

Abstract
AIM
To make comprehensive molecular diagnosis for retinitis pigmentosa (RP) patients in a consanguineous Han Chinese family using next generation sequencing based Capture-NGS screen technology.
METHODS
A five-generation Han Chinese family diagnosed as non-syndromic X-linked recessive RP (XLRP) was recruited, including four affected males, four obligate female carriers and eleven unaffected family members. Capture-NGS was performed using a custom designed capture panel covers 163 known retinal disease genes including 47 RP genes, followed by the validation of detected mutation using Sanger sequencing in all recruited family members.
RESULTS
Capture-NGS in one affected 47-year-old male reveals a novel mutation, c.2417_2418insG:p.E806fs, in exon ORF15 of RP GTPase regulator (RPGR) gene results in a frameshift change that results in a premature stop codon and a truncated protein product. The mutation was further validated in three of four affected males and two of four female carriers but not in the other unaffected family members.
CONCLUSION
We have identified a novel mutation, c.2417_2418insG:p.E806fs, in a Han Chinese family with XLRP. Our findings expand the mutation spectrum of RPGR and the phenotypic spectrum of XLRP in Han Chinese families, and confirms Capture-NGS could be an effective and economic approach for the comprehensive molecular diagnosis of RP.
Keywords: retinitis pigmentosa GTPase regulator, retinitis pigmentosa, next-generation sequencing, genetic diagnosis
INTRODUCTION
Retinitis pigmentosa (RP), the most common inherited retinal degeneration disease, affects millions of people worldwide[1]. RP can be categorized into dominant, recessive and X-linked forms based on the inheritance, as well as a portion of isolated cases[2],[3]. RP is a highly variable disorder, the typical symptoms of RP initiates from night blindness and dark adaption defects, followed by the loss of mid-peripheral vision field due to the dysfunction and loss of rod photoreceptors. As the disease progresses, patients lose their peripheral vision and then central vision subsequently and ultimately develop tubular visual field or complete blindness around the age of 60[3]. The diagnosis of RP mainly based on the characteristic fundus changes including attenuation of the retinal arterioles, bone spicule-like pigment deposit and waxy yellow appearance of the optic disc, as well as the vision field defects and abnormal electroretinograms (ERG) recordings[3],[4]. Currently there is no effective treatment for RP and clinical diagnosed RP cases are most in advanced stages, since the inconspicuous early symptoms such as night blindness and vision field loss may not be noticed until late stages[3].
Genetic diagnosis provides an effective approach for the diagnosis of affected RP patients and the risk evaluation of their descendants. Currently, there are over 50 genes causing non-syndromic RP, in which 23 genes account for autosomal-dominant RP, 36 genes lead to recessive RP, and 3 genes cause X-linked recessive RP (XLRP)[5]. However, most of these causative genes and mutations are reported in European Caucasians with only a few associated with Chinese RP families[6]–[10]. RP is highly heterogeneous as not only the causative genes and mutations can vary among different ethnic groups but also the same mutation may lead to different outcomes among the members in the same family. Since reported mutation spectra of RP genes in Chinese population are dramatically different from those among European Caucasians, identification of causative genes and mutations of RP in Chinese population is of great importance[11]–[15]. Now there are at least 340 000 RP patients in China based on the reported prevalence in the 80s, and the high heterogeneity of RP makes it challenging to make a comprehensive genetic diagnosis using the traditional Sanger sequencing due to the high cost of reagent and labor[16]. Capture-Next generation sequencing (Capture-NGS), a method that coupled NGS with DNA capture technology, provides an effective alternative for its ability to parallel sequence a large panel of known retinal disease genes rapidly and economically. In this study, we report a novel mutation detected by Capture-NGS in a five-generation Han Chinese family with diagnosis of XLRP. In this family, affected males exhibit typical early onset and severe RP phenotypes at the time of diagnosis, while female carriers reveal normal ophthalmic examination results. Capture-NGS in one affected male identified a novel mutation, c.2417_2418insG:p.E806fs, in the exon ORF15 of RP GTPase regulator (RPGR) gene. The mutation was further validated in three of four affected males and two of four female carriers by Sanger sequencing. This mutation results in a premature stop codon due to the frameshift change and ultimately leads to a truncated protein product.
SUBJECTS AND METHODS
Participants and Clinical Diagnosis
A five-generation Han Chinese consanguineous family with non-symdromic XLRP was recruited. All subjects were identified at the First Affiliated Hospital of Gannan Medical University (GMU, Ganzhou, Jiangxi, China). This research was approved by the ethical review board of GMU in accordance with the Declaration of Helsinki and the Guidance on Sample Collection of Human Genetic Diseases by the Ministry of Public Health of China. Written informed consent was obtained from all patients and their relatives participating in this study. The individual in this manuscript has given written informed consent (as outlined in PLOS consent form) to publish these case details. All recruited family members underwent comprehensive ophthalmologic examination including best correct visual acuity (BCVA), slit-lamp biomicroscopy, fundus photography, visual field tests and electroretinograms. Genomic DNA was isolated from peripheral blood using a commercial kit (TIANamp Blood DNA kit, Cat#DP318, TIANGEN, Beijing, China) according to the manufacturer's protocol. The diagnosis of RP was based on the family history and presence of night blindness, retinal pigmentation, vessel attenuation, visual field loss and abnormal ERG recordings.
Genetic Analysis
Capture-NGS was performed as previously described[17],[18]. Briefly, a capture panel that covers 163 known retinal disease genes including 47 RP genes was designed to synthesize the hybridization probes. Patient genomic DNA sample (1 μg) was sheared into 300-500 bp fragments, followed by the end-repair and insertion of an extra “adenine” base to the 3′ end. Eight cycles of PCR amplification were applied to each sample after ligation of Illumina Y-shape index adapters to the ends of the DNA fragments, followed by the quantification of DNA libraries using the PicoGreen assay (Invitrogen). Fifty precapture libraries were pooled together for one capture reaction. After Hybridization and washing, captured libraries were sequenced as 100-bp paired-end reads on the Illumina HiSeq 2000 sequencer. Bioinformatics analysis of sequencing results was performed as previously described for variants calling, filtering and annotation[17]. To validate the mutations detected by capture-NGS, Sanger sequencing was performed as previously described on Applied BioSystems (ABI) 3730×l capillary sequencer[18].
RESULTS
Clinical Evaluation
There are 46 members in this five-generation pedigree, which was compiled and revealed X-linked recessive inheritance (Figure 1). Four individuals in the family passed away, among which one female had night blindness and visual impairment from mid-age as described by her grandchildren. Nineteen family members agreed to participate in our study including four affected males, four obligate female carriers and eleven unaffected family members. All recruited family members were subjected to ophthalmic examination by the Ophthalmologists in the Department of Ophthalmology, the First Affiliated Hospital of GMU. Four affected males were diagnosed as XLRP.
Figure 1. Pedigree of a Han Chinese family with XLRP. Squares and circles are males and females respectively. Filled squares are affected males. A diagonal line through a symbol indicates a deceased family member. The arrow indicates the proband.

The phenotypes of all affected males were described in details in Table 1. Patient No.11, the index patient, is a 53-year-old male who had an onset of night blindness at age of 13. The proband's brother (No.15) and two younger cousins (No.20 and 27) had their onset of night blindness at the age of 15, 12 and 13, respectively. All affected males advanced rapidly into severe RP phenotypes. At the time of diagnosis, the BCVA of four affected males ranged from Hand Motion (HM) to 0.04. Fundus examination revealed vessel attenuation and pigment deposit in the mid-peripheral retina in all affected males (Figure 2). Patients No.11, 15 and 20 showed light yellow appearance of the optic disk and patient No.20 and 27 showed tigroid fundus. Octopus perimetry showed tunnel vision in all affected males, as well as both rod and cone ERG amplitudes were below the normal thresholds. The other six unaffected male family members displayed no abnormity in ophthalmic examinations. All female carriers in this pedigree showed normal visual field with BCVA 20/20 in both eyes, as well as the normal ERG responses. Fundus observation of female carriers found no spicule formation in the peripheral retina.
Table 1. Clinical findings of all affected males.
| Patient No. | Age of onset (a) | Age of diagnosis (a) | BCVA OD/OS | Fundus change | Visual field | ERG recordings |
| 11 | 13 | 53 | HM/HM | Light yellow appearance of optic disk; pigment deposit; retinal vessel attenuation | Tubular visual field | Reduced |
| 15 | 15 | 40 | 0.02/0.02 | Tigroid fundus; light yellow appearance of optic disk; pigment deposit; Retinal vessel attenuation | Tubular visual field | Reduced |
| 20 | 12 | 42 | 0.04/0.04 | Light yellow appearance of optic disk; pigment deposit; retinal vessel attenuation | Tubular visual field | Reduced |
| 27 | 13 | 47 | 0.01/0.01 | Light yellow appearance of optic disk; pigment deposit; retinal vessel attenuation. | Tubular visual field | Reduced |
BCVA: Best correct visual acuity; OD: Right eye; OS: Left eye; HM: Hand motion; ERG: Electroretinograms.
Figure 2. Representative fundus images of affected males.
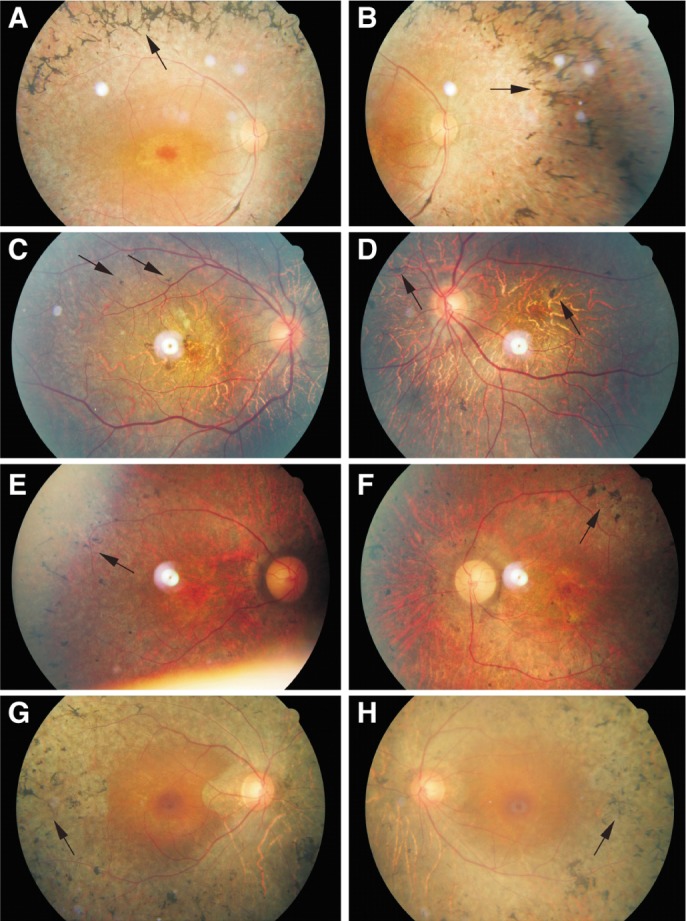
All affected males in this family exhibit characteristic fundus changes. Characteristic fundus image of patient No.11 (A-B), No.15 (C-D), No.20 (E-F) and No.27 (G-H) are present. All affected males revealed pigment deposit (arrows), retinal vessel attenuation and light yellow appearance of optic disk. Patient No.15 showed a tigroid fundus.
Genetic Analysis
One affected male (No.27) was screened by Capture-NGS. Sequence reads were mapped to hg19 human genome reference using Burrows-Wheeler Aligner (BWA version 0.5.9) and variants were called using Atlas toolkit[19],[20]. Raw variants were further filtered against several variants database including 1000 genome, dbSNP, ESP6500 and 997 internal control database using a cutoff of 0.5%[17],[21]. The remaining rare variants were annotated using ANNOVAR software[22]. Finally a novel mutation, c.2417_2418insG:p.E806fs, in exon ORF15 of RPGR was identified. The exon ORF15 was then PCR amplified using primers previously described[23] and validated in all recruited family members using Sanger sequencing. The novel mutation was found in three of four affected males (No.15, 20 and 27) and two of four female carriers (No.13 and 16), but not in other unaffected family members. The insertion of guanine between codons 2417 and 2418 results in the significant change of translational products and brings in a premature stop codon (Figure 3). The mutation in one affected male (No.11) and two female carriers (No.12 and 29) could not be cleanly validated by Sanger sequencing due to the high repetitive feature of exon ORF15.
Figure 3. Identification of c.2417_2418insG:p.E806fs mutation in the RPGR gene.

A: Structure of RPGR gene and its location in the X chromosome. Red line indicates the location of novel mutation in exon ORF15; B: Sanger sequencing reveals the insertion of Guanine between codons 2417 and 2418 (arrow); C: The frameshift change (p.E806fs) (arrow) caused by the c.2417_2418insG mutation results in a premature stop codon and a truncated protein product.
DISCUSSION
To date, more than 3000 mutations have been reported in identified RP genes[5]. Due to the limited resources, only a small set of genes have been accessed by Sanger sequencing, the traditional main approach for RP molecular diagnosis. With the emergence of Next-generation Sequencing and DNA capture technology, Capture-NGS provides a promising alternative for the molecular diagnosis of RP with an approximately diagnostic rate above 50%[17],[24]–[26]. In this study, we reported a novel mutation, c.2417_2418insG:p.E806fs, in exon ORF15 of RPGR by Capture-NGS technology in a Han Chinese family. After screening of one affected male using Capture-NGS, the mutation was identified and then further validated in three of four affected males and two of four female carriers but not in unaffected family members by Sanger sequencing.
XLRP is the most severe form of RP for its early age of onset and rapid progression. Affected males always exhibit night blindness and severely abnormal ERG recordings in early adolescence, followed by the profound loss of peripheral visual fields in the second decade and ultimately tubular visual field or complete blindness in the fourth or fifth decade. Affected males usually exhibit much more severe phenotypes than the affected females. While female carriers may experience variable symptoms and visual impairment in the mid-age[3],[4],[27],[28]. In this study, all four affected males suffered severe RP phenotypes that initiated around the age of 12 to 15 and progressed rapidly. At the time of diagnosis, all affected males showed tubular visual field with barely left central vision, accompany with the severe abnormal ERG recordings (Table 1 and Figure 2). Whereas, there is no ophthalmic abnormality observed in four female carriers.
Most XLRP cases are reported caused by the mutations in RPGR or retinitis pigmentosa 2 gene (RP2), which covers approximately up to 75% and 25% of XLRP cases respectively[29]–[32]. Meanwhile, RPGR mutations are suggested be responsible for up to 20% of all RP cases, higher than any other single genetic locus[33]. RP2 is composed of five exons encoding a ubiquitously expressed polypeptide of 350 amino acids[34]. RP2 suggested being a membrane- and/or tubulin-associated chaperone or signaling protein and most causative mutations are localized in its cofactor C homology domain in the N-terminus[33]. RPGR locates at chromosome Xp21.1. It contains 19 originally identified exons and is thought to encode a cilia-centrosomal protein essential for cilia function and protein trafficking along the photoreceptor cilium[35]–[38]. Alterations of RPGR protein are believed to interrupt the normal protein interactions and leads to photoreceptor degeneration. A number of alternatively spliced isoforms of RPGR have been identified including RPGRex1−19 and RPGRORF15[39]–[41]. RPGRex1−19 was derived from initially identified exon 1 to 19 and mutations within RPGRex1−19 was found in only up to 20% of the families with XLRP[8]. RPGRORF15, which preferentially expressed in retina, was derived from exon 1 to 14 of RPGRex1−19 and a large terminal exon ORF15 that encodes a 567 amino acid polypeptide with a repetitive domain rich in glutamic acid residues[41]. Exon ORF15 is regarded as a mutation hot spot, in which clustered over 60% mutations of RPGRORF15 in XLRP families[33],[40],[43]–[44]. Most mutations in exon ORF15 are frameshift or missense mutations, both of which lead to early translational termination and result in truncated protein products[45],[46]. In this study, we have identified a novel mutation, c.2417_2418insG:p.E806fs, located in the highly repetitive region of exon ORF15. The mutation leads to a frameshift change and results in a truncated protein product lost due to the premature stop codon. To date, more than 100 mutations have been identified within exon ORF15, but only a few are reported in Han Chinese population, and the precise role of ORF15 remains unknown[6],[9],[10],[46]–[48].
In conclusion, our study is the first attempt of comprehensive genetic diagnosis of XLRP in Chinese population using Capture-NGS technology. The identification of novel mutation, c.2417_2418insG:p.E806fs, in exon ORF15 of RPGR associated with XLRP in this Han Chinese family expands the mutation spectrum of RPGR and the phenotypic spectrum of XLRP in Chinese families. Our finding is important for exploring the genotype-phenotype correlation and for elucidating the distribution and contribution of the ORF15 mutations and will be useful for genetic consultation and diagnosis. Further investigation on the mechanism through which this mutation acts will require further study of RPGR protein in both normal and mutant retinas.
Acknowledgments
We thank the members of Department of Ophthalmology in the First Affiliated Hospital of Gannan Medical University for sample collection and Dr. Feng Wang in Human Genome Sequencing Center of Baylor College of Medicine for helpful discussions and technical assistances.
Foundations: Supported by National Natural Science Foundation of China (No. 81060081; 81241124 and 81360155); the Research to Prevent Blindness Challenge Grant to the Department of Ophthalmology at the University of Rochester.
Conflicts of Interest: Hu F, None; Zeng XY, None; Liu LL, None; Luo YL, None; Jiang YP, None; Wang H, None; Xie J, None; Hu CQ, None; Gan L, None; Huang L, None.
REFERENCES
- 1.Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34(5):1659–1676. [PubMed] [Google Scholar]
- 2.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002;(233):1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 3.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 4.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi: 10.1186/1750-1172-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daiger SP, Sullivan LS, Bowne SJ, Birch DG, Heckenlively JR, Pierce EA, Weinstock GM. Targeted high-throughput DNA sequencing for gene discovery in retinitis pigmentosa. Adv Exp Med Biol. 2010;664:325–331. doi: 10.1007/978-1-4419-1399-9_37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang W, Ding Q, Tang Z, Liu P, Jiang F, Ke T, Ren X, Wang Z, Liu J, Wang QK, Liu M. A novel de novo frameshift mutation of RPGR ORF15 is associated with X-linked retinitis pigmentosa in a Chinese family. Mol Vis. 2007;13:1548–1554. [PubMed] [Google Scholar]
- 7.Sheng X, Li Z, Zhang X, Wang J, Ren H, Sun Y, Meng R, Rong W, Zhuang W. A novel mutation in retinitis pigmentosa GTPase regulator gene with a distinctive retinitis pigmentosa phenotype in a Chinese family. Mol Vis. 2010;16:1620–1628. [PMC free article] [PubMed] [Google Scholar]
- 8.Li N, Dai S, Zhang L, Mei H, Wang L. A novel mutation of RPGR gene in an X-linked Chinese family with retinitis pigmentosa. Mol Genet Metab. 2011;102(4):488–493. doi: 10.1016/j.ymgme.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Zhao K, Wang L, Wang L, Wang L, Zhang Q, Wang Q. Novel deletion of the RPGR gene in a Chinese family with X-linked retinitis pigmentosa. Ophthalmic Genet. 2001;22(3):187–194. doi: 10.1076/opge.22.3.187.2221. [DOI] [PubMed] [Google Scholar]
- 10.Liu L, Jin L, Liu M, Wei Y, Wu X, Liu Y, Wang H, Chu R, Chai J. Identification of two novel mutations (E332X and c1536delC) in the RPGR gene in two Chinese families with X-linked retinitis pigmentosa. Hum Mutat. 2000;15(6):584. doi: 10.1002/1098-1004(200006)15:6<584::AID-HUMU26>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 11.Chan WM, Yeung KY, Pang CP, Baum L, Lau TC, Kwok AK, Lam DS. Rhodopsin mutations in Chinese patients with retinitis pigmentosa. Br J Ophthalmol. 2001;85(9):1046–1048. doi: 10.1136/bjo.85.9.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao K, Xiong S, Wang L, Wang L, Cui Y, Wang Q. Novel rhodopsin mutation in a Chinese family with autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 2001;22(3):155–162. doi: 10.1076/opge.22.3.155.2225. [DOI] [PubMed] [Google Scholar]
- 13.Li S, Xiao X, Wang P, Guo X, Zhang Q. Mutation spectrum and frequency of the RHO gene in 248 Chinese families with retinitis pigmentosa. Biochem Biophys Res Commun. 2010;401(1):42–47. doi: 10.1016/j.bbrc.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Sung CH, Davenport CM, Hennessey JC, Maumenee IH, Jacobson SG, Heckenlively JR, Nowakowski R, Fishman G, Gouras P, Nathans J. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88(15):6481–6485. doi: 10.1073/pnas.88.15.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dryja TP, Hahn LB, Cowley GS, McGee TL, Berson EL. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88(20):9370–9374. doi: 10.1073/pnas.88.20.9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu DN. Prevalence and mode of inheritance of major genetic eye diseases in China. J Med Genet. 1987;24(10):584–588. doi: 10.1136/jmg.24.10.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu Q, Wang F, Wang H, Xu F, Zaneveld JE, Ren H, Keser V, Lopez I, Tuan HF, Salvo JS, Wang X, Zhao L, Wang K, Li Y, Koenekoop RK, Chen R, Sui R. Next-generation sequencing-based molecular diagnosis of a Chinese patient cohort with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(6):4158–4166. doi: 10.1167/iovs.13-11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Wang H, Sun V, Tuan H-F, Keser V, Wang K, Ren H, Lopez I, Zaneveld JE, Siddiqui S, Bowles S, Khan A, Salvo J, Jacobson SG, Iannaccone A, Wang F, Birch D, Heckenlively JR, Fishman GA, Traboulsi EI, Li Y, Wheaton D, Koenekoop RK, Chen R. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50(10):674–688. doi: 10.1136/jmedgenet-2013-101558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Challis D, Yu J, Evani US, Jackson AR, Paithankar S, Coarfa C, Milosavljevic A, Gibbs RA, Yu F. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.1000 Genomes Project Consortium. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Churchill JD, Bowne SJ, Sullivan LS, Lewis RA, Wheaton DK, Birch DG, Branham KE, Heckenlively JR, Daiger SP. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(2):1411–1416. doi: 10.1167/iovs.12-11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neveling K, Collin RW, Gilissen C, van Huet RA, Visser L, Kwint MP, Gijsen SJ, Zonneveld MN, Wieskamp N, de Ligt J, Siemiatkowska AM, Hoefsloot LH, Buckley MF, Kellner U, Branham KE, den Hollander AI, Hoischen A, Hoyng C, Klevering BJ, van den Born LI, Veltman JA, Cremers FP, Scheffer H. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat. 2012;33(6):963–972. doi: 10.1002/humu.22045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Sullivan J, Mullaney BG, Bhaskar SS, Dickerson JE, Hall G, O'Grady A, Webster A, Ramsden SC, Black GC. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J Med Genet. 2012;49(5):322–326. doi: 10.1136/jmedgenet-2012-100847. [DOI] [PubMed] [Google Scholar]
- 26.Shanks ME, Downes SM, Copley RR, Lise S, Broxholme J, Hudspith KA, Kwasniewska A, Davies WI, Hankins MW, Packham ER, Clouston P, Seller A, Wilkie AO, Taylor JC, Ragoussis J, Németh AH. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet. 2013;21(3):274–280. doi: 10.1038/ejhg.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fishman GA, Weinberg AB, McMahon TT. X-linked recessive retinitis pigmentosa. Clinical characteristics of carriers. Arch Ophthalmol. 1986;104(9):1329–1335. doi: 10.1001/archopht.1986.01050210083030. [DOI] [PubMed] [Google Scholar]
- 28.Souied E, Segues B, Ghazi I, Rozet JM, Chatelin S, Gerber S, Perrault I, Michel-Awad A, Briard ML, Plessis G, Dufier JL, Munnich A, Kaplan J. Severe manifestations in carrier females in X linked retinitis pigmentosa. J Med Genet. 1997;34(10):793–797. doi: 10.1136/jmg.34.10.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musarella MA, Anson-Cartwright L, Leal SM, Gilbert LD, Worton RG, Fishman GA, Ott J. Multipoint linkage analysis and heterogeneity testing in 20 X-linked retinitis pigmentosa families. Genomics. 1990;8(2):286–296. doi: 10.1016/0888-7543(90)90284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ott J, Bhattacharya S, Chen JD, Denton MJ, Donald J, Dubay C, Farrar GJ, Fishman GA, Frey D, Gal A, et al. Localizing multiple X chromosome-linked retinitis pigmentosa loci using multilocus homogeneity tests. Proc Natl Acad Sci U S A. 1990;87(2):701–704. doi: 10.1073/pnas.87.2.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teague PW, Aldred MA, Jay M, Dempster M, Harrison C, Carothers AD, Hardwick LJ, Evans HJ, Strain L, Brock DJ, et al. Heterogeneity analysis in 40 X-linked retinitis pigmentosa families. Am J Hum Genet. 1994;55(1):105–111. [PMC free article] [PubMed] [Google Scholar]
- 32.Fujita R, Buraczynska M, Gieser L, Wu W, Forsythe P, Abrahamson M, Jacobson SG, Sieving PA, Andréasson S, Swaroop A. Analysis of the RPGR gene in 11 pedigrees with the retinitis pigmentosa type 3 genotype: paucity of mutations in the coding region but splice defects in two families. Am J Hum Genet. 1997;61(3):571–580. doi: 10.1086/515523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breuer DK, Yashar BM, Filippova E, Hiriyanna S, Lyons RH, Mears AJ, Asaye B, Acar C, Vervoort R, Wright AF, Musarella MA, Wheeler P, MacDonald I, Iannaccone A, Birch D, Hoffman DR, Fishman GA, Heckenlively JR, Jacobson SG, Sieving PA, Swaroop A. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70(6):1545–1554. doi: 10.1086/340848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AA, Rosenberg T, Pinckers AJ, Fundele R, Rosenthal A, Cremers FP, Ropers HH, Berger W. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. 1998;19(4):327–332. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- 35.Murga-Zamalloa CA, Swaroop A, Khanna H. RPGR-containing protein complexes in syndromic and non-syndromic retinal degeneration due to ciliary dysfunction. J Genet. 2009;88(4):399–407. doi: 10.1007/s12041-009-0061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murga-Zamalloa CA, Atkins SJ, Peranen J, Swaroop A, Khanna H. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum Mol Genet. 2010;19(18):3591–3598. doi: 10.1093/hmg/ddq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roepman R, van Duijnhoven G, Rosenberg T, Pinckers AJ, Bleeker-Wagemakers LM, Bergen AA, Reinhardt R, Ropers HH, Cremers FP, Berger W. Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology with the guanine-nucleotide-exchange factor RCC1. Hum Mol Genet. 1996;5(7):1035–1041. doi: 10.1093/hmg/5.7.1035. [DOI] [PubMed] [Google Scholar]
- 38.Meindl A, Dry K, Herrmann K, Manson F, Ciccodicola A, Edgar A, Carvalho MR, Achatz H, Hellebrand H, Lennon A, Migliaccio C, Porter K, Zrenner E, Bird A, Jay M, Lorenz B, Wittwer B, D'Urso M, Meitinger T, Wright A. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3) Nat Genet. 1996;13(1):35–42. doi: 10.1038/ng0596-35. [DOI] [PubMed] [Google Scholar]
- 39.Neidhardt J, Glaus E, Barthelmes D, Zeitz C, Fleischhauer J, Berger W. Identification and characterization of a novel RPGR isoform in human retina. Hum Mutat. 2007;28(8):797–807. doi: 10.1002/humu.20521. [DOI] [PubMed] [Google Scholar]
- 40.Vervoort R, Lennon A, Bird AC, Tulloch B, Axton R, Miano MG, Meindl A, Meitinger T, Ciccodicola A, Wright AF. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25(4):462–466. doi: 10.1038/78182. [DOI] [PubMed] [Google Scholar]
- 41.Kirschner R, Rosenberg T, Schultz-Heienbrok R, Lenzner S, Feil S, Roepman R, Cremers FP, Ropers HH, Berger W. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum Mol Genet. 1999;8(8):1571–1578. doi: 10.1093/hmg/8.8.1571. [DOI] [PubMed] [Google Scholar]
- 42.Rozet JM, Perrault I, Gigarel N, Souied E, Ghazi I, Gerber S, Dufier JL, Munnich A, Kaplan J. Dominant X linked retinitis pigmentosa is frequently accounted for by truncating mutations in exon ORF15 of the RPGR gene. J Med Genet. 2002;39(4):284–285. doi: 10.1136/jmg.39.4.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharon D, Bruns GA, McGee TL, Sandberg MA, Berson EL, Dryja TP. X-linked retinitis pigmentosa: mutation spectrum of the RPGR and RP2 genes and correlation with visual function. Invest Ophthalmol Vis Sci. 2000;41(9):2712–2721. [PubMed] [Google Scholar]
- 44.Shu X, Black GC, Rice JM, Hart-Holden N, Jones A, O'Grady A, Ramsden S, Wright AF. RPGR mutation analysis and disease: an update. Hum Mutat. 2007;28(4):322–328. doi: 10.1002/humu.20461. [DOI] [PubMed] [Google Scholar]
- 45.Vervoort R, Wright AF. Mutations of RPGR in X-linked retinitis pigmentosa (RP3) Hum Mutat. 2002;19(5):486–500. doi: 10.1002/humu.10057. [DOI] [PubMed] [Google Scholar]
- 46.Jin ZB, Hayakawa M, Murakami A, Nao-i N. RCC1-like domain and ORF15: essentials in RPGR gene. Adv Exp Med Biol. 2006;572:29–33. doi: 10.1007/0-387-32442-9_5. [DOI] [PubMed] [Google Scholar]
- 47.Sheng X, Li Z, Zhang X, Wang J, Ren H, Sun Y, Meng R, Rong W, Zhuang W. A novel mutation in retinitis pigmentosa GTPase regulator gene with a distinctive retinitis pigmentosa phenotype in a Chinese family. Mol Vis. 2010;16:1620–1628. [PMC free article] [PubMed] [Google Scholar]
- 48.Ji Y, Wang J, Xiao X, Li S, Guo X, Zhang Q. Mutations in RPGR and RP2 of Chinese patients with X-linked retinitis pigmentosa. Curr Eye Res. 2010;35(1):73–79. doi: 10.3109/02713680903395299. [DOI] [PubMed] [Google Scholar]