

Abstract
The lomaiviticins and kinamycins are complex DNA damaging natural products that contain a diazofluorene functional group. Herein, we elucidate the influence of skeleton structure, ring and chain isomerization, D-ring oxidation state, and naphthoquinone substitution on DNA binding and damaging activity. We show that the electrophilicity of the diazofluorene appears to be the most significant determinant of DNA damaging activity. These studies identify the monomeric diazofluorene 11 as a potent DNA cleavage agent in tissue culture. The simpler structure of 11 relative to the natural products establishes it as a useful lead for translational studies.
Keywords: cancer, DNA cleavage, DNA damage, natural products
Lomaiviticins A–E (1–5)[1] and the kinamycins (6–8)[2] are complex antiproliferative agents that contain one (for 3–8) or two (for 1 and 2) diazotetrahydrobenzo[b]fluorene (diazofluorene) functional groups (Figure 1).[3] The kinamycins and lomaiviticins C–E (3–5) display half maximal inhibitory potencies in the ~300 nM range against various cultured human cancer cell lines, while (−)-lomaiviticin A (1) is two–five orders of magnitude more cytotoxic, with IC50 values in the low nanomolar–picomolar range.[1a] We recently established that the superior cytotoxicity of 1 derives from the production of DNA double-strand breaks (dsbs) that are induced by vinyl radicals[4] formed from each diazofluorene.[5],[6] This mode of DNA damage is not recapitulated by (−)-lomaiviticin C (3) or (−)-kinamycin C (6).[5] The laboratories of Melander and Hasinoff–Dmitrienko have demonstrated that kinamycins D (7), F (8), and synthetic analogs nick DNA in vitro and in tissue culture,[6d-f, 6h] but DNA cleavage has not been detected, in accord with our observations using 6. DNA dsbs are the most cytotoxic of all lesions,[7] and these data provide an explanation for the superior potency of 1.
Figure 1.
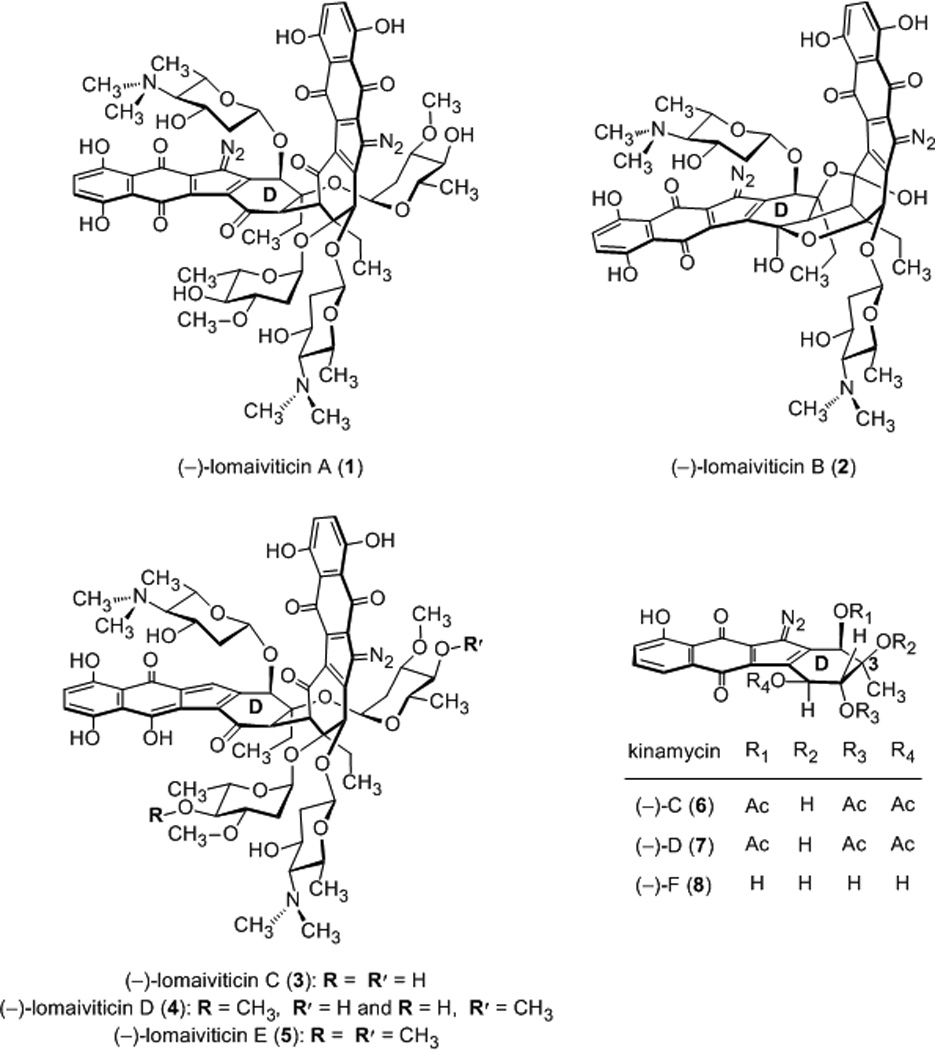
Structures of (−)-lomaiviticins A–E (1–5) and kinamycins C, D, and F (6–8, respectively).
Thermal denaturation and fluorescence intercalator displacement studies using calf thymus DNA and various kinamycins[6c, 6e] have established their DNA-binding ability, but nothing is known about the sequence selectivity of binding or the structural features that enhance or inhibit DNA damaging activity. Such information is central to an understanding of the mechanism of action of these metabolites and the preclinical development of synthetic diazofluorene-based anticancer agents. Accordingly, we report a comprehensive evaluation of the DNA-binding and cleavage activities of a panel of diazofluorenes embodying the essential structural features of the lomaiviticins and kinamycins. We demonstrate that certain synthetic diazofluorenes induce formation of DNA dsbs in tissue culture, including drug-resistant cell lines.[8] We employed kinamycins C (6), F (8), and the synthetic diazofluorenes 9–13[9] in this study (Figure 2). These compounds were chosen because they allow for evaluation of the influence of dimerization, ring and chain isomerization, D-ring oxidation state, and naphthoquinone substitution on activity.
Figure 2.
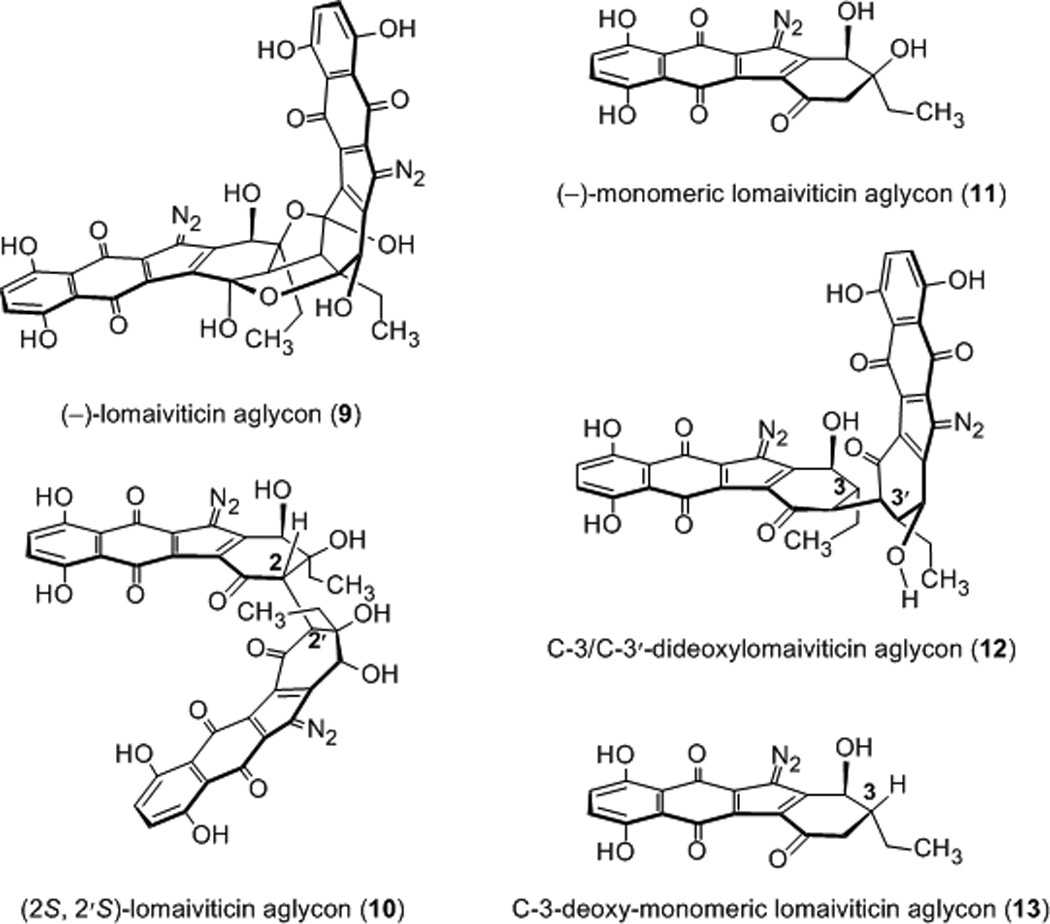
Structures of synthetic dimeric and monomeric diazofluorenes employed in this work.
Our studies began by determining the relative affinities of these diazofluorenes for DNA by a fluorescent intercalator displacement (FID) assay, employing thiazole orange (TO) as the intercalator probe.[10] Among all of the diazofluorenes examined, (−)-lomaiviticin aglycon (9) displayed the highest affinity for dsDNA (30% ± 1.2% decrease in fluorescence, Figure 3) and in general, dimeric diazofluorenes bound DNA with higher affinity than monomeric diazofluorenes (29–22% and 19–12% decrease in fluorescence for dimeric and monomeric diazofluorenes, respectively). We performed FID titration experiments to determine DC50 values (where DC50 corresponds to the amount of ligand required to displace 50% of the bound intercalator).[11] These studies showed that dimeric diazofluorenes bind polynucleotides with low micromolar affinity (Table 1). (−)-Lomaiviticin aglycon (9) displayed a modest preference for GC-rich sequences, while the C-3/C-3′-dideoxy aglycon (12) bound AT-rich sequences with highest affinity. The DC50 values of monomeric diazofluorenes were much higher (>100 µM) than dimeric diazofluorenes, in agreement with the TO displacement assays above. In both FID assays, the C-3/C-3′-dideoxy aglycon (12) bound with higher affinity than the (2S, 2′S)-lomaiviticin aglycon (10), suggesting the (2R, 2′R)-configuration found in 12 and the natural lomaiviticins may be stereochemically-matched with the absolute configuration of DNA.
Figure 3.

FID assays of equimolar concentrations of diazofluorenes (0.88 µM) against thiazole orange (TO, 1.25 µM) using calf thymus DNA as substrate (0.88 µM in base pairs).
Table 1.
FID-based determination of DC50 values (µM) of dimeric diazofluorenes against polynucleotides of increasing GC content.
| polynucleotide (%GC content) |
C. perfringens (32%) |
Calf thymus (42%) |
M. lysodeiktius (75%) |
|---|---|---|---|
| (−)-lomaiviticin aglycon (9) | 14.4 | 7.47 | 9.98 |
| (2S,2′S)-lomaiviticin aglycon (10) | 56.2 | 40.7 | 63.2 |
| C-3/C-3′-dideoxylomaiviticin aglycon (12) | 18.6 | 19.8 | 59.7 |
| (−)-monomeric lomaiviticin aglycon (11) | >100 | >100 | >100 |
| (−)-kinamycin C (6) | >100 | >100 | >100 |
| C-3-deoxy-monomeric lomaiviticin aglycon (13) | >100 | >100 | >100 |
FID titration plots were utilized to determine the binding site size (Figure S1). (−)-Lomaiviticin aglycon (9), the (2S, 2′S)-lomaiviticin aglycon (10), and the C-3/C-3′-dideoxy aglycon (12) showed a binding site size of ~2 base pairs per molecule (1.8, 2.0, 2.0, base pairs per ligand for 9, 10, and 12, respectively). This binding site size is similar to that of well-known intercalators, such as ethidium bromide.[12] Circular dichroism (CD) and linear dichroism (LD) titration experiments using (−)-lomaiviticin aglycon (9) or (−)-kinamycin C (6), and calf thymus DNA, established an intercalative mode of binding. Sequential additions of (−)-lomaiviticin aglycon (9) to DNA led to small changes in the CD signal at ~303 nm and a small positive induced CD at 553 nm (Figure S2A). Serial additions of (−)-kinamycin C (6) resulted in an induced CD at 313 nm and 406 nm, and changes in the DNA absorption region at 280 nm were also discernable (Figure S2B). The small induced CD observed in our studies suggests intercalation of the diazofluorenes into the base stack.[13] An LD titration using (−)-lomaiviticin aglycon (9, ratio of base pairs to drug = 8) led to changes in the intensity of LD signal at 221 nm and 258 nm (Figure 4a). The enhancement in the negative induced LD of DNA bases arises from lengthening of the DNA helix,[14] consistent with intercalation (Figure 4b). LD spectra of (−)-kinamycin C (6) additionally supported intercalation as the primary mode of binding (Figure S3).
Figure 4.

Linear dichroism spectra of (a) (−)-lomaiviticin aglycon (9) and (b) ethidium bromide at rbd 8. Buffer conditions: 10 mM sodium cacodylate, 0.5 mM EDTA, 100 mM KCl at pH 6.8 (T = 22–23 °C).
To evaluate the DNA cleavage ability of these diazofluorenes, we performed a plasmid cleavage assay in the presence of the reducing cofactor dithiothreitol (DTT, Figure 5).[15] (−)-Kinamycin C (6) was inactive at the concentrations up to 500 µM (lane 2). Surprisingly, (−)-lomaiviticin aglycon (9), which bound DNA with the highest affinity (vide supra), was also an ineffective cleavage agent at concentrations up to 500 µM (lanes 3–5). In contrast, the (2S, 2′S)-lomaiviticin aglycon (10) displayed potent levels of DNA nicking (lanes 6–8). Minor amounts of DNA dsbs were observed at 500–250 µM 10 (lanes 6, 7). The (−)-monomeric lomaiviticin aglycon (11) also nicked DNA in a concentration-dependent fashion (lanes 9–11). Identical cleavage activities were observed with NADPH or GSH as reductant (Figure S4).
Figure 5.
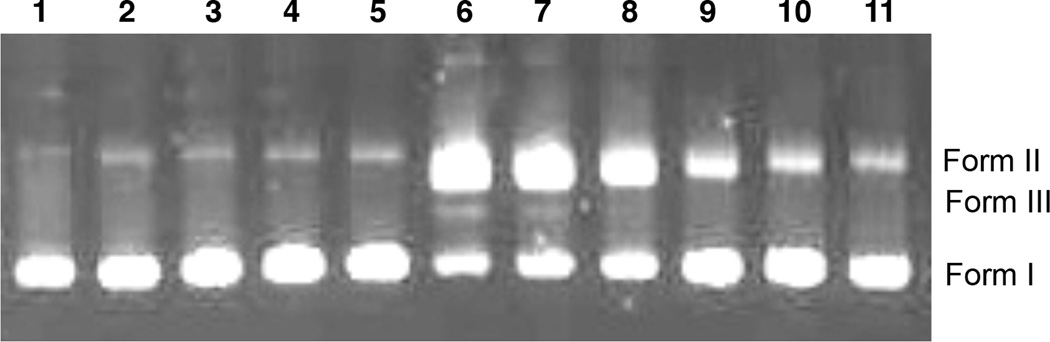
Agarose gel electrophoresis of the DNA cleavage of pBr322 DNA (800 ng) treated with diazofluorenes and DTT (5 mM) cofactor (24 h, 37 °C). Lane 1: pBr322 DNA. Lane 2: [6] = 500 µM. Lane 3: [9] = 500 µM. Lane 4: [9] = 250 µM. Lane 5: [9] = 125 µM. Lane 6: [10] = 500 µM. Lane 7: [10] = 250 µM. Lane 8: [10] = 125 µM. Lane 9: [11] = 500 µM. Lane 10: [11] = 250 µM. Lane 11: [11] = 125 µM. Form I = supercoiled DNA; Form II = nicked DNA; Form III = linearized DNA.
Surprisingly, we observed that the (2S, 2′S)-lomaiviticin aglycon (10) and, to a lesser extent the (−)-monomeric lomaiviticin aglycon (11), nick plasmid DNA in the absence of added reductant (Figure 6). At concentrations of 250 µM, substantial amounts of DNA nicking were observed with 10 (lane 9). Although DNA nicking activity by 11 was considerably lower, an increase in Form II DNA was observed at 500–250 µM 11 (compare lanes 1, 6, and 7). (−)-Kinamycin C (6) and (−)-lomaiviticin aglycon (9) were inactive in this assay, as expected (lanes 2–5).
Figure 6.

Agarose gel electrophoresis of the DNA cleavage of pBr322 DNA (800 ng) treated with diazofluorenes (24 h, 37 °C). Lane 1: pBr322. Lane 2: [6] = 500 µM. Lane 3: [6] = 250 µM. Lane 4: [9] = 500 µM. Lane 5: [9] = 250 µM. Lane 6: [11] = 500 µM. Lane 7: [11] = 250 µM. Lane 8: [10] = 500 µM. Lane 9: [10] = 250 µM. Form I = supercoiled DNA; Form II = nicked DNA; Form III = linearized DNA.
We sought to determine if the in vitro DNA cleavage activity by the (2S, 2′S)-lomaiviticin aglycon (10) was recapitulated in tissue culture. Production of phospho-SER139-H2AX (γH2AX) is a widely-employed marker for detection of DNA dsbs.[16] We evaluated the ability of 10, (−)-kinamycin C (6), the (−)-monomeric lomaiviticin aglycon (11), and the C-3/C-3′-dideoxy lomaiviticin aglycon (12), to induce production of γH2AX in K562 cells. The cells were incubated with the diazofluorenes (1 µM) for 4 h at 37 °C, treated with an anti-γH2AX antibody conjugated to fluorescein isothiocyanate (FITC), and counted by flow cytometry (Table 2). Surprisingly, the (−)-monomeric lomaiviticin aglycon (11) was significantly more potent than any other compound investigated, and upregulated the production of γH2AX by 600% relative to the control. These results point to the existence of an optimal balance of uptake and reactivity within the studied diazofluorenes that is captured by 11. Serial dilutions of the (−)-monomeric lomaiviticin aglycon (11) revealed a lower limit of 50 nM for DNA dsbs production (23% upregulation of γH2AX, Figure S5). Consistent with this, 11 displayed an IC50 = 530 nM against this cell line. The other analogs 9, 10, and 12 were 3–5-fold less potent. By comparison, (−)-lomaiviticin A (1) is still significantly more potent (IC50 = 8 nM). The increased potency of 1 likely arises from an increased efficiency in production of DNA dsbs and the higher solubility of the natural metabolite.[5] Additionally, DNA dsb production by 11 was observed in both a cisplatin-sensitive ovarian cancer cell line (PEO1) and a cisplatin-resistant ovarian cancer cell line (PEO4, Table S1).[8]
Table 2.
H2A.X phosphorylation assay of human leukemia cells (K562) treated with diazofluorenes (1 µM) for 4 h at 37 °C.a.
| compound | geometric mean |
% increase |
|---|---|---|
| (−)-kinamycin C (6) | 1.41E4 | 82% |
| (−)-lomaiviticin aglycon (9) | 8.95E3 | 15% |
| (2S,2′S)-lomaiviticin aglycon (10) | 9.83E3 | 27% |
| C-3/C-3′-dideoxy lomaiviticin aglycon (12) | 1.03E4 | 33% |
| (−)-monomeric lomaiviticin aglycon (11) | 5.43E4 | 600% |
| control | 7.76E3 | – |
Cells were stained for γ-H2AX. Immunological detection was performed by labeling with anti-γH2AX (Ser139) AB–fluorescein isothiocyanate conjugate. Sample analysis was performed on an Accuri flow cytometer using 488 nm excitation laser. Emission detected with the filter/bandpass: 530/30 for FITC
Taken together, several important conclusions emerge from these studies. First, dimeric diazofluorenes bind DNA with higher affinity than monomeric diazofluorenes, and the primary mode of binding appears to be intercalation into the double helix. However, while the dimeric structure of the lomaiviticins increases affinity for dsDNA, this structure is not sufficient for DNA cleavage activity. Instead, our studies suggest that the D-ring carbonyl of the diazofluorenes is critical for DNA damaging activity. We have previously shown[9b] that the (2S, 2′S)-aglycon 10 and the (−)-monomeric lomaiviticin aglycon (11) undergo hydrodediazotization under conditions (1 equiv DTT, methanol, 37 °C) where (−)-kinamycin C (6) and (−)-lomaiviticin aglycon (9) are inert. It is likely that the D-ring carbonyl raises the oxidation potential of the diazofluorene functional group, facilitating nucleophilic addition to the diazo group and the production of vinyl radical intermediates.[5] Interestingly, the activity of the (2S, 2′S)-lomaiviticin aglycon (10), which is the most potent DNA damaging agent in vitro, is not recapitulated in tissue culture. Instead, the (−)-monomeric lomaiviticin aglycon (11) appears to present the optimal balance of reactivity, stability, and cellular uptake. As this compound is readily prepared in 11 steps from 3-ethylphenol,[9b] it provides a useful starting point for translational development. Future studies will focus on increasing the potency of 11 by increasing its solubility and affinity for DNA. Given the structural dissimilarities of 1 and 11, it seems plausible that the nature of the DNA breaks produced by the two compounds are distinct.
Supplementary Material
Acknowledgments
Financial support from the National Institute of General Medical Sciences (5R01GM090000 to S.B.H., 1R41GM097917 to D.P.A.), the National Science Foundation (Graduate Research Fellowship to C.M.W.), the Searle Scholars Program, and Yale University is gratefully acknowledged. We acknowledge the Developmental Therapeutics Program of the National Cancer Institute for a gift of (−)-kinamycin C (6, NSC 138425). S.B.H. is a fellow of the David and Lucile Packard and the Alfred P. Sloan Foundations, is a Cottrell Scholar of the Research Corporation for Science Advancement, and is a Camille Dreyfus Teacher–Scholar.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Christina M. Woo, Department of Chemistry, Yale University, 225 Prospect Street, New Haven, CT 06520-8107 (USA)
Nihar Ranjan, Department of Chemistry, Clemson University, Clemson, SC 29634 (USA).
Dev P. Arya, Department of Chemistry, Clemson University, Clemson, SC 29634 (USA)
Seth B. Herzon, Department of Chemistry, Yale University, 225 Prospect Street, New Haven, CT 06520-8107 (USA).
References
- 1.Isolation of the lomaiviticins: He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J. Am. Chem. Soc. 2001;123:5362. doi: 10.1021/ja010129o. Woo CM, Beizer NE, Janso JE, Herzon SB. J. Am. Chem. Soc. 2012;134:15285. doi: 10.1021/ja3074984.
- 2.Isolation of the kinamycins: Ito S, Matsuya T, Ōmura S, Otani M, Nakagawa A. J. Antibiot. 1970;23:315. doi: 10.7164/antibiotics.23.315. Hata T, Ōmura S, Iwai Y, Nakagawa A, Otani M. J. Antibiot. 1971;24:353. doi: 10.7164/antibiotics.24.353. Ōmura S, Nakagawa A, Yamada H, Hata T, Furusaki A, Watanabe T. Chem. Pharm. Bull. 1971;19:2428. doi: 10.1248/cpb.21.931. Furusaki A, Matsui M, Watanabe T, Ōmura S, Nakagawa A, Hata T. Isr. J. Chem. 1972;10:173. Ōmura S, Nakagawa A, Yamada H, Hata T, Furusaki A. Chem. Pharm. Bull. 1973;21:931. doi: 10.1248/cpb.21.931. Cone MC, Seaton PJ, Halley KA, Gould SJ. J. Antibiot. 1989;42:179. doi: 10.7164/antibiotics.42.179. Seaton PJ, Gould SJ. J. Antibiot. 1989;42:189. doi: 10.7164/antibiotics.42.189.
- 3.For reviews, see: Gould SJ. Chem. Rev. 1997;97:2499. doi: 10.1021/cr9600215. Marco-Contelles J, Molina MT. Curr. Org. Chem. 2003;7:1433. Arya DP. Top. Heterocycl. Chem. 2006;2:129. Nawrat CC, Moody CJ. Nat. Prod. Rep. 2011;28:1426. doi: 10.1039/c1np00031d. Herzon SB. In: Total Synthesis of Natural Products. At the Frontiers of Organic Chemistry. Li JJ, Corey EJ, editors. Springer-Verlag: Berlin Heidelberg; 2012. Herzon SB, Woo CM. Nat. Prod. Rep. 2012;29:87. doi: 10.1039/c1np00052g.
- 4.For the first evidence and proposals for generation of vinyl radicals from the diazofluorene, see: Laufer RS, Dmitrienko GI. J. Am. Chem. Soc. 2002;124:1854. doi: 10.1021/ja0167809. Feldman KS, Eastman KJ. J. Am. Chem. Soc. 2005;127:15344. doi: 10.1021/ja053880w. Feldman KS, Eastman KJ. J. Am. Chem. Soc. 2006;128:12562. doi: 10.1021/ja0642616.
- 5.Colis LC, Woo CM, Hegan DC, Li Z, Glazer PM, Herzon SB. Nat. Chem. 2014;6:504. doi: 10.1038/nchem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For earliers mechanistic proposals and studies of the kinamycins and lomaiviticins, see: Moore HW. Science. 1977;197:527. doi: 10.1126/science.877572. Arya DP, Jebaratnam DJ. J. Org. Chem. 1995;60:3268. Hasinoff BB, Wu X, Yalowich JC, Goodfellow V, Laufer RS, Adedayo O, Dmitrienko GI. Anti-Cancer Drugs. 2006;17:825. doi: 10.1097/01.cad.0000224442.78211.27. Zeng W, Ballard TE, Tkachenko AG, Burns VA, Feldheim DL, Melander C. Bioorg. Med. Chem. Lett. 2006;16:5148. doi: 10.1016/j.bmcl.2006.07.024. O'Hara KA, Wu X, Patel D, Liang H, Yalowich JC, Chen N, Goodfellow V, Adedayo O, Dmitrienko GI, Hasinoff BB. Free Radic. Biol. Med. 2007;43:1132. doi: 10.1016/j.freeradbiomed.2007.07.005. Ballard TE, Melander C. Tetrahedron Lett. 2008;49:3157. Khdour O, Skibo EB. Org. Biomol. Chem. 2009;7:2140. doi: 10.1039/b903844b. Heinecke CL, Melander C. Tetrahedron Lett. 2010;51:1455. O'Hara KA, Dmitrienko GI, Hasinoff BB. Chem. Biol. Interact. 2010;184:396. doi: 10.1016/j.cbi.2010.01.013. Mulcahy SP, Woo CM, Ding WD, Ellestad GA, Herzon SB. Chem. Sci. 2012;3:1070.
- 7.For reviews, see: Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. Nat. Rev. Canc. 2008;8:193. doi: 10.1038/nrc2342. Aziz K, Nowsheen S, Pantelias G, Iliakis G, Gorgoulis VG, Georgakilas AG. Pharmacol. Ther. 2012;133:334. doi: 10.1016/j.pharmthera.2011.11.010.
- 8.Langdon SP, Lawrie SS, Hay FG, Hawkes MM, McDonald A, Hayward IP, Schol DJ, Hilgers J, Leonard RCF, Smyth JF. Canc. Res. 1988;48:6166. [PubMed] [Google Scholar]
- 9.(a) Herzon SB, Lu L, Woo CM, Gholap SL. J. Am. Chem. Soc. 2011;133:7260. doi: 10.1021/ja200034b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Woo CM, Gholap SL, Lu L, Kaneko M, Li Z, Ravikumar PC, Herzon SB. J. Am. Chem. Soc. 2012;134:17262. doi: 10.1021/ja307497h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boger DL, Dechantsreiter MA, Ishii T, Fink BE, Hedrick MP. Bioorg. Med. Chem. 2000;8:2049. doi: 10.1016/s0968-0896(00)00137-1. Boger DL, Fink BE, Hedrick MP. J. Am. Chem. Soc. 2000;122:6382. Boger DL, Lee JK. J. Org. Chem. 2000;65:5996. doi: 10.1021/jo000382r. Boger DL, Fink BE, Brunette SR, Tse WC, Hedrick MP. J. Am. Chem. Soc. 2001;123:5878. doi: 10.1021/ja010041a. For reviews, see: Tse WC, Boger DL. Current Protocols in Nucleic Acid Chemistry. John Wiley & Sons, Inc.; 2001. Tse WC, Boger DL. Acc. Chem. Res. 2004;37:61. doi: 10.1021/ar030113y.
- 11.Kumar S, Xue L, Arya DP. J. Am. Chem. Soc. 2011;133:7361. doi: 10.1021/ja108118v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishimura T, Okobira T, Kelly AM, Shimada N, Takeda Y, Sakurai K. Biochemistry. 2007;46:8156. doi: 10.1021/bi602402m. [DOI] [PubMed] [Google Scholar]
- 13.Eriksson M, Nordén B. In: Methods Enzymol. Jonathan MJW, Chaires B, editors. Vol. 340. Academic Press; 2001. p. 68. [DOI] [PubMed] [Google Scholar]
- 14.(a) Tuite E, Norden B. J. Am. Chem. Soc. 1994;116:7548. [Google Scholar]; (b) Dafforn TR, Rodger A. Curr. Opin. Struct. Biol. 2004;14:541. doi: 10.1016/j.sbi.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Burden DA, Froelich-Ammon SJ, Osheroff N. Methods in Molecular Biology. 2000;95:283. doi: 10.1385/1-59259-057-8:283. [DOI] [PubMed] [Google Scholar]
- 16. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. J. Biol. Chem. 1998;273:5858. doi: 10.1074/jbc.273.10.5858. Muslimovic A, Ismail IH, Gao Y, Hammarsten O. Nat. Protoc. 2008;3:1187. doi: 10.1038/nprot.2008.93. For a review, see: Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. Nat Rev Cancer. 2008;8:957. doi: 10.1038/nrc2523.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.