Cereb. Blood Flow Metab. 1988;8:282–284. doi: 10.1038/jcbfm.1988.59. [DOI] [PubMed] [Google Scholar]
- Walker AK, Rivera PD, Wang Q, Chuang J-C, Tran S, Osborne-Lawrence S, Estill SJ, Starwalt R, Huntington P, Morlock L, et al. The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Mol. Psych. 2014 Apr 22; doi: 10.1038/mp.2014.34. [EPub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, McKnight SL. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014 doi: 10.1016/j.cell.2014.07.040. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney W, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J. Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SJ, Bebarta VS, Bonnett CJ, Bonnett PT, Pons SV, Cantrill SV. Blast injuries. Lancet. 2009;374:405–415. doi: 10.1016/S0140-6736(09)60257-9. [DOI] [PubMed] [Google Scholar]
- Xu P, Grueter BA, Britt JK, McDaniel L, Huntington PJ, Hodge R, Tran S, Mason BL, Lee C, Vong L, et al. Double deletion of melanocortin 4 receptors and SAPAP3 corrects compulsive behavior and obesity in mice. Proc. Natl. Acad. Sci. USA. 2013;110:10769–10764. doi: 10.1073/pnas.1308195110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan T, Feng Y, Zhai Q. Axon degeneration: Mechanisms and implications of a distinct program from cell death. Neurochem. Int. 2010;56:529–534. doi: 10.1016/j.neuint.2010.01.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.