

Abstract
WAVE3 belongs to the WASP/WAVE family of actin cytoskeleton remodeling proteins. These proteins are known to be involved in several biological functions ranging from controlling cell shape and movement, to being closely associated with pathological conditions such as cancer progression and metastasis. Last decade has seen an explosion in the literature reporting significant scientific advances on the molecular mechanisms whereby the WASP/WAVE proteins are regulated both in normal physiological as well as pathological conditions.
The purpose of this review is to present the major findings pertaining to how WAVE3 has become a critical player in the regulation of signaling pathways involved in cancer progression and metastasis. The review will conclude with suggesting options for the potential use of WAVE3 as a therapeutic target to prevent the progression of cancer to the lethal stage that is the metastatic disease.
Keywords: WAVE3, cancer cell invasion, Invasion-Metastasis Cascade, microRNAs, TNBC, Invadopodia
1. Background
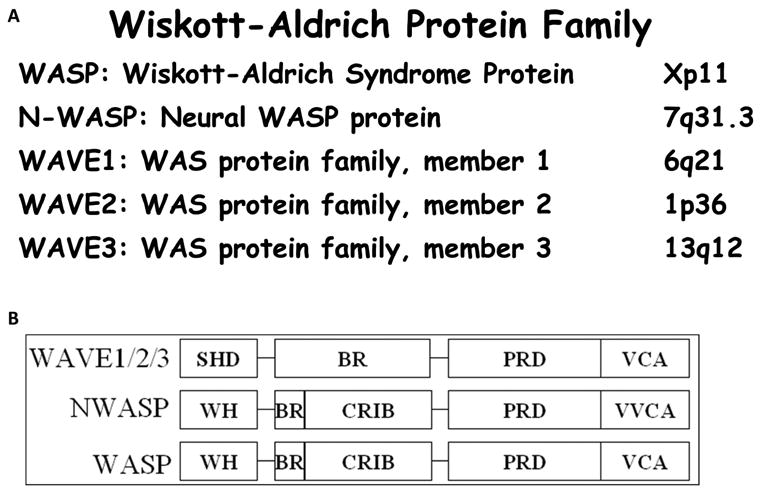
Actin cytoskeleton plays a key role in a number of cellular functions that include cell shape changes, cytokinesis, cell motility, cell proliferation and membrane traffic [1, 2]. Precise control of the assembly, dynamics and organization of the actin filaments that constitute the core of the actin cytoskeleton, is critical for these functions. Defects in proper actin regulation, however, can contribute to human disease including cancer metastasis, and Wiskott-Aldrich syndrome to name a few [3-7]. Several genes were identified to be involved in the regulation of actin-cytoskeleton organization including the genes encoding for the WASP/WAVE family of proteins. This family of proteins is comprised of 5 members (Fig. 1) that form two distinct subfamilies based upon structural homologies [8, 9]. The WASP subfamily includes the WASP protein which is associated with the Wiskott-Aldrich syndrome [4, 10], and its more widely-expressed homologue, NWASP [11]. The WAVE subfamily of proteins contains three members, WAVE 1, 2 and 3 [12, 13].
Figure 1. The WASP/WAVE protein family.
A. Chromosome location in the human genome of each member of the WASP/WAVE family. B. Schematic representation of the functional domains of the WASP/WAVE proteins. SHD: Scar Homology Domain. WH: Wiskott Homology Domain. BR: Basic Region. CRIB: Cdc42- and Rac-Interactive Binding Domain. PRD: Proline Rich Domain. VCA: Verprolin homology, Cofilin homology and Acidic Domain.
The WASP/WAVE proteins which function as effectors downstream of the Rho GTPases play a critical role in several cellular processes, such as cell motility and proliferation, by regulating actin polymerization and cytoskeleton organization. In response to extracellular signals, both the WASP and WAVE subfamilies activate the Arp2/3 complex, leading to the stimulation of actin polymerization and assembly of actin filaments. The involvement of the Arp2/3 complex in the formation of new branched actin filaments is dependent upon interactions with nucleation-promoting factors (NPFs). These NPFs consist of WASP [14], N-WASP [15, 16], WAVE1 [17, 18], WAVE2 [19, 20], WAVE3 [5, 8], and the newly identified WASH [21], WHAMM [22] and JMY [23]. Recent work has demonstrated that Abp1 [24], Pan1 and cortactin [25-27] can directly activate the Arp2/3 complex, while the NPFs need first to be activated by Cdc42 and Rac [28-30].
The activity of WASP and WAVE proteins is controlled by different mechanisms of regulation. Cdc42 is required for the activation of WASP and N-WASP, while the activity of the WAVE proteins is regulated through their association with multiprotein complexes, downstream of Rac.
Despite the considerable efforts that have been expended on the analysis of WASP, N-WASP, WAVE1 and WAVE2, up to recently, very little was known about the role of WAVE3 in the pathologies that are associated with defects in the remodeling of the actin cytoskeleton. Loss of function of either WAVE1 or WAVE2 has been shown to significantly affect mouse embryonic development and survival [17, 19, 20]. Moreover, although the WASP/WAVE proteins are believed to be equally involved in the mechanisms that regulate the actin-cytoskeleton organization, comparatively, little is known about the impact of WAVE3 deregulation on these processes. In this review we present the recent advances in the field about the involvement of WAVE3 in both normal physiology and in pathological conditions, including cancer.
2. Mechanisms of Regulation of WAVE3
2.1. Different mechanisms of regulation for WASPs and WAVEs
WAVE3, as a member of the WASP/WAVE family of structurally and functionally related proteins, plays a critical role in actin polymerization and cytoskeleton organization [1, 31]. The WASP and WAVE proteins share several domains in their protein structures (Fig. 1), which are believed to regulate their activity and sub-cellular localization, in response to a variety of extra-cellular signals [32]. WASP and WAVE proteins function as effectors downstream of the Rho GTPases to regulate the actin cytoskeleton [9]. Members of the WASP subfamily are activated through Cdc42 to induce filopodia, while the WAVE proteins function downstream of Rac to induce the formation of lamellipodia [9]. All members of the WASP and WAVE family of proteins share a tripartite VCA (Verprolin homology, Cofilin homology and Acidic) C-terminal domain. Activation of the WASP and WAVE proteins leads to the exposure of the VCA domain, which can bind to the Arp2/3 complex and initiate the rapid polymerization of actin filaments [33, 34], ultimately leading to cytoskeletal remodeling, which is necessary for cell motility and migration [31].
WASP/WAVE proteins differ both in the signaling inputs that they receive, as well as in their mode of regulation (Fig. 3). A CRIB (Cdc42- and Rac-interactive binding) domain, which is present in the N-terminal half of both WASP and NWASP, and which was shown to specifically mediate binding of active GTP-bound Cdc42, is thought to regulate the activity of these two proteins [35, 36]. In the absence of extra-cellular signals, WASP and NWASP are auto-inhibited through the intramolecular interaction between the CRIB and the VCA domains [37, 38]. The binding of Cdc42 and/or phosphatidylinositol (4,5)P2 (PtdIns(4,5)P2) to the CRIB domain is required for their activation. This allows the release of the VCA domain which becomes exposed, and can then bind to the Arp2/3 complex and initiate actin polymerization [30, 39]. In contrast, the WAVE1 and WAVE2 proteins, which function downstream of Rac to induce actin polymerization [12, 40], were found to be regulated through the formation of multimeric protein complexes with other actin-cytoskeleton remodeling proteins [41-43]. WAVE1 and WAVE2 proteins were shown to be sequestered in an inactive state through the formation of a complex with four other proteins, PIR121, Nap125, HSPC300 and Abi1 [40-43]. WAVE3 has also been shown to be included in a similar multimeric protein complex [44].
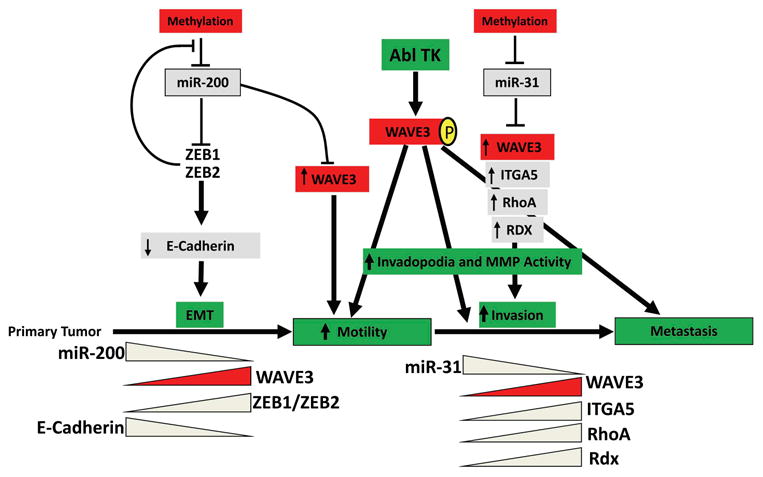
Figure 3. Mechanisms of WAVE3 regulation in cancer metastasis.
WAVE3 is regulated by microRNA miR-200, where Epithelial-to mesenchymal transition (EMT) is critical during the early steps of cancer cell invasion. Loss of miR-31 which results in the re-activation of its metastasis promoter target genes, including WAVE3, is a defining step during the invasion-metastasis cascade. Phosphorylation of WAVE3 by c-Abl provides an additional checkpoint to modulate invadopodia formation and MMP activity.
In addition to the VCA domain, which is critical for the Arp2/3-mediated actin polymerization, the WASP/WAVE proteins have other functional domains (Fig. 1), that mediate interactions with several other proteins to regulate the activity of the WASP/WAVE proteins [9, 32]. Profilin, which has a high affinity to poly-L-proline, binds to the proline-rich domain (PRD) which is conserved in all WASP and WAVE proteins [12], and was shown to play a critical role in the enhancement of Cdc42-WASP-induced nucleation of actin polymerization [45]. On the other hand, the WISH protein, an SH3-containing protein, was found to activate N-WASP independently of either Cdc42 or PtdIns(4,5)P2, by binding to the PRD of N-WASP [46]. Other proteins such as Syndapin, WIP and Nck have been shown to physically interact with the WASP protein through different motifs to induce actin polymerization [47-49]. While the regulation of WASP proteins is well documented, the regulation of the WAVE proteins, and WAVE3 in particular, has not been well elucidated. The best-characterized WAVE protein partner is IRSp53, which has been shown to be an essential intermediate between Rac and WAVE2 in the regulation of actin polymerization and membrane ruffling [50]. In addition to the involvement of WAVE3 in actin polymerization and cytoskeleton organization [1, 13, 44, 51], WAVE3was found to be associated with the development of low grade neuroblastoma [5].
2.2. Independent roles for the WAVE proteins
The expression profiles of the WAVE genes clearly show an overlap in the expression of all three WAVE transcripts in several embryonic and adult tissues [8]. Previous studies have also suggested co-localization of all three WAVE proteins in multimeric proteins complexes [43, 52], which suggests the involvement of the WAVE proteins in similar cellular pathways. However, although WAVE1 and WAVE2 are both expressed in mouse embryonic fibroblasts, they were found to have differential roles in cell migration [53]. WAVE1 was found to be required for the formation of dorsal ruffles, while WAVE2 is required for the formation of peripheral ruffles, two membrane-based actin structures that are necessary for the initiation of cell migration [53], clearly suggesting independent roles for the WAVE proteins
2.3. Effect of Wasp and Wave genes on mouse development and actin-based motility
Except for Wave3, all the other 4 members of the Wasp and Wave family of genes have been disrupted in mouse by independent groups, using gene-targeting mutations [15, 17, 19, 20, 54]. Targeted disruption of the X-linked immunodeficiency Wiskott-Aldrich syndrome protein (WASP), although not leading to embryonic lethality [54], resulted in impairment in the proliferation of blood cells as a consequence of severe defects in the cytoskeleton organization [55]. On the other hand, disruption of Nwasp, Wave1 or Wave2, each resulted in embryonic lethality, associated with marked developmental delay and organ malformations [15, 17, 19, 20]. Analysis of mouse embryonic fibroblasts (MEFs) derived from mice embryos lacking either one of these genes also showed a defect in the actin-based motility, as a consequence of alterations in cytoskeleton organization [15, 17, 19, 20]. In addition to the striking and distinct phenotypes that result from the targeted-disruption of each Wasp/Wave gene, these genes are individually required for the normal development of the mouse embryo, and for normal cell movement. These studies also imply a crucial non-redundant role for each WASP and WAVE gene in mouse embryogenesis, for the formation specific actin-containing structures, and in the actin-based motility. Thus, generating a mouse lacking the expression of WAVE3 may help in revealing the specific role that WAVE3 plays in mouse development and in actin cytoskeleton organization. While several studies have focused on elucidating the involvement of WAVE1 and WAVE2 in remodeling the actin cytoskeleton and in cell motility, little was known about the impact of of WAVE3 in these processes. Our first encounter with WAVE3 more than a decade ago was somehow serendipitous. While we were looking for the causative defective gene in a patient with low-grade neuroblastoma, and a genomic defect described as a constitutive balanced translocation between chromosomes 1 and 13, we found that WAVE3 was truncated and inactivated as a result of this translocation [5]. We subsequently reported on the hypothesis that loss of WAVE3 function might be associated with the development of this type of malignancy, and probably other cancers [5]. Since then more than 30 manuscripts have been published from both our group and others dealing with the mechanisms that involve WAVE3 in both physiological and pathological conditions.
2.4. The WAVE3-mediated remodeling of actin cytoskeleton is driven by PDGF downstream of PI3K
As mentioned above the WASP and WAVE proteins differ in both the signaling inputs that they receive, as well as in their mode of regulation. WASP proteins are auto-inhibited by intramolecular interactions that mask the VCA domain in the absence of signaling molecules such as Cdc42 and phosphatidylinositol 4,5-biphosphate [37, 38]. On the other hand, WAVE1 and WAVE2 proteins are sequestered in a complex with four other proteins, PIR121, Nap125, HSPC300, and Abi1 [40-42, 56]. Extracellular stimuli result in the dissolution of this complex, which subsequently leads to Arp2/3-mediated actin polymerization. Reports on the basal activity of this complex; i.e., in the absence of external stimuli, however, are not concordant. On one hand, the in vitro study reported by Innocenti and colleagues suggested that the WAVE2 complex is constitutively active [42]. On the other hand, a more recent study by Lebensohn et al., has clearly shown that the WAVE2 complex is basally inactive [56]. WAVE3 was later found to be included in the same protein complex as WAVE1 and WAVE2, suggesting a similar mode of regulation [44]. Regardless of which WAVE isoform is involved, the activity and functionality of the WAVE complex in resting conditions have yet to be definitely elucidated.
Platelet-derived growth factor (PDGF) induces a variety of cellular responses in several cell types, including proliferation, migration, invasion, and cell survival, via its receptor [57, 58]. PDGF treatment of MDA-MB-231 breast cancer cells was found to induce the formation of lamellipodia at the edge of migrating cells, and the accumulation of WAVE3 in these lamellipodia [59]. The same study showed that the WAVE3 regulation of cell migration and the formation of lamellipodia that is induced by PDGF, involves PI3K as an upstream modulator, where a direct interaction between WAVE3 and p85, the regulatory subunit of PI3K, is required [59]. Inhibition of PI3K activity with LY294002, in the PDGF-treated cells, resulted in the inhibition of both lamellipodia formation and cell migration, as well as a disruption of the accumulation of WAVE3 at the edge of migrating cells. Similar studies have previously reported that the formation of lamellipodia and cell migration of PDGF-treated fibroblasts, or the HGF-treated myogenic cells, require the production of phosphatidylinositol 3,4,5-triphosphate (PIP3) by PI3K, which subsequently leads to the recruitment of WAVE2 to the polarized plasma membrane [60, 61]. It is also possible that the production of PIP3, as a result of PI3K activity, is essential for WAVE3-mediated lamellipodia formation and cell migration, as it has been shown for WAVE2 [60, 61]. These results support the previously reported data on the involvement of PI3K in the formation of lamellipodia and cell migration in other cell types in response to specific growth factors [1, 20, 53, 60].
2.5. The WAVE3-p85 interaction is required for cell migration
PI3K is a heterodimer consisting of p110, a 110-kDa catalytic subunit and p85, an 85-kDa regulatory subunit. PI3K activity depends upon the production of PIP3, a messenger molecule produced in response to a variety of growth factors. The targeting of PI3K to specific locations within the cell leads to its activation and therefore to the remodeling of the cytoskeleton organization [62]. p85, is a critical component of the actin cytoskeleton regulatory pathway [62]. Although the downstream targets of PI3K are still not completely understood, it has been shown that the binding of the PI3K complex to activated PDGF receptor via the p85 Src homology 2 (SH2) domains leads to the internalization of the complex into endocytotic vesicles, where cellular trafficking is regulated [63]. However, in addition to the SH2 domains located in the C-terminal half of p85, p85 also has additional functional domains in its N-terminal half (Fig. 2), including an SH3 domain and a domain homologous to the breakpoint cluster region (BCR) gene product. These domains may function to target PI3K to distinct regions in the cell in order to initiate specific downstream response cascades or provide a mechanism by which specific protein-protein interactions are mediated [64]. A direct interaction between WAVE3 and p85 proteins was demonstrated by both in vitro binding and co-immunoprecipitation assays [59]. WAVE3 was found to bind to the C-terminal SH2 domain of p85 via the basic region domain (BR) of WAVE3 (Fig. 2). Using immunocytochemistry, an in vivo interaction was demonstrated between these two proteins in the actin-based membrane cytoskeleton, supporting the involvement of p85 in the regulation of the activity of WAVE3 [59]. These observations clearly support the involvement of p85 in the WAVE3-mediated actin polymerization and cytoskeleton organization. However, the mechanisms of p85-mediated regulation of the WAVE3 activity are yet to be identified.
Figure 2. Role of p85 in the WASP- and WAVE-mediated actin-cytoskeleton organization.
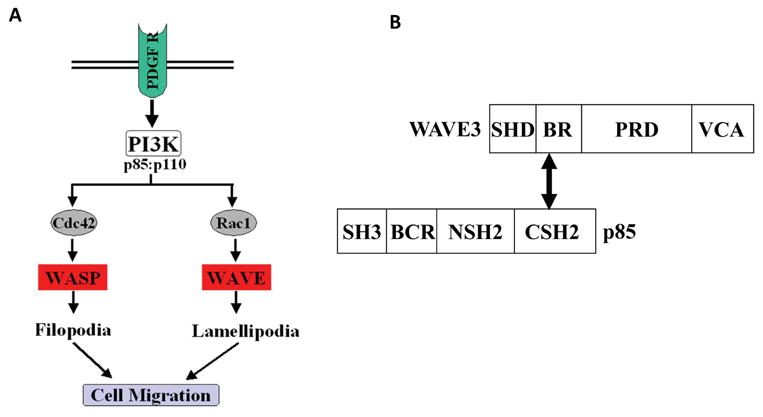
A. Stimulation of the receptor tyrosine kinase, PDGFR, leads to activation of the components of PI3K; p110 and p85. p85. By regulating the activity of Cdc42 and Rac1, PI3K regulates, in turn, the WASP- and WAVE-mediated actin polymerization. B. Schematic representation of the functional domains of WAVE3 and p85. The double-headed arrow points the interacting domains between WAVE3 (BR) and p85 (CSH2).
2.6. Phosphorylation of WAVE3 by c-Abl tyrosine kinase is required for the regulation of cell motility, cancer cell migration and invasion
Phosphorylation is a major mechanism whereby gene function is regulated, and the WASP/WAVE proteins are no exception [65-69]. The non-receptor tyrosine kinase ACK1 phosphorylates and enhances the WASP-mediated actin polymerization [70], while stimulation of cultured cells with growth factors increases the phosphorylation levels of the WAVE proteins [71]. Phosphorylation of WAVE1 by CDK5 [72], or of WAVE2 by the non-receptor tyrosine kinase c-Abl [73], leads to the stimulation of membrane ruffling and cell spreading [74]. Not surprisingly, the kinase activity of c-Abl is critical for the remolding of actin cytoskeleton, since cells lacking c-Abl expression show a severe deficiency in membrane ruffling in response to growth factors [75]. In WAVE3, c-Abl was also found to phosphorylate four tyrosine residues scattered throughout the functional domains of the WAVE3 protein [51]. Among the four tyrosine residues, Tyrosine 151 is conserved among all three WAVE proteins, both in human and mouse [8], and its equivalent in WAVE2 (Y150) is also targeted for phosphorylation by c-Abl [73]. Y337 is specific to WAVE3, whereas Y486 is conserved in both WAVE1 and WAVE3, but not in WAVE2 [8]. Therefore, phosphorylation of different tyrosine residues in the different WAVE proteins might account for the specific and independent roles of the WAVE proteins in the regulation of actin polymerization and cytoskeleton remodeling. We showed that the c-Abl-mediated phosphorylation of WAVE3 also results in the stimulation of lamellipodia formation and cell migration, which clearly supports the hypothesis that WAVE3 is also a downstream effector of c-Abl. Interestingly, because c-Abl can bind all WAVE isoforms, it is possible that it might exert its effect by targeting different tyrosine residues in the different WAVE isoforms to regulate their specific functions the in the regulation of actin cytoskeleton.
Other studies have shown that cell stimulation with PDGF results in an increase in cell motility and lamellipodia formation downstream of PI3K [59]. It was also suggested that the PI3K-mediated activation of WAVE3 required the binding of the regulatory subunit p85 to a tyrosine-phosphorylated WAVE3 [59] to promote lamellipodia formation and cell motility. Accordingly it has been demonstrated that tyrosine phosphorylation levels of WAVE3 were indeed increased after PDGF stimulation of cultured cells [51]. More importantly, phosphorylation of WAVE3 was found to require c-Abl kinase activity, further supporting the hypothesis that c-Abl phosphorylation of WAVE3 might be required to link WAVE3 to the PI3K complex, thus allowing for the PDGF-mediated activation of WAVE3. Those studies have, therefore, provided a novel mechanism by which WAVE3, and probably other members of the WAVE family of proteins, is linked to the PDGF-induced cytoskeleton remodeling downstream of PI3K.
Probing further into the biological significance of WAVE3 phosphorylation by c-Abl it was found that the lack of WAVE3 phosphorylation resulted in a dramatic inhibition of lamellipodia formation and cell migration, even in the presence of extracellular stimuli, such as PDGF. It is, however, still not clear which of the four tyrosine residues is (are) the most critical for lamellipodia formation and cell migration.
3. WAVE3 and Cancer Invasion and Metastasis
3.1. WAVE3 and MMPs
Degradation of the extracellular matrix (ECM) via MMP activity is essential for many normal physiological processes, e.g., during development, cell migration, growth, and wound healing [76]. On the other hand, increased expression and activity of MMPs are also associated with tumor invasion, metastasis, and tumor angiogenesis [77-79]. Expression of most MMPs is normally low in tissues, and only induced when remodeling of the extracellular matrix is required. MMP expression is primarily regulated at the transcriptional level, although stabilization of MMP transcripts in response to growth factors, as well as the influence of cytokines, also plays a role in the regulation of MMP activity [80, 81].
The possible involvement of the WAVE proteins in the regulation of MMPs was initially reported by Suetsugu and colleagues who showed that downregulation of WAVE1, but not WAVE2, affected MMP-2 activity [53]. Conversely, our group, while seeking to investigate the functional consequences of loss of WAVE3 using RNA interference (RNAi), we identified a MAPK signaling axis, where WAVE3 was essential for the regulation of the expression of different MMPs [82]. We showed that knockdown of WAVE3 expression affects the activity of the p38 MAPK pathway, but not that of AKT or ERK1/2. WAVE3-mediated downregulation of p38 activity was found to be independent of both WAVE1 and WAVE2 expression, as WAVE3 knockdown did not alter the transcription levels of either WAVE1 or WAVE2. Loss of WAVE3 resulted in a significant decrease in the expression levels of MMP-1, -3, and -9. The inhibition of these MMPs as a result of WAVE3 loss led to a dramatic inhibition of cell migration and invasion using both in the in vitro wound closure and Matrigel assays [82]. The study provided for the first time a clear evidence for a novel role of WAVE3 in the regulation of MMP activity via the p38 MAPK pathway. In fact, the MAPK signaling pathway plays an important role in regulating many fundamental processes such as cell growth, migration, and differentiation [83-86], which is linked to MMPs. Activation of MAPK pathways results in alterations in the expression levels and activity of MMPs, which in turn, are responsible for the degradation of extracellular matrix, and as such, are required for cell migration [87]. A recent study has also confirmed the important role of WAVE3 in the expression and activity of MMPs [88].
3.2. WAVE3 and microRNAs
MicroRNAs are emerging as important regulators of cellular differentiation, and they are now known to often be dysregulated during carcinogenesis. MicroRNAs are small noncoding RNAs, usually 20- to 22-nucleotides long, which regulate gene expression at the post-transcriptional level. So far, more than 1000 microRNAs have been identified in mammalian cells, and each microRNA has several target genes. The broad spectrum of genes that can be regulated by a single microRNA is attributed to the high level of conservation of the target motifs, known as seed sequences, within the 3′untranslated regions (UTR) of the target genes. micoRNAs are now widely regarded as the most powerful regulators of gene expression in complex cellular processes including cancer cell invasion and metastasis [89-96]. In fact, several microRNAs have been found to function as tumor suppressors, such as miR-15a, miR-16-1, and let-7 [91, 97-101], whereas others were found to possess oncogenic properties, such as miR-155, miR-17-5p, and miR-21 [91, 102-104]. In our previously published work, we reported a highly significant correlation between the expression levels of WAVE3 and advanced stages of breast cancer [105], clearly supporting the function of WAVE3 as a metastasis promoter protein [106, 107]. However, the mechanisms for regulation of WAVE3 expression levels during tumor progression remained unresolved. In an effort to dissect the role of WAVE3 in cancer cell invasion and metastasis, we were able to establish a significant role of microRNAs in the regulation of WAVE3 expression and activity at two distinct stages of cancer progression; (i) during the epithelial to mesenchymal transition (EMT) process downstream of miR200 [108], and (ii) during the invasion-metastasis cascade downstream of miR31 [109-111].
3.2.1. The WAVE3-mediated modulation of cancer cell invasion is regulated by miR200 during EMT
We found that this evolutionary conserved microRNA, miR200, which regulates key differentiation processes during development, is also involved in the regulation of WAVE3 [108]. The key findings from the miR200 study in the regulation of WAVE3 expression and activity in vitro are summarized as follows: (i) Expression of WAVE3 is inversely correlated to miR200 in epithelial versus mesenchymal-type cancer cells; (ii) miR200 targets the 3′UTR of WAVE3 at three different seed sequences and represses its expression; (iii) miR200-mediated downregulation of WAVE3 expression inhibits cancer cell invasion and induces an mesenchymal to epithelial transition (MET)-like phenotype in vitro, which the reverse process of the epithelial-to-mesenchymal transition (EMT); (iv) Re-expression of miR200-resistant WAVE3 reverses the inhibition cancer cell invasion imposed by miR200; and (v) Expression levels of miR200 correlate inversely with human breast cancer progression.
3.2.2. miR31 regulates WAVE3 expression and activity during the invasion-metastasis cascade of breast cancer
We have previously performed in silico analysis [112-114] to search for potential binding sites for microRNAs in WAVE3 [108] and noted that, in addition to miR200, miR31 has a single target site in the 3′UTR of WAVE3, with a perfect match to the seed sequence recognized by miR31 [111]. The potential relationship between miR31 and WAVE3 became particularly noteworthy in view of the recent reports of the major role of miR31 in cancer metastasis [115-117] and the role of WAVE3 in cancer progression and metastasis [82, 105-108, 111]. We therefore investigated whether the influence of miR31 on cancer cell invasion and metastasis is WAVE3 dependent. The key findings from the miR31 study on the regulation of WAVE3 expression and activity in vitro have been published [108-111] and are summarized as follows: (i) Expression of WAVE3 is inversely correlated to miR31 in epithelial versus mesenchymal BC cells. (ii) miR31 targets the 3′UTR of WAVE3 at s specific seed sequence and represses its expression. (iii) miR31-mediated downregulation of WAVE3 expression inhibits cancer cell invasion [111]. (iv) Re-expression of miR31-resistant WAVE3 is sufficient to reverse the inhibition of cancer cell invasion imposed by miR31 [111]. (v) β1 integrins, which we have shown to be involved in WAVE3-mediated regulation of cancer invasion, are specifically targeted and by miR31 in metastatic triple-negative BC (TNBC) [109]. (vi) miR31 expression and activity are regulated by epigenetic modification of its promoter leading to its silencing, and therefore enhancement of aggressiveness of TNBCs [110].
The data from these in vitro analyses were reproduced in other invasive cancer cells from different origins demonstrating that the effect of miR31 is not restricted to a single cell line or a cancer, but appears to be a generalized effect that extends to other cancers. More importantly, our analyses of the non-invasive breast cancer MCF7 cells, which express high levels of miR31 and low levels of WAVE3, have shown that inhibition of endogenous miR31 results in increased invasion associated with increased WAVE3 expression levels [111]. Based on the findings summarized above, we have developed a model whereby the regulation of WAVE3 during cancer progression and metastasis is achieved at the post-transcriptional level by both miR200, during EMT, and by miR31, during the late steps of the invasion-metastasis cascade. WAVE3 is also regulated at the post-translational level by tyrosine phosphorylation downstream of c-Abl (Fig. 3).
Interestingly, we also found that both miR200 and miR31, which modulate the activity of WAVE3 during EMT and the invasion-metastasis cascade of BC, are also regulated by TGF-β, therefore, placing TGF-β-miR200-miR31-WAVE3 in the same signaling axis (unpublished data). As such, we hypothesized that TNBC metastasis comprises TGF-β to downregulate expression of miR200 and miR31, which in turn enhances WAVE3 expression and activity necessary to the acquisition of EMT and metastatic phenotypes in TNBCs.
The body of evidence presented above, which has been published from several independent research groups who used a diverse array of cell culture, in vitro and in vivo assays, convincingly points to the critical role of WAVE3 in cell motility and migration as well as cancer cell invasion and metastasis both in vitro, in vivo in mouse models and in human clinical settings. Surprisingly a recent report from Spence et al. [118], claimed that WAVE3 may not be essential for the invasive ability of MDA-MB-231 breast cancer cells. Interestingly this cancer cell line has been used by our group and other groups as a model to investigate WAVE3 function in cancer invasion [82, 105, 119, 120]. Given that WAVE3 was confirmed to be involved in cancer invasion in many cancer types (Breast, Prostate and Colon) using different cancer cell lines [82, 88, 105, 107, 120-123], we could suggest that cells used by Spence et al. [118] must be different. In support of this possibility, we note that the shape and phenotype of the WAVE3-knockdown cells reported in their study were strikingly different from those we routinely observe as well as from those frequently published by others. Different reagents and assays conducted in different labs, including the knockdown levels of WAVE3, also could account for these differences.
3.3. WAVE3 is required for cancer progression and metastasis in mouse
Given the central role that WAVE3 plays in the regulation of the actin cytoskeleton, cell motility and cancer cell invasion [51, 59, 82, 105], the suggestion that WAVE3 may also play a critical role in cancer progression and metastasis seems evident. In fact, at least one member of the WAVE2, a close relative to WAVE3 has been associated with the metastatic phenotype of murine melanoma cells [124, 125].
One of the early studies provided the first evidence for the role of WAVE3 in tumor cell invasion and metastasis [105]. In the lung colonization assay, also known as experimental metastasis model, where human BC cells were injected the blood circulation of SCID mice via tail vein, WAVE3 was found to be required for BC cells to form tumor colonies in the lungs. Although the BC cells, where WAVE3 was stably knocked-down with shRNAs, were able to be established in the lungs, they formed significantly smaller sized colonies compared to their control counterparts. On the other hand, in the orthotopic xenograft assay, also known as the spontaneous metastasis assay, where cancer cells are implanted in mammary fat pads of SCID mice, the WAVE3-deficient cells were significantly less tumorigenic in SCID mice. Tumor incidence in the mice injected with the WAVE3-knockdown BC cells was reduced by 60 to 80% compared to their control counterparts, and tumors that developed from the shWAVE3 cells grew more slowly and were less angiogenic [105]. Based on the CD31 staining, the tumors derived from the WAVE3-knockdown cells showed a significant reduction in the number of blood vessels and the size of the blood vessel lumens. VEGF expression was also reduced in the shWAVE3 tumors, suggesting that WAVE3 may regulate expression of VEGF and the recruitment of the blood vessel-forming cells. Although these findings suggest a unique role for WAVE3 in tumor cells, one cannot exclude the possibility that all three WAVE isoforms may play a specific role in some aspects of cancer metastasis, given that WAVE2 also contributes to metastasis of melanoma cells [124, 125]. In fact different WAVE isoforms were found to have independent functions in regulating Arp2/3-mediated actin polymerization and cell migration [20, 53, 124].
Two possible molecular mechanisms were suggested to as how WAVE3 may contribute to tumor metastasis. Previous studies suggest that p38 MAPK is a downstream effector molecule of WAVE3 [82]. Coincidently, a blockade of p38 MAPK signaling by dominant-negative p38 MAPK (DN-p38) partially recapitulated the effect of WAVE3 on metastasis. The DN-p38 cells showed a three-fold reduction in lung tumor formation compared with a six-fold reduction from cells expressing shWAVE3. As with the shWAVE3 tumors, the DN-p38 orthotopic tumors were also less angiogenic [105, 126]. Given that Rac1 can activate WAVE3 [71] as well as p38 MAPK in several cell lines [34, 127] including MDA-MB231 [128], it is suggestive that WAVE3 is required for the Rac1-mediated activation of p38 MAPK. It is still not clear, however, how WAVE3 affects p38 MAPK activity or its intermediate effectors. For example, WAVE3 may be involved in the Rac1-dependent activation of PAK1, an upstream kinase in the p38 MAPK cascade [129]. Although p38 MAPK may contribute to some of the WAVE3 effects, other factors such as Arp2/3 can also mediate WAVE3 effects on tumor cell motility, invasiveness, and metastasis. Cell motility is tightly linked to the regulation of the Arp2/3-mediated remodeling of actin cytoskeleton [130, 131], driven by the WASP/WAVE proteins, [9, 82, 132]. Previous studies have indicated that WAVE3 is involved in the regulation of lamellipodia production, actin stress fibers formation, and focal adhesions assembly in MDA-MB-231 cells [59, 82]. Therefore it was suggested that WAVE3 may contribute to anchorage-independent cell growth and cell death associated with detachment from extracellular matrix (anoikis).
Recently more studies have reported on the critical role thatWAVE3 plays in other malignancies such as prostate [107, 121] and colon cancers [133]. WAVE3 was also found to be upregulated under hypoxia which is believed to be required to trigger the invasion and metastatic phenotype of cancer cells [119].
3.4. WAVE3 is a biomarker for breast cancer progression and metastasis
Breast cancer is the most common malignancy diagnosed in women and the second leading cause of cancer mortality after lung cancer [134-137]. Metastasis is responsible for ∼90% of deaths in patients with solid tumors [89, 138-142], including those originating in the breast [143-145]. The risk of developing distant metastasis and therefore prognosis in BC is associated with the presence of a number of pathologic characteristics: positive lymph node status, increasing tumor size and histologic grade. BC is a heterogeneous disease that is characterized by the presence of at least five genetically distinct subtypes. For instance, luminal BCs, which also tend to be estrogen receptor positive (ER+) and low grade, have the lowest risk of developing distant metastases and have the best prognosis. At the other end of the spectrum, the basal BC subtypes, which also include the Triple Negative BCs (TNBCs) exhibit dismal survival rates due to their highly aggressive and metastatic behavior, and to their propensity to rapidly recur [146-152]. Genetically, TNBCs are characterized by lack of expression of hormone receptors (ER-α and PR) and HER2, harbor BRCA1-defects and/or deficiencies, and may remain p53-positive [153], which makes them refractory to hormonal therapy, further contributing to the risk of aggressive relapse and dismal survival rates amongst women bearing TNBCs [138, 141, 142, 154, 155].
Given the clinical characteristics of high-grade breast cancers, WAVE3 was thought to be expressed in higher levels compared to low grade tumors and this elevated expression might contribute to the increased metastatic potential seen in the high-grade tumors compared to low-grade tumors. To answer this question, two retrospective studies were conducted using two different cohorts of BC patients, in an attempt to isolate the effect of the levels of expression of WAVE3 on BC progression and metastasis [123]. In the first study 122 BC patients were identified from two very different groups of patients modified Scarff-Bloom-Richardson (mSBR1) and mSBR3 and assessed for WAVE3 expression levels in the primary tumors using IHC. Correlation of WAVE3 expression levels to the patients' clinicopatholological characteristics and the disease outcome and led to the following findings: (i) WAVE3 was highly expressed in malignant vs. adjacent normal ductal epithelium, (ii) WAVE3 expression was positively correlated with adverse clinicopathologic parameters, (iii) WAVE3 expression was increased in the tumors of patients who developed distant metastases, (iv) WAVE3 expression levels are positively correlated with reduced distant recurrence free survival and with decreased disease specific survival (Fig. 4 and ref [123]).
Figure 4. WAVE3 expression levels are positively correlated with reduced distant-recurrence-free-survival and with decreased disease-specific-survival.
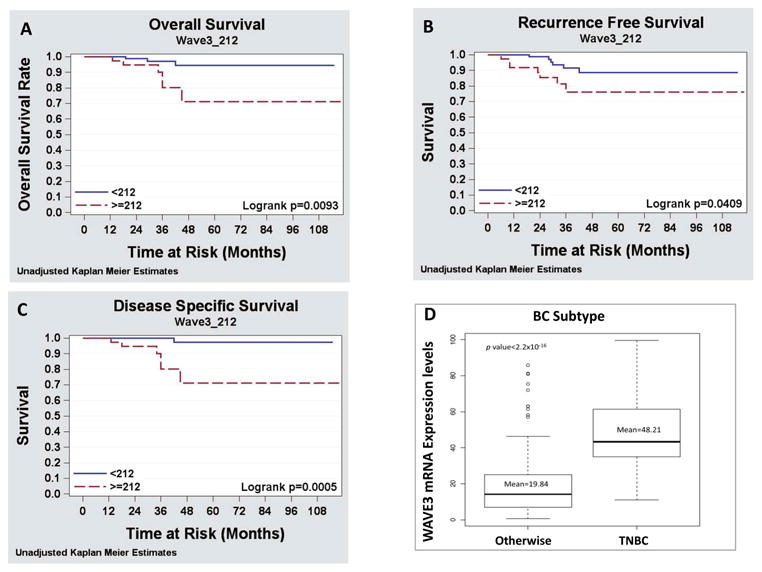
Kaplan-Meier analysis of (A) Overall survival, (B) Distant-recurrence-free-survival and (C) Disease-specific-survival. The WAVE3 score was dichotomized as positive or negative to determine the relative contribution of the WAVE3 score to each of the three disease outcome parameters, respectively. (D) WAVE3 expression levels are associated the TNBC subtype, the most aggressive subtype in breast cancer.
In the second part of the same study the prognostic value of WAVE3 mRNA expression levels was evaluated in the circulating tumor cells in the peripheral blood of women with operable breast cancer, based on the unique characteristic of the lack of WAVE3 expression in the peripheral blood mononuclear cells (PBMCs). Analysis of WAVE3 expression levels in the blood of 200 BC patients and correlation with the patients' clinical data revealed that (i) WAVE3 mRNA was highly expressed in the peripheral blood of patients with metastatic breast cancer, and (ii) WAVE3 expression levels in the blood of BC patients correlates positively with the aggressive TNBC subtype (Fig. 4 and ref. [123]). Other studies have also found significant correlation between WAVE3 expression levels and the pathology of other cancers, notably, prostate and colon cancers [107, 133].
4. Conclusions and perspectives
The last decade has seen significant advances related to the function of WAVE3 in both physiological and pathological setting, therefore closing the gap of lack of knowledge between WAVE3 and its close sisters. Both the mechanistic and the clinical studies have now clearly cemented the position of WAVE3 as a critical player in cancer progression and metastasis. The clinical studies have now characterized WAVE3 as biomarker for BC progression and metastasis, and more importantly, have identified as a driving force behind the pathology of the most aggressive BC subtype, i.e., TNBC. Additionally, WAVE3 has now a potential therapeutic use in a non-invasive liquid bioassay for early detection of TNBCs. Detection of increased levels of WAVE3 in the blood of BC patients after the completion of an adjuvant systemic treatment could help identify those patients who may have a substantial clinical benefit from a ‘secondary’ adjuvant treatment before the occurrence of overt metastasis. Finally, despite these significant advances, several unanswered questions remain to be addressed, pertaining to the molecular mechanisms whereby the function of WAVE3 is being regulated in both physiological and pathological conditions.
Highlights.
Regulation of MMPs by WAVE3 is required for cancer cell invasion.
c-Abl-mediated phosphorylation of WAVE3 is required for lamellipodia formation and cell migration.
Metastasis-suppressor microRNAs regulate WAVE3 during EMT and the invasion-metastasis cascade.
WAVE3, a metastasis promoter gene, regulates invasion and metastasis of breast and prostate cancer cells.
WAVE3 is a biomarker for the triple negative breast cancer subtype.
Acknowledgments
The author is very grateful to all the researchers and collaborators who, during the last decade, contributed to the generation and the publication of the data on WAVE3 which he is associated with this review. Their names are listed in the respective publications. The author would like to thank Dr. Edward Plow for his unconditional support and mentorship. Many thanks to Nancy Fiordalisi for proof-reading the manuscript. This work was supported in part by the following funding sources: Roswell Park Cancer Institute, Cleveland Clinic Foundation, U.S. Department of Defense grant number W81XWH-08-1-0236 to KSA, by National Institutes of Health grants P01HL073311 and P50HL077107, and by pilot funding from the Case Comprehensive Cancer Center (P30 CA043703). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviation list
- NPFs
Nucleating promoter factors
- EMT
Epithelial-to-Mesenchymal transition
- TNBC
Triple negative breast cancer
- ECM
extracellular matrix
- MMP
matrix metalloproteinase
- PDGF
Platelet-derived growth factor
- PI3K
phosphatidylinositol 3-kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Suetsugu S, Takenawa T. Regulation of cortical actin networks in cell migration. Int Rev Cytol. 2003;229:245–86. doi: 10.1016/s0074-7696(03)29006-9. [DOI] [PubMed] [Google Scholar]
- 2.Lee TY, Gotlieb AI. Rho and basic fibroblast growth factor involvement in centrosome redistribution and actin microfilament remodeling during early endothelial wound repair. J Vasc Surg. 2002 Jun;35(6):1242–52. doi: 10.1067/mva.2002.123094. [DOI] [PubMed] [Google Scholar]
- 3.Billadeau DD. Cell growth and metastasis in pancreatic cancer: is Vav the Rho'd to activation? Int J Gastrointest Cancer. 2002;31(1-3):5–13. doi: 10.1385/IJGC:31:1-3:5. [DOI] [PubMed] [Google Scholar]
- 4.Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994 Dec 2;79(5) following. [PubMed] [Google Scholar]
- 5.Sossey-Alaoui K, Su G, Malaj E, Roe B, Cowell JK. WAVE3, an actin-polymerization gene, is truncated and inactivated as a result of a constitutional t(1;13)(q21;q12) chromosome translocation in a patient with ganglioneuroblastoma. Oncogene. 2002 Aug 29;21(38):5967–74. doi: 10.1038/sj.onc.1205734. [DOI] [PubMed] [Google Scholar]
- 6.Kurisu S, Takenawa T. The WASP and WAVE family proteins. Genome Biol. 2009;10(6):226. doi: 10.1186/gb-2009-10-6-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurisu S, Takenawa T. WASP and WAVE family proteins: friends or foes in cancer invasion? Cancer Sci. 2010 Oct;101(10):2093–104. doi: 10.1111/j.1349-7006.2010.01654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sossey-Alaoui K, Head K, Nowak N, Cowell JK. Genomic organization and expression profile of the human and mouse WAVE gene family. Mamm Genome. 2003 May;14(5):314–22. doi: 10.1007/s00335-002-2247-7. [DOI] [PubMed] [Google Scholar]
- 9.Takenawa T, Miki H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci. 2001 May;114(Pt 10):1801–9. doi: 10.1242/jcs.114.10.1801. [DOI] [PubMed] [Google Scholar]
- 10.Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001 Mar;27(3):313–7. doi: 10.1038/85886. [DOI] [PubMed] [Google Scholar]
- 11.Miki H, Miura K, Takenawa T. N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO J. 1996 Oct 1;15(19):5326–35. [PMC free article] [PubMed] [Google Scholar]
- 12.Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998 Dec 1;17(23):6932–41. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suetsugu S, Miki H, Takenawa T. Identification of two human WAVE/SCAR homologues as general actin regulatory molecules which associate with the Arp2/3 complex. Biochem Biophys Res Commun. 1999 Jun 24;260(1):296–302. doi: 10.1006/bbrc.1999.0894. [DOI] [PubMed] [Google Scholar]
- 14.Bosticardo M, Marangoni F, Aiuti A, Villa A, Grazia RM. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood. 2009 Jun 18;113(25):6288–95. doi: 10.1182/blood-2008-12-115253. [DOI] [PubMed] [Google Scholar]
- 15.Snapper SB, Takeshima F, Anton I, Liu CH, Thomas SM, Nguyen D, et al. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat Cell Biol. 2001 Oct;3(10):897–904. doi: 10.1038/ncb1001-897. [DOI] [PubMed] [Google Scholar]
- 16.Lommel S, Benesch S, Rottner K, Franz T, Wehland J, Kuhn R. Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep. 2001 Sep;2(9):850–7. doi: 10.1093/embo-reports/kve197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahl JP, Wang-Dunlop J, Gonzales C, Goad ME, Mark RJ, Kwak SP. Characterization of the WAVE1 knock-out mouse: implications for CNS development. J Neurosci. 2003 Apr 15;23(8):3343–52. doi: 10.1523/JNEUROSCI.23-08-03343.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soderling SH, Langeberg LK, Soderling JA, Davee SM, Simerly R, Raber J, et al. Loss of WAVE-1 causes sensorimotor retardation and reduced learning and memory in mice. Proc Natl Acad Sci U S A. 2003 Feb 18;100(4):1723–8. doi: 10.1073/pnas.0438033100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamazaki D, Suetsugu S, Miki H, Kataoka Y, Nishikawa S, Fujiwara T, et al. WAVE2 is required for directed cell migration and cardiovascular development. Nature. 2003 Jul 24;424(6947):452–6. doi: 10.1038/nature01770. [DOI] [PubMed] [Google Scholar]
- 20.Yan C, Martinez-Quiles N, Eden S, Shibata T, Takeshima F, Shinkura R, et al. WAVE2 deficiency reveals distinct roles in embryogenesis and Rac-mediated actin-based motility. EMBO J. 2003 Jul 15;22(14):3602–12. doi: 10.1093/emboj/cdg350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linardopoulou EV, Parghi SS, Friedman C, Osborn GE, Parkhurst SM, Trask BJ. Human subtelomeric WASH genes encode a new subclass of the WASP family. PLoS Genet. 2007 Dec;3(12):e237. doi: 10.1371/journal.pgen.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campellone KG, Webb NJ, Znameroski EA, Welch MD. WHAMM is an Arp2/3 complex activator that binds microtubules and functions in ER to Golgi transport. Cell. 2008 Jul 11;134(1):148–61. doi: 10.1016/j.cell.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuchero JB, Coutts AS, Quinlan ME, Thangue NB, Mullins RD. p53-cofactor JMY is a multifunctional actin nucleation factor. Nat Cell Biol. 2009 Apr;11(4):451–9. doi: 10.1038/ncb1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goode BL, Rodal AA, Barnes G, Drubin DG. Activation of the Arp2/3 complex by the actin filament binding protein Abp1p. J Cell Biol. 2001 Apr 30;153(3):627–34. doi: 10.1083/jcb.153.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weed SA, Karginov AV, Schafer DA, Weaver AM, Kinley AW, Cooper JA, et al. Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J Cell Biol. 2000 Oct 2;151(1):29–40. doi: 10.1083/jcb.151.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uruno T, Liu J, Zhang P, Fan Y, Egile C, Li R, et al. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat Cell Biol. 2001 Mar;3(3):259–66. doi: 10.1038/35060051. [DOI] [PubMed] [Google Scholar]
- 27.Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, et al. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001 Mar 6;11(5):370–4. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- 28.Georgiou M, Marinari E, Burden J, Baum B. Cdc42, Par6, and aPKC regulate Arp2/3-mediated endocytosis to control local adherens junction stability. Curr Biol. 2008 Nov 11;18(21):1631–8. doi: 10.1016/j.cub.2008.09.029. [DOI] [PubMed] [Google Scholar]
- 29.Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999 Apr 16;97(2):221–31. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 30.Rohatgi R, Ho HY, Kirschner MW. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4, 5-bisphosphate. J Cell Biol. 2000 Sep 18;150(6):1299–310. doi: 10.1083/jcb.150.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Millard TH, Sharp SJ, Machesky LM. Signalling to actin assembly via the WASP (Wiskott-Aldrich syndrome protein)-family proteins and the Arp2/3 complex. Biochem J. 2004 May 15;380(Pt 1):1–17. doi: 10.1042/BJ20040176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burns S, Cory GO, Vainchenker W, Thrasher AJ. Mechanisms of WASp-mediated haematological and immunological disease. Blood. 2004 Aug 12; doi: 10.1182/blood-2004-04-1678. [DOI] [PubMed] [Google Scholar]
- 33.Machesky LM, Mullins RD, Higgs HN, Kaiser DA, Blanchoin L, May RC, et al. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci U S A. 1999 Mar 30;96(7):3739–44. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci U S A. 1998 May 26;95(11):6181–6. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdul-Manan N, Aghazadeh B, Liu GA, Majumdar A, Ouerfelli O, Siminovitch KA, et al. Structure of Cdc42 in complex with the GTPase-binding domain of the ‘Wiskott-Aldrich syndrome’ protein. Nature. 1999 May 27;399(6734):379–83. doi: 10.1038/20726. [DOI] [PubMed] [Google Scholar]
- 36.Aspenstrom P, Lindberg U, Hall A. Two GTPases, Cdc42 and Rac, bind directly to a protein implicated in the immunodeficiency disorder Wiskott-Aldrich syndrome. Curr Biol. 1996 Jan 1;6(1):70–5. doi: 10.1016/s0960-9822(02)00423-2. [DOI] [PubMed] [Google Scholar]
- 37.Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature. 2000 Mar 9;404(6774):151–8. doi: 10.1038/35004513. [DOI] [PubMed] [Google Scholar]
- 38.Weaver AM, Young ME, Lee WL, Cooper JA. Integration of signals to the Arp2/3 complex. Curr Opin Cell Biol. 2003 Feb;15(1):23–30. doi: 10.1016/s0955-0674(02)00015-7. [DOI] [PubMed] [Google Scholar]
- 39.Higgs HN, Pollard TD. Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J Cell Biol. 2000 Sep 18;150(6):1311–20. doi: 10.1083/jcb.150.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002 Aug 15;418(6899):790–3. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- 41.Gautreau A, Ho HY, Li J, Steen H, Gygi SP, Kirschner MW. Purification and architecture of the ubiquitous Wave complex. Proc Natl Acad Sci U S A. 2004 Mar 30;101(13):4379–83. doi: 10.1073/pnas.0400628101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, et al. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004 Apr;6(4):319–27. doi: 10.1038/ncb1105. [DOI] [PubMed] [Google Scholar]
- 43.Oda A, Miki H, Wada I, Yamaguchi H, Yamazaki D, Suetsugu S, et al. WAVE/Scars in Platelets. Blood. 2004 Aug 3; [Google Scholar]
- 44.Stovold CF, Millard TH, Machesky LM. Inclusion of Scar/WAVE3 in a similar complex to Scar/WAVE1 and 2. BMC Cell Biol. 2005;6(1):11. doi: 10.1186/1471-2121-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang C, Huang M, DeBiasio J, Pring M, Joyce M, Miki H, et al. Profilin enhances Cdc42-induced nucleation of actin polymerization. J Cell Biol. 2000 Sep 4;150(5):1001–12. doi: 10.1083/jcb.150.5.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukuoka M, Suetsugu S, Miki H, Fukami K, Endo T, Takenawa T. A novel neural Wiskott-Aldrich syndrome protein (N-WASP) binding protein, WISH, induces Arp2/3 complex activation independent of Cdc42. J Cell Biol. 2001 Feb 5;152(3):471–82. doi: 10.1083/jcb.152.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qualmann B, Roos J, DiGregorio PJ, Kelly RB. Syndapin I, a synaptic dynamin-binding protein that associates with the neural Wiskott-Aldrich syndrome protein. Mol Biol Cell. 1999 Feb;10(2):501–13. doi: 10.1091/mbc.10.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramesh N, Anton IM, Hartwig JH, Geha RS. WIP, a protein associated with wiskott-aldrich syndrome protein, induces actin polymerization and redistribution in lymphoid cells. Proc Natl Acad Sci U S A. 1997 Dec 23;94(26):14671–6. doi: 10.1073/pnas.94.26.14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rivero-Lezcano OM, Marcilla A, Sameshima JH, Robbins KC. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol Cell Biol. 1995 Oct;15(10):5725–31. doi: 10.1128/mcb.15.10.5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miki H, Yamaguchi H, Suetsugu S, Takenawa T. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature. 2000 Dec 7;408(6813):732–5. doi: 10.1038/35047107. [DOI] [PubMed] [Google Scholar]
- 51.Sossey-Alaoui K, Li X, Cowell JK. c-Abl-mediated phosphorylation of WAVE3 is required for lamellipodia formation and cell migration. J Biol Chem. 2007 Sep 7;282(36):26257–65. doi: 10.1074/jbc.M701484200. [DOI] [PubMed] [Google Scholar]
- 52.Westphal RS, Soderling SH, Alto NM, Langeberg LK, Scott JD. Scar/WAVE-1, a Wiskott-Aldrich syndrome protein, assembles an actin-associated multi-kinase scaffold. EMBO J. 2000 Sep 1;19(17):4589–600. doi: 10.1093/emboj/19.17.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suetsugu S, Yamazaki D, Kurisu S, Takenawa T. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev Cell. 2003 Oct;5(4):595–609. doi: 10.1016/s1534-5807(03)00297-1. [DOI] [PubMed] [Google Scholar]
- 54.Snapper SB, Rosen FS, Mizoguchi E, Cohen P, Khan W, Liu CH, et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998 Jul;9(1):81–91. doi: 10.1016/s1074-7613(00)80590-7. [DOI] [PubMed] [Google Scholar]
- 55.Lacout C, Haddad E, Sabri S, Svinarchouk F, Garcon L, Capron C, et al. A defect in hematopoietic stem cell migration explains the nonrandom X-chromosome inactivation in carriers of Wiskott-Aldrich syndrome. Blood. 2003 Aug 15;102(4):1282–9. doi: 10.1182/blood-2002-07-2099. [DOI] [PubMed] [Google Scholar]
- 56.Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009 Nov 13;36(3):512–24. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Betsholtz C. Biology of platelet-derived growth factors in development. Birth Defects Res C Embryo Today. 2003 Nov;69(4):272–85. doi: 10.1002/bdrc.10030. [DOI] [PubMed] [Google Scholar]
- 58.Funa K, Uramoto H. Regulatory mechanisms for the expression and activity of platelet-derived growth factor receptor. Acta Biochim Pol. 2003;50(3):647–58. [PubMed] [Google Scholar]
- 59.Sossey-Alaoui K, Li X, Ranalli TA, Cowell JK. WAVE3-mediated cell migration and lamellipodia formation are regulated downstream of phosphatidylinositol 3-kinase. J Biol Chem. 2005 Jun 10;280(23):21748–55. doi: 10.1074/jbc.M500503200. [DOI] [PubMed] [Google Scholar]
- 60.Kawamura K, Takano K, Suetsugu S, Kurisu S, Yamazaki D, Miki H, et al. N-WASP and WAVE2 acting downstream of phosphatidylinositol 3-kinase are required for myogenic cell migration induced by hepatocyte growth factor. J Biol Chem. 2004 Dec 24;279(52):54862–71. doi: 10.1074/jbc.M408057200. [DOI] [PubMed] [Google Scholar]
- 61.Oikawa T, Yamaguchi H, Itoh T, Kato M, Ijuin T, Yamazaki D, et al. PtdIns(3,4,5)P3 binding is necessary for WAVE2-induced formation of lamellipodia. Nat Cell Biol. 2004 May;6(5):420–6. doi: 10.1038/ncb1125. [DOI] [PubMed] [Google Scholar]
- 62.Okkenhaug K, Vanhaesebroeck B. New responsibilities for the PI3K regulatory subunit p85 alpha. Sci STKE. 2001 Jan 16;2001(65):e1. doi: 10.1126/stke.2001.65.pe1. [DOI] [PubMed] [Google Scholar]
- 63.Kapeller R, Chakrabarti R, Cantley L, Fay F, Corvera S. Internalization of activated platelet-derived growth factor receptor-phosphatidylinositol-3′ kinase complexes: potential interactions with the microtubule cytoskeleton. Mol Cell Biol. 1993 Oct;13(10):6052–63. doi: 10.1128/mcb.13.10.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hill KM, Huang Y, Yip SC, Yu J, Segall JE, Backer JM. N-terminal domains of the class ia phosphoinositide 3-kinase regulatory subunit play a role in cytoskeletal but not mitogenic signaling. J Biol Chem. 2001 May 11;276(19):16374–8. doi: 10.1074/jbc.M006985200. [DOI] [PubMed] [Google Scholar]
- 65.Cory GO, Garg R, Cramer R, Ridley AJ. Phosphorylation of tyrosine 291 enhances the ability of WASp to stimulate actin polymerization and filopodium formation. Wiskott-Aldrich Syndrome protein. J Biol Chem. 2002 Nov 22;277(47):45115–21. doi: 10.1074/jbc.M203346200. [DOI] [PubMed] [Google Scholar]
- 66.Park SJ, Suetsugu S, Takenawa T. Interaction of HSP90 to N-WASP leads to activation and protection from proteasome-dependent degradation. EMBO J. 2005 Apr 20;24(8):1557–70. doi: 10.1038/sj.emboj.7600586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suetsugu S, Hattori M, Miki H, Tezuka T, Yamamoto T, Mikoshiba K, et al. Sustained activation of N-WASP through phosphorylation is essential for neurite extension. Dev Cell. 2002 Nov;3(5):645–58. doi: 10.1016/s1534-5807(02)00324-6. [DOI] [PubMed] [Google Scholar]
- 68.Torres E, Rosen MK. Contingent phosphorylation/dephosphorylation provides a mechanism of molecular memory in WASP. Mol Cell. 2003 May;11(5):1215–27. doi: 10.1016/s1097-2765(03)00139-4. [DOI] [PubMed] [Google Scholar]
- 69.Wu X, Suetsugu S, Cooper LA, Takenawa T, Guan JL. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J Biol Chem. 2004 Mar 5;279(10):9565–76. doi: 10.1074/jbc.M310739200. [DOI] [PubMed] [Google Scholar]
- 70.Yokoyama N, Lougheed J, Miller WT. Phosphorylation of WASP by the Cdc42-associated kinase ACK1: dual hydroxyamino acid specificity in a tyrosine kinase. J Biol Chem. 2005 Dec 23;280(51):42219–26. doi: 10.1074/jbc.M506996200. [DOI] [PubMed] [Google Scholar]
- 71.Miki H, Fukuda M, Nishida E, Takenawa T. Phosphorylation of WAVE downstream of mitogen-activated protein kinase signaling. J Biol Chem. 1999 Sep 24;274(39):27605–9. doi: 10.1074/jbc.274.39.27605. [DOI] [PubMed] [Google Scholar]
- 72.Kim Y, Sung JY, Ceglia I, Lee KW, Ahn JH, Halford JM, et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006 Aug 17;442(7104):814–7. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- 73.Leng Y, Zhang J, Badour K, Arpaia E, Freeman S, Cheung P, et al. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proc Natl Acad Sci U S A. 2005 Jan 25;102(4):1098–103. doi: 10.1073/pnas.0409120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stuart JR, Gonzalez FH, Kawai H, Yuan ZM. c-Abl interacts with the WAVE2 signaling complex to induce membrane ruffling and cell spreading. J Biol Chem. 2006 Oct 20;281(42):31290–7. doi: 10.1074/jbc.M602389200. [DOI] [PubMed] [Google Scholar]
- 75.Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999 Sep 15;13(18):2400–11. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shapiro SD. Matrix metalloproteinase degradation of extracellular matrix: biological consequences. Curr Opin Cell Biol. 1998 Oct;10(5):602–8. doi: 10.1016/s0955-0674(98)80035-5. [DOI] [PubMed] [Google Scholar]
- 77.Basset P, Okada A, Chenard MP, Kannan R, Stoll I, Anglard P, et al. Matrix metalloproteinases as stromal effectors of human carcinoma progression: therapeutic implications. Matrix Biol. 1997 Mar;15(8-9):535–41. doi: 10.1016/s0945-053x(97)90028-7. [DOI] [PubMed] [Google Scholar]
- 78.Vihinen P, Kahari VM. Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer. 2002 May 10;99(2):157–66. doi: 10.1002/ijc.10329. [DOI] [PubMed] [Google Scholar]
- 79.Vihinen P, Ala-aho R, Kahari VM. Matrix metalloproteinases as therapeutic targets in cancer. Curr Cancer Drug Targets. 2005 May;5(3):203–20. doi: 10.2174/1568009053765799. [DOI] [PubMed] [Google Scholar]
- 80.Sasaki M, Kashima M, Ito T, Watanabe A, Izumiyama N, Sano M, et al. Differential regulation of metalloproteinase production, proliferation and chemotaxis of human lung fibroblasts by PDGF, interleukin-1beta and TNF-alpha. Mediators Inflamm. 2000;9(3-4):155–60. doi: 10.1080/09629350020002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reunanen N, Li SP, Ahonen M, Foschi M, Han J, Kahari VM. Activation of p38 alpha MAPK enhances collagenase-1 (matrix metalloproteinase (MMP)-1) and stromelysin-1 (MMP-3) expression by mRNA stabilization. J Biol Chem. 2002 Aug 30;277(35):32360–8. doi: 10.1074/jbc.M204296200. [DOI] [PubMed] [Google Scholar]
- 82.Sossey-Alaoui K, Ranalli TA, Li X, Bakin AV, Cowell JK. WAVE3 promotes cell motility and invasion through the regulation of MMP-1, MMP-3, and MMP-9 expression. Exp Cell Res. 2005 Aug 1;308(1):135–45. doi: 10.1016/j.yexcr.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 83.Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 84.Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F, et al. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995 Apr 14;268(5208):286–90. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 85.Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995 Jun 30;81(7):1147–57. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 86.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997 Apr;9(2):180–6. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 87.Westermarck J, Li SP, Kallunki T, Han J, Kahari VM. p38 mitogen-activated protein kinase-dependent activation of protein phosphatases 1 and 2A inhibits MEK1 and MEK2 activity and collagenase 1 (MMP-1) gene expression. Mol Cell Biol. 2001 Apr;21(7):2373–83. doi: 10.1128/MCB.21.7.2373-2383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Teng Y, Liu M, Cowell JK. Functional interrelationship between the WASF3 and KISS1 metastasis-associated genes in breast cancer cells. Int J Cancer. 2011 Dec 15;129(12):2825–35. doi: 10.1002/ijc.25964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Berx G, Raspe E, Christofori G, Thiery JP, Sleeman JP. Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin Exp Metastasis. 2007;24(8):587–97. doi: 10.1007/s10585-007-9114-6. [DOI] [PubMed] [Google Scholar]
- 90.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008 Jun;9(6):582–9. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006 Apr;6(4):259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 92.Korpal M, Kang Y. The emerging role of miR-200 family of microRNAs in epithelialmesenchymal transition and cancer metastasis. RNA Biol. 2008 Jul;5(3):115–9. doi: 10.4161/rna.5.3.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008 May 30;283(22):14910–4. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008 Apr 1;22(7):894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peter ME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle. 2009 Mar 15;8(6):843–52. doi: 10.4161/cc.8.6.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Spaderna S, Brabletz T, Opitz OG. The miR-200 family: central player for gain and loss of the epithelial phenotype. Gastroenterology. 2009 May;136(5):1835–7. doi: 10.1053/j.gastro.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 97.Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006 May;29(5):903–6. doi: 10.1248/bpb.29.903. [DOI] [PubMed] [Google Scholar]
- 98.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002 Nov 26;99(24):15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007 Aug 15;67(16):7713–22. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 100.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004 Jun 1;64(11):3753–6. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 101.Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006 Mar;9(3):189–98. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 102.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005 Jun 9;435(7043):828–33. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Voorhoeve PM, le SC, Schrier M, Gillis AJ, Stoop H, Nagel R, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006 Mar 24;124(6):1169–81. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 104.Taylor MA, Sossey-Alaoui K, Thompson C, Danielpour D, Schiemann WP. TGF-beta-mediated upregulation of miR-181a promotes breast cancer metastasis by targeting the pro-apoptotic protein Bim. J Clin Invest. 2012 doi: 10.1172/JCI64946. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sossey-Alaoui K, Safina A, Li X, Vaughan MM, Hicks DG, Bakin AV, et al. Down-regulation of WAVE3, a metastasis promoter gene, inhibits invasion and metastasis of breast cancer cells. Am J Pathol. 2007 Jun;170(6):2112–21. doi: 10.2353/ajpath.2007.060975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004 Dec 1;64(23):8585–94. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- 107.Fernando HS, Sanders AJ, Kynaston HG, Jiang WG. WAVE3 is associated with invasiveness in prostate cancer cells. Urol Oncol. 2010 May;28(3):320–7. doi: 10.1016/j.urolonc.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 108.Sossey-Alaoui K, Bialkowska K, Plow EF. The miR200 family of microRNAs regulates WAVE3-dependent cancer cell invasion. J Biol Chem. 2009 Nov 27;284(48):33019–29. doi: 10.1074/jbc.M109.034553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Augoff K, Das M, Bialkowska K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 is a broad regulator of beta1-integrin expression and function in cancer cells. Mol Cancer Res. 2011 Nov;9(11):1500–8. doi: 10.1158/1541-7786.MCR-11-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Augoff K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol Cancer. 2012;11:5. doi: 10.1186/1476-4598-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sossey-Alaoui K, Downs-Kelly E, Das M, Izem L, Tubbs R, Plow EF. WAVE3, an actin remodeling protein, is regulated by the metastasis suppressor microRNA, miR-31, during the invasion-metastasis cascade. Int J Cancer. 2011 Sep 15;129(6):1331–43. doi: 10.1002/ijc.25793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007 Jul 6;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003 Dec 26;115(7):787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 114.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005 Jan 14;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 115.Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szasz AM, Wang ZC, et al. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009 Jun 12;137(6):1032–46. doi: 10.1016/j.cell.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 116.Valastyan S, Benaich N, Chang A, Reinhardt F, Weinberg RA. Concomitant suppression of three target genes can explain the impact of a microRNA on metastasis. Genes Dev. 2009 Nov 15;23(22):2592–7. doi: 10.1101/gad.1832709. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 117.Yamagishi M, Nakano K, Miyake A, Yamochi T, Kagami Y, Tsutsumi A, et al. Polycomb-mediated loss of miR-31 activates NIK-dependent NF-kappaB pathway in adult T cell leukemia and other cancers. Cancer Cell. 2012 Jan 17;21(1):121–35. doi: 10.1016/j.ccr.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 118.Spence HJ, Timpson P, Tang HR, Insall RH, Machesky LM. Scar/WAVE3 contributes to motility and plasticity of lamellipodial dynamics but not invasion in 3D. Biochem J. 2012 Aug 21; doi: 10.1042/BJ20112206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ghoshal P, Teng Y, Lesoon LA, Cowell JK. HIF1A induces expression of the WASF3 metastasis-associated gene under hypoxic conditions. Int J Cancer. 2012 Sep 15;131(6):E905–E915. doi: 10.1002/ijc.27631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Teng Y, Ngoka L, Mei Y, Lesoon L, Cowell JK. HSP90 and HSP70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J Biol Chem. 2012 Mar 23;287(13):10051–9. doi: 10.1074/jbc.M111.335000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Teng Y, Ren MQ, Cheney R, Sharma S, Cowell JK. Inactivation of the WASF3 gene in prostate cancer cells leads to suppression of tumorigenicity and metastases. Br J Cancer. 2010 Sep 28;103(7):1066–75. doi: 10.1038/sj.bjc.6605850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Condeelis J, Singer RH, Segall JE. The great escape: when cancer cells hijack the genes for chemotaxis and motility. Annu Rev Cell Dev Biol. 2005;21:695–718. doi: 10.1146/annurev.cellbio.21.122303.120306. [DOI] [PubMed] [Google Scholar]
- 123.Kulkarni S, Augoff K, Rivera L, McCue B, Khoury T, Groman A, et al. Increased Expression Levels of WAVE3 are Associated with the Progression and Metastasis of Triple Negative Breast Cancer. PLOS ONE. 2012;7(8):e42895. doi: 10.1371/journal.pone.0042895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kurisu S, Suetsugu S, Yamazaki D, Yamaguchi H, Takenawa T. Rac-WAVE2 signaling is involved in the invasive and metastatic phenotypes of murine melanoma cells. Oncogene. 2005 Feb 17;24(8):1309–19. doi: 10.1038/sj.onc.1208177. [DOI] [PubMed] [Google Scholar]
- 125.Park SJ, Kim YT, Jeon YJ. Antioxidant dieckol downregulates the Rac1/ROS signaling pathway and inhibits Wiskott-Aldrich syndrome protein (WASP)-family verprolinhomologous protein 2 (WAVE2)-mediated invasive migration of B16 mouse melanoma cells. Mol Cells. 2012 Apr;33(4):363–9. doi: 10.1007/s10059-012-2285-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Safina A, Vandette E, Bakin AV. ALK5 promotes tumor angiogenesis by upregulating matrix metalloproteinase-9 in tumor cells. Oncogene. 2007 Apr 12;26(17):2407–22. doi: 10.1038/sj.onc.1210046. [DOI] [PubMed] [Google Scholar]
- 127.Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, et al. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995 Jun 30;81(7):1137–46. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 128.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002 Aug 1;115(Pt 15):3193–206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- 129.Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, et al. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem. 1995 Oct 13;270(41):23934–6. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- 130.Lambrechts A, Van TM, Ampe C. The actin cytoskeleton in normal and pathological cell motility. Int J Biochem Cell Biol. 2004 Oct;36(10):1890–909. doi: 10.1016/j.biocel.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 131.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003 Feb 21;112(4):453–65. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 132.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003 May;3(5):362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 133.Zhang Y, Guan XY, Dong B, Zhao M, Wu JH, Tian XY, et al. Expression of MMP-9 and WAVE3 in colorectal cancer and its relationship to clinicopathological features. J Cancer Res Clin Oncol. 2012 Jul 18; doi: 10.1007/s00432-012-1274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 135.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000 Aug 17;406(6797):747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 136.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001 Sep 11;98(19):10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003 Jul 8;100(14):8418–23. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med. 2008 Dec 25;359(26):2814–23. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 2001 Oct;23(10):912–23. doi: 10.1002/bies.1132. [DOI] [PubMed] [Google Scholar]
- 140.Spaderna S, Schmalhofer O, Hlubek F, Jung A, Kirchner T, Brabletz T. Epithelialmesenchymal and mesenchymal-epithelial transitions during cancer progression. Verh Dtsch Ges Pathol. 2007;91:21–8. [PubMed] [Google Scholar]
- 141.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007 May;8(5):341–52. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 142.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009 Apr;9(4):274–84. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 143.May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011 Feb 8;13(1):202. doi: 10.1186/bcr2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010 Jun;15(2):169–90. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008 Jun;14(6):818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 146.Anders C, Carey LA. Understanding and treating triple-negative breast cancer. Oncology (Williston Park) 2008 Oct;22(11):1233–9. [PMC free article] [PubMed] [Google Scholar]
- 147.Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009 Jun;9(Suppl 2):S73–S81. doi: 10.3816/CBC.2009.s.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 2010 Dec;7(12):683–92. doi: 10.1038/nrclinonc.2010.154. [DOI] [PubMed] [Google Scholar]
- 149.Finnegan TJ, Carey LA. Gene-expression analysis and the basal-like breast cancer subtype. Future Oncol. 2007 Feb;3(1):55–63. doi: 10.2217/14796694.3.1.55. [DOI] [PubMed] [Google Scholar]
- 150.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010 Nov 11;363(20):1938–48. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 151.Jiang Z, Jones R, Liu JC, Deng T, Robinson T, Chung PE, et al. RB1 and p53 at the crossroad of EMT and triple negative breast cancer. Cell Cycle. 2011 May 15;10(10) doi: 10.4161/cc.10.10.15703. [DOI] [PubMed] [Google Scholar]
- 152.Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, Richardson A, et al. Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res. 2008 Dec 15;14(24):8010–8. doi: 10.1158/1078-0432.CCR-08-1208. [DOI] [PubMed] [Google Scholar]
- 153.Rakha EA, Elsheikh SE, Aleskandarany MA, Habashi HO, Green AR, Powe DG, et al. Triple-negative breast cancer: distinguishing between basal and nonbasal subtypes. Clin Cancer Res. 2009 Apr 1;15(7):2302–10. doi: 10.1158/1078-0432.CCR-08-2132. [DOI] [PubMed] [Google Scholar]
- 154.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006 Nov 17;127(4):679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 155.Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009 Jan;19(1):89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]