

Abstract
(−)-P7C3-S243 is a neuroprotective aminopropyl carbazole with improved druglike properties compared with previously reported compounds in the P7C3 class. It protects developing neurons in a mouse model of hippocampal neurogenesis and protects mature neurons within the substantia nigra in a mouse model of Parkinson’s disease. A short, enantioselective synthesis provides the neuroprotective agent in optically pure form. It is nontoxic, orally bioavailable, metabolically stable, and able to cross the blood–brain barrier. As such, it represents a valuable lead compound for the development of drugs to treat neurodegenerative diseases and traumatic brain injury.
Neurodegenerative diseases currently affect millions of people worldwide, and the incidence of disease is rapidly increasing as the aging population expands. The magnitude and trend of this problem places a growing human and financial strain on healthcare systems, which is exacerbated by the absence of effective treatments for many of the most common afflictions.1 Neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD), traumatic brain injury (TBI), and normal age-related cognitive decline feature, by definition, neuronal cell death. Accordingly, we have searched for small molecules that could prevent the death of neurons in a variety of in vivo contexts. We hypothesize that such neuroprotective agents could possess general utility for treating disorders associated with neuron cell death.
Rather
than focusing on predefined molecular targets thought to
be related to specific neurodegenerative diseases, we have searched
for small molecules with in vivo neuroprotective properties. In 2010
we reported the results of an unbiased in vivo screen designed to
identify such agents. This assay involved administration of compounds
concurrent with exposure to the thymidine analogue bromodeoxyuridine
(BrdU), which marked newly born neurons through incorporation into
newly synthesized DNA. The neuroprotective efficacy of compounds was
gauged by evaluating their protective effect on newborn neurons within
the hippocampus over a 1 week period. Under the assay conditions,
around 40% of newborn neural precursor cells in the hippocampus of
untreated or vehicle-treated mice underwent apoptotic cell death within
1 week of their birth. Our screen revealed an aminopropyl carbazole,
which we named P7C3 (1),2 that
approximately doubled the number of surviving newborn hippocampal
cells at this time point. Additional experiments demonstrated that 1 and derivatives thereof increased the number of new neurons
by blocking apoptotic cell death rather than by increasing neural
stem cell proliferation. Subsequent chemical optimization yielded
P7C3-A20 (2),3 a derivative
with improved potency and toxicity profile that is available on a
large scale.4 Recently, we and others have
shown that the P7C3 class of compounds are broadly protective of mature
neurons, with potent efficacy in multiple models of neurodegenerative
disease. For instance, we discovered that 1 and 2 protect neurons in the substantia nigra (SNc) in a mouse
model of PD involving the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP).5 Similarly, we found that they
minimize motor neuron cell death in the spinal cord of G93A SOD1 transgenic
mice, a common model of ALS.6 More recently, 2 was found to protect rat cerebral cortical neurons in the
moderate fluid percussion model of TBI and to exert antidepressant
effects by increasing hippocampal neurogenesis.7,8 Finally, 1 demonstrated neuroprotective effects in a rat model of age-related
cognitive decline2 and a zebrafish model
of retinal degeneration.9 While the antidepressant
activity of 1 and 2 is correlated with protection
of immature neurons, in all other cases, 1 and/or 2 protected mature neurons from cell death.
Moreover, in cases where neuron death was associated with behavioral
or learning deficits, neuroprotective efficacy was reflected in an
improvement in motor function and/or learning and memory.
While 2 is active, orally available, brain-penetrant, and nontoxic, even at doses in excess of the minimally efficacious dose, we recognized the opportunity to design a molecule with improved druglike properties. In particular, we sought to replace the aniline moiety of 2 with an alternative heterocycle, increase the polarity of the compound, and synthesize the drug lead as a single enantiomer. Few marketed drugs contain aniline moieties, and most chiral drugs are produced in optically active form. Here we describe the discovery and evaluation of (−)-P7C3-S243 (15), a neuroprotective agent with improved physicochemical properties. It appears to be nontoxic and more efficacious than 1 and 2 in animal models of PD. It therefore represents an optimized neuroprotective agent emerging from the P7C3 class of compounds.
Results
Prior and ongoing work in our groups indicates that the P7C3 class of compounds exert their neuroprotective effects by stabilizing mitrochondria against the depolarization events that are known to trigger apoptosis.2 Nonetheless, our most robust assay involves monitoring neuroprotection of newborn hippocampal neurons in mice because this assay gives a simultaneous readout of efficacy, acute toxicity, and cell permeability. For this reason, all analogues of 1 were evaluated initially by infusing them directly into the left lateral ventricle of environmentally deprived10 live mice using a subcutaneously implanted osmotic minipump. Compounds were delivered intracerebroventricularly (ICV) over a 7 day period at a concentration of 10 μM (12 μL/day).11 Mice were additionally dosed intraperitoneally (IP) daily with BrdU (50 mg/kg) to mark newly born hippocampal neural precursor cells. At the end of the 1 week dosing, animals were sacrificed and transcardially perfused. Brain slices were stained with antibodies to BrdU, and the number of surviving neural precursor cells was quantified by counting the number of BrdU+ cells in the hippocampal dentate gyrus. To avoid complications arising from the surgical implantation of the pump, we quantified neurogenesis in the hemisphere opposite to compound infusion. Finally, to obtain consistent data across many animals, the number of BrdU+ cells was normalized to the volume of the dentate gyrus.
Our initial objective was to replace the aniline ring with a heterocyclic ring. Previous studies had shown that the N-phenyl ring of 1 could be replaced with a 2-pyridyl group (giving 3), but not a 3- or 4-pyridyl group, while maintaining activity in the in vivo hippocampal neuroprotection assay.3 Moreover, we had discovered that fluorination of the central methylene and substitution of the N-aryl ring improved the activity, but neither change alone had substantial impact. For example, 4 (i.e., methoxy-1) and 5 (i.e., fluoro-1) showed similar activity as 1. However, fluorinating the anisidine congener 4 to provide 2 substantially improved the activity in the hippocampal neuroprotection assay. In this context, we targeted 15, a fluorinated compound containing a methoxy-substituted 2-aminopyridine. Following the approach we described for the large-scale synthesis of 2,4 terminal epoxide 7 was opened with 4-nitrobenzenesulfonyl (4-nosyl, 4-Ns)-protected 2-methoxy-6-aminopyridine (Scheme 1). Surprisingly, when the resultant amino alcohol 8 was subjected to fluorination conditions with MorphoDast, HPLC/MS and 1H NMR analyses indicated the formal loss of methanol. We speculated that the pyridine nitrogen displaced an activated alcohol and the cyclized pyridinium salt (not shown) was demethylated to yield aminopyridone 9.
Scheme 1.

We next considered an approach involving amination of a primary amine. To this end, epoxide 7 was opened with sodium azide. The resulting secondary alcohol 10 was fluorinated to provide azido fluoride 11, and the azide was reduced with triphenylphosphine to yield fluoroamine 12. Reaction of this amine with iodopyridine 13(12) in the presence of CuI and diketone ligand 14 provided the desired analogue 15 in high yield on a multigram scale.13 Alternative conditions featuring Pd catalysts or lacking the diketone ligand provided less than 20% yield of the intended product. In a similar manner, amino alcohol 16 was arylated to provide 17, the hydroxy congener of 15. Amino alcohol 16 proved to be much less reactive than fluoroamine 12, resulting in incomplete conversion. Moreover, in this case adding the diketone ligand improved the yield by only around 10%, suggesting that the amino alcohol itself can serve as a ligand for Cu.14
As described above, compounds were administered to mice ICV over a 1 week period concurrent with dosing of BrdU IP. Figure 1 shows that aminopyridine 15 approximately doubled the number of surviving newborn hippocampal neurons over this time period and displayed an activity superior to that of 1 and comparable to that of 2. Several of the synthetic intermediates also displayed neuroprotective activity in this assay. While azide 11 was inactive under the conditions of this experiment, the primary amines 12 and 16 as well as the N-pyridyl amino alcohol 17 showed an increase in the number of BrdU+ cells compared with vehicle. However, these intermediates were less potent than 15 and even slightly less active than our initial hit 1. We originally adopted intracerebroventricular delivery to avoid complications related to brain penetration. Nonetheless, ICV is clearly not an ideal method of dosing. Fortunately, the optimized lead 15 showed a smooth dose–response curve upon intraperitoneal (IP) administration. It was active at a dose of 1 mg kg–1 day–1 and reached its ceiling of efficacy at 10 mg kg–1 day–1, results which parallel those found in the MPTP model of Parkinson’s disease (see below). Control experiments demonstrated that the increase in BrdU+ neurons within the hippocampus was due to neuroprotection rather than increased neural stem cell proliferation (Figure S1 in the Supporting Information).
Figure 1.

Efficacy of 15 in a mouse model of neurogenesis. Compounds were administered using the indicated dose and method for 7 days, during which time mice were dosed IP daily with BrdU (50 mg kg–1 day–1) to label newborn hippocampal neurons. Data are shown as mean ± standard error of the mean (SEM).
Previous work had demonstrated that the preponderance of neuroprotective activity resided in a single enantiomer of the P7C3 class of compounds. The enantiomers of 18, 19, and 20 that are shown in Scheme 2 were the more active of each pair, suggesting a specific protein-binding partner. We also determined that tertiary alcohol 21 was more active than the initial hit 1, suggesting the presence of a hydrophobic binding site into which the methyl group could project.3 These observations prompted the question of which, if either, enantiomer of fluorinated analogues such as 2 and 15 would display higher potency. In particular, we sought to determine whether fluorine would act as a polar group and occupy the binding site favored by the hydroxyl group of 18, 19, and 20 or act as a hydrophobic group and bind similarly to 21. Unfortunately, previous attempts to fluorinate an optically active precursor to 2 provided material with 0–20% ee, so developing an enantioselective synthesis remained a high priority.
Scheme 2. Stereoselective Synthesis of (−)-15.

Condensation of 3,6-dibromocarbazole (6) with the 3-Ns-protected glycidol derivative 22 provided the epoxide (−)-7 in 97% yield with 96% ee.15 Epoxide ring opening with azide was quantitative and provided a substrate for fluorination, (+)-10. Comparison with the material formed from a Mitsunobu reaction with (S)-glycidol demonstrated that, as described by Sharpless and co-workers,16 sulfonate 22 reacted by direct displacement of the leaving group rather than by epoxide opening/epoxide closure. Consistent with previous results, we found that reaction with MorphoDast was not stereospecific, as azidofluoride 11 was recovered with only 63% ee. We speculate that the common invertive mechanistic pathway was compromised by an intramolecular displacement with the carbazole nitrogen, yielding an aziridinium ion intermediate (not shown). This pathway would occur with overall retention and lower the observed optical purity. In contrast, we discovered that the reagent combination perfluorobutanesulfonyl fluoride and tetrabutylammonium difluorotriphenylsilicate (TBAT) yielded the desired fluoride with clean inversion.17 Subsequent processing as before provided optically active (−)-15 with 95% ee. Recrystallization from isopropanol enriched the optical purity to 99% ee. The (+) enantiomer was similarly prepared starting from ent-22.
The pure enantiomers of 15 were first evaluated in the mouse model of hippocampal neuroprotection after they were dosed ICV. As shown in Figure 2, (−)-15 is highly active, whereas its enantiomer shows no neuroprotective activity in this assay. The more active enantiomer shows roughly the same activity as the racemic mixture at identical doses. We interpret this observation to mean that this in vivo assay cannot resolve the 2-fold difference in concentration between, for example, a single enantiomer at 10 μM and a racemic mixture at 10 μM (5 μM each enantiomer). X-ray crystallographic analysis of a single crystal of (−)-15 revealed it to be the S enantiomer (Figure S2 in the Supporting Information). Interestingly, the more active fluoride is enantiomeric to the more active enantiomer in the hydroxyl series (18–20). This observation implies that the binding partner for the P7C3 class of neuroprotective chemicals contains both a hydrophobic pocket, which can be occupied by the fluoride or methyl group, and a polar pocket, which preferentially accommodates a hydroxyl group.
Figure 2.
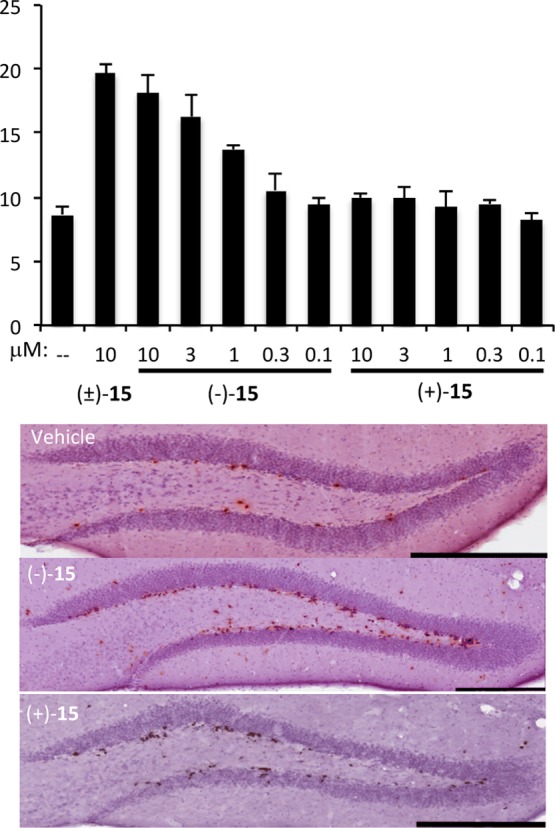
Enantiomer-specific efficacy of 15 in a mouse model of neurogenesis. Compound was administered ICV at the indicated concentration (0.5 μL/h) for 7 days, during which time mice were dosed IP daily with BrdU (50 mg kg–1 day–1) to label newborn hippocampal neurons. The graph shows the mean ± SEM for each group (N = 4 or 5). Images are of representative stained sections. Scale bars = 0.3 mm.
The mouse model of neurogenesis involves protecting newborn hippocampal neurons from premature apoptotic cell death. We also sought to determine whether 15 could protect mature neurons from cell death. In this context, we had previously demonstrated that 1 and 2 could protect mature dopaminergic neurons within the SNc from toxicity associated with MPTP.5 This mouse model of PD entails treating mice for 5 days with a toxic dose of MPTP (30 mg kg–1 day–1) to initiate cell death within the SNc. In the brain, MPTP is oxidized to 1-methyl-4-phenylpyridinium cation (MPP+), which is selectively transported into dopaminergic neurons within the SNc.18 Within these cells, MPP+ acts as a mitochondrial poison, generating a pathology that resembles that of Parkinsonian patients.19 Under our assay conditions, treatment with drug was initiated a full 24 h after the final dose of MPTP. This dosing regime ensured that any effect of the P7C3 class of compounds could be attributed to neuroprotective activity rather than to simply blocking the uptake or metabolism of MPTP.20 Mice were administered drug twice daily for 21 days, after which time they were sacrificed by transcardial perfusion. Fixed brains were sectioned through the SNc at 30 μM intervals. Every fourth section was stained with antibodies specific to the enzyme tyrosine hydroxylase (TH). TH catalyzes the oxidation of l-tyrosine to l-3,4-dihydroxyphenylalanine (l-DOPA), which is the first step in the biosynthesis of dopamine. Accordingly, the number of TH+ cells provides a quantification of the neuroprotective efficacy of test compounds following MPTP exposure. Importantly, all microscopic analyses were performed blinded to the treatment group.
As shown in Figure 3, MPTP effects an approximately 50% reduction in the number of TH+ neurons under the chronic dosing protocol described above. Consistent with our earlier reports, 2 exhibits marked neuroprotective activity, showing a significant neuroprotective effect when administered at 5 mg kg–1 day–1 and protection resulting in around 75% rescue at a dose of 10 mg kg–1 day–1.5 Encouragingly, 15 demonstrated even higher neuroprotective activity than 2, with the former showing significant activity at a dose of 1 mg kg–1 day–1 and rescuing over 85% of the TH+ neurons at the highest dose tested (10 mg kg–1 day–1). Moreover, the enantiomeric specificity observed in the MPTP model of PD mirrored that observed in the mouse model of neurogenesis. Specifically, (−)-(S)-15 was strongly protective, while the (+)-R enantiomer was indistinguishable from vehicle treatment under the conditions of the assay. In this side-by-side experiment, 15 appeared to be at least as effective as 2, a compound that has proven efficacy in several animal models of neurodegenerative disease.
Figure 3.

Efficacy of 15 in the MPTP model of Parkinson’s disease. Mice were administered MPTP (30 mg kg–1 day–1) for 5 days. On the sixth day, treatment with drug was initiated at the indicated dose (IP, BID, 21 days) . Bars indicate numbers of tyrosine hydroxylase-positive (TH+) cells detected by immunohistochemical staining of the substantia nigra (mean ± SEM). Asterisks indicate p < 0.01 (*), p < 0.001 (**), or p < 0.0001 (***) relative to the vehicle (Veh) + MPTP group. The number of mice in each group is shown within the corresponding bar. Representative immunohistochemical pictures of TH staining in the SNc are shown for 5 mg kg–1 day–1 treatment groups. Scale bars = 300 μM.
The pharmacokinetic and toxicity profiles of 15 were evaluated using both in vivo and in vitro methods. We observed nearly complete metabolic stability in the presence of murine hepatocytes (t1/2 > 240 min) and good stability in the presence of liver microsomes (t1/2 values: mouse, 49 min; rat, >240 min; dog, 45 min; monkey, 81 min; human, 117 min). We then monitored plasma and brain levels of the compound following intravenous, oral, or intraperitoneal dosing. As shown in Table 1, 15 exhibited a dose-proportional increase in brain levels when dosed IP. Plasma bioavailability was higher when the compound was dosed IP as opposed to per os (PO) by oral gavage, but partitioning into the brain was superior when the compound was administered by oral gavage. We noted the same effect with 2, and thus, comparable brain levels are achieved either IP or by oral gavage. By all routes of administration, 15 was metabolically stable. While full pharmacokinetic profiling was performed with (±)-15, we additionally observed indistinguishable plasma and brain concentrations for racemic and (−)-15 at a single time point following intraperitoneal injection of 1, 3, 10, or 30 mg/kg doses (Figure S3 in the Supporting Information). These data are consistent with similar ADME properties for the racemic and optically active forms of 15.
Table 1. Pharmacokinetic Properties of (±)-15.
| administration | dose (mg/kg) | AUC(brain) (μg·min/g)a | %F (plasma/brain) | AUCbrain/AUCplasma | t1/2 (h) |
|---|---|---|---|---|---|
| IV | 10 | 671 ± 14 | – | 1.1 | 7.4 |
| IP | 10 | 189 ± 35 | 66/28 | 0.45 | >24 |
| IP | 20 | 319 ± 68 | 90/48 | 0.28 | 15 |
| PO | 20 | 388 ± 29 | 38/29 | 0.81 | 9.6 |
Presented as mean ± SEM.
(−)-15 did not show significant toxicity toward the three human cancer cell lines tested (IC50 > 10 μM vs H2122, H2009, and HCC44). Likewise, we observed <15% inhibition of the hERG channel at 50 μM using a patch clamp assay. Several cytochrome P450s (CYP3A4, -2C9, -3D6, -2C8, -2B6) were insensitive to (−)-15, while CYP1A2 and CYP2C19 showed modest IC50 values of 20 and 1.9 μM, respectively. On the basis of the high protein binding of 15 (>99%21), free concentrations of the drug are unlikely to reach those concentrations. Finally, (−)-15 was screened through the NIH’s Psychoactive Drug Screening Program, and no compelling binding partners were identified. Of 47 neuronal receptors and channels tested, only the μ-opioid receptor (IC50 = 8.2 μM) and the peripheral benzodiazepine receptor [also known as translocator protein (TSPO)] (IC50 = 0.35 μM) showed any ligand displacement. The binding affinity of the former is clearly too weak to be causative for the biological activity we observed, provided that the in vitro data are predictive of binding in vivo. The relevance of binding to TSPO is less clear, however. While the total brain concentration remains in the low micromolar range for several hours after dosing, the free concentration is much lower. Moreover, while we observed enantiomeric specificity in the neurogenesis (Figure 2) and MPTP assays (Figure 3), both enantiomers of 15 bind TSPO with similarly weak affinities (0.35 and 0.68 μM). Finally, 2, which shows similar in vivo activity as (−)-15, binds TSPO more weakly (IC50 = 2.0 μM). A wide structural range of chemicals have been found to bind TSPO, from the eponymous benzodiazapines to cholesterol to porphyrins,22 suggesting that it might represent a nonspecific binding partner for small molecules. Moreover, a tight-binding inhibitor of this protein showed no toxicity in phase I and II clinical trials for ALS and is currently in phase II trials for spinal muscular atrophy.23 Overall, the clean receptor binding results, which are shown in full in Table S1 in the Supporting Information, are consistent with our observation that long-term treatment with 2 (up to 40 mg kg–1 day–1 for 21 days) or 15 (up to 10 mg kg–1 day–1 for 21 days) has no negative impact on the weight, behavior, or appearance of the treated animals.
Conclusions
We have discovered a novel class of potent neuroprotective agents known as the P7C3 series of aminopropyl carbazoles. We have now synthesized and characterized an optimized member of this series that lacks an aniline moiety. Moreover, it exhibits improved polarity and is readily available as a single enantiomer. These characteristics improve its druglike properties compared with previously reported analogues. Here we have demonstrated that (−)-15 potently blocks neuron cell death under two conditions: apoptosis of newborn hippocampal neurons and MPTP-mediated killing of dopaminergic neurons in the substantia nigra. The latter effect is notable for almost complete protection in this model of Parkinson’s disease. Future reports will describe the effect of this neuroprotective chemical in a rat model of PD and mouse models of TBI. Likewise, we are actively seeking to validate the molecular target of the P7C3 class of compounds and will report the results of those studies in due course. Taken together, the data suggest that (−)-15 is a promising candidate for treating some of the most devastating neurological disorders—disorders that are likely to increase in prevalence and currently lack effective therapies.
Experimental Section
General Methods
Unless otherwise stated, commercially available materials were used without further purification, and reactions were performed under a nitrogen atmosphere using freshly purified solvents. Solvents were purified using solvent purification columns purchased from Glass Contour (Laguna Beach, CA). All of the reactions were monitored by thin-layer chromatography (TLC) using E. Merck silica gel 60 F254 precoated plates (0.25 mm). Flash chromatography was performed with the indicated solvents using silica gel (particle size 0.032–0.063 μm) purchased from Sorbent Technologies. 1H and 13C NMR spectra were recorded on a Varian Inova 400 or 500 spectrometer. Chemical shifts are reported relative to internal solvent. Electrospray ionization mass spectrometry (ESI-MS) was performed on an Agilent 2010 LC–MS instrument. Optical rotations were measured at 20 °C on a Rudolph Research Analytical Autopol IV polarimeter. Compounds (±)-7, 10, and 16 were synthesized as previously described.3,4 All of the synthetic compounds were confirmed to be ≥95% pure by reversed-phase HPLC and 1H NMR analyses.
N-(6-Methoxypyridin-2-yl)-4-nitrobenzenesulfonamide
A solution of 6-methoxypyridin-2-ylamine24 (206.8 mg, 1.66 mmol) and pyridine (0.2 mL) in methylene chloride (8.0 mL) was stirred for 30 min before being cooled in an ice bath. 4-Nitrobenzenesulfonyl chloride (383.9 mg, 1.73 mmol) was added slowly to the cold stirring solution, and the ice bath was removed 10 min later. The reaction mixture was stirred overnight and then condensed, and the dark-colored mixture was purified on an automated flash chromatography system in 0–10% MeOH/dichloromethane (DCM) to provide the nosylate as a yellow liquid in 55% yield. 1H NMR (CDCl3, 400 MHz): δ 8.34 (d, J = 8.9 Hz, 2H), 8.14 (d, J = 8.9 Hz, 2H), 7.53 (t, J = 8.0 Hz, 1H), 7.35–7.29 (m, 1H), 6.81 (d, J = 7.7 Hz, 1H), 6.46 (d, J = 8.2 Hz, 1H), 3.74 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ 53.9, 103.7, 106.7, 124.5, 141.1, 145.4, 147.6, 163.7. ESI-MS (m/z): 309.9 [M + 1]+; C12H11N3O5S requires 309.04.
(−)-(S)-3,6-Dibromo-9-(oxiran-2-ylmethyl)-9H-carbazole [(−)-(S)-7]
In a modification of a published method,17 a stirred solution of 3,6-dibromocarbazole (6.27 g, 19.3 mmol) and powdered potassium hydroxide (1.30 g, 23.2 mmol) in dimethylformamide (DMF) (100 mL) was cooled in ice before the dropwise addition of (R)-glycidyl-3-nitrobenzenesulfonate (22)18 (5.01 g, 19.3 mmol). The ice bath was removed, and the reaction mixture was stirred overnight at ambient temperature, diluted with water, and washed several times with water and then brine. The organic layer was dried over Na2SO4, filtered, and concentrated to give epoxide (−)-7 in quantitative yield, which was used without additional purification. 1H NMR (CDCl3, 400 MHz): δ 8.13 (d, J = 2.0 Hz, 2H), 7.57 (dd, J = 8.7, 1.8 Hz, 2H), 7.39–7.31 (m, 2H), 4.65 (dd, J = 16.0, 2.5 Hz, 1H), 4.28 (dd, J = 16.1, 5.0 Hz, 1H), 3.32 (ddd, J = 5.2, 2.6, 1.3 Hz, 1H), 2.82 (t, J = 4.3 Hz, 1H), 2.50 (dd, J = 4.7, 2.6 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ 45.0, 45.1, 50.6, 110.7, 112.8, 123.4, 123.8, 129.5, 139.8. Poor ionization precluded analysis by mass spectrometry. HPLC (ChiralPak AD-H, 1.0 mL/min 1% iPrOH/hexanes): tR(major) = 24.3–34.0 min; tR(minor) = 48.4–58.6 min; ee = 95.9%. [α]D20 = −5.74 (c = 0.244, THF).
N-(3-(3,6-Dibromo-9H-carbazol-9-yl)-2-hydroxypropyl)-N-(6-methoxypyridin-2-yl)-4-nitrobenzenesulfonamide (8)
n-BuLi (0.65 mL of a 2.5 M solution in hexanes) was added dropwise to a solution of N-(6-methoxypyridin-2-yl)-4-nitrobenzenesulfonamide (278.7 mg, 0.90 mmol) in DMF (3.0 mL) at 0 °C. The solution was stirred for 15 min before the addition of 3,6-dibromo-9-(oxiran-2-ylmethyl)-9H-carbazole (7) (265.1 mg, 0.70 mmol). The reaction mixture was heated to 80 °C for 4 h, after which more epoxide 7 (113.0 mg, 0.3 mmol) was added. The reaction mixture was stirred overnight at 80 °C, cooled, diluted with EtOAc, and washed twice with water, twice with 1 N NaOH, twice more with water, and finally brine. The crude mixture was purified on an automated flash chromatography system in 0–10% MeOH/DCM to provide a light-orange solid in 12.5% yield. 1H NMR (CDCl3, 400 MHz): δ 8.17 (d, J = 1.9 Hz, 2H), 8.04 (d, J = 9.2 Hz, 2H), 7.56 (dd, J = 8.7, 2.0 Hz, 2H), 7.44 (t, J = 7.9 Hz, 1H), 7.38 (d, J = 8.7 Hz, 2H), 7.05–6.77 (m, 2H), 6.48 (d, J = 7.7 Hz, 1H), 6.40 (d, J = 8.1 Hz, 1H), 4.43 (ddd, J = 15.5, 13.8, 7.5 Hz, 3H), 4.22–4.03 (m, 1H), 3.92 (dd, J = 15.3, 8.9 Hz, 1H), 3.49 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ 163.9, 155.5, 140.9, 140.0, 129.4, 126.1, 125.7, 123.7, 123.4, 120.2, 114.6, 112.8, 111.2, 107.4, 105.0, 69.1, 68.1, 53.7, 25.8. ESI-MS (m/z): 732.6 [M + HCOO]−; C27H22Br2N4O6S requires 688.0.
3-((3,6-Dibromo-9H-carbazol-9-yl)methyl)-1-((4-nitrophenyl)sulfonyl)-2,3-dihydroimidazo[1,2-a]pyridin-5(1H)-one (9)
MorphoDast (100 μL, 0.82 mmol) was added to a solution of alcohol 8 (60.2 mg, 0.087 mmol) in methylene chloride (2.0 mL). The reaction mixture was stirred overnight, and the reaction was quenched by the addition by an equivalent volume of saturated aqueous sodium bicarbonate. The mixture was extracted three times with methylene chloride. The combined organic layers were dried over Na2SO4, filtered, concentrated, and purified by preparative TLC in 5% MeOH/DCM to provide the pyridone as a yellow solid in 21% yield. 1H NMR (CDCl3, 400 MHz): δ 8.26–8.13 (m, 2H), 8.06 (d, J = 1.8 Hz, 2H), 7.48 (dd, J = 8.7, 1.9 Hz, 2H), 7.39 (d, J = 8.7 Hz, 2H), 7.35 (dd, J = 9.1, 7.5 Hz, 1H), 7.01–6.80 (m, 2H), 6.22 (dd, J = 9.1, 0.8 Hz, 1H), 5.87 (dd, J = 7.5, 0.8 Hz, 1H), 5.31 (s, 1H), 5.28 (dd, J = 7.4, 3.2 Hz, 1H), 4.88 (dd, J = 15.2, 3.1 Hz, 1H), 4.70 (dd, J = 15.1, 7.5 Hz, 1H), 4.15 (dd, J = 10.2, 8.3 Hz, 1H), 3.87 (dd, J = 10.2, 1.3 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ 161.3, 146.3, 144.9, 142.4, 139.4, 129.8, 125.7, 123.9, 123.6, 117.8, 113.2, 111.4, 110.3, 95.2, 86.3, 53.7, 52.0. ESI-MS (m/z): 700.6 [M + HCOO]−; C26H18Br2N4O5S requires 655.9.
(+)-(S)-1-Azido-3-(3,6-dibromo-9H-carbazol-9-yl)propan-2-ol [(+)-(S)-10]
A 20% aqueous solution of sodium azide (75 mL) was added to a mixture of (S)-(−)-7 (7.79 g, 20.4 mmol) and tetrahydrofuran (THF) (65 mL), and the mixture was heated to 75 °C. A further 20 mL of a 20% aqueous solution of sodium azide was added after 24 h to ensure complete conversion of the epoxide. The biphasic mixture was condensed under reduced pressure, diluted with EtOAc, and washed twice with water and then brine. The organic layer was dried over Na2SO4, filtered, and concentrated to give the azido alcohol in 91% yield, which was used without further purification. 1H NMR (CDCl3, 400 MHz): δ 8.06 (d, J = 1.9 Hz, 2H), 7.53 (dd, J = 8.7, 2.0 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 4.26 (dd, J = 6.1, 1.6 Hz, 2H), 4.23–4.14 (m, 1H), 3.44 (dd, J = 12.6, 4.3 Hz, 1H), 3.30 (dd, J = 12.6, 5.6 Hz, 1H), 2.48 (s, 1H). 13C NMR (CDCl3, 100 MHz): δ 46.4, 54.1, 69.5, 110.6, 110.7, 112.7, 123.3, 123.3, 123.6, 129.4, 139.5. ESI-MS (m/z): 466.6 [M + HCOO]−; C15H12Br2N4O requires 421.9. [α]D20 = +3.18 (c = 0.26, THF).
(−)-(R)-9-(3-Azido-2-fluoropropyl)-3,6-dibromo-9H-carbazole [(−)-(R)-11]
Following a published procedure, diisopropylethylamine (7.75 mL, 44.5 mmol) and toluene (140 mL) were added alternatingly (ca. 20 portions each) to a mixture of tetrabutylammonium difluorotriphenylsilicate (TBAT) (9.50 g, 17.6 mmol) and (S)-1-azido-3-(3,6-dibromo-9H-carbazol-9-yl)propan-2-ol (7.41 g, 17.5 mmol) at room temperature.19 Perfluoro-1-butanesulfonyl fluoride (PBSF) (6.91 mL, 38.5 mmol) was added dropwise, and the reaction mixture was stirred overnight at ambient temperature. The mixture was concentrated and purified by automated chromatography (SiO2, 50% DCM/hexanes) to provide the fluoro azide in 70% yield as a white solid. 1H NMR (CDCl3, 400 MHz): δ 8.14 (t, J = 2.3 Hz, 2H), 7.64–7.52 (m, 2H), 7.32 (dd, J = 8.7, 1.8 Hz, 2H), 5.12–4.85 (m, 1H), 4.54 (ddt, J = 17.9, 5.4, 2.4 Hz, 2H), 3.58 (ddd, J = 17.7, 13.5, 4.5 Hz, 1H), 3.40 (ddd, J = 24.1, 13.5, 4.7 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ 44.2 (d, JC–F = 23.9 Hz), 51.6 (d, JC–F = 23.9 Hz), 90.0 (d, JC–F = 177.6 Hz), 110.5 (d, JC–F = 1.3 Hz), 113.1, 123.5 (d, JC–F = 2.2 Hz), 123.9, 129.6, 139.5. ESI-MS (m/z): 468.7 [M + HCOO]−; C15H11Br2FN4 requires 423.9. HPLC (ChiralPak AD-H, 1.0 mL/min 0.5% iPrOH/hexanes): tR(minor) = 25.0–29.1 min; tR(major) = 29.4–40.7 min; ee = 96.2%. [α] D20 = −5.90 (c = 0.217, THF).
(+)-(S)-3-(3,6-Dibromo-9H-carbazol-9-yl)-2-fluoropropan-1-amine [(S)-(+)-12]
Triphenylphosphine (3.24, 12.3 mmol) was added to a solution of azide (−)-(R)-11 (5.10 g, 12.0 mmol) in THF (80 mL). The cloudy mixture was heated at 65 °C overnight. Water was added to the cooled reaction mixture, and the resulting mixture was stirred for 6 h before volatile solvents were removed under reduced pressure. The viscous mixture was diluted with EtOAc and washed twice with water and then brine. The organic layer was dried over Na2SO4, filtered, and concentrated to give a yellow waxy residue. Sufficient cold methylene chloride was added to dissolve the waxy residue (ca. 100 mL), and then at 0 °C 1 M HCl was added dropwise to precipitate the hydrochloride salt. The slurry was stirred for about 20 min before it was filtered and then washed with cold DCM several times and finally with hexanes. The powdery solid (5.29 g, quantitative) was dried on a fritted funnel and contained less than 1% PPh3O byproduct. The salt was freebased by adding a saturated solution of sodium bicarbonate (90.0 mL) to a milky mixture of the amine HCl salt (5.29 g) in methylene chloride (90.0 mL) with Et3N (3.0 mL). The solution was stirred for 1 h until the organic layer was translucent. The organic layer was separated, and the aqueous phase was extracted with methylene chloride. The combined organic layers were dried over Na2SO4, filtered, and concentrated to give the free amine as an off-white solid. 1H NMR (CDCl3, 400 MHz): δ 8.15 (d, J = 2.0 Hz, 2H), 7.57 (dd, J = 8.8, 1.9 Hz, 2H), 7.36 (d, J = 8.7 Hz, 2H), 4.85 (dtt, J = 47.9, 6.1, 4.4 Hz, 1H), 4.67–4.39 (m, 2H), 3.22–2.78 (m, 2H). 13C NMR (CD3OD, 100 MHz): δ 141.1, 130.5, 125.1, 124.3, 113.8, 112.4, 112.4, 91.2 (d, JC–F = 172.4 Hz), 45.5 (d, JC–F = 20.4 Hz), 42.1 (d, JC–F = 20.4 Hz). [α]D20 = +8.21 (c = 0.268, MeOH). ESI-MS (m/z): 398.7 [M + 1]+; C15H13Br2FN2 requires 397.94.
1-(3,6-Dibromo-9H-carbazol-9-yl)-3-((6-methoxypyridin-2-yl)amino)propan-2-ol (17)
1-Amino-3-(3,6-dibromo-9H-carbazol-9-yl)propan-2-ol (16) (60.1 mg, 0.15 mmol), 2-iodo-6-methoxypyridine (13) (35.9 mg, 0.15 mmol), copper(I) iodide (2.3 mg, 0.012 mmol), and cesium carbonate (100.9 mg, 0.31 mmol) were combined in an oven-dried vial under nitrogen. Dimethyl sulfoxide (DMSO) (0.3 mL) and 2,2,6,6-tetramethyl-3,5-heptanedione (14) (3.5 μL, 0.017 mmol) were then added. The reaction mixture was stirred overnight at ambient temperature, diluted with EtOAc, and washed several times with water and then brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The well-dried solid was dissolved in 80% DCM/hexanes, passed through a short silica plug, and washed with DCM to remove unreacted amine. The filtrate was condensed to give a residue in 30% yield. 1H NMR (CDCl3, 500 MHz): δ 8.15 (d, J = 2.0 Hz, 2H), 7.57 (dd, J = 8.6, 2.0 Hz, 2H), 7.36 (m, 3H), 6.09 (d, J = 7.9 Hz, 1H), 6.02 (d, J = 7.8 Hz, 1H), 4.55 (bs, 1H), 4.45–4.22 (m, 3H), 3.76 (s, 3H), 3.50 (d, J = 14.7 Hz, 1H), 3.26 (dt, J = 14.1, 5.6 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ 163.3, 157.4, 140.2, 139.2, 128.8, 123.2, 122.9, 112.0, 110.5, 99.9, 97.4, 70.2, 53.3, 46.5, 46.3. ESI-MS (m/z): 503.7 [M + H]+; C21H19Br2N3O2 requires 503.0.
(S)-N-(3-(3,6-Dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(−)-(S)-15
First, (S)-12 (72.2 mg, 0.18 mmol), 2-iodo-6-methoxypyridine14 (45.4 mg, 0.19 mmol), copper(I) iodide (2.9 mg, 0.015 mmol), and cesium carbonate (119.4 mg, 0.36 mmol) were added to an oven-dried vial, which was then purged with nitrogen for 10 min. Then DMSO (0.36 mL) and 14 (4 μL, 0.019 mmol) were added. The reaction mixture was heated at 40 °C for 3 h. The cooled mixture was diluted with EtOAc and washed three times with a 9:1 saturated NH4Cl/NH4OH solution, three times with water, and then brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The well-dried solid was dissolved in methylene chloride and passed through a short silica plug, which removed the small amount of unreacted amine. The filtrate was condensed to give a white solid, which was purified on an automated flash chromatography system in 50–80% DCM/hexanes to provide (−)-(S)-15 as a white solid in 61% yield with 96% ee, which was stored under nitrogen. Recrystallization to enhance the enantiomeric excess was carried out by heating a solution of 15.8 mg of the purified product in 1.0 mL of isopropanol. Thin needles formed over 24 h at room temperature. The overall yield from (S)-12 was 59% with 98.5% ee. Single crystals suitable for X-ray diffraction were grown by diffusion of hexanes into a concentrated solution of (−)-15 in dichloroethane (see Figure S2 in the Supporting Information). 1H NMR (CDCl3, 400 MHz): δ 8.16 (d, J = 1.9 Hz, 2H), 7.56 (d, J = 1.9 Hz, 2H), 7.35 (t, J = 7.8 Hz, 1H), 7.30 (d, J = 8.7 Hz, 1H), 6.04 (dd, J = 32.7, 8.0 Hz, 2H), 5.29–5.02 (dm, 1H), 4.65–4.46 (m, 3H), 3.87–3.74 (m, 1H), 3.70 (s, 3H), 3.66–3.49 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 163.7, 156.9, 140.3, 139.7, 129.5, 123.9, 123.4, 112.8, 110.7 (d, JC–F = 1.8 Hz), 99.6, 98.2, 91.4 (d, JC–F = 174.9 Hz), 45.31 (d, JC–F = 23.9 Hz), 43.6 (d, JC–F = 22.0 Hz). ESI-MS (m/z): 505.8 [M + 1]+; C22H19Br2FN2O requires 505.0. HPLC (ChiralPak IA, 1.0 mL/min 100% MeOH): tR(major) = 8.37–10.02 min; tR(minor) = 10.26–11.26 min. [α]D20 = −9.5 (c = 0.46, THF).
Animal Studies
All of the animal studies were performed in accordance with the ethical guidelines established by UT Southwestern and the University of Iowa.
In Vivo Neuroprotection Assay
Test compounds were evaluated as described previously.2 Compounds were infused ICV into the left lateral ventricle of four adult (12 week old) wild-type C57BL/J6 mice by means of surgically implanted Alzet osmotic minipumps that delivered solution into the animals at a constant rate of 0.5 μL/h for 7 days. Alternatively, compounds were administered IP twice daily. Bromodeoxyuridine (BrdU) was injected IP at 50 mg kg–1 day–1 for 6 days during pump infusion. Twenty-four hours after the final BrdU administration, mice were sacrificed by transcardial perfusion with 4% paraformaldehyde at pH 7.4, and their brains were processed for immunohistochemical detection of incorporated BrdU in the subgranular zone (SGZ). Dissected brains were immersed in 4% paraformaldehyde overnight at 4 °C and then cryoprotected in sucrose before being sectioned into 40 μm thick free-floating sections. Unmasking of BrdU antigen was achieved by incubation of tissue sections for 2 h in 50% formamide/2× saline sodium citrate (SSC) at 65 °C followed by a 5 min wash in 2× SSC and subsequent incubation for 30 min in 2 M HCl at 37 °C. Sections were processed for immunohistochemical staining with mouse monoclonal anti-BrdU (1:100). The number of BrdU+ cells in the entire dentate gyrus SGZ in the contralateral hemisphere (opposite side of the surgically implanted pump) was quantified by counting BrdU+ cells within the SGZ and dentate gyrus in every fifth section throughout the entire hippocampus and then normalizing for dentate gyrus volume.
MPTP Assay
As previously reported,5 adult male C57Bl/6 mice were individually housed for 1 week and then injected IP daily for 5 days with 30 mg kg–1 day–1 free-base MPTP (Sigma). On day 6, 24 h after the mice received the fifth and final dose of MPTP, daily treatment with compound was initiated. Mice received twice-daily doses of each compound (or vehicle) for the following 21 days, after which mice were sacrificed by transcardial perfusion with 4% paraformaldehyde. The brains were dissected, fixed overnight in 4% paraformaldehyde, and cryoprotected in sucrose for freezing by standard procedures. Frozen brains were sectioned through the striatum and SNc at 30 μM intervals, and every fourth section (spaced 120 μM apart) was stained with antibodies directed against tyrosine hydroxylase (TH) (Abcam, rabbit anti-TH, 1:2500). Diaminobenzidine was used as a chromagen, and tissue was counter-stained with hematoxylin to aid in visualization of the neuroanatomy. Images were analyzed with a Nikon Eclipse 90i motorized research microscope with Plan Apo lenses coupled with Metamorph Image Acquisition software (Nikon). TH+ neurons were counted with ImageJ software (NIH) in every section by two blinded investigators, and the results were averaged and multiplied by the sectioning interval to determine the total number of TH+ neurons per SNc.
Pharmacokinetic Analysis
15 was formulated in 5% DMSO/10% cremophor EL (Sigma, St. Louis, MO)/85% D5W (5% dextrose in water, pH 7.2). Adult mice were dosed IV, IP or via oral gavage (PO) in a total volume of 0.2 mL. Animals were sacrificed at varying times after dosing by inhalation overdose of CO2. Whole blood was collected with a syringe and needle coated with anticoagulant citrate dextrose (ACD) solution (sodium citrate). The blood was subsequently centrifuged at 9300g for 10 min to isolate plasma. Plasma was stored at −80 °C until analysis. Brains were isolated from mice immediately after sacrifice, rinsed extensively with phosphate-buffered saline (PBS), blotted dry, weighed, and snap-frozen in liquid nitrogen. Lysates were prepared by homogenizing the brain tissue in a 3-fold volume of PBS (weight of brain in g × 3 = volume in mL of added PBS). The total lysate volume was estimated as the volume of added PBS plus the volume of brain. Either plasma or brain (100 μL) was processed by addition of 200 μL of acetonitrile containing 0.15% formic acid to precipitate plasma or tissue protein and release bound drug. This mixture was vortexed for 15 s and then centrifuged at 13120g for 5 min, and the supernatant was analyzed directly by HPLC/MS/MS. Standard curves were prepared by addition of compound to blank plasma or blank brain lysate. A value 3-fold above the signal obtained from blank plasma or brain lysate was designated as the limit of detection (LOD). The limit of quantitation (LOQ) was defined as the lowest concentration at which back-calculation yielded a concentration within 20% of the theoretical value. Pharmacokinetic parameters were calculated using the noncompartmental analysis tool of WinNonLin (Pharsight). Bioavailability was calculated as (AUCoral/AUCIV) × (DoseIV/Doseoral) × 100%, where AUC denotes the area under the concentration–time curve). The brain:blood ratio was calculated using AUC values.
Plasma Protein Binding
Protein binding of compound 15 was determined using a recently described ultracentrifugation method;23 100% compound recovery was achieved during the assay, indicating no loss due to absorption to plastic.
Acknowledgments
We thank Prof. Jef De Brabander (UT Southwestern) for helpful discussions regarding Cu-catalyzed amination and the NIH’s Psychoactive Drug Screening Program at the University of North Carolina. Funding was provided by the Edward N. and Della C. Thome Memorial Foundation and the Welch Foundation (I-1612) (to J.M.R.) and the NSF (2012140236-02) (to H.D.J.-C.). Additional support was received from the NIH (R01 MH087986) to A.A.P. and an unrestricted endowment provided to Steven L. McKnight by an anonymous donor.
Glossary
Abbreviations Used
- ALS
amyotrophic lateral sclerosis
- PD
Parkinson’s disease
- TBI
traumatic brain injury
- BrdU
bromodeoxyuridine
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- SOD
superoxide dismutase
- ICV
intracerebroventricularly
- IP
intraperitoneally
- PO
per os (by mouth)
- TH
tyrosine hydroxylase
- SNc
substantia nigra
- AUC
area under the curve.
Supporting Information Available
Table of screening results from the Psychoactive Drug Screening Program, X-ray crystal structure of (−)-15, comparison of plasma and brain levels for rac-15 and (−)-15, and spectra of synthetic materials. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ J.N. and H.D.J.-C. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Lees A. J.; Hardy J.; Revesz T. Parkinson’s Disease. Lancet 2009, 373, 2055–2066. [DOI] [PubMed] [Google Scholar]; b Hebert L. E.; Scherr P. A.; Bienias J. L.; Bennett D. A.; Evans D. A. State-Specific Projections through 2025 of Alzheimer Disease Prevalence. Neurology 2004, 62, 1645. [DOI] [PubMed] [Google Scholar]
- a Pieper A. A.; Xie S.; Capota E.; Estill S. J.; Zhong J.; Long J. M.; Becker G. L.; Huntington P.; Goldman S. E.; Shen C.-H.; Capota M.; Britt J. K.; Kotti T.; Ure K.; Brat D. J.; Williams N. S.; MacMillan K. S.; Naidoo J.; Melito L.; Hsieh J.; De Brabander J.; Ready J. M.; McKnight S. L. Discovery of a Proneurogenic, Neuroprotective Chemical. Cell 2010, 142, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McKnight S. L.; Pieper A. A.; Ready J. M.; De Brabander J.. Proneurogenic Compounds. U.S. Patent 8,604,074, 2013.; c McKnight S. L.; Pieper A. A.; Ready J. M.; De Brabander J.. Proneurogenic Compounds. U.S. Patent 8,362,277, 2013.
- MacMillan K. S.; Naidoo J.; Liang J.; Melito L.; Williams N. S.; Morlock L.; Huntington P. J.; Estill S. J.; Longgood J.; Becker G. L.; McKnight S. L.; Pieper A. A.; De Brabander J. K.; Ready J. M. Development of Proneurogenic, Neuroprotective Small Molecules. J. Am. Chem. Soc. 2011, 133, 1428–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo J.; Bemben C. J.; Allwein S. R.; Liang J.; Pieper A. A.; Ready J. M. Development of a Scalable Synthesis of P7C3-A20, a Potent Neuroprotective Agent. Tetrahedron Lett. 2013, 54, 4429–4431. [Google Scholar]
- De Jesus-Cortes H.; Xu P.; Drawbridge J.; Estill S. J.; Huntington P.; Tran S.; Britt J.; Tesla R.; Morlock L.; Naidoo J.; Melito L. M.; Wang G.; Williams N. S.; Ready J. M.; McKnight S. L.; Pieper A. A. Neuroprotective Efficacy of Aminopropyl Carbazoles in a Mouse Model of Parkinson Disease. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17010–17015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesla R.; Wolf H. P.; Xu P.; Drawbridge J.; Estill S. J.; Huntington P.; McDaniel L.; Knobbe W.; Burket A.; Tran S.; Starwalt R.; Morlock L.; Naidoo J.; Williams N. S.; Ready J. M.; McKnight S. L.; Pieper A. A. Neuroprotective Efficacy of Aminopropyl Carbazoles in a Mouse Model of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17016–17021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaya M. O.; Bramlett H.; Nadoo J.; Pieper A. A.; Dietrich W. D. III. Neuroprotective Efficacy of a Proneurogenic Compound after Traumatic Brain Injury. J. Neurotrauma 2014, 31, 476–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A. K.; Rivera P. D.; Wang Q.; Chuang J.-C.; Tran S.; Osborne-Lawrence S.; Estill S. J.; Starwalt R.; Huntington P.; Morlock L.; Naidoo J.; Williams N.; Ready J. M.; Eisch A. J.; Pieper A. A.; Zigman J. M.. The P7C3-class of Neuroprotective Compounds Exerts Antidepressant Efficacy in Mice by Increasing Hippocampal Neurogenesis. Mol. Psychiatry 2014, in press [DOI] [PMC free article] [PubMed]
- Asai-Coakwell M.; March L.; Dai X. H.; DuVal M.; Lopez I.; French C. R.; Famulski J.; De Baere E.; Francis P. J.; Sundaresan P.; Sauve Y.; Koenekoop R. K.; Berry F. B.; Allison W. T.; Waskiewicz A. J.; Lehmann O. J. Contribution of Growth Differentiation Factor 6-Dependent Cell Survival to Early-Onset Retinal Dystrophies. Hum. Mol. Genet. 2013, 22, 1432–1442. [DOI] [PubMed] [Google Scholar]
- Mice housed alone and without objects such as nesting material show decreased hippocampal neurogenesis, magnifying the effect of neuroprotective compounds.
- Rough calculations suggested that the test compounds would reach a concentration of approximately 100 nM by the end of the testing period if all of the material remained in the brain.
- Struk L.; Sosnicki J. G. Noncryogenic Synthesis of Functionalized 2-Methoxypyridines by Halogen–Magnesium Exchange Using Lithium Dibutyl(Isopropyl)Magnesate and Lithium Chloride. Synthesis 2012, 44, 735–746. [Google Scholar]
- Das P.; De Brabander J. K. A Room Temperature Copper Catalyzed N-Selective Arylation of β-Amino Alcohols with Iodoanilines and Aryl Iodides. Tetrahedron 2013, 69, 7646–7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Similar observations have been made by the De Brabander group with regard to the work published in ref (13) (De Brabander J. K. Personal communication).; Recently this work was retracted because of variability of the results with some substrates, although we have reproduced the results in Schemes 1 and 2 on a >2 g scale. See:Das P.; De Brabander J. K. Retraction Notice to “A Room Temperature Copper Catalyzed N-Selective Arylation of β-Amino Alcohols with Iodoanilines and Aryl Iodides” [Tetrahedron 69 (2013) 7646–7652]. Tetrahedron 2014, 70, 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombrun A.; Gerber P.; Casi G.; Terradillos O.; Antonsson B.; Halazy S. 3,6-Dibromocarbazole Piperazine Derivatives of 2-Propanol as First Inhibitors of Cytochrome C Release Via Bax Channel Modulation. J. Med. Chem. 2003, 46, 4365–4368. [DOI] [PubMed] [Google Scholar]
- Klunder J. M.; Onami T.; Sharpless K. B. Arenesulfonate Derivatives of Homochiral Glycidol: Versatile Chiral Building Blocks for Organic Synthesis. J. Org. Chem. 1989, 54, 1295–1304. [Google Scholar]
- Zhao X.; Zhuang W.; Fang D.; Xue X.; Zhou J. A Highly Efficient Conversion of Primary or Secondary Alcohols into Fluorides with N-Perfluorobutanesulfonyl Fluoride–Tetrabutylammonium Triphenyldifluorosilicate. Synlett 2009, 779–782. [Google Scholar]
- a Javitch J. A.; D’Amato R. J.; Strittmatter S. M.; Snyder S. H. Parkinsonism-Inducing Neurotoxin, N-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine: Uptake of the Metabolite N-Methyl-4-phenylpyridine by Dopamine Neurons Explains Selective Toxicity. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 2173–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]; b D’Amato R. J.; Lipman Z. P.; Snyder S. H. Selectivity of the Parkinsonian Neurotoxin Mptp: Toxic Metabolite Mpp+ Binds to Neuromelanin. Science 1986, 231, 987–989. [DOI] [PubMed] [Google Scholar]
- Fukuda T. Neurotoxicity of MPTP. Neuropathology 2001, 21, 323–332. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V.; Przedborski S. Protocol for the MPTP Mouse Model of Parkinson’s Disease. Nat. Protoc. 2007, 2, 141–151. [DOI] [PubMed] [Google Scholar]
- Wang C.; Williams N. S. A Mass Balance Approach for Calculation of Recovery and Binding Enables the Use of Ultrafiltration as a Rapid Method for Measurement of Plasma Protein Binding for Even Highly Lipophilic Compounds. J. Pharm. Biomed. Anal. 2013, 75, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht R.; Papadopoulos V.; Rammes G.; Baghai T. C.; Fan J.; Akula N.; Groyer G.; Adams D.; Schumacher M. Translocator Protein (18 KDa) (TSPO) as a Therapeutic Target for Neurological and Psychiatric Disorders. Nat. Rev. Drug Discovery 2010, 9, 971–988. [DOI] [PubMed] [Google Scholar]
- a Bordet T.; Berna P.; Abitbol J.-L.; Pruss R. M. Olesoxime (Tro19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals 2010, 3, 345–368. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sunyach C.; Michaud M.; Arnoux T.; Bernard-Marissal N.; Aebischer J.; Latyszenok V.; Gouarne C.; Raoul C.; Pruss R. M.; Bordet T.; Pettmann B. Olesoxime Delays Muscle Denervation, Astrogliosis, Microglial Activation and Motoneuron Death in an ALS Mouse Model. Neuropharmacology 2012, 62, 2346–2353. [DOI] [PubMed] [Google Scholar]
- Elmkaddem M. K.; Fischmeister C.; Thomas C. M.; Renaud J.-L. Efficient Synthesis of Aminopyridine Derivatives by Copper Catalyzed Amination Reactions. Chem. Commun. 2010, 46, 925–927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.