

Abstract
Human granulocytic anaplasmosis (HGA) is caused by the obligate intracellular bacterium, Anaplasma phagocytophilum. The proinflammatory cytokine, IFN-γ is necessary for innate immunity and plays an important role in the induction of severe histopathology in A. phagocytophilum-infected mice and disease in horses and humans. In this study, we examined the role of activation of signal transducer and activator of transcription (Stat) 1 phosphorylation with A. phagocytophilum infection, and found it to be markedly increased at day 7 post infection compared to mock-infected controls. This increase in phosphorylated Stat1 (pStat1) was significantly correlated with IFN-γ production and inflammatory tissue injury. Since pStat1 operates as a transcription factor central to the generation of effectors of inflammatory injury, these data suggest that Stat1 signaling is involved in IFN-γ-mediated immunopathologic lesions and disease in A. phagocytophilum infection and could be an important target for intervention of disease.
Keywords: Anaplasma phagocytophilum, IFN-γ, Stat1
INTRODUCTION
Anaplasma phagocytophilum is an obligate intracellular bacterium that causes human granulocytic anaplasmosis (HGA) that induces neutrophil vacuoles in which the bacterium propagates. Features of human infection range from mild to severe disease with inflammatory complications such as septic and toxic shock-like syndromes, respiratory distress syndrome, immune compromise, opportunistic infections, and death (2,12). While the majority of human infections caused by A. phagocytophilum do not result in severe disease, HGA severity in murine models is closely linked to macrophage-activation by IFN-γ, following increased NO production, among other inflammatory mediators of tissue injury (5,12).
Interferon gamma mediates biological actions such as macrophage activation, antimicrobial effector mechanisms, and the production of proinflammatory cytokines, chemokines, and reactive oxygen species (18). Studies in the murine model underscore an important role of IFN-γ in A. phagocytophilum infection. Deletion of the genes encoding IFN-γ or IFN-γ receptor in mice infected with A. phagocytophilum significantly reduces histopathologic inflammation, unrelated to pathogen numbers (9,18,21). Moreover, suppression of IFN-γ and its effects, as illustrated in IL-10 knockout mice, in humans, and in horses with A. phagocytophilum infection, also greatly impacts inflammatory tissue injury and disease severity (9,10,17). The underlying mechanism of IFN-γ involvement in pathogenesis of A. phagocytophilum infection is perhaps the main determinant of inflammatory disease outcome (19). Therefore, this study was performed to investigate the signal transduction pathways associated with this phenomenon and to demonstrate the role of Stat1 in proinflammatory responses and tissue injury with A. phagocytophilum infection.
MATERIALSAND METHODS
A. phagocytophilum infection
Female C57BL6 mice (6 wk) were purchased from The Jackson Laboratories (Bar Harbor, Maine). All animals were maintained under specific pathogen-free conditions and used in strict accordance with the guidelines and protocols approved by the Johns Hopkins University School of Medicine Animal Care and Use Committee.
A. phagocytophilum was maintained in RPMI 1640 medium supplemented with 1% FBS and 2 mM L-glutamine until >95% of cells contained morulae. On the day of inoculation, 106 infected HL-60 cells were centrifuged (3,000 × g, 10 min) to concentrate the cells, cell-free A. phagocytophilum were isolated by syringe lysis, cellular debris removed by centrifugation at 2,000 × g for 10 min, and the cell-free A. phagocytophilum were harvested from the supernatant by centrifugation at 12,000 × g for 20 min, and finally resuspended in PBS for infection. This method was validated in pilot experiments to reproduce the histopathologic changes and bacteremia observed when using whole infected HL-60 cells as inoculum, but avoids the exposure of animals to xenogeneic human cells that could affect immune and inflammatory response. Challenge was performed by intraperitoneal injection with 0.4 mL of either A. phagocytophilum or PBS.
Necropsy
At each interval post infection, 4 infected and 1 mock-infected mice were euthanized for harvest of plasma, liver, and spleen. Plasma was collected by centrifugation of EDTA-anticoagulated blood obtained after cardiac puncture; the cellular fraction was used for analysis of A. phagocytophilum bacteremia by quantitative PCR, whereas the plasma was used for analysis of cytokine concentrations. Splenic tissue was harvested for splenocyte preparations used in direct analysis of phosphorylated Stat1 (pStat1), as a reflection of systemic immune activation. Liver removed at necropsy was fixed in zinc fixation solution (BD Pharmingen, San Diego, CA) and paraffin-embedded for hematoxylin and eosin (H&E) stains and hepatic histopathologic assessment as a surrogate measurement of generalized inflammatory tissue injury observed in the mouse and horse models, and reflecting the histopathology of human disease (4,16,17). Histopathologic changes in infected and mock-infected mice were investigated and ranked for severity as previously described (22). All evaluations were conducted by 3 microscopists to assure fidelity of ranking. Statistical analysis to show significant changes in pathologic severity over the course of the infection was performed using two-sided Kruskal-Wallis nonparametric test, comparing median ranks of time intervals; p values <0.05 were considered significant.
Quantitative PCR for A. phagocytophilum
The A. phagocytophilum blood load at 4 h, and 4, 7, and 14 days was determined by preparing DNA (Qiagen DNA blood kit, Valencia, CA) from the cellular fraction of EDTA anticoagulated blood from each infected mouse. qPCR was performed as previously described targeting multicopy msp2, using DNA extracted from known numbers of A. phagocytophilum-infected HL-60 cells as standards (3). The analytical sensitivity of this assay is regularly as low as 1 infected cell/mL blood, approximately equivalent to 10 bacteria/mL blood.
Interferon gamma and interleukin-10 analysis in plasma
The concentrations of IFN-γ and IL-10 in the plasma from mice were analyzed using a multibead (BioPlex, Bio-Rad, Hercules, CA) method. This experiment was performed with each separate sample. Briefly, 50 μL of plasma was mixed with IFN-γ capture beads and quantitation was achieved by comparison with standards as per the manufacturer’s instructions. All samples were tested in duplicate. Differences in IFN-γ and IL-10 concentrations and their ratios were examined between days and mock-infected animals by ANOVA, with REGWQ correction where a p value <0.05 was considered significant. Correlations between plasma cytokine concentrations and A. phagocytophilum bacteremia load were determined using Spearman rank correlation, where a p value <0.05 was considered significant.
Western blotting
Four A. phagocytophilum-infected mice and 1 mock-infected mouse were necropsied on days 4, 7 and 14. Spleens from individual mice (5 at each time point) were minced, and dispersed into a single cell suspension. Splenocytes from infected mice were pooled for the first experiment, and 3 aliquots were separately suspended in lysis buffer (Pierce, Rockford, IL), and assayed for protein content (bicinchoninic acid protein assay; Pierce). Cell lysates were resolved in 10% SDS-PAGE and transferred to nitrocellulose membranes. After blocking, the membranes were incubated with Phospho-Stat Antibody Sampler Kit (Cell Signaling Technology Inc., Danvers, MA), and anti-actin (Sigma, St. Louis, MO) antibodies. The nitrocellulose blots were then washed, incubated with alkaline phosphatase-conjugated goat anti-rabbit antibody (KPL, Gaithersburg, MD), and visualized using an ECL (Bio-Rad). In each case, the blots were stripped of the phospho-specific antibody and reprobed with anti-actin to allow normalization of protein loading. A standard defined area comprising the entire band(s) of interest for each blot was scanned and analyzed for pixel density (Adobe Photoshop CS5) to determine relative quantity for total Stat1, phosphorylated Stat1 and Stat3. Stat expression was normalized to actin control expression for each experiment or animal and then results were calculated as a ratio of Stat1 or pStat1 quantity after digitization in infected animals to the same measures in mock-infected controls. Based on the observation that infection leads to increased phosphorylated Stat1 among pooled splenocytes at day 7 post-infection, 3 subsequent experiments were conducted in which splenocytes were harvested from individual animals for examination on day 7 post-infection only.
RESULTS
Immunopathology, quantitative bacteremia, IFN-γ and IL-10 expression in A. phagocytophilum infection
Hepatic histopathology was examined over a 14-day period. None of the mice exhibited clinical signs with infection. As anticipated, A. phagocytophilum-infected mice showed modest to severe hepatic lobular inflammatory lesions, with occasional apoptotic cells and necrosis that were not observed in mock-infected mice (not shown). Hepatic histopathology was already severely abnormal at 4 hr, peaked on day 7, and remained abnormal even on day 14; the difference in severity after day 4 was statistically significant compared to at 4 hr (P = 0.011; Kruskal–Wallis test; Fig. 1). By qPCR, low level A. phagocytophilum bacteremia (34 infected cells/mL) was detected in one animal at 4 h post infection, but only detected again on days 7 and 14. Bacterial load peaked on day 7 (2.0 × 104 ± 1.2 × 104 [sem] infected cells/mL), and was still detectable on day 14 (6.5 × 102 ± 3.9 × 102 [sem] infected cells/mL) (Fig. 2). IL-10 plasma concentrations were generally low throughout a 14-day period and varied only two-fold at most (p=0.172, ANOVA) over the course of the experiment. The plasma concentration of IFN-γ was at low levels at 4 h and day 4, but peaked at 983 pg/mL on day 7 (p<0.049), higher than other days and in mock infected animals (Fig. 2). Largely owing to the elevations in IFN-γ plasma concentrations on day 7, the IFN-γ:IL-10 ratio also peaked on day 7 at 6.3 (p=0.028, ANOVA) (not shown). A. phagocytophilum bacteremia load was significantly correlated with both plasma IFN-γ concentrations (p=0.008) and IFN-γ:IL-10 ratio (p=0.006; Spearman rank correlation).
Figure 1.
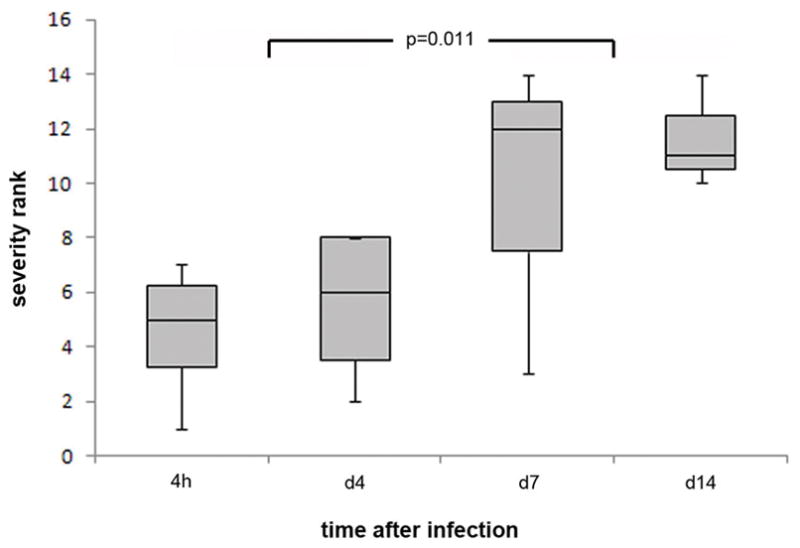
Hepatic histopathologic severity in A. phagocytophilum-infected mice significantly increases after day 4 of infection. The severity scores reflect the degree of histopathologic inflammatory infiltration and underlying tissue injury in the liver, as a surrogate for systemic inflammatory injury, and as normalized to mock-infected controls. The horizontal bar represents the median severity rank, the box borders represent the 1st and 3rd rank quartiles, and the bars represent the minimum and maximum rank scores at each time interval.
Figure 2.
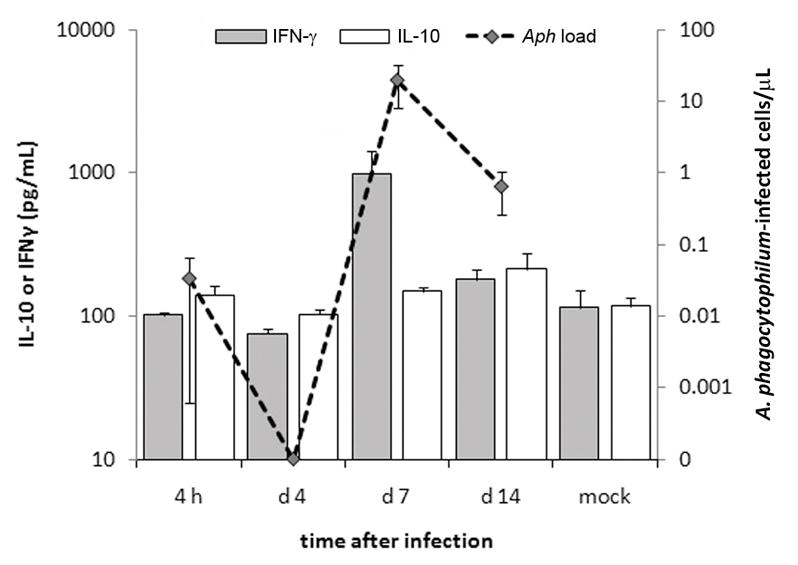
IFN-γ expression, not compensated by increased IL-10 expression, peaks concurrently (p=0.038, ANOVA) and significantly correlates (p≤0.008, Spearman rank correlation) with A. phagocytophilum blood loads at day 7 post-infection when histopathologic severity is also high.
Stat signaling in the murine model of A. phagocytophilum infection
We previously demonstrated that A. phagocytophilum infection results in high IFN-γ levels in murine plasma (7,19). To clarify the role of the Jak/Statpathway in IFN-γ signaling during A. phagocytophilum infection, we examined phosphorylation of Stats 1, 2, 3, 5, and 6 in splenocytes of infected and mock-infected mice at 4, 7 and 14 days after infection. Based on significant changes in phosphorylated Stat1 at day 7 post infection in this experiment, a subsequent experiment examined Stat1 expression in splenocytes from 3 individual mice at day 7 post infection. No changes were observed in expression or phosphorylation of Stats 2, 5 or 6. Stat3 was also increased on days 4–14, although only minimally above baseline (data not shown). As shown in Fig. 3, when normalized to actin expression, A. phagocytophilum infection induced a 1.6 (±0.24 s.e.m.)-fold increase in phosphorylated Stat1 at day 7 post infection compared to mock-infected controls (p<0.014, Mann-Whitney test), whereas total Stat1 expression remained unchanged over the course of the experiment. These results demonstrate phosphorylation of Stat1 in vivo at times concurrent with peak hepatic inflammatory injury and plasma IFN-γ concentrations.
Figure 3.
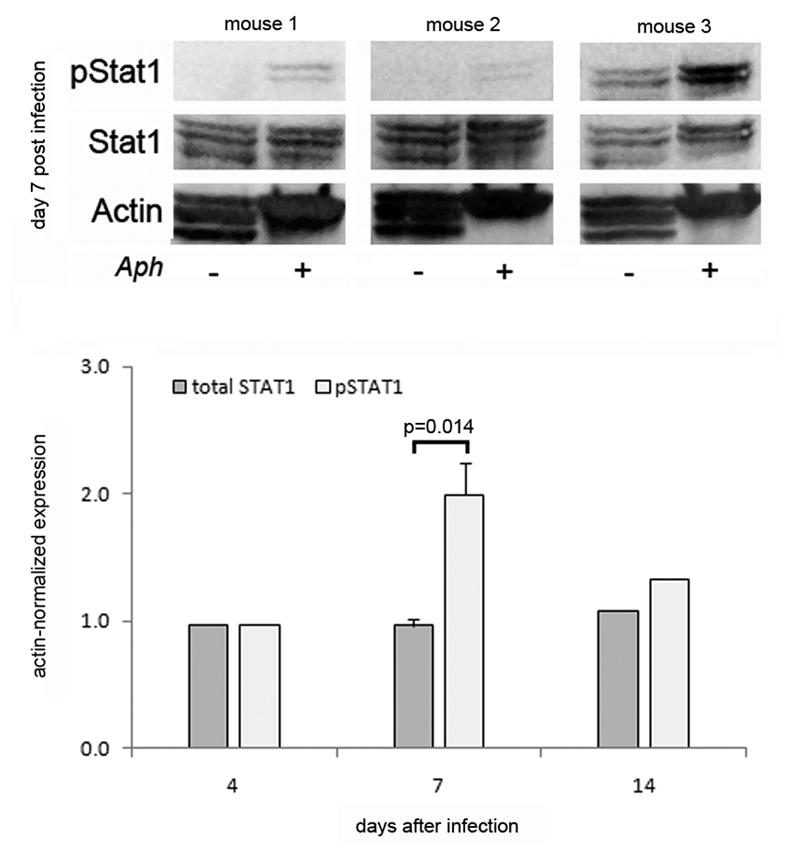
Phosphorylation of Stat1 in splenocytesis increased in A. phagocytophilum-infected mice. C57BL6 mice were infected with cell free A. phagocytophilum and sacrificed at 4,7 and 14 days after infection. The expression of pStat1, total Stat1, and actin in pooled splenocytes from 4 animals and from 3 individual mice were analyzed by immunoblotting. The top panel shows the results of 3 individual mice (1 experiment representative of 4 total experiments); note the increase in pStat1 in each animal, despite the lack of increased total Stat1. The bottom panel shows the results of the normalized densitometric quantitative analysis. Error bars are shown only for day 7 that reflects the average expression in 4 pooled mice and 3 individual mice, whereas the results on days 4 and 14 reflect the expression only in the splenocytes pooled from 4 mice.
DISCUSSION
We show that Stat1 signaling occurs simultaneously with increased IFN-γ production and in the context of hepatic inflammatory injury in A. phagocytophilum infection. This is significant because in murine models A. phagocytophilum infection repeatedly show discrepancies between pathogen load and histopathologic injury, implying that the histopathologic lesions and disease manifestations are likely the result of immune pathology, not infection (20,22). This concept is supported by the observation that IFN-γ knockout mice have a markedly increased pathogen load, yet substantial abrogation of histopathologic inflammatory lesions, whereas IL-10 knockout mice have an unchanged pathogen load but markedly worse inflammatory injury (6). We also recently showed a similar process in the horse model that mimics clinical disease as observed in humans, where the ratio of IL-10:IFN-γ or IL-10:IL-12 is altered to favor an anti-inflammatory or Th2 response by treatment with corticosteroids during active infection (9). Since inflammatory hepatic lesions in mice, horses and humans reflect systemic rather than local processes (4,16,17), it is likely that the increased phosphorylation observed among splenocytes reflects similar signaling among cells such as Kupffer cells resident in the liver, although this has not been directly tested. In addition, the identity of the splenocytes in which Stat1 phosphorylation occurs has not been directly examined, but using the same argument, this likely reflects signaling among resident and circulating macrophages known to be systemically activated after A. phagocytophilum infection (10,13). Given the well-known relationship between IFN-γ receptor ligation and Stat1 signaling, it is a reasonable conjecture that these surrogate analyses reflect the generalized response to A. phagocytophilum infection, although this will require confirmation as well as non-Stat-related signaling with IFN-γ production (14). These studies indicate that tissue histopathology in mice and clinical disease in horses, and likely humans, tracks more closely with IFN-γ and its regulation than with pathogen load (10,11).
Interferon gamma is the major cytokine implicated as driving macrophage activation and is closely linked to the immunopathogenesis of A. phagocytophilum infection. Humans, horses, and mice demonstrate a substantial increase in the IFN-γ expression during active infection (1,9–11,13). In horses, disease is ameliorated by corticosteroid treatment during active infection that impacts IFN-γ effects by altering the transcription of genes involved in polarization of immune response, largely the ratio of IL-10 and IFN-γ or IL-12 (9). The majority of IFN-γ-mediated effects occur via its ligation of IFN-γ receptors followed by phosphorylation of Stat1 and are counter balanced by the effects of IL-10 stimulation of Stat3 signaling (19). These data demonstrate that increased phosphorylation of Stat1 but not Stat3 occurred in splenocytes of A. phagocytophilum-infected mice at a time of peak IFN-γ plasma concentrations and IFN-γ:IL-10 plasma ratios, providing evidence that IFN-γ and Stat1 signaling largely govern the proinflammatory activation of macrophages implicated in the severe inflammatory sequelae of A. phagocytophilum infection, as reflected by hepatic histopathologic lesions. Therefore, Stat1 is an important transcription program factor involved in the genesis of immunopathologic responses in A. phagocytophilum infection in the murine model.
Because most histopathologic injury in mice occurs before induction of adaptive immune responses and is related to IFN-γ production likely by innate immune NK and NKT cells, Stat1 signaling is likely an important component of tissue injury and disease manifestations with A. phagocytophilum infection (7,8). It therefore also represents a potential target for therapeutic intervention and subversion by intracellular infection by the pathogen, because enhanced inflammation clearly benefits an organism that requires inflammatory cells for growth and transmission (6,20). A. phagocytophilum can impair IFN-γ signaling in infected neutrophils, including inhibition of IP-10/CXCL10 and MIG/CXCL9 expression, IFN-γ receptor α-chain CD119, and phosphorylated Stat1, among other factors that promote intracellular survival (5). Likewise, the related Ehrlichia chaffeensis infection of THP-1 monocytes blocks phosphorylation of Stat1, JAK1, and JAK2, suggesting a broad activity of these agents directly targeting the infected host cells (15). The current observations differ because the majority of the ex vivo splenocytes examined are neither infected, nor potential hosts for A. phagocytophilum. Rather, the responses likely reflect the roles that uninfected cells serve in inflammatory and immune recognition, and contribute not only to pathogen control but to disease immunopathology. As anticipated, Stat1 plays a key role in IFN-γ-mediated innate immunity and ultimately tissue damage by activated macrophages in A. phagocytophilum infection. The importance of this work is the in vivo evidence of this signaling pathway that demonstrates that it could be a potentially important target for interventions with severe disease in HGA.
Acknowledgments
The work was supported by grants R01 AI41213 and R21AI096062 to JSD and KSC was supported by grant NRF-2011-013-E00055. The authors would like to recognize the contributions of Sara Trembley, DVM and Diana Scorpio, DVM MPH, National University of Singapore.
List of Abbreviations
- A. phagocytophilum
Anaplasma phagocytophilum
- ANOVA
analysis of variance
- HGA
human granulocytic anaplasmosis
- IFN-γ
interferon gamma
- IL-10
interleukin 10
- pStat1
phosphorylated signal transducer and activator of transcription 1
- qPCR
quantitative polymerase chain reaction
- REGWQ
Ryan-Einot-Gabriel-Welsch Q
- Stat1
signal transducer and activator of transcription 1
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The authors declare that they have no competing interests.
References
- 1.Akkoyunlu M, Fikrig E. Gamma interferon dominates the murine cytokine response to the agent of human granulocytic ehrlichiosis and helps to control the degree of early rickettsemia. Infect Immun. 2000;68:1827–33. doi: 10.1128/iai.68.4.1827-1833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakken JS, Dumler JS. Human granulocytic ehrlichiosis. Clin Infect Dis. 2000;31:554–60. doi: 10.1086/313948. [DOI] [PubMed] [Google Scholar]
- 3.Browning MD, Garyu JW, Dumler JS, Scorpio DG. Role of reactive nitrogen species in development of hepatic injury in a C57BL/6 mouse model of human granulocytic anaplasmosis. Comp Med. 2006;56:55–62. [PubMed] [Google Scholar]
- 4.Bunnell JE, Trigiani ER, Srinivas SR, Dumler JS. Development and distribution of pathologic lesions are related to immune status and tissue deposition of human granulocytic ehrlichiosis agent-infected cells in a murine model system. J Infect Dis. 1999;180:546–50. doi: 10.1086/314902. [DOI] [PubMed] [Google Scholar]
- 5.Bussmeyer U, Sarkar A, Broszat K, Ludemann T, Moller S, Van Zandbergen G, Bogdan C, Behnen M, Dumler JS, Von Loewenich FD, Solbach W, Laskay T. Impairment of gamma interferon signaling in human neutrophils infected with Anaplasma phagocytophilum. Infect Immun. 2010;78:358–63. doi: 10.1128/IAI.01005-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlyon JA, Fikrig E. Mechanisms of evasion of neutrophil killing by Anaplasma phagocytophilum. Curr Opin Hematol. 2006;13:28–33. doi: 10.1097/01.moh.0000190109.00532.56. [DOI] [PubMed] [Google Scholar]
- 7.Choi KS, Dumler JS. Mitogenic component in polar lipid-enriched Anaplasma phagocytophilum membranes. Clin Vaccine Immunol. 2007;14:1260–65. doi: 10.1128/CVI.00204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi KS, Scorpio DG, Barat NC, Stephen Dumler J. Msp2 variation in Anaplasma phagocytophilum in vivo does not stimulate T cell immune responses or interferon-gamma production. FEMS Immunol Med Microbiol. 2007;49:374–86. doi: 10.1111/j.1574-695X.2007.00214.x. [DOI] [PubMed] [Google Scholar]
- 9.Davies RS, Madigan JE, Hodzic E, Borjesson DL, Dumler JS. Dexamethasone-induced cytokine changes associated with diminished disease severity in horses infected with Anaplasma phagocytophilum. Clin Vaccine Immunol. 2011;18:1962–68. doi: 10.1128/CVI.05034-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dumler JS, Barat NC, Barat CE, Bakken JS. Human granulocytic anaplasmosis and macrophage activation. Clin Infect Dis. 2007;45:199–204. doi: 10.1086/518834. [DOI] [PubMed] [Google Scholar]
- 11.Dumler JS, Choi KS, Garcia-Garcia JC, Barat NS, Scorpio DG, Garyu JW, Grab DJ, Bakken JS. Human granulocytic anaplasmosis and Anaplasma phagocytophilum. Emerg Infect Dis. 2005;11:1828–34. doi: 10.3201/eid1112.050898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dumler JS, Madigan JE, Pusterla N, Bakken JS. Ehrlichioses in humans: epidemiology, clinical presentation, diagnosis, and treatment. Clin Infect Dis. 2007;45(Suppl 1):S45–51. doi: 10.1086/518146. [DOI] [PubMed] [Google Scholar]
- 13.Dumler JS, Trigiani ER, Bakken JS, Aguero-Rosenfeld ME, Wormser GP. Serum cytokine responses during acute human granulocytic ehrlichiosis. Clin Diagn Lab Immunol. 2000;7:6–8. doi: 10.1128/cdli.7.1.6-8.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev. 2008;19:383–94. doi: 10.1016/j.cytogfr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Lee EH, Rikihisa Y. Protein kinase A-mediated inhibition of gamma interferon-induced tyrosine phosphorylation of Janus kinases and latent cytoplasmic transcription factors in human monocytes by Ehrlichia chaffeensis. Infect Immun. 1998;66:2514–20. doi: 10.1128/iai.66.6.2514-2520.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lepidi H, Bunnell JE, Martin ME, Madigan JE, Stuen S, Dumler JS. Comparative pathology, and immunohistology associated with clinical illness after Ehrlichia phagocytophila-group infections. Am J Trop Med Hyg. 2000;62:29–37. doi: 10.4269/ajtmh.2000.62.29. [DOI] [PubMed] [Google Scholar]
- 17.Martin ME, Bunnell JE, Dumler JS. Pathology, immunohistology, and cytokine responses in early phases of human granulocytic ehrlichiosis in a murine model. J Infect Dis. 2000;181:374–78. doi: 10.1086/315206. [DOI] [PubMed] [Google Scholar]
- 18.Paludan SR. Synergistic action of pro-inflammatory agents: cellular and molecular aspects. J Leukoc Biol. 2000;67:18–25. doi: 10.1002/jlb.67.1.18. [DOI] [PubMed] [Google Scholar]
- 19.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 20.Scorpio DG, Akkoyunlu M, Fikrig E, Dumler JS. CXCR2 blockade influences Anaplasma phagocytophilum propagation but not histopathology in the mouse model of human granulocytic anaplasmosis. Clin Diagn Lab Immunol. 2004;11:963–68. doi: 10.1128/CDLI.11.5.963-968.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scorpio DG, Von Loewenich FD, Gobel H, Bogdan C, Dumler JS. Innate immune response to Anaplasma phagocytophilum contributes to hepatic injury. Clin Vaccine Immunol. 2006;13:806–9. doi: 10.1128/CVI.00092-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scorpio DG, von Loewenich FD, Bogdan C, Dumler JS. Innate immune tissue injury and murine HGA: tissue injury in the murine model of granulocytic anaplasmosis relates to host innate immune response and not pathogen load. Ann NY Acad Sci. 2005;1063:425–28. doi: 10.1196/annals.1355.077. [DOI] [PubMed] [Google Scholar]