

Abstract
Hydrated membranes with cocontinuous hydrophilic and hydrophobic phases are needed to transport protons in hydrogen fuel cells. Herin we study the water uptake and proton conductivity of a model fuel cell membrane comprising a triblock copolymer, polystyrenesulfonate-block-polyethylene-block-polystyrenesulfonate (S-SES), as a function of water activity in both humid air and liquid water. We demonstrate that the water uptake and proton conductivity of S-SES membranes equilibrated in liquid water are fundamentally different from values obtained when they were equilibrated in humid air. Using synchrotron small angle X-ray scattering and cryogenic scanning transmission electron microscopy, we discover the morphological underpinnings of our observations. A discontinuous increase in conductivity when nearly saturated humid air is replaced with liquid water coincides with the emergence of heterogeneity in the hydrated channels: a water-rich layer is sandwiched between two polymer-rich brushes. While the possibility of obtaining heterogeneous hydrated channels in polymer electrolyte membranes has been discussed extensively, this is, to our knowledge, the first time that direct evidence for the formation of water-rich subdomains is presented.
Keywords: proton exchange membranes, fuel cells, block copolymer electrolytes, morphology, water uptake, proton conductivity
There is continuing interest in proton exchange membrane fuel cells, due to their potential as zero-pollutant emitting, eco-friendly power sources for transportation and personal electronic devices.1–3 The ability of fuel cells to deliver power depends crucially on the rate of proton transport through a polymer electrolyte membrane (PEM) that is placed between the electrodes.4 The PEM in commercial fuel cells is Nafion, which is a random copolymer comprising ion-containing hydrophilic perfluorinated sulfonic acid groups and non-ionic hydrophobic tetrafluoroethylene groups.5 In the dry state, the ionic groups are clustered and Nafion is an insulator. In the wet state, a percolating network of hydrated channels emerges by self-assembly, transforming Nafion from an insulator to a proton conductor. Although numerous papers have been written on this transformation,6–20 there is still considerable debate surrounding the morphology of the hydrated channels. Differential scanning calorimetric (DSC) experiments suggest the presence of three states of water, often referred to as nonfreezing water, freezable loosely bound water and free water.21, 22 One possible explanation for the DSC results is that the hydrated channels are heterogeneous. It is, however, difficult to answer questions regarding heterogeneity when the overall morphology of the hydrated channels is not well-established.
Model PEMs based on ion-containing block copolymers, comprising ionic and non-ionic blocks, are better suited for fundamental investigation of morphology-conductivity relationships.23–30 The morphology of the ion-containing domains in both dry and hydrated states can be studied by X-ray scattering, neutron scattering, and electron microscopy. Consistency between scattering and microscopy data in these systems enables unambiguous determination of the morphology. In spite of this, questions concerning the internal structure of hydrated channels and the relationship between the hydrated structure and proton transport in block copolymer electrolyte membranes remain unanswered.
Herein we study the water uptake and proton conductivity of a triblock copolymer, polystyrenesulfonate-block-polyethylene-block-polystyrenesulfonate (S-SES) as a function of water activity, aw,31 in both humid air and liquid water. We demonstrate that the water uptake and proton conductivity of S-SES membranes equilibrated in liquid water are fundamentally different from values obtained when they were equilibrated in humid air. We use humidity-controlled synchrotron small angle X-ray scattering (SAXS) and cryogenic scanning transmission electron microscopy (STEM) to identify the morphological underpinnings of our observations. In particular, the discontinuity observed from humid air to liquid water coincides with the emergence of heterogeneous hydrated channels in water-equilibrated samples.
The molecular weights of the polystyrenesulfonate (PSS) and polyethylene (PE) blocks of S-SES used in this study are 21.0 kg/mol and 37.4 kg/mol, respectively. The sulfonation level of PSS is 67 mol %. The synthesis and characterization of this polymer are described in the Supporting Information. We begin by examining the water uptake and proton transport properties of S-SES as a function of aw, shown in Figure 1. The experimental protocol used to obtain these data is described in the Supporting Information. All of the experiments reported in this paper (water uptake, conductivity, SAXS and STEM) were conducted on samples annealed at 25 °C.
Figure 1.
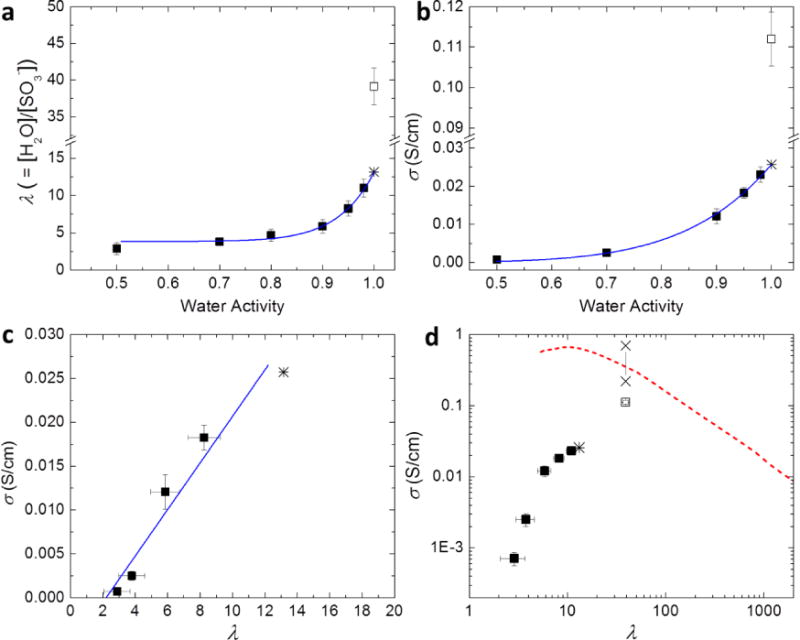
Water uptake and proton transport properties of hydrated S-SES. (a) Hydration number, λ, and (b) proton conductivity, σ, as a function of water activity, aw. (c) Proton conductivity, σ, as a function of λ, on a linear scale. The solid line represents a linear fit. (d) Proton conductivity, σ, as a function of λ, on a log-log plot. Solid squares represent measurements conducted in a humidity-controlled environmental chamber. Open squares represent measurements performed in liquid water. Solid curves in (a) and (b) are power law fits of data points with aw ≤ 0.98. The extrapolation of the power law to aw = 1 yields λfit and σfit, shown by stars in (a), (b), and (c). Two × data points in (d) represent the intrinsic conductivity range of the hydrated microphase of the S-SES copolymer equilibrated in liquid water. Also shown in (d) is σ as a function of λ of aqueous HCl solutions, dashed curve, data taken from ref 36.
The hydration number, λ, specified as the number of water molecules per sulfonic acid group, as a function of aw is shown in Figure 1a. The measurements in the range 0.50 ≤ aw ≤ 0.98 were taken in a humidity-controlled environmental chamber (solid squares). These data are consistent with a power law shown by the solid curve in Figure 1a. Extrapolating the power law fit to aw = 1 yields λfit = 13.2 (star). The data obtained in humid air suggest that if the membrane were equilibrated in either air with a relative humidity (RH) of 100% or liquid water, the resulting hydration number would be 13.2. We did not attempt to equilibrate samples in air with RH = 100% due to the lack of appropriate equipment. However, measurements in liquid water yielded a value of λ = 39.1 (open square), almost a factor of three larger than λfit.
The dependence of proton conductivity, σ, on aw, obtained using the same protocol that was used to obtain Figure 1a, is plotted in Figure 1b. In the range 0.50 ≤ aw ≤ 0.98 (solid squares), the relationship between σ and aw follows a power law (solid curve in Figure 1b). Extrapolation of the power law fit to aw = 1 yields σfit = 0.026 S/cm (star). The measured value of proton conductivity of the membrane equilibrated in water is σ = 0.11 S/cm (open square), a factor of four higher than σfit. The dependence of λ and σ on aw is similar with smooth dependences when aw < 1, and a discontinuous increase in the vicinity of aw = 1. This discontinuity is often referred to as Schroeder’s Paradox.32, 33
In Figure 1c, we plot σ as a function of λ obtained in humid air on a linear scale. It is evident that σ vs λ data obtained from sample equilibrated in humid air (solid squares) can be approximated by a straight line (solid line in Figure 1c), σ = −0.00583 + 0.00265λ. Extrapolation of conductivity to σ = 0, we find that the onset of finite conductivity occurs at λ = 2.2. In their study of conductivity through hydrated Nafion, Hsu and Gierke suggest that percolated proton conducting pathways are formed in the vicinity of λ = 2.8, 34 Our results are in good agreement with theirs. σ as a function of λ obtained in both humid air and liquid water is depicted on a log-log plot in Figure 1d. Also shown in Figure 1d is the dependence of the proton conductivity of aqueous HCl solutions as a function of λ.35 The conductivity of the water-equilibrated membrane is close to the curve representing HCl solutions, while the conductivities of S-SES membranes equilibrated in humid air lie well below the curve. It is perhaps remarkable that the ionic conductivity of a hydrated single-ion conductor is only a factor of three lower than that of free HCl at the same concentration. The two data points marked by “×”s in Figure 1d will be discussed shortly.
The data described above define two outstanding questions: (1) Why do λ and σ change discontinuously when nearly saturated humid air is replaced by liquid water? (2) Why is conductivity of S-SES equilibrated in liquid water similar to that of an aqueous HCl solution with the same concentration? We attempted to answer these questions by studying the morphology of S-SES as a function of hydration, using humidity-controlled SAXS and STEM. The protocol used in these experiments is described in the Supporting Information.
SAXS intensity is plotted as a function of the magnitude of the scattering vector, q, for S-SES samples equilibrated at different aw values in Figure 2a. Measurements with aw ≤ 0.95 were acquired using an in situ humidity-controlled chamber.36 Measurements at aw = 1 were performed in liquid water. In the dry state, the SAXS profile exhibited a single broad peak at q = q*, indicating the presence of a periodic structure. The characteristic length of the periodic structure, d, is given by d = 2π/q*. The absence of higher order peaks at aw ≤ 0.95 indicates that long-range order is limited in our samples. As aw increases, the peak gradually shifts to lower q* values, indicating a gradual increase in d. In liquid water, the SAXS profile showed two peaks, a primary peak at q = q* = 0.108 nm−1 and a secondary peak at q = 1.8q*. In Figure 2b, we plot d as a function of aw. As was the case with the data shown in Figure 1, d values obtained from humid air are consistent with a power law (solid curve in Figure 2b), and extrapolation of the power law fit to aw = 1 yields dfit = 43.8 nm (star). In contrast, d obtained from liquid water is 58.0 nm. The discontinuous increase in d and the appearance of a secondary SAXS peak in liquid water suggest a discontinuous change in the sample morphology. In many cases, the appearance of a secondary peak indicates an increase in long-range order. However, the increase in long-range order is also accompanied by a decrease in peak width. There is no qualitative difference in peak widths of SAXS profiles obtained in humid air and liquid water. In Figure 2c we show wide angle X-ray scattering (WAXS) profiles obtained from a dry and a water-equilibrated S-SES sample. The WAXS peaks reflecting crystallinity of the PE blocks are unaffected by hydration.
Figure 2.
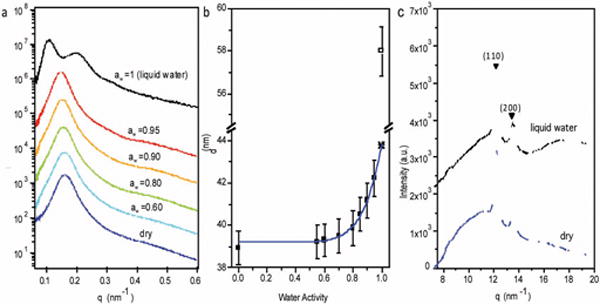
(a) Small angle X-ray scattering (SAXS) intensity as a function of the magnitude of the scattering vector, q, of S-SES at different aw values. When aw ≤ 0.95, SAXS were performed using an in situ humidity-controlled chamber. Measurements at aw = 1 were done in liquid water. (b) Domain spacing, d, as a function of aw. Solid squares represent values obtained in humid air, whereas the open square represents the value obtained in liquid water. Solid curve is a power law fit of data points with aw ≤ 0.95. The extrapolation of the power law to aw = 1 yields dfit, shown by a star. (c) Wide angle X-ray scattering (WAXS) profiles of S-SES showing the (110) and (200) peaks of the polyethylene (PE) crystals, in dry state (blue profile) and equilibrated in liquid water (black profile).
Cryogenic STEM was also used to examine the morphology of S-SES (Figure 3 and 4). Thin sections (about 70 nm thick) of dry S-SES were prepared by cryomicrotoming. The sections were placed on a lacey carbon supported copper grid. Water-containing samples were annealed in a humidity-controlled environment using an FEI Vitrobot before plunging into liquid ethane. STEM experiments were performed on a Tecnai F20 UT FEG, equipped with a high angle annular dark field (HAADF) detector, using 200 keV acceleration voltage. Dry samples were imaged at room temperature. Hydrated samples were imaged at −184 °C. All the samples were unstained. Figure 3a shows the morphology of S-SES in the dry state. It is evident that the sample has a lamellar morphology with relatively poor long-range order. For clarity, a small portion of the STEM image was magnified and shown in the bottom left corner of the figure. Since sulfur is the heaviest atom in our system, regions that appear bright in HAADF reflect the locations of the PSS-rich lamellae. The dark regions represent the PE-rich lamellae. The average thickness of the PSS-rich lamellae, dPSS, and that of the PE-rich lamellae, dPE, determined from line scans, are 20.0 nm and 19.3 nm, respectively. In the dry state, dPSS and dPE are similar, as expected. Fast Fourier transform (FFT) of this image was performed and the result is shown in Figure 3b. The 2D FFT pattern contains a ring (inset of Figure 3b). Radial integration of the 2D FFT is shown by squares in Figure 3b. The 1D profile contains a maximum at q = 0.153 nm−1. The SAXS profile of S-SES in the dry state was plotted on top of the 1D FFT for direct comparison (dashed profile in Figure 3b). Both the FFT and SAXS profiles indicate the presence of a periodic structure with little long-range order. There are small quantitative disagreements between SAXS and STEM results. The characteristic length scales obtained from FFT, dSTEM = 43.8 nm, and line scans through STEM images, dPSS + dPE = 39.3 nm, are slightly larger than d measured by SAXS, 38.9 nm. Such minor discrepancies are likely to arise from STEM sample preparation protocol. Nevertheless, all of the SAXS and STEM data indicate the presence of alternating PSS-rich and PE-rich lamellae, as shown schematically in Figure 3c. For concreteness, we also give the thicknesses of the lamellae determined from line scans through the micrographs.
Figure 3.
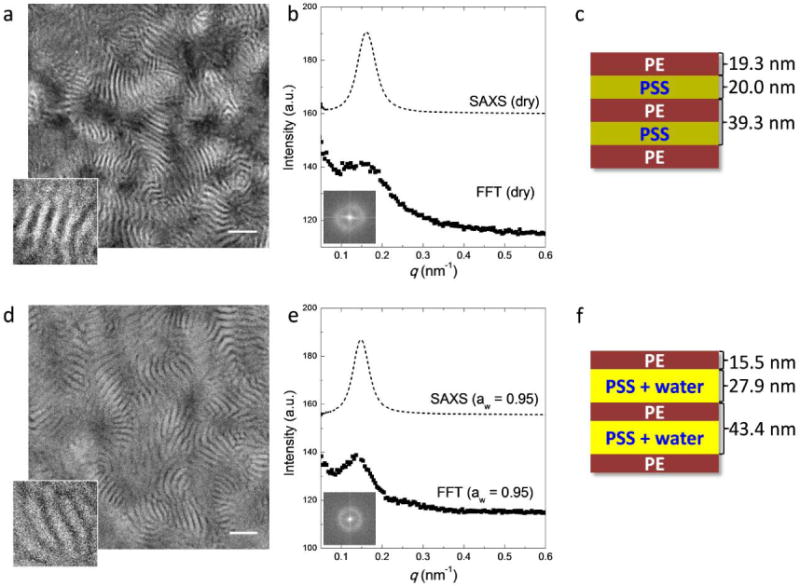
(a) and (d) Scanning transmission electron microscopy (STEM) of S-SES in the dry state (a) and equilibrated at aw ≈ 0.95 (d). For clarity, a small portion of the images was magnified and shown on the bottom left corner of each image. Scale bar represents 200 nm. The corresponding 2D and radially-integrated FFT patterns are shown in (b) and (e) as insets and solid squares, respectively. Dashed lines in (b) and (e) depict the SAXS profiles of S-SES bulk membranes in the dry state and equilibrated at aw = 0.95. (c) and (f) Schematics showing the morphologies of S-SES in the dry state and equilibrated at aw ≈ 0.95.
Figure 4.
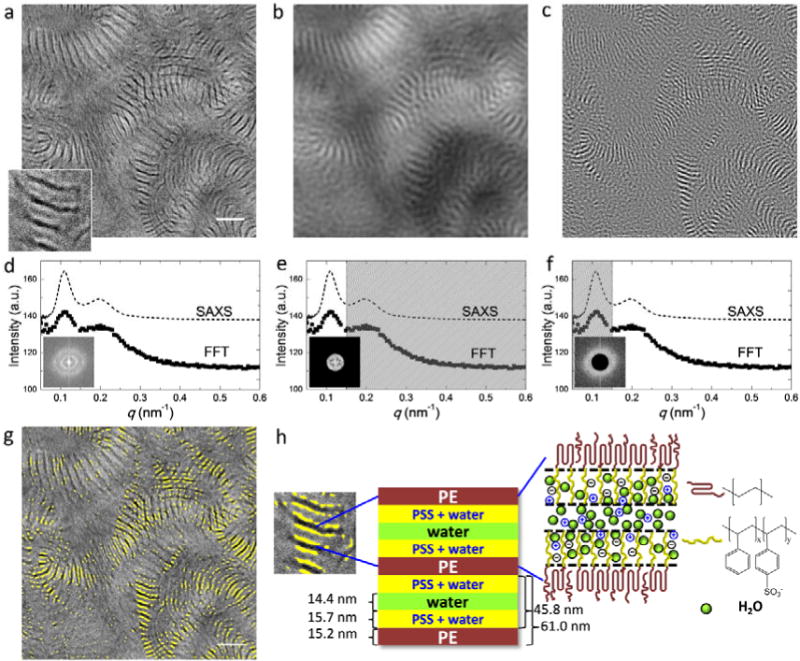
(a) Cryogenic STEM of S-SES equilibrated in liquid water. For clarity, a small portion of the image was magnified and shown on the bottom left corner. Scale bar represents 200 nm. The corresponding 2D and radially-integrated FFT patterns are shown in (d) as inset and solid squares. SAXS profile of S-SES bulk membrane equilibrated in liquid water is depicted by the dashed line in (d). Low pass and high pass frequency filters with a cutoff frequency of 0.15 nm−1 were applied to the STEM image in (a) and the resulting images are shown in (b) and (c), respectively. The actual frequency filters are shown in (e) and (f). Frequency filtering enables the identification of the structures responsible for the primary and secondary FFT and SAXS peaks. Figure (g) is the superposition of (a) and (c). The bright regions in (c) are highlighted in yello0w in (g). A schematic of the morphology of S-SES equilibrated in liquid water is shown in (h). In particular, a heterogeneous channel comprising hydrated PSS-rich brushes sandwiching a water-rich layer is depicted.
Figure 3d–f show the morphology of S-SES equilibrated in humid air with aw ≈ 0.95. The analysis used above to analyze dry S-SES was repeated. Both SAXS and 1D FFT data show a single broad peak (Figure 3e), indicating the presence of alternating hydrated PSS-rich lamellae and PE-rich lamellae, as shown in Figure 3d. The characteristic length scale obtained from FFT is dSTEM = 45.9 nm, slightly larger than d measured by SAXS, 42.2 nm. Compared to the dry state, d of S-SES equilibrated at aw = 0.95 only increased by 2.1 nm measured by FFT and 3.3 nm measured by SAXS. However, quantitative analysis of line scans gives dPSS = 27.9 nm, a 7.9 nm increase compared to dPSS in the dry state, and dPE = 15.5 nm, a 3.8 nm decrease compared to dPE in the dry state. A schematic of the morphology of S-SES equilibrated in humid air is shown in Figure 3f. At aw ≈ 0.95, the thickness of the hydrated PSS-rich lamellae is about a factor of two larger than that of PE-rich lamellae. It is well-known that the addition of a selective solvent to a block copolymer leads to an expansion of the well-solvated microphase and a contraction of the poorly solvated microphase.37–41
The morphology of S-SES equilibrated in liquid water is depicted in Figure 4. At first glance (Figure 4a), the morphology appears similar to S-SES equilibrated in humid air (Figure 3d). Alternating lamellae of expanded hydrated PSS and contracted PE with little long-range order are evident. An expanded view of the lamellar structure, shown in the bottom left corner of Figure 4a, however, reveals an interesting new feature. The lamellae appear to have three distinct gray scales. The darkest lamellae correspond to dry PE-rich domains (similar to Figure 3). The region between two adjacent PE-rich lamellae, which must contain primarily PSS and water, comprises three sub-domains, two bright stripes adjacent to the PE-rich lamellae, and one gray stripe in the middle of the region. Since the brightest regions reflect the presence of sulfur, we conclude that the two bright stripes reflect the presence of PSS-rich (or water-poor) domains. It is then logical to conclude that the gray domains have a low concentration of PSS, i.e., they are the water-rich domains. Interestingly, the FFT of the micrograph, shown in Figure 4d, shows two peaks. The locations of these peaks are in remarkable agreement with the SAXS profile obtained from the sample equilibrated in liquid water. The origin of the secondary peak seen in both FFT and SAXS is, however, unclear at this point.
In order to identify the structures that are responsible for the primary and secondary peaks in FFT and SAXS, frequency filters were applied to the STEM image in Figure 4a. Firstly, a low pass filter with a cutoff frequency of 0.15 nm−1 was applied and the resulting image is shown in Figure 4b. The corresponding frequency filter is shown in Figure 4e. Thus Figure 4b reveals the periodicity responsible for the primary peak in SAXS and STEM, which is an unremarkable lamellar phase. Secondly, a high pass filter with a cutoff frequency of 0.15 nm−1 was applied to Figure 4a and the resulting image is shown in Figure 4c. The corresponding frequency filter is shown in Figure 4f. Figure 4c reveals the periodicity that delineates the secondary peak in SAXS and STEM. It is perhaps not surprising that the periodic structure seen Figure 4c is much finer than that seen in Figure 4b.
In Figure 4g we superimpose Figure 4c on the original micrograph, Figure 4a. The yellow color in Figure 4g represents the bright regions in Figure 4c. Most of the gray water-rich channels are located between two yellow stripes. It is clear that the secondary FFT and SAXS peaks arise due to the heterogeneous nature of the hydrated PSS phase, shown schematically in Figure 4h. When S-SES is equilibrated in liquid water, there is a water-rich channel with a thickness of dw = 14.4 nm in the center of the hydrated PSS phase. The PSS-rich region formed two brushes sandwiching the water-rich channel. The average brush thickness is dbrush = 15.7 nm. The thickness of the PE-rich phase, dPE = 15.2 nm. Evidently dPE remains approximately the same as that in S-SES equilibrated at aw ≈ 0.95, and the thickness of the two PSS-rich brushes, 2 × dbrush = 31.4 nm, is only a slight increase from dPSS in S-SES equilibrated at aw ≈ 0.95. This suggests that in liquid water, the PSS-rich brushes are saturated with water molecules and reached maximum extension. This further implies that the water-rich channels in the center of the PSS domains may be devoid of PSS chains.
It is perhaps worth noting that our conclusions on the morphology of hydrated S-SES are based on both reciprocal space scattering measurements (Figure 2) and position space electron micrographs (Figures 3 and 4). It would have been impossible to derive a unique morphology at a given state of hydration based on scattering alone.
We conclude by answering the two questions that were posed above:
Why do λ and σ change discontinuously when nearly saturated humid air is replaced by liquid water? The discontinuous changes in λ and σ when nearly saturated humid air is replaced by liquid water are due to a morphological transition of the hydrated channel from a homogeneous hydrated phase to a heterogeneous channel comprising water-poor (or PSS-rich) and water-rich (or PSS-poor) lamellar domains. Our tentative hypothesis is that this unbinding transition42 is a first-order phase transition that occurs between aw = 0.98 and aw = 1. Further work is needed to confirm this hypothesis. In the absence of a first-order phase transition, λ and σ do not change discontinuously when nearly saturated air is replaced by liquid water. While we do not have definitive proof, it is likely that the PSS chains are excluded from water-rich domains. To our knowledge, this is the first time that direct evidence for the formation of water-rich subdomains is presented.
Why is conductivity of S-SES equilibrated in liquid water similar to that of an aqueous HCl solution with the same concentration? The conductivity of S-SES equilibrated in liquid water is similar to that of an aqueous HCl solution at the same concentration because of the presence of water-rich domains in the hydrated channels. It is well-known that proton transport in aqueous HCl solutions is extremely efficient due to the Grotthus mechanism. Newman and coworkers have argued that the Grotthus mechanism cannot apply to proton conduction when λ < 14, i.e., within the PSS brush.34, 43 This suggests in liquid water, although there is parallel proton transport in both hydrated PSS-rich brushes and water-rich domains, the majority of the proton transport occurs in the water-rich domains. Conductivity through randomly oriented alternating insulating and conductive lamellae is given by σ = 2/3 ϕcσc, where ϕc is the volume fraction of the conducting phase and σc is the intrinsic conductivity of the conductive lamellae.44 The volume fraction of the water-rich lamellae, ϕc,w, calculated from the STEM analysis shown in Figure 4h, is ϕc,w = 0.24. This gives an intrinsic conductivity σc,w = 0.70 S/cm. The volume fraction of the entire conducting domains, including both water-rich and PSS-rich lamellae, calculated from the STEM analysis shown in Figure 4h, is ϕc,w+PSS = 0.75. This gives an intrinsic conductivity σc,w+PSS = 0.22 S/cm. The two × data points in Figure 1c indicate values of σc,w and σc,w+PSS. The fact that the conductivity of aqueous HCl at the same value of when λ lies between σc,w and σc,w+PSS suggests that most of the proton transport occurs by the Grotthus mechanism in the water-rich lamellae. The sharp discontinuity in either water uptake or conductivity when highly humid air is replaced by liquid water is, therefore, not necessarily paradoxical.
Supplementary Material
Acknowledgments
Primary funding for the work was provided by the Electron Microscopy of Soft Matter Program from the Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. SAXS experiments were performed at the Advanced Light Source (ALS). The STEM experiments were performed as user projects at the National Center for Electron Microscopy, Lawrence Berkeley National Laboratory. The ALS and NCEM are DOE national user facilities and are supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under the same contract. X.C.C thanks Dr. Roseann Csencsits for help with cryo-plunging and cryo-transfer of STEM samples.
Footnotes
Supporting Information Available. Details on the synthesis of material, water uptake and conductivity measurements, SAXS and STEM. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.EG&G Technical Services, I. Fuel Cell Handbook. 7. U.S. Department of Energy; West Virginia: 2004. [Google Scholar]
- 2.Steele BCH, Heinzel A. Nature. 2001;414(6861):345–352. doi: 10.1038/35104620. [DOI] [PubMed] [Google Scholar]
- 3.Costamagna P, Srinivasan S. Journal of Power Sources. 2001;102(1–2):242–252. [Google Scholar]
- 4.Kreuer KD. Chemistry of Materials. 1996;8(3):610–641. [Google Scholar]
- 5.Srinivasan S, Ticianelli EA, Derouin CR, Redondo A. Journal of Power Sources. 1988;22(3–4):359–375. [Google Scholar]
- 6.Yeo SC, Eisenberg A. Journal of Applied Polymer Science. 1977;21(4):875–898. [Google Scholar]
- 7.Gierke TD, Munn GE, Wilson FC. Journal of Polymer Science: Polymer Physics Edition. 1981;19(11):1687–1704. [Google Scholar]
- 8.Hsu WY, Gierke TD. Journal of Membrane Science. 1983;13(3):307–326. [Google Scholar]
- 9.Xue T, Trent JS, Osseo-Asare K. Journal of Membrane Science. 1989;45(3):261–271. [Google Scholar]
- 10.Zawodzinski TA, Neeman M, Sillerud LO, Gottesfeld S. The Journal of Physical Chemistry. 1991;95(15):6040–6044. [Google Scholar]
- 11.Zawodzinski TA, Derouin C, Radzinski S, Sherman RJ, Smith VT, Springer TE, Gottesfeld S. Journal of The Electrochemical Society. 1993;140(4):1041–1047. [Google Scholar]
- 12.Zawodzinski TA, Davey J, Valerio J, Gottesfeld S. Electrochimica Acta. 1995;40(3):297–302. [Google Scholar]
- 13.Gebel G. Polymer. 2000;41(15):5829–5838. [Google Scholar]
- 14.Elliott JA, Hanna S, Elliott AMS, Cooley GE. Macromolecules. 2000;33(11):4161–4171. [Google Scholar]
- 15.James PJ, McMaster TJ, Newton JM, Miles MJ. Polymer. 2000;41(11):4223–4231. [Google Scholar]
- 16.Haubold HG, Vad T, Jungbluth H, Hiller P. Electrochimica Acta. 2001;46(10–11):1559–1563. [Google Scholar]
- 17.Rubatat L, Gebel G, Diat O. Macromolecules. 2004;37(20):7772–7783. [Google Scholar]
- 18.Kim MH, Glinka CJ, Grot SA, Grot WG. Macromolecules. 2006;39(14):4775–4787. [Google Scholar]
- 19.Galperin DY, Khokhlov AR. Macromolecular Theory and Simulations. 2006;15(2):137–146. [Google Scholar]
- 20.Schmidt-Rohr K, Chen Q. Nat Mater. 2008;7(1):75–83. doi: 10.1038/nmat2074. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura K, Hatakeyama T, Hatakeyama H. Polymer. 1983;24(7):871–876. [Google Scholar]
- 22.Kim YS, Dong L, Hickner MA, Glass TE, Webb V, McGrath JE. Macromolecules. 2003;36(17):6281–6285. [Google Scholar]
- 23.Shi Z, Holdcroft S. Macromolecules. 2005;38(10):4193–4201. [Google Scholar]
- 24.Elabd YA, Napadensky E, Walker CW, Winey KI. Macromolecules. 2005;39(1):399–407. [Google Scholar]
- 25.Park MJ, Downing KH, Jackson A, Gomez ED, Minor AM, Cookson D, Weber AZ, Balsara NP. Nano Letters. 2007;7(11):3547–3552. doi: 10.1021/nl072617l. [DOI] [PubMed] [Google Scholar]
- 26.Rubatat L, Li C, Dietsch H, Nykänen A, Ruokolainen J, Mezzenga R. Macromolecules. 2008;41(21):8130–8137. [Google Scholar]
- 27.Chen L, Hallinan DT, Elabd YA, Hillmyer MA. Macromolecules. 2009;42(16):6075–6085. [Google Scholar]
- 28.Park MJ, Kim S, Minor AM, Hexemer A, Balsara NP. Advanced Materials. 2009;21(2):203–208. [Google Scholar]
- 29.Wang X, Yakovlev S, Beers KM, Park MJ, Mullin SA, Downing KH, Balsara NP. Macromolecules. 2010;43(12):5306–5314. [Google Scholar]
- 30.Bae B, Miyatake K, Watanabe M. Macromolecules. 2010;43(6):2684–2691. [Google Scholar]
- 31.Zhao Q, Majsztrik P, Benziger J. The Journal of Physical Chemistry B. 2011;115(12):2717–2727. doi: 10.1021/jp1112125. [DOI] [PubMed] [Google Scholar]
- 32.Schroeder P. Z Phys Chem. 1903;45:57. [Google Scholar]
- 33.Choi P, Datta R. Journal of The Electrochemical Society. 2003;150(12):E601–E607. [Google Scholar]
- 34.Weber AZ, Newman J. Journal of The Electrochemical Society. 2003;150(7):A1008–A1015. [Google Scholar]
- 35.Cukierman S. Biophysical Journal. 2000;78(4):1825–1834. doi: 10.1016/S0006-3495(00)76732-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson A, Beers KM, Chen XC, Hexemer A, Pople JA, Kerr JB, Balsara NP. Review of Scientific Instruments. 2013;84(7):075114–7. doi: 10.1063/1.4815981. [DOI] [PubMed] [Google Scholar]
- 37.Winey KI, Thomas EL, Fetters LJ. Macromolecules. 1991;24(23):6182–6188. [Google Scholar]
- 38.Shull KR, Winey KI. Macromolecules. 1992;25(10):2637–2644. [Google Scholar]
- 39.Uneyama T, Doi M. Macromolecules. 2005;38(13):5817–5825. [Google Scholar]
- 40.Chen XC, Yang H, Green PF. Macromolecules. 2011;44(14):5758–5763. [Google Scholar]
- 41.Chen XC, Yang H, Green PF. Macromolecules. 2012;45(9):3993–4000. [Google Scholar]
- 42.Nagle JF, Tristram-Nagle S. Biochimica et Biophysica Acta (BBA) – Reviews on Biomembranes. 2000;1469(3):159–195. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber AZ, Newman J. Journal of The Electrochemical Society. 2004;151(2):A311–A325. [Google Scholar]
- 44.Panday A, Mullin S, Gomez ED, Wanakule N, Chen VL, Hexemer A, Pople J, Balsara NP. Macromolecules. 2009;42(13):4632–4637. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.