

Abstract
Adaptor protein FADD forms the death inducing signaling complex (DISC) by recruiting the initiating caspases-8 and -10 through homotypic death effector domain (DED) interactions. Cellular FLICE-inhibitory protein (c-FLIP) is an inhibitor of death ligand-induced apoptosis downstream of death receptors, and FADD competes with procaspase-8/10 for recruitment for DISC. However, the mechanism of action of FADD and c-FLIP proteins remain poorly understood at the molecular level. In this study, we provide evidence indicating that the death effector domain (DED) of FADD interacts directly with the death effector domain of human c-FLIP. In addition, we use homology modeling to develop a molecular docking model of FADD and c-FLIP proteins. We also find that four structure-based mutants (E80A, L84A, K169A and Y171A) of c-FLIP DEDs disturb the interaction with FADD DED, and that these mutations lower the stability of the c-FLIP DED. [BMB Reports 2014; 47(9): 488-493]
Keywords: c-FLIP, Death effector domain, FADD, Interaction, Structure-based mutations
INTRODUCTION
The assembly of activated receptor, FADD (Fas-associated death domain), and procaspases is commonly known as a death-inducing signaling complex, or DISC. FADD, which is the nucleus of this assembly, is responsible for sensing the death stimulus at a receptor, and recruiting the procaspases into the nascent DISC (1). The death inducing signaling complex is formed by FADD recruiting the initiating caspases-8 and -10 through homotypic death effector domain (DED) interactions (2, 3).
Cellular FLICE inhibitory protein (c-FLIP) is expressed in various cancers, and is an inhibitor of death ligand-induced apoptosis downstream of death receptors and FADD (4). c-FLIP protein is recruited to the DISC by its DEDs, and competes with the initiator caspase for the FADD binding site. The long form (c-FLIPL) is highly homologous to procaspases-8 and -10, and possesses tandem DEDs at its amino-terminus, and a catalytically inactive protease domain at its carboxy-terminus. The shorter forms (c-FLIPS and c-FLIPR) consist of only the amino-terminal tandem DEDs, followed by a short carboxy-terminal stretch, and are similar in architecture to the viral FLIPs (5, 6). c-FLIPS and c-FLIPR both inhibit death signaling at the DISC, by inhibiting caspase-8 activation. FADD is constructed from two ∼90 amino acid motifs that are structurally similar to one another (7-9), but that can be readily distinguished by amino acid sequence. The N-terminal DED and the C-terminal death domain (DD) each adopt a six α-helical bundle structure that is characteristic of a structural family of “death motifs”; these include the caspase-recruitment domain (CARD), and the pyrin domain (PYD) (10). The FADD DD is responsible for receptor engagement, while the DED contains the binding site for procaspase-8 and/or -10 (11, 12).
The DDs of Fas and FADD appear to be extensively charged at their surface, and mutagenesis data suggest that they associate with each other through electrostatic interactions (13, 14). In contrast, the DED of FADD possesses a key surface-exposed hydrophobic patch that appears to be conserved in virtually all other DEDs, including the tandem DEDs of procaspase-8 and FLIP proteins. This region is thought to be critical in mediating DED-DED interactions (8). To elucidate the mutual relationships between human FADD and c-FLIP in their respective signal transduction pathways, we investigated the interactions between the recombinant DED of human FADD, and the DED of c-FLIP. Using several biochemical and biophysical assays, we demonstrated a direct protein-protein interaction between FADD and c-FLIP DEDs. Structure-based mutagenesis studies were performed for FADD and c-FLIP DED interactions. The FADD/c-FLIP structures that are described provide a structural basis for the observed changes in the site-specific variants. Overall, the results of this study provide additional insights into the mechanism of the apoptotic signaling pathways of FADD and c-FLIPs proteins.
RESULTS AND DISCUSSION
Domain structure and predicted secondary structure of FADD and c-FLIP
Cellular FLICE-like inhibitory protein (c-FLIP) potently blocks FADD-mediated cell death, by interfering with caspase-8 activation. While a great deal of progress has been made in structural and functional c-FLIP research, the direct interaction between FADD and c-FLIP DEDs has not yet been determined in vitro at the molecular level (15, 16). Achievement of this goal may help interpret and explain the complicated biological actions of FADD DED and c-FLIP DEDs. The domain structures of FADD and c-FLIP are shown in Fig. 1A. The FADD protein (amino acids (aa) 1-208) have an N-terminal region comprising the protein death effector domain (DED, aa 3-81), and a C-terminal death domain (DD, aa 97-181). Full-length c-FLIP protein contains two N-terminal death effector domains (DEDs, 1-73 and 92-170), and a C-terminal caspase domain (aa 263-358). The death effector domains (DEDs) are the core domains that are involved in interactions.
Fig. 1. (A) Schematic representation of the full-length FADD and c-FLIP domains. (B) Co-expressions of FADD DED and c-FLIP DEDs are shown by SDS-PAGE. (C) Analysis of FADD and structure-based mutations (E80A, L84A, K169A and Y171A) of c-FLIP DEDs, as revealed by a GST-tag pull-down. (D) Analysis of FADD and structure-based mutations (M120A, L143A and Q148A) of c-FLIP DEDs. (E) c-FLIP mutants (F23G and F114G/L115G) binding to FADD. The protein interactions were detected by Western blot. (F) Input controls of FADD and c-FLIP are shown.
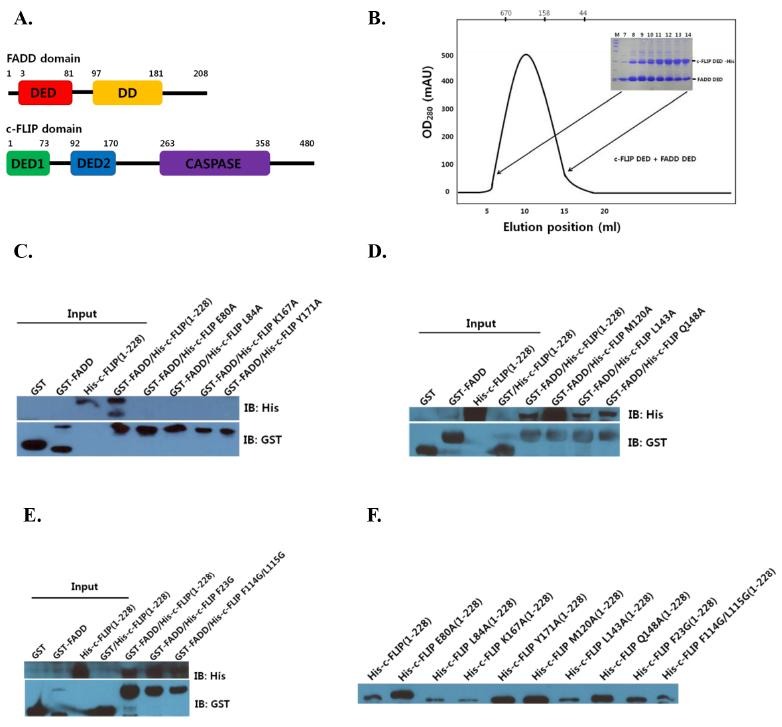
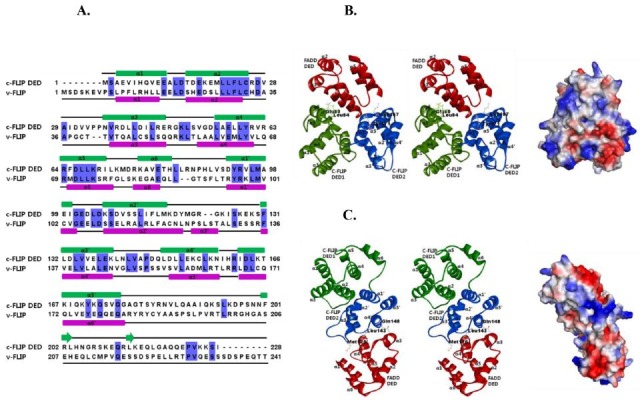
To determine if FADD DED interacts with the c-FLIP DEDs, recombinant FADD (aa 1-83) and c-FLIP (aa 1-228) were isolated by the co-expression method. The soluble FADD and c-FLIP proteins were purified to homogeneity, and analyzed (Fig. 1B). In this study, the FADD and c-FLIP complex band from the gel-filtration showed the formation of a high oligomeric DED complex. Relative to v-FLIP (33%), c-FLIP DED was not highly conserved (Fig. 2A). The structure of c-FLIP is still unknown, but that of v-FLIP has been elucidated (17). The secondary structure of the c-FLIP DEDs in comparison with v-FLIP was also predicted; and c-FLIP DED1 and DED2 were shown to be composed of six and five α-helices, respectively.
Fig. 2. (A) Sequence alignment and predicted secondary structure of the c-FLIP DED and v-FLIP are shown. α-helices are shown as green and purple ellipses, loops as black lines, and β-sheets as green arrows. Residues conserved across two species are colored in blue. (B) The first model of the FADD DED and c-FLIP DEDs complex structure is shown as ribbon representations (left panel). Predicted binding sites between FADD and c-FLIP DEDs are illustrated. The modeled FADD and c-FLIP DEDs complex structure is shown as surface representation. Blue and red represent positive and negative electrostatic potential, respectively (right panel). (C) The second model of the FADD and c-FLIP DEDs complex is shown as ribbon (left panel), and surface representation (right panel).
Binding between FADD DED and c-FLIP DEDs and their modeled structures
We conducted a GST pull-down, and investigated the binding between FADD DED and c-FLIP DEDs by Western blot analysis (Figs. 1C-F). Structural studies of their interaction have been difficult. The prediction of protein-protein interactions is an important step toward the elucidation of protein functions, and understanding the molecular mechanisms inside the cell.
To evaluate structural aspects of the interaction between FADD (1-83) and c-FLIP (1-228) DEDs, we modeled the FADD DED (2-82) structure, using the known structure of human FADD DED/DD (PDB ID: 2GF5, 1-191). Next, we modeled a c-FLIP DEDs (2-172) structure, using the v-FLIP DEDs (PDB ID: 3CL3, 1-183). We selected the 10 lowest-energy levels created in 1,000 docking run structures, and focused on two models of the ten complexes. In the first model, FADD DED was docked to the interface between DED1 and DED2 in the c-FLIP. The α1 and α4 of the FADD, and α5’ and α6 of the c-FLIP regions, may be interaction regions between FADD and c-FLIP. In the first model, FADD DED interacted strongly with DED1 and DED2 in the c-FLIP (Fig. 2B). Conversely, in the second model, FADD DED only interacted with DED2 in the c-FLIP (Fig. 2C). To determine whether both DED1 and DED2 of c-FLIP are dependent on the interaction, we examined the effects of mutants of c-FLIP on complex formation. As predicted, the four single site mutants of c-FLIP DEDs (E80A, L84A, K167A and Y171A) exhibited very weak interaction with FADD DED (Fig. 1C). These results indicate that the predicted interaction sites in the modeled complex structures play important roles in the structural stability of the FADD and c-FLIP protein complexes.
In the second model, three single mutants (M120A, L143A and Q148A) in DED2 of c-FLIP exhibited strong interactions with FADD DED (Fig. 1D). The loop regions of α2’, α3’ and α4’ on c-FLIP DED2 interacted with the α3 and α4 helices on FADD DED. In contrast with the requirement for the first model, the three mutants investigated in the second model had no effect on the ability to form a complex between FADD and c-FLIP. These results suggest that the first model is more reasonable for FADD-c-FLIP complex conformation. We predicted interaction sites using the modeled complex structures, including the mutations (Fig. 2). For comparison, we examined other c-FLIP mutants (F23G and F114G/L115G) binding to FADD by the Schmitz group (17). The two mutants in DED2 of c-FLIP exhibited strong interaction with FADD DED (Fig. 1E).
The secondary structure of FADD and c-FLIP
Loop regions are often flexible, and adopt several different conformations. The loops are involved in the function of the protein, and can switch from an open conformation. In FADD DED, loop residues between α1 and α5 had negative charges; whereas, most residues of the FADD DED had positive charges in a globular fold (Fig. 2). These charges may promote formation of the FADD-FLIP complex. Because the surface features of DED proteins dictate their modes of interactions with partners, the electrostatic surface differential is critical to interactions with partner proteins.
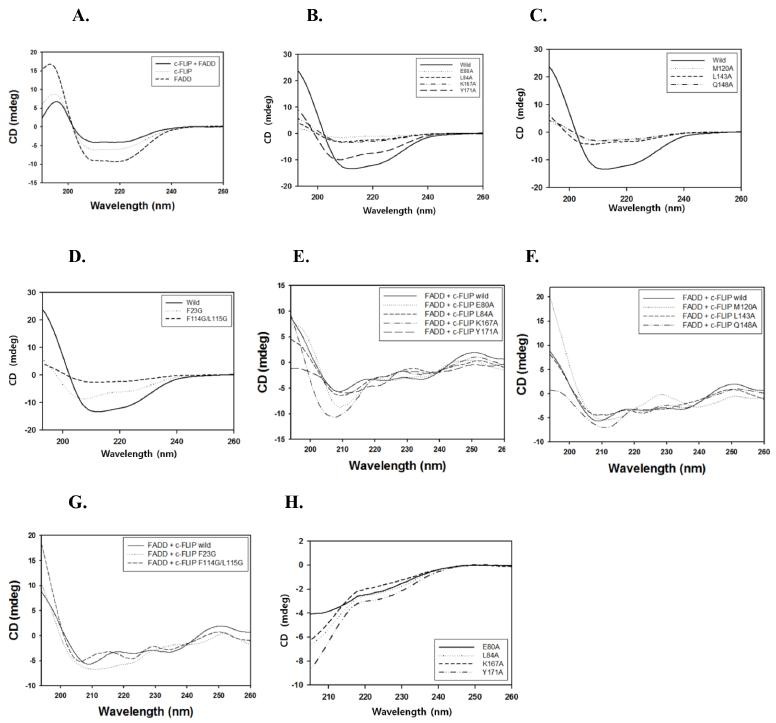
To determine the nature of the secondary structural elements of FADD and c-FLIP, far-UV CD spectra were recorded and analyzed (Fig. 3A). The CD spectra of purified FADD and c-FLIP DEDs exhibited two negative maxima at 208 and 223 nm, and were presumed to predominantly have α-helical structures. To investigate the effects of the mutations on the structural integrity of the c-FLIP protein, CD spectra were obtained for each mutant, as well as for wild-type c-FLIP (Figs. 3B-G). The spectrum of the wild type produced the greatest negative peak; whereas, the curves of the other nine mutations (E80A, L84A, Y171A, K167A, M120A, L143A, Q148A, F23G and F114G/L115G) were less negative. Next, we performed CD measurements of the mutants in the presence of urea, to determine whether the proteins were denatured, or not. The spectra of the mutants were completely different from those of denatured proteins (Fig. 3H). The CD spectra of the c-FLIP mutants showed that each mutation affected the conformation of FADD to a different extent. The CD study showed that due to their mutations, the secondary structures of the nine mutants were more or less lost. This loss of secondary structure in each protein was a result of either its local structural perturbation, or the increase in the unfolded fraction of the protein.
Fig. 3. (A) Far-UV CD spectra of the FADD-c-FLIP complex, and of each individual protein. (B-D) Far-UV CD spectra of the wild-type c-FLIP, and its mutants are represented. (E-G) Far-UV CD spectra of the c-FLIP mutants with FADD protein are shown. (H) Far-UV CD spectra of the c-FLIP mutants are shown in the presence of urea.
FADD DED interacts with c-FLIP DED
To further investigate the interaction between FADD and c-FLIP DEDs, the fluorescence emission spectra of purified FADD DED and c-FLIP DEDs were measured, based on λmax curve detection at 330 nm (Fig. 4A). The spectra of both FADD-c-FLIP complexes were of lower intensity, than those observed when simply combining FADD and c-FLIP. The fluorescence intensities were approximately 1,500 N for FADD, and 800 N for c-FLIP. The interaction was most likely accompanied by significant conformational changes in either one or both proteins, and was likely mediated, as the residues of the aromatic groups were buried within the 3D protein structure. A less rigid hydrophobic environment required for the conformational changes associated with FADD and c-FLIP was observed, as a decrease in fluorescence intensity. Based on our overall findings, the FADD DED region interacts with c-FLIP DEDs.
Fig. 4. (A) Fluorescence analysis of FADD DED binding to c-FLIP DEDs. Fluorescence spectra of the FADD-c-FLIP complex, and of each individual protein are shown. (B) DSC analysis of FADD and c-FLIP. Temperatures were set from 0 to 90℃, and the heat flow was 1℃/min. (C) Thermal unfolding of FADD DED protein was measured by UV-Vis spectrophotometer. (D) Thermal unfolding of c-FLIP DEDs protein was measured. Absorbance was monitored in the 285-340 nm range, under various pH conditions. (E) Co-immunoprecipitation (IP) assay for FADD and c-FLIP interaction. HEK293T cell lysates transfected with plasmids encoding FADD (pCMV-Tag2B) and c-FLIP (pCMV-Tag3B) were immunoprecipitated with control IgG, anti-Flag antibodies, and subjected to immunoblot analysis with the indicated antibodies.
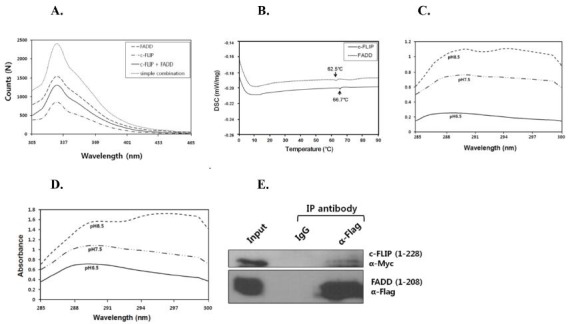
Differential scanning calorimetry (DSC) was used to analyze the effects of heat on protein stability. DSC scans were performed for conditions under which the protein was irreversibly unfolded. The protein showed thermal transitions or denaturation at 0oC, up to 90oC. FADD DED and c-FLIP DEDs showed endothermic peaks of temperature (Td) at 62.5 and 66.7oC, respectively (Fig. 4B). To examine the changes in the protein’s secondary or tertiary structure under different pH conditions, UV absorption spectra of the FADD and c-FLIP DEDs proteins were measured. The intensity of the FADD and c-FLIP proteins increased gradually, as the pH increased from 6.5 to 8.5. The peaks in the difference spectra of the proteins were related to the FADD and c-FLIP conformational changes. FADD and c-FLIP were more soluble under high pH (Figs. 4C and D).
To determine whether FADD interacts with c-FLIP in vivo, co-immunoprecipitation assays were carried out in HEK293T cells transfected with expression vectors encoding FADD (pCMV-Tag2B) and c-FLIP (pCMV-Tag3B). FADD was co-immunoprecipitated with c-FLIP, indicating that FADD interacts with c-FLIP (Fig. 4E). We carried out the purification and biological characterization of FADD and c-FLIP DEDs protein in E. coli. Furthermore, by performing a series of biochemical and biophysical measurements, we confirmed that FADD DED interacts with c-FLIP DEDs. Interestingly, four structure-based mutations (E80A, L84A, K169A and Y171A) disturbed the interaction with FADD DED; and these mutations lowered the stability of the c-FLIP DED. Overall, this study provided important data regarding the FADD DED and c-FLIP DED structures, the binding activity of FADD DED to the c-FLIP death effector domains, and the protein-protein interactions involving the regulation of both apoptosis and necrosis.
MATERIALS AND METHODS
Materials and Methods were provided in Supplementary data: http://www.bmbreports.org/jbmb_by_volume.html?vol=47.
Acknowledgments
This study was supported by the Basic Science Research Program, through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2013-054737) to S.B.J., and (2013-054754) to M.S.J.
References
- 1.Carrington P. E., Sandu C., Wei Y., Hill J. M., Morisawa G., Huang T., Gavathiotis E., Wei Y., Werner M. H. The structure of FADD and its mode of interaction with procaspase-8. Mol. Cell. (2006);22:599–610. doi: 10.1016/j.molcel.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 2.Kischkel F. C., Lawrence D. A., Tinel A., LeBlanc H., Virmani A., Schow P., Gazdar A., Blenis J., Arnott D., Ashkenazi A. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. Biol. Chem. (2001);49:46639–46646. doi: 10.1074/jbc.M105102200. [DOI] [PubMed] [Google Scholar]
- 3.Wang J., Chun H. J., Wong W., Spencer D. M., Lenardo M. J. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. U.S.A. (2001);98:13884–13888. doi: 10.1073/pnas.241358198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krueger A., Baumann S., Krammer P. H., Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol. Cell. Biol. (2001);21:8247–8254. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golks A., Brenner D., Fritsch C., Krammer P. H., Lavrik I. N. c-FLIPR, a new regulator of death receptor-induced apoptosis. J. Biol. Chem. (2005);280:14507–14513. doi: 10.1074/jbc.M414425200. [DOI] [PubMed] [Google Scholar]
- 6.Krueger A., Schmitz I., Baumann S., Krammer P. H., Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem. (2001);276:20633–20640. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 7.Berglund H., Olerenshaw D., Sankar A., Federwisch M., McDonald N. Q., Driscoll P. C. The three-dimensional solution structure and dynamic properties of the human FADD death domain. J. Mol. Biol. (2000);302:171–188. doi: 10.1006/jmbi.2000.4011. [DOI] [PubMed] [Google Scholar]
- 8.Eberstadt M., Huang B. H., Chen Z. H., Meadows R. P., Ng S. C., Zheng L. X., Lenardo M. J., Fesik S. W. NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature. (1998);392:941–945. doi: 10.1038/31972. [DOI] [PubMed] [Google Scholar]
- 9.Jeong E. J., Bang S., Lee T. H., Park Y. I., Sim W. S., Kim K. S. The solution structure of FADD death domain. Structural basis of death domain interactions of Fas and FADD. J. Biol. Chem. (1999);274:16337–16342. doi: 10.1074/jbc.274.23.16337. [DOI] [PubMed] [Google Scholar]
- 10.Fesik S. W. Insights into programmed cell death through structural biology. Cell. (2000);103:273–282. doi: 10.1016/S0092-8674(00)00119-7. [DOI] [PubMed] [Google Scholar]
- 11.Chinnaiyan A. M., O’Rourke K., Tewari M., Dixit V. M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. (1995);81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 12.Chinnaiyan A. M., Tepper C. G., Seldin M. F., O’Rourke K., Kischkel F. C., Hellbardt S., Krammer P. H., Peter M. E., Dixit V. M. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J. Biol. Chem. (1996);276:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- 13.Bang S., Jeong E. J., Kim I. K., Jung Y. K., Kim K. S. Fas- and tumor necrosis factor-mediated apoptosis uses the same binding surface of FADD to trigger signal transduction. A typical model for convergent signal transduction. J. Biol. Chem. (2000);275:36217–36222. doi: 10.1074/jbc.M006620200. [DOI] [PubMed] [Google Scholar]
- 14.Hill J. M., Morisawa G., Kim T., Huang T., Wei Y., Werner M. H. Identification of an expanded binding surface on the FADD death domain responsible for interaction with CD95/Fas. J. Biol. Chem. (2004);279:1474–1481. doi: 10.1074/jbc.M304996200. [DOI] [PubMed] [Google Scholar]
- 15.Day T. W., Huang S., Safa A. R. c-FLIP knockdown induces ligand-independent DR5-, FADD-, Caspase-8-, and Caspase-9-dependent apoptosis in breast cancer cells. Biochem. Pharmacol. (2008);76:1694–1704. doi: 10.1016/j.bcp.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li F. Y., Jeffrey P. D., Yu J. W., Shi Y. Crystal structure of a viral FLIP: insights into FLIP-mediated inhibition of death receptor signaling. J. Biol. Chem. (2006);281:2960–2968. doi: 10.1074/jbc.M511074200. [DOI] [PubMed] [Google Scholar]
- 17.Ueffing N., Keil E., Freund C., Kühne R., Schulze-Osthoff K., Schmitz I. Mutational analyses of c-FLIPR, the only murine short FLIP isoform, reveal requirements for DISC recruitment. Cell Death Differ. (2008);15:773–782. doi: 10.1038/sj.cdd.4402314. [DOI] [PubMed] [Google Scholar]
- 18.Guex N., Peitsch M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. (1997);18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 19.Bagnéris C., Ageichik A. V., Cronin N., Wallace B., Collins M., Boshoff C., Waksman G., Barrett T. Crystal structure of a vFlip-IKKgamma complex: insights into viral activation of the IKK signalosome. Mol. Cell. (2008);30:620–631. doi: 10.1016/j.molcel.2008.04.029. [DOI] [PubMed] [Google Scholar]