

Abstract
The structure of the stable Pd(0) precatalyst [(1,5-cyclooctadiene)(L•Pd)2] (L = AdBrettPhos) for the Pd-catalyzed fluorination of aryl triflates has been further studied by solid state NMR and X-ray cystrallography of the analogous N-phenylmaleimide complex. The reactivity of this complex with CDCl3 to form a dearomatized complex is also presented. In addition, studies suggest that related bulky biaryl phosphine ligands form similar complexes, although the smaller ligand BrettPhos forms a monomeric [(1,5-cyclooctadiene)(L•Pd)] species instead.
Keywords: Pd(0) complex, precatalyst, biaryl phosphine, oxidative addition
1. Introduction
Generation of the catalytically active L•Pd(0) species is a necessary but often overlooked first step in most Pd-catalyzed cross-coupling processes. We recently found that this step was problematic in the Pd-catalyzed fluorination of aryl triflates using the biaryl phosphine ligand AdBrettPhos (1, Figure 1) [1]. Traditionally, Pd(0) species are accessed by combining the desired ligand (e.g. 1) with either a Pd(II) precursor such as PdCl2, Pd(OAc)2, or [(cinnamyl)PdCl]2 (Figure 1, Left) or a stable Pd(0) precursor such as Pd2(dba)3 or Pd(dba)2 (Figure 1, Top). However, both of these methods have drawbacks: the use of Pd(II) precursors requires in situ reduction to Pd(0), which is inefficient in many cases and generates potentially reactive byproducts such as Cl− and AcO−, while commercially available samples of Pdn(dba)m species are of variable quality [2] and release dba upon activation, which is known to have an inhibitory effect on some cross-coupling reactions [3]. In addition, both methods require an excess of the desired ligand relative to Pd. As an alternative to these methods, our laboratory has developed stable pre-ligated Pd(II) species that activate in the presence of base to release the desired L•Pd(0) species without the requirement for additional ligand relative to Pd [4]. However, our third generation precatalyst 1a for 1 (Figure 1, Right) [5] generates one equivalent of both carbazole and acid upon activation; these byproducts were shown to dramatically decrease the yield of Pd-catalyzed fluorination when using this precatalyst [1]. A less explored alternative to these pathways would be a precatalyst strategy using a L•Pd(0) species instead of a L•Pd(II) species, which would preclude the necessity of base for precatalyst activation. Although many biaryl phosphine-ligated Pd(II) complexes have been isolated, there exist only a few examples of related Pd(0) complexes [6]. This is due to the high reactivity of electron-rich phosphine-ligated Pd(0) complexes and their tendency to decompose via oxidation or Pd nanoparticle formation. Nonetheless, we recently reported that combining equimolar quantities of 1 and [(1,5-COD)Pd(CH2TMS)2] (1,5-COD = 1,5-cyclooctadiene) in pentane resulted in precipitation of what we propose to be 2 (Figure 1, bottom), which despite containing two Pd(0) centers is indefinitely stable at room temperature under N2 [1]. Most importantly, 2 readily activates by dissociation of the otherwise innocent 1,5-COD ligand. For example, when exposed to 4-nBuPhOTf 2 converts to 1•Pd(4-nBuPh)OTf in less than 10 min at room temperature. This property makes 2 an ideal precatalyst for the Pd-catalyzed fluorination of aryl triflates [1]. Unfortunately, 2 is highly insoluble or unstable in most organic solvents, making its characterization difficult. We describe herein further investigation of the structure of 2 by solid-state NMR and X-ray cystallographic studies of a related trigonal planar Pd(0) complex. In addition, we report the ability of 2 to dissociate 1,5-COD and react with CDCl3 to produce a dearomatized complex and to undergo oxidative addition to aryl bromides at room temperature. Lastly, we report our attempts to synthesize [(1,5-COD)(L•Pd)2] species for other biaryl phosphine ligands, which may prove useful as precatalysts for reactions that are inhibited by the byproducts generated from activation of other Pd sources and/or reactions that do not require the addition of base.
Figure 1.

Generation of 1•Pd(0) using conventional Pd sources in comparison with 2.
2. Experimental
2.1 Materials
Pentane was purchased from Aldrich in Sure-Seal™ bottles and sparged with argon before use. CD2Cl2, CDCl3, and toluene-d8 were purchased from Cambridge Isotopes in sealed ampules and used without further purification. Preparations of AdBrettPhos (1) [7], tBuBrettPhos (6) [8], RockPhos (7) [8], 2 [1], 5 [9], 12 [10], 13 [9], 14 [10], and 17 [11] have been previously described. The tBuXPhos (8) used in this work was purchased from Sigma Aldrich. The BrettPhos (15) used in this work was received as a gift from Sigma Aldrich, for which we are grateful. [(1,5-cyclooctadiene)Pd(CH2TMS)2] was prepared according to the literature [12] or using a modification of this procedure [1] and was stored at −20 °C in a nitrogen-filled glovebox when not in use. All other reagents were purchased from commercial sources and used without further purification.
2.2 Physical Methods
New compounds were analyzed by 1H, 13C, and 31P NMR, and by IR spectrometry. Air sensitive compounds 2, 9–11, and 16 could not be successfully characterized by elemental analysis. 1H and 13C NMR spectra were recorded on a Varian XL 300 MHz or Varian Inova 500 MHz spectrometers and were calibrated using residual solvent as an internal reference. 31P{1H} spectra were recorded on a Varian XL 300 MHz or Varian Inova 500 MHz spectrometer and were calibrated to an external standard of concentrated H3PO4 (δ 0.0 ppm). The following abbreviations were used to explain multiplicities: s = singlet, d = doublet, t = triplet, pt = pseudotriplet, q = quartet, p = pentet, m = multiplet. IR spectra were recorded on a Thermo Scientific Nicolet iS5 Fourier Transform IR Spectrometer. Solid-state NMR spectra were recorded at 750 MHz 1H frequency using a home-built spectrometer courtesy of David Rubin (MIT) and a triple channel Bruker 1.3 mm triple resonance probe (Bruker Biospin, Billerica, MA). Spectra acquired at 750 MHz were rotated at a MAS frequency of 60 kHz. 15 kHz TPPM proton decoupling was used in all experiments. Protons were suppressed prior to cross polarization to minimize the contributions from protons not bonded to 13C. The pulse sequence used to acquire 1H-13C spectra is as follows: polarization is transferred from 1H to 13C by cross polarization (CP), which then evolves on 13C for a time t1 with 15 kHz of 1H decoupling. The 13C signal is then stored along z and proton signal is saturated to minimize the signal from protons not bonded to 13C. A second CP transfers signal from 13C to 1H, which is then detected. The cross polarization contact time is 1.5 ms for the transfer from 1H to 13C and contact times of 0.2 ms and 0.5 ms for the transfer from 13C to 1H, respectively [13]. The indirect 13C dimension was sampled to 12.5 ms, and the 1H dimension for 40 ms. The 1H dimension was apodized with a 90 Hz gaussian filter, and the 13C dimension was apodized with a 50 Hz gaussian filter. All spectra were referenced to the water peak of an external sample at 4.8 ppm. 13C shifts are reported on the DSS scale. Spectra were processed, displayed and assigned using the NMRPipe software package (Goddard and Kneller, University of California, San Francisco).
2.3 X-ray crystallographic methods
X-ray crysallographic studies were carried out by Dr. Stacey Smith (MIT) using a Siemens three-circle Platform diffractometer coupled to a Bruker-APEX CCD detector. Yellow crystals of 3 (C44H61DCl3O2PPd) were grown by vapor diffusion of a solution of 3 in TMBE using pentane. The structure of 3 has unit cell dimensions of 21.400 Å × 10.397 Å × 18.224 Å. The structure of 3 was highly disordered. A large fraction of the molecule (which contained multiple interconnected ligands) was refined as a two-part disorder, which produced better results than using a larger unit cell with two separate molecules in the asymmetric unit. Only half of the top ring of the ligand was refined as a two-part disorder; the thermal ellipsoids were significantly better-behaved as such than if the entire ligand was split. Yellow crystals of 4 (C53H68NO4PPd) were grown by vapor diffusion of a solution of 4 in 1:1 TBME:CH2Cl2 using pentane. The structure of 4 has unit cell dimensions of 15.853 Å × 14.116 Å × 20.129 Å.
2.4 Chemical syntheses
3: In a nitrogen-filled glovebox, 2 was dissolved in CDCl3 (0.5 mL). The resulting suspension was vigorously stirred for 10 min to give a dark yellow solution. The solvent was then removed under high vacuum. Pentane (1 mL) was added to the resulting brown oil, and the mixture was vigorously stirred for 1 min at which time the solvent was removed under vacuum. The process was repeated a total of three times to yield 3 as bright yellow powder. 1H NMR (500 MHz, CDCl3): δ 6.88 (s, 2H), 5.31 (s, 1H), 5.29 (s, 1H), 3.83 (s, 3H), 3.69 (s, 3H), 2.12–2.22 (m, 12H), 1.92–1.95 (m, 6H), 1.79–1.86 (m, 3H), 1.58–1.70 (m, 12H), 1.32 (d, J = 7 Hz, 6H), 1.13 (d, J = 7 Hz, 6H), 1.04 (d, J = 7 Hz, 6H) ppm; 13C NMR (125 MHz, CDCl3): δ 154.7, 151.0, 150.8, 139.8, 139.6, 137.9, 137.9, 132.0, 131.8, 113.1, 110.8, 103.6, 103.5, 87.8, 87.8, 54.4, 54.1, 53.7, 53.7, 53.7, 43.0, 41.5, 37.7, 36.6, 31.0, 29.2, 29.1, 24.7, 23.9, 22.4, 19.3, 14.2 ppm (observed complexity is due to C-P coupling); 31P NMR (202 MHz, CDCl3): δ 86.5 ppm.
4: In a nitrogen-filled glovebox, N-phenylmaleimide (19 mg, 0.11 mmol, 2.2 eq.) was suspended in pentane (10 mL). To this suspension was added 2 (80 mg, 0.050 mmol, 1.0 eq.). The non-homogenous solution was allowed to vigorously stir at room temperature for 48 h, during which time a color change from pale to bright yellow was observed. The mixture was cooled to −20 °C and kept at this temperature for 1 h, at which time it was filtered through a sintered glass frit. The filter cake was washed thoroughly with pentane (3 × 5 mL) to yield 4 (80 mg, 84%) as a bright yellow solid. 1H NMR (500 MHz, CD2Cl2): δ 7.27–7.33 (m, 2H), 7.17–7.21 (m, 3H), 7.00–7.50 (bs, 2H), 6.94 (d, J = 9 Hz, 1H), 6.86 (d, J = 9 Hz, 1H), 4.17–4.47 (bs, 2H), 3.86 (s, 3H), 3.39 (s, 3H), 3.13 (septet, J = 9 Hz, 1H), 2.03–2.30 (m, 13H), 1.92 (bs, 6H), 1.70 (bs, 12H), 1.42 (bs, 6H), 1.17–1.35 (m, 7H), 0.68–0.95 (m, 7H) ppm; 13C NMR (125 MHz, CD2Cl2): δ 155.3, 155.3, 152.4, 152.3, 148.4, 140.2, 139.9, 134.7, 128.4, 128.3, 128.2, 126.8, 126.2, 116.3, 116.3, 113.0, 110.3, 110.3, 60.1, 54.5, 54.5, 42.9, 42.3, 37.1, 34.1, 31.7, 29.7, 25.8, 24.9 ppm (observed complexity is due to C-P coupling); 31P NMR (202 MHz, CD2Cl2): δ 82.5 ppm. HSQC and HMBC spectra (CD2Cl2, 500 MHz) were collected to aid in structural assignment. The 13C resonances at δ 134.7 ppm and δ 128.3 ppm are for quaternary carbons (by HSQC) that show no long range coupling to any 1H signals on the phosphine ligand but do show correlation to the 1H signals for the 5H on the Ph group (HSQC). These are the most likely signals for the C=O carbonyl and the N-bound carbon of the Ph group. IR (neat): 2904, 2847, 1727, 1681, 1597, 1579, 1497, 1459, 1419, 1352, 1259, 1202, 1120, 1091, 1045, 1016, 946, 873, 807, 791, 753, 696, 619 cm−1.
General procedure for preparation of 9–11
In a nitrogen-filled glovebox, phosphine ligand (0.21 mmol, 1.00 eq.) and [(1,5-cyclooctadiene)Pd(CH2TMS)2] (80 mg, 0.21 mmol, 1.00 eq.) were suspended in pentane (5.0 mL). The reaction mixture was stirred vigorously at room temperature, during which time a solid precipitated from solution. After 48 h, the non-homogenous mixture was filtered though a sintered glass frit. The filter cake was washed with pentane (10.0 mL) to yield the desired complex.
9: Yellow-green solid (Yield: 106 mg, 79%). IR (neat): 2931, 2850, 1580, 1456, 1419, 1376, 1359, 1293, 1252, 1170, 1155, 1087, 1044, 1013, 929, 870, 851, 798, 746, 715 cm−1.
10: Yellow-green solid (Yield: 95 mg, 75%). IR (neat): 2960, 2931, 2894, 2865, 1583, 1564, 1452, 1423, 1377, 1358, 1277, 1250, 1170, 1088, 1019, 931, 865, 807, 747, 717 cm−1.
11: Dark yellow solid (Yield: 113 mg, 98%). IR (neat): 2959, 2931, 2849, 1583, 1460, 1423, 1376, 1360, 1248, 1174, 1047, 1017, 925, 871, 851, 801, 769, 748 cm−1.
16: Pale yellow solid (Yield: 149 mg, 96%). IR (neat): 2957, 2926, 2846, 1581, 1459, 1421, 1375, 1355, 1248, 1172, 1123, 1088, 1043, 1013, 929, 887, 868, 849, 798, 745, 723, 715 cm.
General procedure for reaction of 2, 9–11 with 4-nBuPhBr
In a nitrogen-filled glovebox, the precatalyst (0.010 mmol, 1.0 eq.) was suspended in toluene-d8 (1.0 mL) in an oven-dried screw-cap NMR tube. 4-nBuPhBr (3.9 μL, 0.022 mmol, 2.2 eq.) was then added, and the NMR tube was capped and removed from the glovebox. After <10 min. at room temperature, the homogenous reaction mixture was analyzed by 31P NMR (121 MHz) and 1H (300 MHz, toluene-d8) and compared to the spectra for the corresponding oxidative addition complex reported in the literature. In every case, 0.5 eq. of 1,5-COD could be detected by 1H NMR integration (300 MHz, toluene-d8).
Procedure for reaction of 16 with 4-nBuPhBr
In a nitrogen-filled glovebox, 16 (15.0 mg, 0.020 mmol, 1.0 eq.) was suspended in toluene-d8 (1.0 mL) in an oven-dried screw-cap NMR tube. 4-nBuPhBr (3.9 μL, 0.022 mmol, 1.1 eq.) was then added, and the NMR tube was capped and removed from the glovebox. After <10 min. at room temperature, the homogenous reaction mixture was analyzed by 31P NMR (121 MHz) and 1H (300 MHz, tol-d8) and compared to the spectra for the corresponding oxidative addition complex reported in the literature. In this case, 1.0 eq. of 1,5-COD could be detected by 1H NMR integration (300 MHz, toluene-d8).
3. Results and Discussion
3.1 Solid state NMR studies of 2
To circumvent the problem of the poor solubility of 2 in organic solvents, it was characterized by solid-state 1H, 13C and 1H-13C correlation NMR experiments (Figure 2). The number of observed signals in the 13C NMR spectrum suggests that 2 is symmetrical, which is in agreement with our proposed C2-symmetric structure. Although the solid-state 1H NMR spectrum of 2 was not detailed enough to yield clear structural information, no signals downfield of 10 ppm or upfield of 0 ppm were observed. Complex 2 was also analyzed by cross polarization based 1H-13C correlation NMR using two different length second cross polarization steps (“short” and “long”) to detect 1H-13C contacts. These spectra are also consistent with the proposed structure, as most of the contacts are between aromatic 1H and 13C signals, and aliphatic 1H and 13C signals. The unusual signal observed in the 13C NMR dimension at δ 83 ppm does not correspond to any signals in isolated samples of 1 or 1,5-cyclooctadiene, and thus likely corresponds to the alkene carbons of 1,5-COD ligand bound to the Pd center of 2. Indeed, this signal only shows 1H-13C contacts with signals in the aliphatic region (δ <5 ppm) in the 1H NMR dimension, as would be expected for the alkene carbons of the central 1,5-COD ligand. The signal for this carbon is shifted significantly upfield from where it is normally observed for free1,5-cyclooctadiene (δ ~130 ppm), an effect likely due to significant π-backbonding from the metal center. In all, the solid-state NMR spectra presented here are wholly consistent with the proposed structure of 2. However, at this time the preferred conformation of the central 1,5-COD ligand in the solid state remains unclear.
Figure 2.

Solid state 13C NMR (left) and cross polarization based 1H-13C correlation NMR spectra (right) with 1.5 ms contact time for the initial cross polarization and 0.2 ms and 0.5 ms for the second cross polarization step.
3.2 Preparation of 3–5 from 2
We also investigated processes in which 2 loses 1,5-COD to act as a source of 1•Pd(0). Although 2 is insoluble in most organic solvents, it readily dissolves in CDCl3 to generate 0.5 equiv. of 1,5-cyclooctadiene relative to Pd and what X-ray crystallographic analysis revealed to be dearomatized Pd(II) complex 3 (Figure 3). A nearly identical species bearing a related ligand and also possessing η3-binding of the dearomatized ring to the Pd center has been previously reported [14]. This species likely does not form by oxidative addition of 1•Pd(0) to CDCl3 followed by a dearomative rearrangement similar to that reported for other Pd(II) complexes of 1 [7,9] because the CDCl2 and Pd center are bound to opposite faces of the lower ring of the ligand. Instead, a carbene mechanism is proposed to be involved in the formation of 3 [14].
Figure 3.
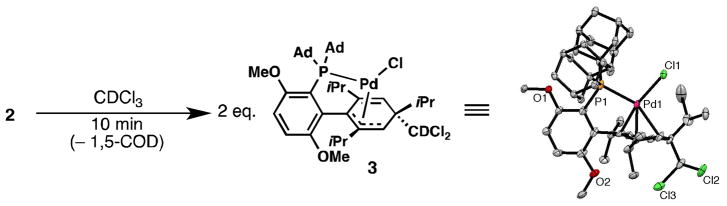
Rapid reaction of 2 with CDCl3 to form 3. Ellipsoids at 50% probability.
The 1,5-COD ligand in 2 could also be exchanged with the more π-acidic olefin N-phenylmaleimide to form 4 (Figure 4), which was significantly more soluble in organic solvents than 2. Thus, 4 could be characterized by solution-state NMR and X-ray crystallography (Figure 4). Complex 4 possesses a trigonal planar structure at the Pd(0) center, which is coordinated to the phosphine and the ipso carbon [15] of the lower ring of 1, as well as to the N-phenylmaleimide ligand. As expected, the Pd-C2 (2.13(6) Å) and Pd-C3 (2.11(6) Å) bond lengths are similar. This complex also possesses a much shorter Pd-ipso distance (2.03(4) Å) than that reported for 1•Pd(4-CNPh)Br (2.49(6) Å) [7], indicating stronger binding of the Pd center to the lower ring of the ligand in the case of the Pd(0) complex. The improved stability and solubility of 4 compared to 2 is likely due to the stronger π-backbonding ability of N-phenylmaleimide compared to 1,5-COD. Consistent with this hypothesis, the C2-C3 bond length in 4 is elongated (1.41(7) Å) relative to that of a typical double bond (1.34 Å), and the 13C NMR (125 MHz, CD2Cl2) resonance for the C=O carbon in 4 is shifted 35 ppm upfield from where it is normally observed. Due to these effects, 4 is significantly less reactive than 2, and does not as readily undergo oxidative addition or behave as a precatalyst for the fluorination of aryl triflates. Nonetheless, this finding suggests that the solubility and reactivity of Pd(0) complexes of 1 can be tuned by choosing the appropriate auxiliary alkene ligand.
Figure 4.
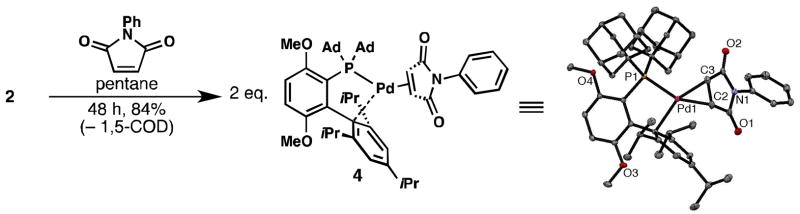
Conversion of 2 to related Pd(0) complex 4. Ellipsoids at 50% probability.
Lastly, we have found that 2 can also oxidatively add to 4-nBuPhBr to produce 1•Pd(4-nBuPhBr) (5) in less than 10 min at room temperature [9], generating 0.5 equiv. of 1,5-cyclooctadiene relative to Pd (Figure 5). Thus, the application of 2 as a precatalyst for Pd-catalyzed cross-coupling reactions should not be limited to reactions of aryl triflates. Indeed, we have recently employed 2 as a precatalyst for the Pd-catalyzed fluorination of aryl bromides and iodides [16].
Figure 5.
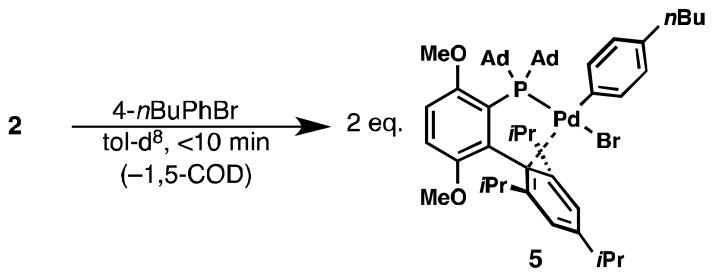
Rapid reaction of 2 with 4-nBuPhBr to produce 5.
3.3 Preparation of other [(1,5-COD)(L•Pd)n] (n = 1–2) species
We next investigated how general this precatalyst framework is by preparing analogous Pd(0) complexes of other biaryl phosphine ligands. When combined with [(1,5-COD)Pd(CH2TMS)2] in pentane, the bulky di-tBu ligands tBuBrettPhos (6), RockPhos (7), and tBuXPhos (8) also form isolable complexes 9–11 with concomitant loss of 0.5 equiv. of 1,5-COD (Figure 6). Like 2, these complexes are nearly insoluble in most organic solvents and rapidly undergo oxidative addition to 4-nBuPhBr in toluene-d8 to produce the corresponding previously reported oxidative addition complexes 12–14 [9–11], generating 0.5 equiv. of 1,5-cyclooctadiene relative to Pd in the process (Figure 6). Based on these results, we expect that 9–11 should possess similar properties to 2.
Figure 6.
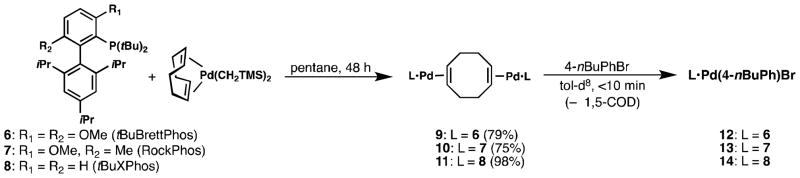
Reaction of 6–8 with [(1,5-COD)Pd(CH2TMS)2] to produce [(1,5-COD)(L•Pd)2] species 9–11, which react with 4-nBuPhBr to produce 12–14, respectively.
In contrast, when the smaller di-cyclohexyl ligand BrettPhos (15) was combined with [(1,5-COD)Pd(CH2TMS)2], only trace amounts of 1,5-COD could be detected in the mother liquor of the reaction, suggesting that the generated species 16 possesses a 1:1:1 ratio of 15:Pd:1,5-COD instead of the 1:1:0.5 ratio possessed by 2 and 9–11 (Figure 7). Indeed, when 16 was exposed to 4-nBuPhBr in toluene-d8, full conversion to the previously reported complex 17 [11] was observed in less than 10 min with concomitant generation of 1 equiv. of 1,5-cyclooctadiene (Figure 7). Instead of the general structure of [(1,5-COD)(L•Pd)2] shared by 2 and 9–11, 16 is likely monomeric in nature, with only one Pd center coordinated to each molecule of 1,5-COD. Nonetheless, 16 still effectively behaves as a highly reactive source of 15•Pd(0).
Figure 7.
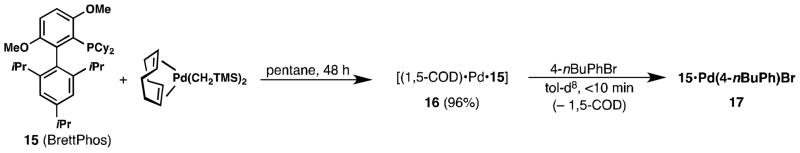
Reaction of 15 with [(1,5-COD)Pd(CH2TMS)2] to produce [(1,5-COD)(15•Pd)] species 16, which reacts with 4-nBuPhBr to produce 17.
4. Conclusion
We have provided further evidence for our proposed symmetric structure of precatalyst 2 (Section 3.1). Further insight into the principal reactivity of 2 as a source of 1•Pd(0) (Section 3.2) reveals that dissociation of 1,5-COD from this complex occurs readily at room temperature. Despite its high reactivity, 2 is thermally stable at room temperature and reasonably air/moisture stable, making it an ideal precatalyst for Pd-catalyzed cross-coupling reactions using 1 as the supporting ligand. We have also found that the general strategy of developing stable Pd(0) precatalysts for other biaryl phosphine ligands, such as 6–8 and 15, is feasible (Section 3.3). In the future we will focus on applying our new highly active precatalyst towards Pd-catalyzed reactions that are inhibited by the byproducts resulting from activation of other Pd sources, as well as those reactions for which the addition of base is not necessary.
Supplementary Material
Highlights.
Solid state NMR spectra of [(1,5-cyclooctadiene)(1•Pd)2]
Reactivity of [(1,5-cyclooctadiene)(1•Pd)2] with CDCl3
X-ray structure of Pd(0) species [(N-phenylmaleimide)Pd•1]
Synthesis and reactivity of [(1,5-cyclooctadiene)(L•Pd)2] complexes (L = 6–8)
Synthesis and reactivity of [(1,5-cyclooctadiene)(15•Pd)]
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health under award numbers GM46059 (H.G.L., P.J.M), EB003151 (M.T.C), and EB001960 (L.A.), and by EB002026 for the MIT-Harvard Center for Magnetic Resonance. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. P.J.M. additionally thanks the National Science Foundation for a pre-doctoral fellowship (2010094243) and Amgen for an educational donation. We gratefully acknowledge the assistance and financial support of M.T.C. and L.A. by Prof. Robert G. Griffin (MIT). The X-Ray diffractometer used in this work was purchased with funds from the National Science Foundation (CHE 0946721). We also thank Dr. Stacey Smith for collecting and solving the X-ray structures of 3 and 4, Ms. Elizabeth Kelley (MIT) for assistance with 2D-NMR experiments, and Dr. J. Robb DeBergh for assistance with this manuscript. MIT has patents on the ligands used in this work from which S.L.B. and former coworkers receive royalty payments.
Footnotes
Dedicated to Professor T. Don Tilley in recognition of his 60th birthday.
Spectroscopic data for new compounds 3 and 4 are provided.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee HG, Milner PJ, Buchwald SL. Org Lett. 2013;15:5602. doi: 10.1021/ol402859k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zelesskiy SS, Ananikov VP. Organometallics. 2012;31:2302. [Google Scholar]
- 3.Amatore C, Broeker G, Jutand A, Khalil F. J Am Chem Soc. 1997;119:5176. [Google Scholar]
- 4.Bruno NC, Tudge MT, Buchwald SL. Chem Sci. 2013;4:916. doi: 10.1039/C2SC20903A. and the references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruno NC, Buchwald SL. Org Lett. 2013;15:2879. doi: 10.1021/ol401208t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Tschan MJL, García-Suárez EJ, Freixa Z, Launay H, Hagen H, Benet-Buchholz J, van Leeuwen PWNM. J Am Chem Soc. 2012;132:6463. doi: 10.1021/ja100521m. [DOI] [PubMed] [Google Scholar]; b) Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]; c) Walker SD, Barder TE, Martinelli JR, Buchwald SL. Angew Chem Int Ed. 2004;43:1871. doi: 10.1002/anie.200353615. [DOI] [PubMed] [Google Scholar]; d) Andreu MG, Zapf A, Beller M. M Chem Commun. 2000:2475. [Google Scholar]
- 7.Su M, Buchwald SL. Angew Chem Int Ed. 2012;51:4710. doi: 10.1002/anie.201201244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoshiya N, Buchwald SL. Adv Synth Catal. 2012;354:2031. doi: 10.1002/adsc.201200398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milner PJ, Maimone TJ, Su M, Chen J, Müller P, Buchwald SL. J Am Chem Soc. 2012;134:19922. doi: 10.1021/ja310351e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maimone TJ, Milner PJ, Kinzel T, Zhang Y, Takase MK, Buchwald SL. J Am Chem Soc. 2011;133:18106. doi: 10.1021/ja208461k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McAtee JR, Martin SES, Ahneman DT, Johnson KA, Watson DA. Angew Chem Int Ed. 2012;51:3663. doi: 10.1002/anie.201200060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou DH, Rienstra CM. J Mag Res. 2008;192:167. doi: 10.1016/j.jmr.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allgeier AM, Shaw BJ, Hwang TL, Milne JE, Tedrow JS, Wilde CN. Organometallics. 2012;31:519. [Google Scholar]
- 15.This “ipso interaction” between C1 of the lower ring of the ligand and the Pd center is commonly observed in Pd(II) complexes of biaryl phosphine ligands. See Barder TE, Biscoe MR, Buchwald SL. Organometallics. 2007;26:2183.
- 16.Lee HG, Milner PJ, Buchwald SL. J Am Chem Soc. 2014;136:3792. doi: 10.1021/ja5009739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.