

Abstract
The defining feature of Parkinson’s disease is a progressive and selective demise of dopaminergic neurons. A recent report on Parkinson’s disease animal model demonstrates that poly (ADP-ribose) (PAR) dependent cell death, also named parthanatos, is accountable for selective dopaminergic neuronal loss. Parthanatos is a programmed necrotic cell death, characterized by PARP1 activation, apoptosis inducing factor (AIF) nuclear translocation, and large scale DNA fragmentation. Besides cell death regulation via interaction with AIF, PAR molecule mediates diverse cellular processes including genomic stability, cell division, transcription, epigenetic regulation, and stress granule formation. In this review, we will discuss the roles of PARP1 activation and PAR molecules in the pathological processes of Parkinson’s disease. Potential interaction between PAR molecule and Parkinson’s disease protein interactome are briefly introduced. Finally, we suggest promising points of therapeutic intervention in the pathological PAR signaling cascade to halt progression in Parkinson’s disease. [BMB Reports 2014; 47(8): 424-432]
Keywords: Dopaminergic neurons, Neurodegeneration, Parkinson’s disease, Parthanatos, Poly (ADP-ribose)
INTRODUCTION
Parkinson’s disease (PD) is characterized by age-dependent and progressive dopaminergic neurodegeneration in the substantia nigra pars compacta, and it can clinically be described as the subsequent progressive loss of fine motor control in patients (1, 2). Despite the wealth of information on the disease-causing mutations in several PD linked genes (e.g., SNCA, parkin, DJ-1, PINK1, and LRRK2 etc.) and a deep understanding of neuropathological features in the affected brain regions, it is still largely unknown about this disorder and active investigations are going on about how dopaminergic neurons degenerate and why they are selectively more vulnerable to progressive cell death.
Poly (ADP-ribose)-dependent cell death, also named parthanatos, is characterized by excessive poly (ADP-ribose) (PAR) synthesis by PAR polymerase 1 (PARP1) (3). Parthanatos is implicated in cell death of diverse disease conditions including neurodegeneration (4). Although parthanatos is largely viewed as a type of cell death causing loss of neuronal viability following an acute insult (i.e., NMDA, or MPTP) or stroke (5-7), recently, it has become a novel focus of research on progressive neurodegenerative disorders including PD (8-11).
This review features the role of PAR-dependent cell death in neurodegeneration of PD. In addition, the potential interactions of PAR and PD signaling network are described. Finally, therapeutic potential of parthanatic signaling pathways in PD are addressed.
STRUCTURE AND METABOLISM OF POLY (ADP-RIBOSE)
Poly (ADP-ribose) is a heterogeneous macromolecule of repeating ADP-ribose units that are linked ribose to ribose by (1→2) O-glycosidic bonds. Chain length varies ranging from 1 to about 200 monomer units. The PAR chain is attached to D, E, and K residues of target proteins, and it is elongated by members of the PAR synthesizing enzyme family such as PARP family (4, 12-14). There are 18 different PARPs that show diverse regional expression, subcellular distribution (i.e. nucleus or/and cytoplasm), and domain composition. Each PARP enzyme exhibits differential PAR synthesizing activity which is induced by different stimuli (15). For instance, the best characterized PARP1 nuclear protein is exclusively present in the nucleus and robustly activated by DNA single strand or double strand break which can be sensed via a DNA binding motif in its N-terminus. It is known that PARP1 contributes to the synthesis of 90% of the total PAR generated following a DNA damage (11).
All PARP members have been identified based upon structural homology to PARP1, and they all contain a PAR-synthesizing catalytic domain. Using NAD+ as a substrate, PARP can elongate ADP-ribose chains via a (1→2) O-glycosidic bond. Besides a simple extension of ADP-ribose, branching also occurs on a regular basis (i.e. once in every 20 to 50 ADP-ribose) when glycosidic bonds are formed between two nicotinamide adjacent to ribose moieties. Through branching and elongation, the structure of PAR can be quite extensive and huge, retaining certain regular patterns which provide a rich environment for protein or DNA interaction (13). PAR can have diverse conformation depending on the extent of PARP activation, unlike macromolecules such as single stranded RNA or double stranded DNA which have regular ribose or deoxyribose backbone structure.
As the distinct structure of PAR suggests, an extensive interaction network has been revealed between PAR or PAR-conjugated proteins and proteins of diverse cellular functions (12). The well-studied PARsylated acceptor proteins are histones, p53, p21, and PARP itself. Extensive studies indicated that the proteins containing the PAR binding motif (16, 17) interact with PAR-conjugated proteins or free PAR. Since PAR is bulky and highly negatively charged, interaction with or post-translational modification by PAR can cause significant conformational changes of the target protein or interacting proteins. Recently published structural analysis illustrated how PAR binding can affect protein structure to a large extent, in the process of regulating its enzymatic activity (18).
PAR conjugating with target proteins including PARP can efficiently be degraded by poly (ADP-ribose) glycohydrolase (PARG) enzyme (19). PARG-mediated hydrolysis of PAR eliminates PAR through exo- and endo-glycosidic activity of the enzyme, and it terminates PAR-related signaling pathways initiated by PARP activation. The endo-glycosidic cleavage of PAR by PARG can generate non-conjugated free PAR which functions in diverse cellular processes including cell death. As expected from the subcellular localization of PARP (See (15)), isoforms of PARG are present in the cytoplasm, mitochondria, and nucleus for its associated action (20). One of the most striking features of PAR metabolism is the dynamic nature of PARsylation. A robust protein PARsylation by PARP is often canceled by rapid PAR degradation through PARG activation. Moreover, orchestrated actions of PARP and PARG can regulate free PAR formation.
FUNCTION OF POLY (ADP-RIBOSE)
PAR regulates diverse cellular functions including DNA repair, maintenance of genomic stability, transcriptional regulation, telomere dynamics and centromere function, trafficking of endosomal vesicles, inflammation, and cell death. For example, the dynamics and precise regulation of PAR levels can be seen in the cell division process (15, 20). Consistent with this notion, cells cannot survive when PAR cycle is disrupted through either deletion of PARP1 and PARP2 or deletion of PARG (21, 22). Moreover, DNA damage repair and genomic stability are among the most studied functions that are mediated by target protein PARsylation through PARPs, especially the PARP1 and PARP2. Since this review is centered on understanding the potential PAR functions in neurodegenerative processes, previous review articles are being referred (4, 23, 24) for descriptions on detailed roles of PARPs in diverse cellular functions.
Cells can die due to excessive generation of PAR (11) which is considered as a cell death messenger. This type of cell death is termed parthanatos, as previously mentioned, by combining “PAR” and the word “Thanatos” which refers to the god of death in Greek mythology (25). According to a cell death nomenclature committee, parthanatos is defined by two main criteria. Firstly, excessive PAR synthesis should accompany cell death. Secondly, the cell death should be completely or partially prevented by PARP deletion or inhibitor treatment. The characteristic features include large-scale DNA fragmentation and chromatin condensation (3). PAR-dependent cell death is not widely conceived compared to apoptosis. However, parthanatos definitely contributes to several types of cell death including neurodegeneration. PARP mediates its cell death signaling in response to DNA damage, through rapid activation followed by a transient production of PAR using Nicotinamide adenine dinucleotide (NAD+) as a substrate. Parthanatos, the PAR mediated cell death, occurs due to acute insults such as stroke, in several intoxication models of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced mitochondrial dysfunction, or in N-methyl-D-aspartate (NMDA)-induced neuronal over-excitation. Interestingly, a recent study using PD mouse models suggested that PAR-dependent cell death is dominant in slowly progressing dopaminergic neurodegeneration in vivo (26).
EXECUTION OF CELL DEATH BY EXCESS PAR SYNTHESIS
As discussed briefly above, excessive PAR synthesis following PARP1 activation via stresses including DNA damage is sufficient to signal parthanatic cell death. Although PAR elevation is transient due to rapid degradation by PARG, the parthanatos signal is irreversible once activated. In the cell death pathway, PARP1 is the major PAR synthesizing enzyme which accounts for about 90% of the total PAR synthesis (11). Deletion of PARP1 is sufficient to provide protection against DNA damage-induced parthanatic cell death. Resistance to parthanatos inducing stresses has been well characterized in PARP1 null mice (6, 7, 27).
Different from other PARPs which exhibit cytoplasmic and nuclear localization, PARP1 is exclusively present in the nucleus and contains the domain which can detect breakage in single or double strand DNA. PARP2 can also be activated by DNA damage via its DNA binding zinc finger domain (28). Due to PARP redundancy, PARP1 null mice are viable and can develop normally. However, deletion of both PARP1 and PARP2 results in embryonic lethality (21). Therefore, basal PAR levels are important for maintaining genomic stability through DNA damage repair via recruitment of PAR binding repair protein complexes. However, if the DNA damage is beyond the level of repair, excessive PAR synthesis (up to 200 fold of basal levels) by PARP1 will release free PAR from the nucleus into the cytoplasm, where PAR signals translocate apoptosis inducing factor (AIF) from the outer membrane of mitochondria into the nucleus (11). As AIF translocates into the nucleus, it brings the associated endonuclease (yet to be identified) to produce large-scale DNA fragmentation and condensation which are the characteristics of parthanatos. The ATP and NAD+ depletion accompany the strong activation of PARP1. Since ATP and NAD+ are the energy sources and fuel enzymatic processes for survival, they have been considered to be parts of the mechanisms by which parthanatos kills the cells. However, injection of poly(ADP-ribose) is sufficient to induce mitochondrial AIF release and large amount of DNA fragmentation, which are the characteristic features of parthanatos (29). Being consistent with the notion that the levels of PAR itself are the key determinants for parthanatos (Fig. 1), even without excessive PARP1 activaactivation, PARG deletion and resulting elevation of PAR levels can lead to neurodegeneration in drosophila (30).
Fig. 1. Illustration of potential involvement of PAR in the pathophysiology of PD. Excessive PAR synthesis can cause cell death via release of mitochondrial AIF. Subsequent nuclear translocation of AIF is responsible for PAR dependent cell death, or parthanatos. PAR is synthesized in response to PARP1 overactivation. In PD animal models, mitochondrial toxins can lead to calcium influx which is induced by energy imbalance and glutamate release. Intracellular calcium activates nitric oxide (NO) synthase which produces NO. The NO reacts with reactive oxygen species and is converted to peroxynitrite, a potent DNA damaging reagent. DNA damage can stimulate PARP1 activation, leading to dopamine cell loss. Even in the absence of DNA damage, E3 ubiquitin ligase parkin dysfunction and resulting accumulation of the substrate AIMP2 can also enhance PARP1 activity via direct binding. PAR overproduction may affect not only the AIF mediated cell death, but other cellular functions as well, by its interaction with proteins containing the PAR binding motif.
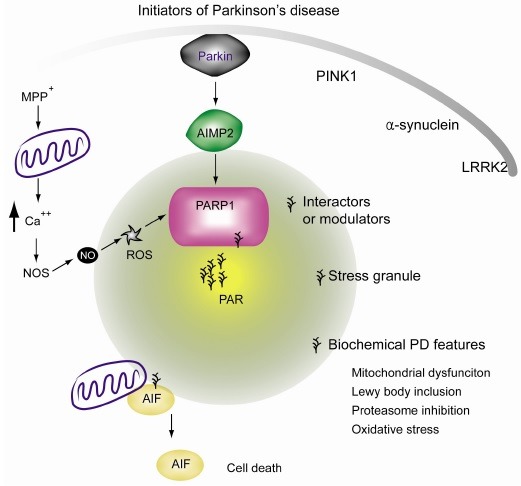
PARP1 STIMULATING SIGNALS
Alkylating agents such as anti-cancer drugs (e.g. temozolomide [TMZ], N-methyl-N-nitro-N-nitrosoguanidine [MNNG], and dacarbazine), gamma irradiation, oxidative stress, and excitatory stimulation followed by NO synthase activation commonly lead to PARP1 activation via DNA breakage. For instance, MPTP intoxication or NMDA excitation converge in an intracellular calcium influx which in turn activates NO synthase (Fig. 1). NO can react and produce peroxynitrite which induces damage to DNA and PARP1 activation. In this scenario, overactivation of PARP1 is directly linked to neuronal death.
Although PARP1 activation in response to DNA damage is extensively studied, there is also a DNA-damage independent path to PARP1 activation. Firstly, PARP1 can be activated by sensing irregular DNA structure to regulate gene expression (13). Secondly, during a long term memory formation, PARP1 is activated and this event is critically important for immediate early gene expression to consolidate synaptic modulation (31). In this context, phosphorylated ERK binding to PARP1 is sufficient to lead to PARP1 activation and PAR synthesis. The association of pERK and activated PARP1 in turn regulates immediate early genes expression such as c-fos, via histone acetylation (32). Interestingly, PARP1 inhibition in mice leads to defective long term memory formation (33), indicating the role of PARP1 in neuronal excitation induced gene expression. Thirdly, CCCTC-binding factor (zinc finger protein) (CTCF) can also bind to PARP1 and stimulate PARP1 activation (34). CTCF is known to function in insulator process or gene imprinting. CTCF induced PARP1 PARsylation recruits DNMT1 to CTCF targeted locus. DNMT1 interaction with PAR on PARP1 inhibits DNMT1 function to block methylation of DNA, ultimately keeping those regions safe from DNA methylation. Although aforementioned DNA damage-independent PARP1 activation is not directly associated with cell death, gene regulation by PAR metabolism might be involved in and intersect with disease or aging processes.
Finally, another illustration of DNA damage independent mechanism of PARP1 activation is that a parkin substrate, AIMP2, mediates dopaminergic neurodegeneration via PARP1 activation in PD mouse model in vivo (26). Different from PARP1 activation by pERK or CTCF, which regulate gene expression or epigenetic information, association of AIMP2 with PARP1 directly leads to parthanatos. Dopaminergic neuronal loss induced by AIMP2 accumulation accompanies elevation of PAR levels. AIMP2-stimulated cell death is completely blocked by either PARP1 gene deletion or PARP inhibitor treatment, thus fulfilling the definition of parthanatos.
Although our discussion is mainly focused on how PARP1 can be activated by various stress signals or protein interactions, there are a lot of unknown stresses or contexts by which other PARP family members are specifically activated. Future studies on PAR and its role in diverse cellular compartments and signal transduction are required for better understanding of pathophysiological roles of PARP families (Fig. 1).
DOPAMINERGIC NEURON SELECTIVITY OF PARTHANATOS
Similar to the extensive involvement of PARP1 activation and excessive PAR synthesis in tumor cell death, acute neurological disorders, and chronic neurodegenerative disease, parthanatos is quite nonspecific and generally occurs in many different cell types including neurons. For example, stroke or NMDA excitotoxicity kills neurons when stresses are applied via PARP1 activation, while MPTP toxin kills dopaminergic neurons in the substantia nigra (Fig. 1). The question remains on how this nonspecific type of cell death translates to PD characterized by progressive and selective dopaminergic neuron degeneration. One answer can be found by looking at the expression levels of PARP1 in various cell types. According to CAGE (Cap Analysis of Gene Expression) analysis developed and updated as a part of functional annotation of the mammalian genome (FANTOM) project by RIKEN (35, 36), PARP1 is exceptionally abundant in nigra dopaminergic neuronal cell types compared to others (i.e., approximately three times more abundant in nigra dopaminergic neurons compared to other neurons or astrocytes). Although PARP1 is quite an abundant nuclear protein in general, the additional pool present in dopaminergic neurons may provide a broader window of PAR regulation. It is conceivable that dopaminergic neurons may be more keenly susceptible to parthanatic cell death, in response to the same PARP1 activating stimuli.
Secondly, the specificity of parthanatos in PD could be understood in pathways upstream of PARP1 activation, since once PARP1 is overactivated, it leads to neuronal toxicity in general. In this respect, the pacemaking potential of dopaminergic neurons places this neuron’s mitochondria under constant burden to buffer calcium (37, 38). Combined with genetic predisposition and aging, mitochondrial derived oxidant stress can ultimately sensitize dopaminergic neurons to PARP1 activation, more substantially compared to other types of neurons. This can simply lead to a cell death program involving DNA damage-induced PARP1 activation. A long standing speculation is that mitochondrial dysfunction and oxidative stress underlie dopaminergic cell loss in PD. It is interesting that PD-linked proteins, especially Parkin and PINK1, are genetically linked to the maintenance of mitochondrial function and structure through mitophagy and quality control (39-41). Dysfunctional mitochondria and subsequently impaired mitochondrial respiration due to parkin or PINK1 inactivation can follow a series of signaling pathways leading to PARP1 activation, in a manner similar to that induced by MPTP mediated complex I inhibition.
Aging is the single most important risk factor in PD (42). However, it is largely unknown how aging may influence PAR metabolism in dopaminergic neurons. Proper basal activities of PARP1 and PARP2 are critical in maintaining genomic stability through sub-lethal DNA damage repair. Age-related progressive damage accumulation and constant PARP1 activation could result in unregulated PAR metabolism, especially when efficient PAR degradation by PARG or other enzymes becomes dysfunctional. It would be instructive to investigate the tissue-specific and age-dependent alteration of expression or activities in PAR regulating enzymes.
Lastly, AIMP2 accumulation is a characteristic feature in many cases of PD, especially of Parkin inactivation (43-46). A recent report on AIMP2-stimulated PARP1 activation indicated that AIMP2 accumulation can activate PARP1 in certain selective contexts; for instance, AIMP2 buildup in cortical neurons does not activate PARP1 (26). The vulnerability of dopaminergic neurons to AIMP2 toxicity seems to stem from the fact that AIMP2 nuclear translocation is facilitated in dopaminergic neurons, thus enabling interaction with the nuclear enzyme PARP1. It is also conceivable that post-translational modifications that are exceptionally active in dopaminergic neurons occur on AIMP2 to influence its trafficking through the cellular compartment. It would be of importance to determine differential AIMP2-PARP1 signaling pathways in dopaminergic vs. non-dopaminergic neurons.
POTENTIAL ROLE OF POLY(ADP-RIBOSE) IN THE PATHOGENESIS OF PD
Research on PD has been largely accelerated via discovery of PD-linked genetic mutations (47, 48). Studies on PD genes and cellular processes in which they are involved has provided new insight on how dopaminergic neurons degenerate in PD. Likewise, to better understand the potential roles of PARP1 activation and PAR elevation in PD, we need to compare and integrate potential PAR regulated signaling cascades with characterized and established signaling pathways involving PD genes (Fig. 1).
In biology, where macromolecules are interconnected to mediate cellular homeostasis, functions of certain molecules are determined by its interactors (i.e., regulator, substrate, or effector). Given that PAR presents with an extensive interactome network (16), it is conceivable that elevated PAR can modulate its interacting proteins to affect the pathophysiology of PD. There are several detailed discussions on the impact of PAR on cell death. Apart from dopaminergic neurodegeneration, patients with PD have additional characteristics such as Lewy body intracellular protein inclusion, proteasome inhibition, mitochondrial dysfunction, and neuronal dysfunction (47), which could potentially interact with PAR molecule. There is, however, no evidence yet that the PAR molecule is involved in the development of PD pathology.
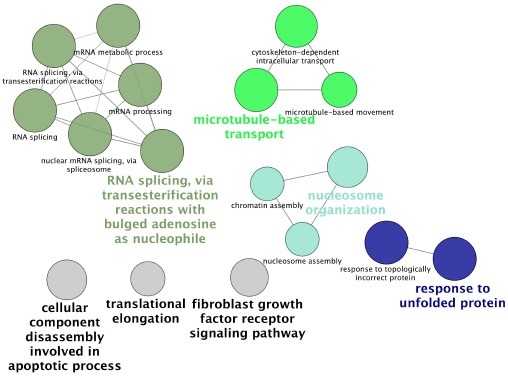
In this section, in order to examine the possibility of interconnection of PAR and PD genetics, we performed a simple comparison of PAR interacting proteins (listed in the reference (16)) and interacting proteins of PD-associated proteins (i.e., Parkin, PINK1, DJ-1, LRRK2, α-synuclein, and AIMP2. From BioGrid public database (49)). The common interacting proteins are listed on Table 1. The functional annotation of these common interactors presents high enrichment of cellular functions such as mRNA splicing, microtubule-based transport, nucleosome organization, and response to unfolded protein, followed by minor enrichment of cellular functions such as translational elongation, cellular component disassembly in apoptotic process, and fibroblast growth factor receptor signaling pathway (Fig. 2, Cytoscape (50) ClueGo plug-in (51)). These functional networks may serve as the communicating points for PD proteins and PAR to ultimately contribute to the pathophysiology of PD.
Table 1. List of common interacting proteins that associate with both PAR and PD proteins.
| Disease Protein | Proteins commonly interacting with both PAR and disease proteins | ||||
|---|---|---|---|---|---|
|
| |||||
| AIMP2 | DARS | EPRS | HNRNPA1 | IARS | IQGAP1 |
| KIAA1967 | KLC2 | LARS | MARS | MATR3 | |
| SNCA | AP1B1 | Ap2a1 | Ap2b1 | Ap2m1 | Atp5c1 |
| Hk1 | Hnrnpa2b1 | IARS | Immt | MAP1B | |
| MAPK1 | Myl12b | Myo5a | Ndufa9 | Ndufb10 | |
| Ndufs1 | Ndufs3 | Ndufv2 | |||
| PARK2 | DNAJA1 | H1F0 | HIST1H1C | HK1 | HK2 |
| HNRNPA1 | HNRNPA2B1 | HNRNPA3 | HNRNPC | HNRNPD | |
| HNRNPF | HNRNPH1 | HNRNPK | HNRNPM | HNRNPR | |
| HNRNPU | HNRPDL | HSP90AA1 | HSP90AB1 | HSP90B1 | |
| IMPDH2 | LMNA | MAPK1 | MATR3 | MCM7 | |
| MRPS22 | MRPS23 | NACA | NDUFS3 | OPA1 | |
| DJ-1 | EFCAB6 | MAP1B | ndufs3 | nonO | sfpq |
| UBC | |||||
| PINK1 | HSP90AA1 | HSP90AB1 | MAP1B | RICTOR | UBC |
| LRRK2 | ARPC4 | EEF1A1 | HSP90AA1 | HSP90AB1R | MDN1 |
| MYL12B | MYL6 | MYO1B | MYO1C | MYO1D | |
| PLEC | PRKDC | RAI14 | RPL23 | RPS8 | |
| UBC | YWHAQ | ||||
Interacting proteins for each PD related proteins (i.e., AIMP2, SNCA, PARK2, PINK1, DJ-1, and LRRK2) were obtained from BioGrid public domain.
Common interactors that are present in both interactomes are presented.
Fig. 2. Gene ontology analysis of PAR and PD protein interactome. By using Cytoscape ClueGo, each common interactor in the Table 1 was functionally annotated. Functional annotations which are highlighted by at least three proteins are presented.
Besides protein interaction network, it is noteworthy to understand that PAR can interact with several transcription factors and can modulate chromatin structure for differential gene expression. Since PAR elevation progresses chronically in PD, especially when AIMP2 is accumulated (26), it would be informative to compare gene expression profiling changes that are dependent on PAR elevation. Gene dosage of disease proteins such as α-synuclein or LRRK2 has been implicated in PD pathogenesis, as such similar pathogenic changes of gene expression by aberrant PAR alteration can be disease-related. Potential gene regulation by PAR could be at the posttranscriptional level because microRNA regulation machinery is sequestered in the stress granule. The function and structure of stress granule is modulated by cytosolic PARP and possibly by free PAR molecules (52-54). MicroRNA pathways have been implicated in PD pathogenesis, especially from the studies of LRRK2 disease mutant in drosophila (55). It is important to understand microRNA profile changes on nuclear PARP1 activation, or in association with LRRK2 disease variants and elevation of free PAR polymers in the stress granule.
Overall, PAR synthesis by PARP1 activation can interact with the PD gene interactome, suggesting wide crosstalk and modulation between these two signaling pathways. Considering extensive interaction of PAR with enzyme like kinases and E3 ligases, there could be unknown PAR signaling cascades which may play an important role in PD pathogenesis.
THERAPEUTIC OPTIONS
AIMP2-PARP1 signaling pathway in vivo suggests that parthanatos could be involved in slowly progressing dopaminergic neurodegeneration in PD. Speculation on how to prevent dopaminergic cell loss in PD may now be expanded to specifically target the parthanatic cell death cascade. It is important to understand the precise upstream and downstream processes of PARP1 activation to find suitable drug targets to halt disease progression in PD.
First of all, PARP1 inhibition could be a promising point of therapeutic intervention. Indeed, there are several PARP inhibitors that are in clinical trials for cancer treatment (24). Since brain permeable PARP inhibitors are also available, these drugs can be candidates to block excessive PARP1 activation, thus preventing parthanatos in dopaminergic neurons in PD. As a proof of concept, in an animal model on a prolonged drug trial, a PARP1 inhibitor effectively prevented AIMP2-induced dopaminergic cell loss (26). Potential detrimental drug effects especially from a prolonged treatment cannot be excluded, because this type of PARP inhibitor can block many other PARP family members. Considering the diverse known functions including the basal protective role of PARP1 and 2 against DNA damage and unknown functions of other PARP family members, targeting upstream of PARP1 activation could be safer and more reasonable.
AIMP2 nuclear localization and subsequent PARP1 activation is critical in the execution of cell death. Determination of signaling pathways mediating AIMP2 nuclear translocation or drug screening for inhibitors of AIMP2 - PARP1 binding are also promising targets of cell death signaling in PD.
Subsequent to PARP1 hyperactivation in the nucleus, high levels of PAR or PARsylated proteins like histones or p53 are likely to mediate transport of PAR out of the nucleus into the cytoplasm for its action. Some members of the histone family are shown to localize to mitochondria upon stress from DNA damage (56). Although further studies are required, identification of PAR carriers will provide additional points of therapeutic intervention to specifically target PARP1 activated crosstalk with mitochondrial AIF release. Approaches to sequester cytosolic PAR can be attempted, but considering the physiological roles of PAR and locally restricted cytosolic PARPs, these attempts may affect yet unknown physiological functions of PARPs.
CONCLUSION AND FUTURE DIRECTION
The structural and functional diversity exerted by 18 PARP family members through synthesis of poly(ADP-ribose) molecule at distinct temporal and spatial trigger points is somewhat challenging to fully understand. This review sheds light on the potential contribution of PARP1 mediated parthanatic cell death in the selective loss of dopaminergic neurons in PD. Although increases in PARsylated proteins have been determined by western blots in the ventral midbrain of AIMP2 PD animal models and postmortem PD patient brains, real time monitoring of PAR synthesis is required to better understand the disease progression. This is especially true because robust PAR synthesis is temporal, and once the level of PAR reaches the threshold required to release AIF from the mitochondria, the cell is destined for parthanatos. Development of fluorescence resonance energy transfer (FRET) based on PAR sensors and incorporating it into mouse genetics may allow us to monitor PARP1 activity during aging and disease progression in vivo.
Using a simple comparison of reported interactions of proteins for PAR and PD related proteins, we were able to identify several proteins that are involved in cellular processes including trafficking and RNA processing. The comparative extent of parthanatos in different models of PD still requires further study. So far, the AIMP2 transgenic mouse model only provided evidence of parthanatos in dopaminergic neurons. AIMP2 is a pathologic substrate of parkin, which is an E3 ubiquitin ligase whose mutation causes autosomal recessive PD. Since PINK1 and parkin are genetically linked to the regulation of dopaminergic cell survival as well as mitochondria function (39, 40), it would be of interest to determine whether AIMP2 protein level or PARP1 activity is enhanced in conditional parkin or PINK1 mouse models. As emphasized in many reviews, PD is a complex neurological disorder characterized by dopaminergic neuronal cell death and functional deficits of multiple cell types including those in the gastrointestinal system. Thus, potential dysregulation of PAR metabolism in animal models of autosomal dominant PD genes can be examined not only in the ventral midbrain, but also in the other regions affected by PD. Additional studies will provide necessary information on the extent of parthanatos in different cases of PD; these will undoubtedly be facilitated by the development of sensitive tools to monitor PAR levels for the accurate monitoring of parthanatos in animal models.
Despite PARP1’s sole known action is mediating neuronal loss, other PARP family members will likely become the subject of future studies. Members of the PARP family that are involved in stress granule formation and regulation of microRNA response could be associated with translation deficits elicited by LRRK2 dysfunction. Overall, the expansion of our knowledge including diverse PAR function and PARP family members will eventually allow us to select appropriate therapeutic options to halt or mitigate pathology in PD.
Acknowledgments
This research was supported by the grants of the NRF (NRF-2012R1A1A1012435) funded by Korea Ministry of Science, ICT & Future Planning (MSIP) and also supported by Samsung Biomedical Research Institute grant (SBRI, SMX1132521).
References
- 1.Lang A. E., Lozano A. M. Parkinson's disease. Second of two parts. N. Engl. J. Med. (1998);339:1130–1143. doi: 10.1056/NEJM199810153391607. [DOI] [PubMed] [Google Scholar]
- 2.Lang A. E., Lozano A. M. Parkinson's disease. First of two parts. N. Engl. J. Med. (1998);339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L., Aaronson S. A., Abrams J., Alnemri E. S., Andrews D. W., Baehrecke E. H., Bazan N. G., Blagosklonny M. V., Blomgren K., Borner C., Bredesen D. E., Brenner C., Castedo M., Cidlowski J. A., Ciechanover A., Cohen G. M., De Laurenzi V., De Maria R., Deshmukh M., Dynlacht B. D., El-Deiry W. S., Flavell R. A., Fulda S., Garrido C., Golstein P., Gougeon M. L., Green D. R., Gronemeyer H., Hajnoczky G., Hardwick J. M., Hengartner M. O., Ichijo H., Jaattela M., Kepp O., Kimchi A., Klionsky D. J., Knight R. A., Kornbluth S., Kumar S., Levine B., Lipton S. A., Lugli E., Madeo F., Malomi W., Marine J. C., Martin S. J., Medema J. P., Mehlen P., Melino G., Moll U. M., Morselli E., Nagata S., Nicholson D. W., Nicotera P., Nunez G., Oren M., Penninger J., Pervaiz S., Peter M. E., Piacentini M., Prehn J. H., Puthalakath H., Rabinovich G. A., Rizzuto R., Rodrigues C. M., Rubinsztein D. C., Rudel T., Scorrano L., Simon H. U., Steller H., Tschopp J., Tsujimoto Y., Vandenabeele P., Vitale I., Vousden K. H., Youle R. J., Yuan J., Zhivotovsky B., Kroemer G. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death. Differ. (2009);16:1093–1107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schreiber V., Dantzer F., Ame J. C., de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. (2006);7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 5.Szabo C., Dawson V. L. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends. Pharmacol. Sci. (1998);19:287–298. doi: 10.1016/S0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 6.Wang H., Shimoji M., Yu S. W., Dawson T. M., Dawson V. L. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson's disease. Ann. N. Y. Acad. Sci. (2003);991:132–139. doi: 10.1111/j.1749-6632.2003.tb07471.x. [DOI] [PubMed] [Google Scholar]
- 7.Yu S. W., Wang H., Dawson T. M., Dawson V. L. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol. Dis. (2003);14:303–317. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Strosznajder J. B., Czapski G. A., Adamczyk A., Strosznajder R. P. Poly(ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer's disease. Mol. Neurobiol. (2012);46:78–84. doi: 10.1007/s12035-012-8258-9. [DOI] [PubMed] [Google Scholar]
- 9.Martire S., Fuso A., Rotili D., Tempera I., Giordano C., De Zottis I., Muzi A., Vernole P., Graziani G., Lococo E., Faraldi M., Maras B., Scarpa S., Mosca L., d M. PARP-1 modulates amyloid beta peptide-induced neuronal damage. PLoS One. 2013;8:e72169. doi: 10.1371/journal.pone.0072169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mashimo M., Kato J., Moss J. ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc. Natl. Acad. Sci. U. S. A. (2013);110:18964–18969. doi: 10.1073/pnas.1312783110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fatokun A. A., Dawson V. L., Dawson T. M. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. (2014);171:2000–2016. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burkle A. Poly(ADP-ribose). The most elaborate metabolite of NAD+. FEBS J. (2005);272:4576–4589. doi: 10.1111/j.1742-4658.2005.04864.x. [DOI] [PubMed] [Google Scholar]
- 13.Gagne J. P., Rouleau M., Poirier G. G. Structural biology. PARP-1 activation--bringing the pieces together. Science. (2012);336:678–679. doi: 10.1126/science.1221870. [DOI] [PubMed] [Google Scholar]
- 14.Luo X., Kraus W. L. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. (2012);26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vyas S., Chesarone-Cataldo M., Todorova T., Huang Y. H., Chang P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. (2013);4:2240. doi: 10.1038/ncomms3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gagne J. P., Isabelle M., Lo K. S., Bourassa S., Hendzel M. J., Dawson V. L., Dawson T. M., Poirier G. G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. (2008);36:6959–6976. doi: 10.1093/nar/gkn771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gagne J. P., Pic E., Isabelle M., Krietsch J., Ethier C., Paquet E., Kelly I., Boutin M., Moon K. M., Foster L. J., Poirier G. G. Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress. Nucleic Acids Res. 2012;40:7788–7805. doi: 10.1093/nar/gks486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z., Michaud G. A., Cheng Z., Zhang Y., Hinds T. R., Fan E., Cong F., Xu W. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl) ation-dependent ubiquitination. Genes. Dev. (2012);26:235–240. doi: 10.1101/gad.182618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slade D., Dunstan M. S., Barkauskaite E., Weston R., Lafite P., Dixon N., Ahel M., Leys D., Ahel I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature. (2011);477:616–620. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim M. Y., Zhang T., Kraus W. L. Poly(ADPribosyl)ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signal. Genes. Dev. (2005);19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 21.Menissier de Murcia J., Ricoul M., Tartier L., Niedergang C., Huber A., Dantzer F., Schreiber V., Ame J. C., Dierich A., LeMeur M., Sabatier L., Chambon P., de Murcia G. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. (2003);22:2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cortes U., Tong W. M., Coyle D. L., Meyer-Ficca M. L., Meyer R. G., Petrilli V., Herceg Z., Jacobson E. L., Jacobson M. K., Wang Z. Q. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol. Cell Biol. (2004);24:7163–7178. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herceg Z., Wang Z. Q. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. (2001);477:97–110. doi: 10.1016/S0027-5107(01)00111-7. [DOI] [PubMed] [Google Scholar]
- 24.Virag L., Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. (2002);54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 25.Andrabi S. A., Dawson T. M., Dawson V. L. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. N. Y. Acad. Sci. (2008);1147:233–241. doi: 10.1196/annals.1427.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee Y., Karuppagounder S. S., Shin J. H., Lee Y. I., Ko H. S., Swing D., Jiang H., Kang S. U., Lee B. D., Kang H. C., Kim D., Tessarollo L., Dawson V. L., Dawson T. M. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. (2013);16:1392–1400. doi: 10.1038/nn.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandir A. S., Przedborski S., Jackson-Lewis V., Wang Z. Q., Simbulan-Rosenthal C. M., Smulson M. E., Hoffman B. E., Guastella D. B., Dawson V. L., Dawson T. M. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc. Natl. Acad. Sci. U. S. A. (1999);96:5774–5779. doi: 10.1073/pnas.96.10.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai P., Canto C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. (2012);16:290–295. doi: 10.1016/j.cmet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 29.Andrabi S. A., Kim N. S., Yu S. W., Wang H., Koh D. W., Sasaki M., Klaus J. A., Otsuka T., Zhang Z., Koehler R. C., Hurn P. D., Poirier G. G., Dawson V. L., Dawson T. M. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. U. S. A. (2006);103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanai S., Kanai M., Ohashi S., Okamoto K., Yamada M., Takahashi H., Miwa M. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. (2004);101:82–86. doi: 10.1073/pnas.2237114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen-Armon M., Visochek L., Katzoff A., Levitan D., Susswein A. J., Klein R., Valbrun M., Schwartz J. H. Long-term memory requires polyADP-ribosylation. Science. (2004);304:1820–1822. doi: 10.1126/science.1096775. [DOI] [PubMed] [Google Scholar]
- 32.Cohen-Armon M., Visochek L., Rozensal D., Kalal A., Geistrikh I., Klein R., Bendetz-Nezer S., Yao Z., Seger R. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol. Cell. (2007);25:297–308. doi: 10.1016/j.molcel.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg S., Visochek L., Giladi E., Gozes I., Cohen-Armon M. PolyADP-ribosylation is required for long-term memory formation in mammals. J. Neurochem. (2009);111:72–79. doi: 10.1111/j.1471-4159.2009.06296.x. [DOI] [PubMed] [Google Scholar]
- 34.Guastafierro T., Cecchinelli B., Zampieri M., Reale A., Riggio G., Sthandier O., Zupi G., Calabrese L., Caiafa P. CCCTC-binding factor activates PARP-1 affecting DNA methylation machinery. J. Biol. Chem. (2008);283:21873–21880. doi: 10.1074/jbc.M801170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Severin J., Lizio M., Harshbarger J., Kawaji H., Daub C. O., Hayashizaki Y., Consortium F., Bertin N., Forrest A. R. Interactive visualization and analysis of large-scale sequencing datasets using ZENBU. Nat. Biotechnol. (2014);32:217–219. doi: 10.1038/nbt.2840. [DOI] [PubMed] [Google Scholar]
- 36.Ohmiya H., Vitezic M., Frith M. C., Itoh M., Carninci P., Forrest A. R., Hayashizaki Y., Lassmann T., Consortium F. RECLU: a pipeline to discover reproducible transcriptional start sites and their alternative regulation using capped analysis of gene expression (CAGE). BMC Genomics. (2014);15:269. doi: 10.1186/1471-2164-15-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guzman J. N., Sanchez-Padilla J., Chan C. S., Surmeier D. J. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. (2009);29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guzman J. N., Sanchez-Padilla J., Wokosin D., Kondapalli J., Ilijic E., Schumacker P. T., Surmeier D. J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. (2010);468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. (2006);441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 40.Park J., Lee S. B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J. M., Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. (2006);441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 41.Ziviani E., Tao R. N., Whitworth A. J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. U. S. A. (2010);107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collier T. J., Kanaan N. M., Kordower J. H. Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non-human primates. Nat. Rev. Neurosci. (2011);12:359–366. doi: 10.1038/nrn3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corti O., Hampe C., Koutnikova H., Darios F., Jacquier S., Prigent A., Robinson J. C., Pradier L., Ruberg M., Mirande M., Hirsch E., Rooney T., Fournier A., Brice A. The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: linking protein biosynthesis and neurodegeneration. Hum. Mol. Genet. 2003;12:1427–1437. doi: 10.1093/hmg/ddg159. [DOI] [PubMed] [Google Scholar]
- 44.Ko H. S., von Coelln R., Sriram S. R., Kim S. W., Chung K. K., Pletnikova O., Troncoso J., Johnson B., Saffary R., Goh E. L., Song H., Park B. J., Kim M. J., Kim S., Dawson V. L., Dawson T. M. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J. Neurosci. (2005);25:7968–7978. doi: 10.1523/JNEUROSCI.2172-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ko H. S., Lee Y., Shin J. H., Karuppagounder S. S., Gadad B. S., Koleske A. J., Pletnikova O., Troncoso J. C., Dawson V. L., Dawson T. M. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc. Natl. Acad. Sci. U.S.A. (2010);107:16691–16696. doi: 10.1073/pnas.1006083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imam S. Z., Zhou Q., Yamamoto A., Valente A. J., Ali S. F., Bains M., Roberts J. L., Kahle P. J., Clark R. A., Li S. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease. J. Neurosci. (2011);31:157–163. doi: 10.1523/JNEUROSCI.1833-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore D. J., West A. B., Dawson V. L., Dawson T. M. Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. (2005);28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 48.Martin I., Dawson V. L., Dawson T. M. Recent advances in the genetics of Parkinson's disease. Annu. Rev. Genomics. Hum. Genet. (2011);12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stark C., Breitkreutz B. J., Reguly T., Boucher L., Breitkreutz A., Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. (2006);34:D535–539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. (2003);13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W. H., Pages F., Trajanoski Z., Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. (2009);25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leung A. K., Calabrese J. M., Sharp P. A. Quantitative analysis of Argonaute protein reveals microRNA-dependent localization to stress granules. Proc. Natl. Acad. Sci. U. S. A. (2006);103:18125–18130. doi: 10.1073/pnas.0608845103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson P., Kedersha N. Stress granules: the Tao of RNA triage. Trends. Biochem. Sci. (2008);33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 54.Leung A. K., Vyas S., Rood J. E., Bhutkar A., Sharp P. A., Chang P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol. Cell. (2011);42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gehrke S., Imai Y., Sokol N., Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. (2010);466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okamura H., Yoshida K., Amorim B. R., Haneji T. Histone H1.2 is translocated to mitochondria and associates with Bak in bleomycin-induced apoptotic cells. J. Cell Biochem. (2008);103:1488–1496. doi: 10.1002/jcb.21537. [DOI] [PubMed] [Google Scholar]