

Abstract
Chronic inflammation predisposes patients with inflammatory bowel disease (IBD) to the risk of developing colitis-associated cancer (CAC). Growing evidence strongly suggests that CAC development is multifactorial, and is attributed to concurrent, dynamic dysregulations in host immunity, enteric microbiota and epithelial restitution during the course of chronic inflammation. This review discusses the recent advances in understanding of the different forms of CAC that may develop in IBD patients and animal models, as well as molecular alterations and other processes that orchestrate the development of CAC.
Keywords: Inflammatory bowel disease, epithelial dysplasia, guts microbiome
Introduction
A major complication for patients with inflammatory bowel disease (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), is the risk of developing colitis-associated cancer (CAC) that was first described in 1925 by Crohn and Rosenberg.(1) Subsequent meta-analysis in 2001 has shown that UC patients have a 2% risk of developing CAC by the 10th year of UC diagnosis, 8% by 20 years and 18% by 30 years.(2) The risk levels and pathological features of CD-derived CAC were proposed to be comparable to those of UC patients, making CAC one of the classical inflammation-driven cancers.(3-5) In fact, inflammation-mediated CAC is considered as one of the 3 major conditions associated with colorectal cancer, along with familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC).(6) CAC often exhibits rapid progression and resistance to therapeutic agents, resulting in high mortality.(7) In addition to the genetic predisposition, CAC development requires 1) preceding inflammation that dysregulates the coordinated balance between mucosal immune cells and the soluble factors which are actively released into the inflamed region; 2) an altered gut microbiome dynamic during colitis that leads to the production (secretion) of additional genotoxins creating a niche environment for tumor cells; 3) dysregulated wound healing and epithelial regeneration that results in uncontrolled proliferation. In this review, we will discuss all aspects of aforementioned factors that contribute to the development of CAC.
Pathogenesis and forms of CAC
It is well known that CAC develops through a complex multi-step and multi-factorial process “inflammation-dysplasia-carcinoma sequence” (Figure 1). The normal colon is first affected by inflammation, and an influx of immune cellular infiltrates results in colitis through the secretion of proinflammatory cytokines and chemokines, as well as oxygen and nitrogen reactive species that increases the probability of inducing genomic mutations and DNA damage.(8) During mucosal injury associated with inflammation, epithelial cell proliferation is enhanced while the tissue regenerates; proliferation and regeneration subside after the inflammation is controlled or the tissue is repaired. In contrast, proliferating cells that sustain genomic mutations and/or DNA damage continue to proliferate. Thus, longstanding and/or repeated bouts of inflammation, as seen during chronic colitis, increase the risk of the epithelial cells to become dysplastic and eventually give rise to invasive adenocarcinoma.
Figure 1. The inflammation-dysplasia-carcinoma pathogenic sequence in CAC.
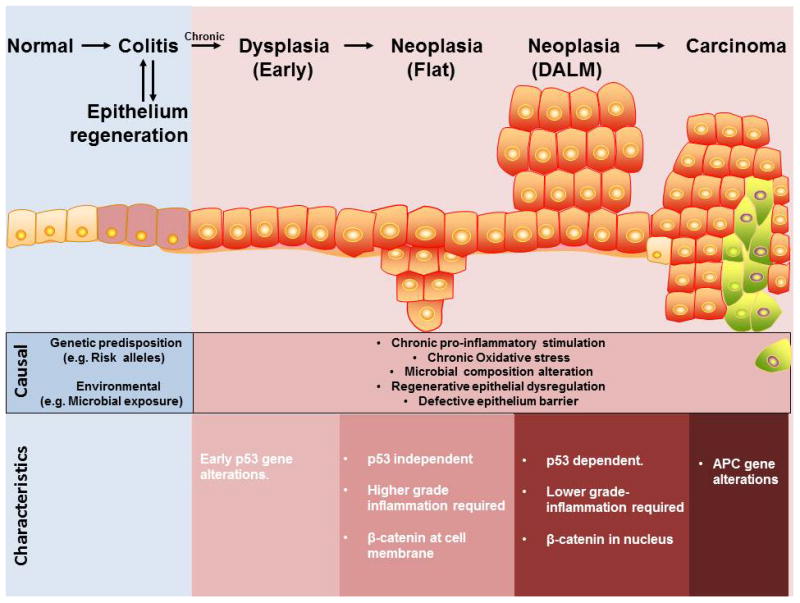
CAC follows an inflammation-dysplasia-carcinoma progression track. Chronic inflammation and repeated epithelium regeneration establish a potential primary tumor-initiating milieu through high production of potent pro-inflammatory cytokines and release of mutagenic reactive oxygen species and carcinogens by immune cells. Survival of early dysplasia often acquires aberrations in the p53 gene, which then progress into the two forms of neoplasia, flat and dysplastic associated lesion of mass (DALM), each exhibiting their own unique morphological and molecular characteristics. Neoplastic cells can transform into carcinoma, whereby aberrations in the APC gene are more frequently observed.
There are two forms of colitis-associated dysplasia, elevated or flat, which can either be localized in one area or scattered throughout the colon.(9) The dysplasia is usually classified as low-grade or high-grade. Low-grade dysplasia is characterized by nuclear elongation, hyperchromasia and stratification, as well as mucin depletion, while high-grade dysplasia shows more prominent cytologic atypia with round nuclei and conspicuous nucleoli, as well as additional architectural changes such as crypt crowding and budding. Loss of nuclear polarity and atypical mitoses are also seen.
Rutters et al reported that 89.3% of the studied CAC patients had elevated polypoid dysplasia.(10) Growing evidence suggests that the difference in morphology between flat and polypoid lesions is attributed to the difference in molecular mechanisms. Firstly, it was demonstrated by Fujimori et al in 2004 that p53 knockout (KO) mice treated with two cycles of dextran sulphate sodium (DSS) developed predominately flat dysplasia, whereas the majority of dysplastic lesions that developed in wildtype (WT) mice with the same treatment were polypoid.(11) Similarly, the study by Chang et al. using 3-4 cycles of DSS treatment showed that flat lesions were associated with p53 KO genotype and polypoid lesions were associated with WT genotype, indicating that loss of p53 may give rise to flat dysplasia in the inflamed colon.(12) Secondly, it has been shown by immunohistochemistry that the nuclear translocation of β-catenin, a molecule regulating cell-cell adhesion and intracellular gene transduction in the Wnt signaling pathway, can be detected only in polypoid lesions (91.3%) but not in flat lesions (0%).(12, 13) Thirdly, an expression array analysis of 550 cancer genes revealed that sixteen genes (PMS1, nm23, p21, FAK, Smad4, c-jun, ECGF-1, Erk1, GAK, GSTP1, IGF-2, Laminin β-3, MMP-15, Mucin3, Rho GDIβ, and TIMP-1) were up-regulated and eleven genes (HLA-DQ, Cdc42, Egr-2, Eph, Galectin-1, gp130, GST-II, MDR1, p120, Ras-GAP, and Rho 8) were down-regulated in flat lesions compared to polypoid lesions.(14) Fourth, flat dysplasia exhibits an inflammation magnitude that is 10-fold higher than that of polypoid dysplasia, suggesting that low-grade inflammation may promote DALMs whereas high-grade inflammation results in flat dysplasia.(13, 15) Finally, both flat and polypoid lesions respond differently to 5-aminosalicylic acid (5-ASA).(15) Treatment with 5-ASA in drinking water was sufficient to decrease the number of flat lesions that developed in Swiss Webster mice secondary to chronic DSS treatment, but it failed to reduce the size of tumor.(15) Conversely, the multiplicity of polypoid lesions was not affected but the size was significantly reduced by the same treatment.
The histological differentiation of dysplasia from normal epithelium undergoing regenerative changes may be difficult in some cases. In general, the healing epithelium has regenerative characteristics at the crypt base whereas the luminal surface of the crypt shows some degree of maturation. Conversely, dysplastic crypts typically exhibit distortion at both the surface and base of the crypts. Furthermore, it is also challenging to distinguish between CAC (especially DALM) and sporadic colorectal cancer (CRC) that may develop independently of inflammation in IBD patients. In general, CRC is seen in a region proximal to the inflamed colonic segments in UC patients. Many clinical, genetic and molecular characteristics are distinct between CAC and sporadic CRC. For instances, p53 mutations in CAC occur during an early stage of neoplastic transformation, whereas p53 mutations mostly occur at later stages in sporadic CRC.(16) In fact, the presence of p53 mutations in the adjacent, morphologically non-dysplastic epithelium in CAC suggests that chronic inflammation can induce genomic changes in the colonic mucosa and carcinoma develops within the genetically altered mucosa, supporting the theory of “field cancerization”.(17) In addition, alterations in the adenomatous polyposis coli (APC) gene are considered as the putative gate-keeping event in sporadic CRC, but occur later and much less frequently in CAC. Histologically, sporadic CRC typically exhibits a top-to-down growth pattern whereby the dysplastic transformation first involves the luminal surface of crypts while the crypt base is relatively normal.(18) In contrast, CAC exhibits a bottom-to-up growth pattern starting from the crypt base.
The immunity orchestra in CAC
Preceding inflammation is a prerequisite for CAC to develop. The influx of immune cells and the variety of cytokines that they secrete, including TNF-α, TGFβ, IL-10 and IL-12, are considered to be one of the major causal factors in cultivating the tumor-promoting milieu (Figure 2).(19-25) In addition to cytokines, these immune cells also produce other signaling molecules, as well as oxidative compounds, that have detrimental effects in promoting tumorigenesis. Immune cells that play a pivotal role in CAC include both innate and adaptive cells such as neutrophils, macrophages, dendritic cells, myeloid suppressor cells and T cells (Figure 3).
Figure 2. Effects of cytokine on CAC.
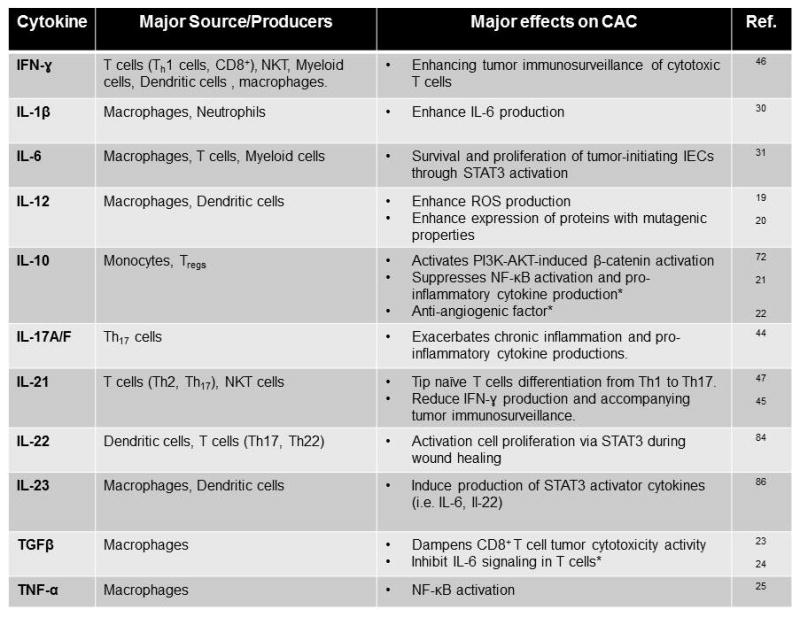
The varieties of cytokines present in the colonic inflammatory-tumor environment are listed in alphabetical order. *Anti-tumor effects.
Figure 3. Effects of colonic infiltrating immune cells on CAC.
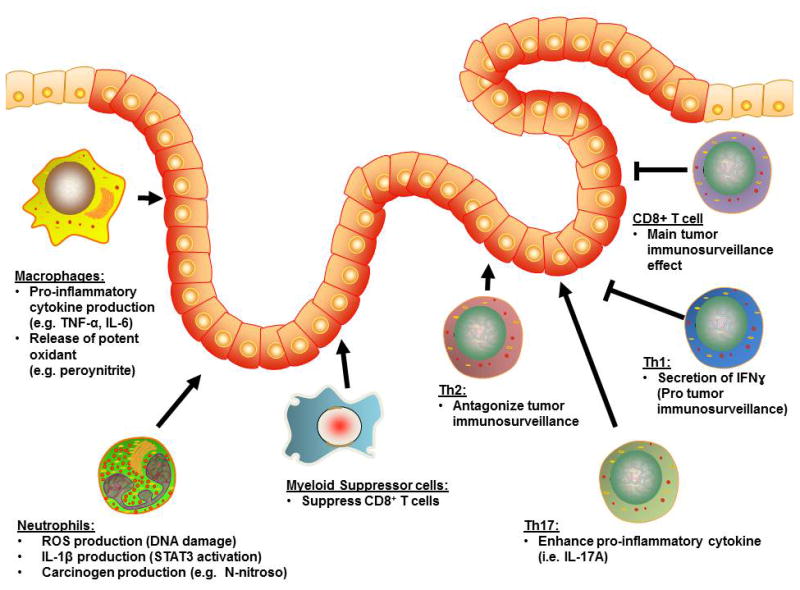
Major infiltrating innate immune cells (e.g. macrophages, neutrophils) contribute to tumor progression through the release of pro-inflammatory cytokines, reactive oxygen species and carcinogens. Infiltrating adaptive immune cells includes Th2 and Th17 and exerts pro-tumor properties through cytokine production and interference with the tumor-immunosurveillance system. Conversely, anti-tumor effects of the adaptive immunity is exerted by Th17 cells through release of IFNɣ and enhancing tumor-immunosurveillance, and cytotoxic CD8+ T cells that mediate direct killing of tumor cells.
The innate immune system provides immediate response and a first-line of defense mechanism for invasive pathogens and/or endogenous danger signals in the small and large intestines. During the course of IBD development, the infiltrating innate immune cells, in particular neutrophils and macrophages, produce large amounts of reactive oxygen species (ROS).(26) Thus, with the environment overloaded with oxygen radicals, this increases intestinal oxidative stress and is capable of inducing DNA damage and genetic mutations.(27) It was shown that colonic epithelial cells (CECs) exposed to activated neutrophils and the soluble factors that they produced harbored increased numbers of replication errors.(28) Co-culturing of CECs with activated neutrophils also showed an accumulation of cells in the G2/M phase of the cell cycle, which is consistent with the enhancement of a DNA-damage checkpoint.(28) This cell cycle arrest allows repair of damaged or incompletely replicated chromosomes, or otherwise allows the induction of apoptosis to eliminate seriously damaged cells.(29) In addition to contributing high amounts of ROS, neutrophils also secrete a pro-tumorigenic factor, interleukin (IL)-1β.(30) IL-1β can induce expression of IL-6, which is one of the other critical components in mediating CAC via the signal transducer and activator of transcription (STAT) 3 signaling pathway.(30, 31) Furthermore, neutrophils can also directly produce carcinogenic N-nitroso compounds during intestinal inflammation that implies a further risk of colonic carcinogenesis in chronic inflammatory conditions.(32)
Activated macrophages (M1) can inhibit cell proliferation and potentially cause tissue damage, while alternatively activated macrophages (M2) promote cell proliferation and tissue repair.(33) M1 macrophages are characterized by their microbicidal properties and secretion of pro-inflammatory cytokines. M1 also produce a large amount of free radicals, such as peroynitrite, that is a particularly strong oxidant ROS with low specificity.(34) In contrast, M2 have reduced cytotoxic response and pro-inflammatory influence.(35) The balance between M1 and M2 populations can be a key component in determining the transition from inflammation to tumorigenesis in CAC. It was shown that during the transition from colonic hyperplasia to dysplasia or from dysplasia to carcinoma, there is a consistent depletion in M1, whereas M2 remain stable.(36) Interestingly, tumor-associated macrophages (TAMs) exhibit a M2-macrophage phenotype with similar functions and have pro-tumor properties.(37) TAMs highly express the glycoprotein 96 (gp96), a chaperone that maintains proper folding of most Toll-like receptors (TLRs).(38) The absence of gp96 results in loss of responsiveness to TLRs ligands.(39) Macrophage-specific deletion of gp96 protects azoxymethane (AOM)/DSS treated mice from colitis and tumorigenesis, accompanied with a decrease in the production of IL-17A/F and IL-23, both of which are associated with the development/maintenance of T helper 17 (Th17) cells.(40) Interestingly, it has been shown that the crosstalk between TAMs and other innate cells (e.g. mast cells and CECs) are dependent on PI3K/AKT signaling.(41)
While the innate immune system initiates the intestinal inflammation, it is the adaptive immune system that perpetuates chronic inflammatory conditions. Both CD4+ and CD8+ T cells play pathogenic as well as protective roles in CAC development, depending on their subsets. While both CD4+ Th1 and Th2 signatures are the main drivers of CD and UC, respectively, it appears that Th2 cell-derived factors are the main culprit for tumor development, whereas Th1 cell-derived factors are protective against CAC. The notion is supported by the in vivo study by Osawa et al, in which AOM/2,4,6-trinitrobenzenesulfonic acid (TNBS) treatment was used to induce CAC in the interferon gamma (IFNɣ) KO mice (i.e. Th2-dominant mice), or IL-4 KO mice (i.e. Th1-dominant mice).(42) They found that IFNɣ KO mice developed significantly higher numbers of tumors compared to IL-4 KO mice, indicating that the Th2 response plays a pivotal role in CAC. In addition to the inflammatory microenvironment, the cytokine milieu is important in determining naïve T cell differentiation in response to antigenic exposure. When the adaptive immune system fails to resolve the inflammatory immune response, as seen in most IBD patients, an increase in IL-21 appears to have two major impacts. First, the increase in IL-21 causes naïve T cells to preferentially differentiate into Th17 rather than Th1 cells.(43) This enhances the increased production of Th17 cytokine, including IL-17A and IL-17F, that has been shown to have a great influence on inflammation and tumorigenesis.(44) Secondly, the transition from Th1 to Th17, as a consequence of the IL-21 production, also results in loss of Th1-mediated IFNɣ production and reduces the cytotoxic effector function of CD8+ T cells.(44-47) Since tumor immunosurveillance is predominately exerted by cytotoxic CD8+ T cells, the Th1-Th17 transition results in reduction of their tumor suppressing functions. When CD8+ T cells encounter a tumor-associated antigen, they become activated and express high levels of IFNɣ and FasL. This membrane bound FasL then induces apoptosis of dysplastic cells that express high levels of surface bound Fas.(48) Furthermore, it has been show that IFNɣ further enhances the expression of Fas in HT29 human CEC line, augmenting the killing process.(49)
Indeed, the suppression of cytotoxic T cells results in cancer immune evasion. Such suppression can be exerted by myeloid-derived suppressor cells (MDSC). High numbers of blood MDSCs correlate well with clinical cancer stages and metastatic burdens in CRC and other cancers.(50, 51) These MDSCs express large amounts of a chemokine receptor, C-X-C chemokine receptor type 2 (CXCR2), whereas the inflamed colonic mucosa and tumor cells express high levels of CXCR2 ligands.(52) The overexpression of both the receptor and its ligand induces chemotactic attractions, which in turn cause the infiltration of MDSC into the colonic mucosa and tumors. MDSC then suppress the colonic cytotoxic CD8+ T cells through the production of superoxide and nitric oxide, depletion of L-arginine, alteration of signaling molecules and surface receptors of T cells.(53) This MDSC-mediated suppression of CD8+ cytotoxic T cells compromises tumor immunosurveillance effects and promotes CAC development. (52)
Microbial contributions to CAC
The intestine harbors over 1013-14 commensal bacteria, composing of up to 36,000 different species.(54, 55) These commensal microbes closely contact with the apical surface of the intestinal epithelium, and are separated only by a protective epithelial barrier (including CECs and mucin layers) from the immune system present in the lamina propria. Thus, the commensal organisms can have an enormous impact not only on intestinal inflammation, but also on tumorigenesis through the production of tumor-promoting genotoxins that can also induce chromosomal instability.(56)
Indeed, it has been widely reported that translocation of commensal microflora, such as Citrobacter rodentium, Bacteroides fragilis, adherent-invasive Escherichia coli (AIEC) into CEC, and DSS treatment that induces epithelial injury and exposes the lamina propria cells including macrophages to the luminal microflora, can result in intestinal inflammation.(57) Association between selected commensal microbiota and development of CAC has also been studied in animal models of spontaneous colitis. For instance, IL-10 KO mice develop spontaneous colitis in the conventional (CV) or specific pathogen free (SPF), but not under germ-free (GF), environment. It was shown that IL-10 KO mice pre-injected with AOM and housed in the CV environment developed colorectal carcinomas, and the number of tumors positively correlated with the severity of colitis.(58) In contrast, AOM-injected IL-10 KO mice that were housed in the GF environment showed intact colonic mucosa without tumor formation. Interestingly, high throughput sequencing in IL-10 KO mice revealed that chronic inflammation can alter the composition of gut microbiota and induce expansion of certain microorganisms that have genotoxic effects.(59) Data from IBD and CAC patient specimens showed increased numbers of colonic AIEC strains that highly express the long polar fimbriae (lpfA) that is associated with the translocation across M cells of the follicle-associated epithelium, as well as the polyketide synthase (pks) that is responsible in producing the genotoxin colibactin.(60) Colonization of mice with AIEC NC101 strain, which contains deletion of the pks genotoxic island, does not alter the severity of intestinal inflammation, but showed remarkable reduction in tumor-multiplicity and -invasion.(59)
The sensing and communication of bacteria with the host intestine depends on pattern recognition receptors (PRRs) including TLRs and nucleotide-binding oligomerization domain receptors (Nod)-like receptors, which are both up-regulated in inflammation-associated colorectal neoplasia. Deletion of TLR4 that senses bacterial lipopolysaccharides (LPS) protects mice from CAC development.(61) Deletion of MyD88, the major adaptor protein for most TLR signaling, also protects IL-10 KO mice from AOM/DSS-induced CAC development.(58) Similarly, Nod2 deficiency creates a pro-inflammatory environment that promotes epithelial dysplasia. Interestingly, the dysplasia can be suppressed by anti-biotic treatment.(62) Transfer of the dysregulated microbiota that developed in Nod2 KO mice into WT mice leads to an increased risk of CAC development in the recipient mice.(62)
Interestingly, Couturier-Maillard et al demonstrated that the magnitude of CAC risk is dependent on the composition of maternally transmitted microbiota.(62) In addition to the parental-inherited effects of the microbiota, translocation of indigenous microbiota from the other areas of the body to the intestine can also promote intestinal tumorigenesis. Kostic et al has reported an enrichment of Fusobacterium, a bacterium that is indigenous to the human oral cavity, in colorectal carcinomas.(63) APCMin/+ mice, a murine model of multiple intestinal adenomas, exposed to Fusobacterium showed a significant increase in colonic tumors when compared to APCMin/+ mice that were exposed to Streptococcus, or control mice.(64) In addition, treatment with antibiotics in mice can confer some degree of protection against CAC, supporting the pivotal role of the gut microbiota during tumorigenesis.(65)
Dysplasia of epithelial cells: The victim of chronic inflammation
The harsh dynamic inflammatory environment during chronic colitis exerts a strong influence on the intestinal epithelial cells (IECs) that may eventually become dysplastic. First, danger molecular signals released from exogenous (pathogen associated molecular patterns; PAMPs) or host-derived (danger associated molecular pattern; DAMPs) can activate intracellular molecular pathways in the IECs. Concurrently, potent factors, such as ROS, released into the environment to ward off invading pathogens may induce genomic mutations and/or DNA damage in IECs. Finally, during the restitution (regeneration) of the epithelium in the mucosa that was damaged by the inflammation, the IECs harboring genomic mutations and/or DNA damage likely undergo uncontrolled proliferation. Collectively, all these processes promote CAC tumorigenesis.
Under inflammatory conditions, the intestinal epithelium expresses pattern recognition receptors (PRR) to sense the changes in its immediate environment. When these PRRs detect the presence of 1) host-derived DAMPs, such as S100A family molecules and the High-Mobility Group Box (HMGB), and/or 2) conserved microbial PAMPs (i.e. bacterial and fungal cell wall components and viral nucleic acids), they gets activated and transmits danger signals.(66) For instance, the Receptor for Advance Glycation End product (RAGE) is a receptor that can sense endogenous DAMP signals. RAGE receptor expression is low in the intestinal epithelium but is inducibly expressed on the IECs upon inflammatory stimulation.(67) RAGE that are expressed on colonic tumor cells were found to be post-translationally modified with carboxylated N-glycans, which are recognized and subsequently bound by host-secreted DAMPs ligands including the S100A family molecules (e.g. S100A8, S100A9, S100A12) released by both neutrophils and IECs. S100A9 and RAGE ligation specifically activates downstream NF-κB signaling, which promotes cell proliferation and exacerbates tumor progression in AOM-DSS treated mice.(68) On the other hand, TLRs are one of the widely studied PRRs that recognize PAMPs as described above.(69) Importantly, TLR2 and TLR4 can also sense DAMPs, such as HMGB and S100A proteins. TLR4 KO mice-derived bone marrow chimera mice receiving AOM/DSS treatment had much more increased dysplastic lesions, as well as more significant neutrophil and macrophage infiltrations, compared to TLR4 KO mice transplanted with WT mice-derived bone marrow.(70) The results indicate that CEC specific TLR4 response is pro-tumorigenic. Interestingly, the villin-TLR4 mice, which have increased TLR4 signaling in the intestinal epithelium, can cause spontaneous intestinal dysplasia.(71) Subsequently, the activation of TLR4 triggers β-catenin signaling in CECs in a PI3K-dependent manner.(71)
Indeed, β-catenin signaling activation in CECs plays a major role during colonic tumorigenesis. During intestinal inflammation, the activation of PI3K/AKT pathway also collaborates with WNT/Frizzled pathway to further enhance β-catenin activation in the intestinal stem cells that may be critical for the CAC progression.(72) Many colonic factors that are expressed at a low level in the homeostatic state, but are highly inducible and up-regulated in IBD and CAC, have been reported to efficiently activate β-catenin. These factors include the indoleamine 2,3 dioxygenase-1 (IDO1), chitinase 3-like 1 (CHI3L1) and PCNA-associated factor (PAF) that has been shown to be highly associated with tumor cell proliferation via β-catenin activation.(73-75) 5-ASA, the most common anti-inflammatory therapy for UC patients, can inhibit β-catenin signaling in the CECs of patients with CAC, and protect IL-10 KO mice from the development of dysplasia.(76) The inhibition of β-catenin signaling by 5-ASA is the most significant in the middle and upper parts of colonic crypts.
Another prominent signaling activation seen in the IECs during CAC development is IL-6-STAT3 pathway. IL-6, an NF-κB-regulated cytokine, is extremely important for the survival of normal and premalignant IECs, preventing them from undergoing apoptosis during intestinal inflammation.(31) However, IL-6 activation of the transcription factor STAT3 in pre-cancerous IECs not only enhances their survival but also promotes tumor proliferation.(31) It has been reported that the lamina propria macrophages are the source of IL-6 which exerts the trans-signaling effects on the CECs.(77) Mechanistically, one of the factors that can induce the production of IL-6 during colitis and CAC is the sphingosine-1-phosphate (S1P) that is a pleiotropic bioactive sphingolipid metabolite produced by sphingosine kinases. The subsequent activation of S1P receptor, S1PR, by S1P leads to up-regulation of IL-6 expression.(78) Once IL-6 is up-regulated, it then binds to the IL-6 receptor gp130 on the IECs that activates STAT3 and alters cell-cycle progression at the G1 and G2/M phases.(79) STAT3 activation, which is predominately detected in the CECs, was found to be more prominent in low-grade CACs than high-grade CACs.(80) Conversely, STAT6 activation is found to be more common in the normal CECs than those of CAC, suggesting a preferential shift from STAT6 to STAT3 activity profile during the disease progression.(80)
In addition to altering cell-cycle progression, STAT3 activation is also linked to mucosal wound healing following intestinal epithelial injury. However, IEC specific STAT3 activation, which enhances wound healing processes, is not induced by IL-6, but rather by IL-22 that is produced by CD11c+ cells as well as Th17 cells.(81) It has also been reported that a dysregulated IL-22/STAT3 pathway is present in CAC patients.(82) Leukocytes infiltrating tumor cells produced large amounts of IL-22 compared to those infiltrating the adjacent non-neoplastic mucosa.(83) The IL-22 produced by the immune cells then binds to the IL-22 receptor (IL22RA1) that is expressed on IECs, and the receptor-ligand interaction subsequently activates STAT3 that may promote tumor growth.(83) Physiological regulation and neutralization of IL-22 function can be achieved by its ligation with soluble IL-22 binding protein (IL-22BP). IL-22BP is a soluble IL-22 receptor and a natural inhibitor of IL-22 that prevents IL-22 from binding to membrane bound IL-22RA1. It was reported that IL-22BP deficiency in mice was strongly associated with accelerated tumor development with an increase in tumor size and number.(84) IL-22BP/IL-22 double KO mice showed similar tumor development as IL-22 KO mice, in which both the tumor number and load was lower than IL-22BP KO mice, confirming that the IL-22-IL-22BP axis critically regulates intestinal mucosal repair and tumorigenesis in the colon.(84)
Other important factors that may prevent the CECs from developing tumorigenesis during the process of wound healing and repair is the proper restoration of epithelium barrier through the recoating of the epithelium with the protective mucus layer. The mucus barrier is composed of secreted and cell-surface mucins that are made up of large glycoproteins with dense arrays of complex O-linked glycans and function as a physical barrier to limit the access of microbes and toxins to the underlying intestinal tissue.(85) It was shown that intestinal epithelium involved by tumor showed the absence of mucin producing cells as well as markedly depleted production and coating of mucin 2 (Muc2) when compared to the adjacent normal epithelium.(86) Consequently, microbes and their genotoxic products can easily penetrate the tumor region but not the adjacent normal tissue, creating a tumor-promoting microenvironment.(86)
Therefore, the IECs are extremely vulnerable since they receive all sorts of signaling cues present in the inflammatory environment that are necessary to complete inflammatory episodes but have potential tumor-promoting effects when dysregulated (Figure 4).
Figure 4. Signaling pathway transduction on intestinal epithelial cells in CAC.
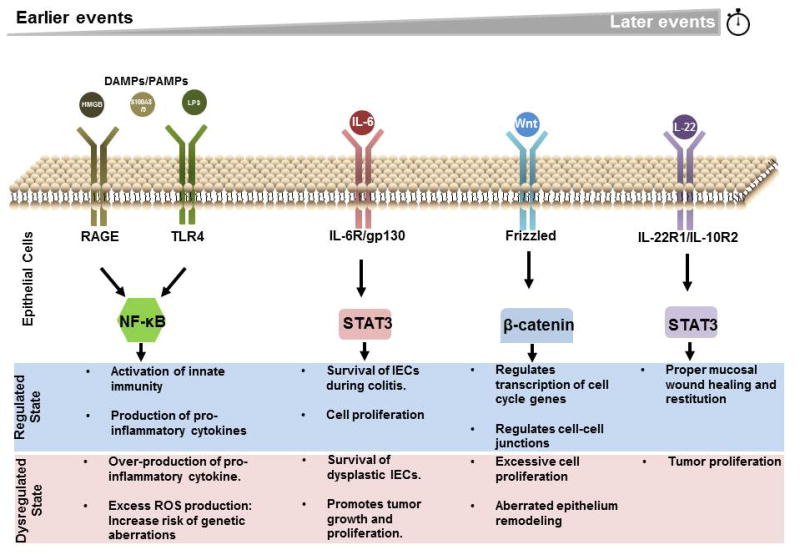
Signaling molecules, including cytokines and pattern recognition molecules, are important for intestinal epithelial cells (IECs) responses during inflammation. At regulated state, these signals allows IECs to activate innate immunity and produce pro-inflammatory cytokines to ward off the invading pathogen, activate pro-survival genes to survive the harsh inflammatory environment, remodel the epithelium for regeneration and to initiate cell proliferation for restitution. In a dysregulated state, these signals may allow survival and proliferation of dysplastic cells and allow epithelium remodeling for further tumor growth.
Conclusion
Patients with IBD are susceptible to colorectal cancer development as a result of chronic inflammation, in addition to any genetic predisposition. To develop effective interventions against the development of CAC, careful consideration to all causal factors is required. These include host immunity, commensal microbial influences and epithelial homeostasis.
Acknowledgments
The authors are grateful to Mr. Alan Kamba for his excellent assistance in preparing this manuscript.
Grant Supports: This work was supported by National Institutes of Health (DK 80070, DK74454, and DK64289), and grants from the Eli and Edythe L. Broad Medical Foundation and American Gastroenterological Association Foundation to EM. DL was awarded the Singapore A*STAR International Fellowship. M.M.-K. was supported by National Cancer Institute grants P50 CA127003 and R01 CA169086.
Abbreviations
- 5-ASA
5-aminosalicylic acid
- AOM
azoxymethane
- CAC
colitis-associated cancer
- CD
Crohn's disease
- CEC
colonic epithelial cells
- CRC
colorectal cancer
- CV
conventional
- DALM
dysplasia associated lesion or mass
- DSS
dextran sulphate sodium
- GF
germ-free
- IBD
inflammatory bowel disease
- IECs
intestinal epithelial cells
- IFN
interferon
- IL
interleukin
- KO
knockout
- LP
lamina propria
- LPS
lipopolysaccharide
- MDSC
myeloid-derived suppressor cells
- Nod
nucleotide-binding oligomerization domain receptors
- PRR
pattern recognition receptors SPF, specific pathogen-free
- STAT
signal transducer and activator of transcription
- TAM
tumor-associated macrophages Th, T helper
- TLR
toll-like receptors
- UC
ulcerative colitis
- WT
wildtype
References
- 1.Crohn B, Rosenberg H. The sigmoidoscopic picture of chronic ulcerative colitis (non-specific) The American Journal of the Medical Sciences. 1925;170:220–228. [Google Scholar]
- 2.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubio CA, Befrits R. Colorectal adenocarcinoma in Crohn's disease: a retrospective histologic study. Dis Colon Rectum. 1997;40:1072–1078. doi: 10.1007/BF02050932. [DOI] [PubMed] [Google Scholar]
- 4.Maykel JA, Hagerman G, Mellgren AF, et al. Crohn's colitis: the incidence of dysplasia and adenocarcinoma in surgical patients. Dis Colon Rectum. 2006;49:950–957. doi: 10.1007/s10350-006-0555-9. [DOI] [PubMed] [Google Scholar]
- 5.Friedman S, Rubin PH, Bodian C, et al. Screening and surveillance colonoscopy in chronic Crohn's colitis. Gastroenterology. 2001;120:820–826. doi: 10.1053/gast.2001.22449. [DOI] [PubMed] [Google Scholar]
- 6.Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- 7.Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol. 2009;6:297–305. doi: 10.1038/nrgastro.2009.44. [DOI] [PubMed] [Google Scholar]
- 8.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Odze RD. Pathology of dysplasia and cancer in inflammatory bowel disease. Gastroenterol Clin North Am. 2006;35:533–552. doi: 10.1016/j.gtc.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Rutter MD, Saunders BP, Wilkinson KH, et al. Most dysplasia in ulcerative colitis is visible at colonoscopy. Gastrointest Endosc. 2004;60:334–339. doi: 10.1016/s0016-5107(04)01710-9. [DOI] [PubMed] [Google Scholar]
- 11.Fujii S, Fujimori T, Kawamata H, et al. Development of colonic neoplasia in p53 deficient mice with experimental colitis induced by dextran sulphate sodium. Gut. 2004;53:710–716. doi: 10.1136/gut.2003.028779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang WC, Coudry RA, Clapper ML, et al. Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis. 2007;28:2375–2381. doi: 10.1093/carcin/bgm134. [DOI] [PubMed] [Google Scholar]
- 13.Cooper HS, Murthy S, Kido K, et al. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis. 2000;21:757–768. doi: 10.1093/carcin/21.4.757. [DOI] [PubMed] [Google Scholar]
- 14.Nosho K, Yamamoto H, Adachi Y, et al. Gene expression profiling of colorectal adenomas and early invasive carcinomas by cDNA array analysis. Br J Cancer. 2005;92:1193–1200. doi: 10.1038/sj.bjc.6602442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang WC, Zenser TV, Cooper HS, et al. Differential response of flat and polypoid colitis-associated colorectal neoplasias to chemopreventive agents and heterocyclic amines. Cancer Lett. 2013 doi: 10.1016/j.canlet.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brentnall TA, Crispin DA, Rabinovitch PS, et al. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology. 1994;107:369–378. doi: 10.1016/0016-5085(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 17.Matkowskyj KA, Chen ZE, Rao MS, et al. Dysplastic lesions in inflammatory bowel disease: molecular pathogenesis to morphology. Arch Pathol Lab Med. 2013;137:338–350. doi: 10.5858/arpa.2012-0086-RA. [DOI] [PubMed] [Google Scholar]
- 18.Shih IM, Wang TL, Traverso G, et al. Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A. 2001;98:2640–2645. doi: 10.1073/pnas.051629398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang J, Seki T, Tsukamoto T, et al. Protection from inflammatory bowel disease and colitis-associated carcinogenesis with 4-vinyl-2,6-dimethoxyphenol (canolol) involves suppression of oxidative stress and inflammatory cytokines. Carcinogenesis. 2013;34:2833–2841. doi: 10.1093/carcin/bgt309. [DOI] [PubMed] [Google Scholar]
- 20.Takai A, Marusawa H, Minaki Y, et al. Targeting activation-induced cytidine deaminase prevents colon cancer development despite persistent colonic inflammation. Oncogene. 2012;31:1733–1742. doi: 10.1038/onc.2011.352. [DOI] [PubMed] [Google Scholar]
- 21.Schottelius AJ, Mayo MW, Sartor RB, et al. Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J Biol Chem. 1999;274:31868–31874. doi: 10.1074/jbc.274.45.31868. [DOI] [PubMed] [Google Scholar]
- 22.Huang S, Xie K, Bucana CD, et al. Interleukin 10 suppresses tumor growth and metastasis of human melanoma cells: potential inhibition of angiogenesis. Clin Cancer Res. 1996;2:1969–1979. [PubMed] [Google Scholar]
- 23.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 24.Becker C, Fantini MC, Schramm C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 25.Mukaida N, Sasakki S, Popivanova BK. Tumor Necrosis Factor (TNF) and Chemokines in Colitis-Associated Cancer. Cancers (Basel) 2011;3:2811–2826. doi: 10.3390/cancers3032811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grisham MB. Oxidants and free radicals in inflammatory bowel disease. Lancet. 1994;344:859–861. doi: 10.1016/s0140-6736(94)92831-2. [DOI] [PubMed] [Google Scholar]
- 27.Roessner A, Kuester D, Malfertheiner P, et al. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol Res Pract. 2008;204:511–524. doi: 10.1016/j.prp.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Campregher C, Luciani MG, Gasche C. Activated neutrophils induce an hMSH2-dependent G2/M checkpoint arrest and replication errors at a (CA)13-repeat in colon epithelial cells. Gut. 2008;57:780–787. doi: 10.1136/gut.2007.141556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barzilai A, Yamamoto K. DNA damage responses to oxidative stress. DNA Repair (Amst) 2004;3:1109–1115. doi: 10.1016/j.dnarep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Wang K, Han GC, et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol. 2014 doi: 10.1038/mi.2013.126. [DOI] [PubMed] [Google Scholar]
- 31.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vermeer IT, Henderson LY, Moonen EJ, et al. Neutrophil-mediated formation of carcinogenic N-nitroso compounds in an in vitro model for intestinal inflammation. Toxicol Lett. 2004;154:175–182. doi: 10.1016/j.toxlet.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 33.Engstrom A, Erlandsson A, Delbro D, et al. Conditioned media from macrophages of M1, but not M2 phenotype, inhibit the proliferation of the colon cancer cell lines HT-29 and CACO-2. Int J Oncol. 2014;44:385–392. doi: 10.3892/ijo.2013.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 35.Martinez FO, Sica A, Mantovani A, et al. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 36.Wang W, Li X, Zheng D, et al. Dynamic changes of peritoneal macrophages and subpopulations during ulcerative colitis to metastasis of colorectal carcinoma in a mouse model. Inflamm Res. 2013;62:669–680. doi: 10.1007/s00011-013-0619-y. [DOI] [PubMed] [Google Scholar]
- 37.Sica A, Schioppa T, Mantovani A, et al. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3:891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 39.Liu B, Yang Y, Qiu Z, et al. Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat Commun. 2010;1:79. doi: 10.1038/ncomms1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morales C, Rachidi S, Hong F, et al. Immune Chaperone gp96 Drives the Contributions of Macrophages to Inflammatory Colon Tumorigenesis. Cancer Res. 2014;74:446–459. doi: 10.1158/0008-5472.CAN-13-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan MW, Keshavarzian A, Gounaris E, et al. PI3K/AKT signaling is essential for communication between tissue-infiltrating mast cells, macrophages, and epithelial cells in colitis-induced cancer. Clin Cancer Res. 2013;19:2342–2354. doi: 10.1158/1078-0432.CCR-12-2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osawa E, Nakajima A, Fujisawa T, et al. Predominant T helper type 2-inflammatory responses promote murine colon cancers. Int J Cancer. 2006;118:2232–2236. doi: 10.1002/ijc.21639. [DOI] [PubMed] [Google Scholar]
- 43.Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. 2008;26:57–79. doi: 10.1146/annurev.immunol.26.021607.090316. [DOI] [PubMed] [Google Scholar]
- 44.Hyun YS, Han DS, Lee AR, et al. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis. 2012;33:931–936. doi: 10.1093/carcin/bgs106. [DOI] [PubMed] [Google Scholar]
- 45.Danese S, Malesci A, Vetrano S. Colitis-associated cancer: the dark side of inflammatory bowel disease. Gut. 2011;60:1609–1610. doi: 10.1136/gutjnl-2011-300953. [DOI] [PubMed] [Google Scholar]
- 46.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 47.Jauch D, Martin M, Schiechl G, et al. Interleukin 21 controls tumour growth and tumour immunosurveillance in colitis-associated tumorigenesis in mice. Gut. 2011;60:1678–1686. doi: 10.1136/gutjnl-2011-300612. [DOI] [PubMed] [Google Scholar]
- 48.Itoh N, Yonehara S, Ishii A, et al. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66:233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 49.Xu X, Fu XY, Plate J, et al. IFN-gamma induces cell growth inhibition by Fas-mediated apoptosis: requirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 1998;58:2832–2837. [PubMed] [Google Scholar]
- 50.Mandruzzato S, Solito S, Falisi E, et al. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. 2009;182:6562–6568. doi: 10.4049/jimmunol.0803831. [DOI] [PubMed] [Google Scholar]
- 51.Diaz-Montero CM, Salem ML, Nishimura MI, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katoh H, Wang D, Daikoku T, et al. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell. 2013;24:631–644. doi: 10.1016/j.ccr.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ostanin DV, Bhattacharya D. Myeloid-derived suppressor cells in the inflammatory bowel diseases. Inflamm Bowel Dis. 2013;19:2468–2477. doi: 10.1097/MIB.0b013e3182902b11. [DOI] [PubMed] [Google Scholar]
- 54.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 55.Blaser MJ. Harnessing the power of the human microbiome. Proc Natl Acad Sci U S A. 2010;107:6125–6126. doi: 10.1073/pnas.1002112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, Huycke MM. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology. 2007;132:551–561. doi: 10.1053/j.gastro.2006.11.040. [DOI] [PubMed] [Google Scholar]
- 57.Low D, Nguyen DD, Mizoguchi E. Animal models of ulcerative colitis and their application in drug research. Drug Des Devel Ther. 2013;7:1341–1357. doi: 10.2147/DDDT.S40107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uronis JM, Muhlbauer M, Herfarth HH, et al. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4:e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arthur JC, Perez-Chanona E, Muhlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prorok-Hamon M, Friswell MK, Alswied A, et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut. 2013 doi: 10.1136/gutjnl-2013-304739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fukata M, Chen A, Vamadevan AS, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133:1869–1881. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Couturier-Maillard A, Secher T, Rehman A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123:700–711. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kostic AD, Gevers D, Pedamallu CS, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kostic AD, Chun E, Robertson L, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klimesova K, Kverka M, Zakostelska Z, et al. Altered gut microbiota promotes colitis-associated cancer in IL-1 receptor-associated kinase M-deficient mice. Inflamm Bowel Dis. 2013;19:1266–1277. doi: 10.1097/MIB.0b013e318281330a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Srikrishna G, Freeze HH. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia. 2009;11:615–628. doi: 10.1593/neo.09284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zen K, Chen CX, Chen YT, et al. Receptor for advanced glycation endproducts mediates neutrophil migration across intestinal epithelium. J Immunol. 2007;178:2483–2490. doi: 10.4049/jimmunol.178.4.2483. [DOI] [PubMed] [Google Scholar]
- 68.Turovskaya O, Foell D, Sinha P, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008;29:2035–2043. doi: 10.1093/carcin/bgn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pradere JP, Dapito DH, Schwabe RF. The Yin and Yang of Toll-like receptors in cancer. Oncogene. 2013 doi: 10.1038/onc.2013.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fukata M, Hernandez Y, Conduah D, et al. Innate immune signaling by Toll-like receptor-4 (TLR4) shapes the inflammatory microenvironment in colitis-associated tumors. Inflamm Bowel Dis. 2009;15:997–1006. doi: 10.1002/ibd.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Santaolalla R, Sussman DA, Ruiz JR, et al. TLR4 activates the beta-catenin pathway to cause intestinal neoplasia. PLoS One. 2013;8:e63298. doi: 10.1371/journal.pone.0063298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee G, Goretsky T, Managlia E, et al. Phosphoinositide 3-kinase signaling mediates beta-catenin activation in intestinal epithelial stem and progenitor cells in colitis. Gastroenterology. 2010;139:869–881. 881, e861–869. doi: 10.1053/j.gastro.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jung HY, Jun S, Lee M, et al. PAF and EZH2 induce Wnt/beta-catenin signaling hyperactivation. Mol Cell. 2013;52:193–205. doi: 10.1016/j.molcel.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thaker AI, Rao MS, Bishnupuri KS, et al. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology. 2013;145:416–425. e411–414. doi: 10.1053/j.gastro.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee IA, Kamba A, Low D, et al. Novel methylxanthine derivative-mediated anti-inflammatory effects in inflammatory bowel disease. World J Gastroenterol. 2014;20:1127–1138. doi: 10.3748/wjg.v20.i5.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brown JB, Lee G, Managlia E, et al. Mesalamine inhibits epithelial beta-catenin activation in chronic ulcerative colitis. Gastroenterology. 2010;138:595–605. 605, e591–593. doi: 10.1053/j.gastro.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matsumoto S, Hara T, Mitsuyama K, et al. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol. 2010;184:1543–1551. doi: 10.4049/jimmunol.0801217. [DOI] [PubMed] [Google Scholar]
- 78.Liang J, Nagahashi M, Kim EY, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–120. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 80.Wick EC, LeBlanc RE, Ortega G, et al. Shift from pStat6 to pStat3 predominance is associated with inflammatory bowel disease-associated dysplasia. Inflamm Bowel Dis. 2012;18:1267–1274. doi: 10.1002/ibd.21908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu LZ, Wang HY, Yang SP, et al. Expression of interleukin-22/STAT3 signaling pathway in ulcerative colitis and related carcinogenesis. World J Gastroenterol. 2013;19:2638–2649. doi: 10.3748/wjg.v19.i17.2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jiang R, Wang H, Deng L, et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer. 2013;13:59. doi: 10.1186/1471-2407-13-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huber S, Gagliani N, Zenewicz LA, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9:874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grivennikov SI, Wang K, Mucida D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]