

Abstract
Ruthenium-based compounds have intriguing anti-cancer properties and some of these novel compounds are currently in clinical trials. To continue the development of new metal-based drug combinations, we coupled ruthenium (Ru) with the azole compounds ketoconazole (KTZ) and clotrimazole (CTZ), which are well-known antifungal agents that also display anticancer properties. We report the activity of a series of twelve Ru-KTZ and Ru-CTZ compounds against three prostate tumor cell lines with different androgen sensitivity, as well as cervical cancer and lymphoblastic lymphoma cell lines. In addition, human cell lines were used to evaluate the toxicity against non-transformed cells and to establish selectivity indexes. Our results indicate that the combination of ruthenium and KTZ/CTZ in a single molecule results in complexes that are more cytotoxic than the individual components alone, displaying in some cases low micromolar CC50 values and high selectivity indexes. Additionally, all compounds are more cytotoxic against prostate cell lines with lower cytotoxicity against non-transformed epidermal cell lines. Some of the compounds were found to primarily induce cell death via apoptosis yet weakly interact with DNA. Our studies also demonstrate that the cytotoxicity induced by our Ru-based compounds is not directly related to their ability to interact with DNA.
Keywords: ruthenium, ketoconazole, clotrimazole, apoptosis, prostate cancer cell lines
Introduction
The increasing understanding of the biochemistry of cancer has resulted in the generation of a wide variety of compounds with potential antitumor properties. Many of those compounds are often used in combination chemotherapy, as each component may elicit a different mechanism of action by reaching diverse biological targets. In line with this, the combination of a number of organic drugs with well-known metal-based antitumor agents, notably cisplatin and carboplatin, has been widely explored(Lippert, 2006; Sweetman, 2010). Significant examples include combinations of 5-fluorouracil with carboplatin, cisplatin and oxaliplatin in the treatment of gastrointestinal, head and neck, colorectal, and pancreatic cancer (Lorch et al. 2011; Posner et al. 2007; Van et al. 2006). Combination therapies, however, may present pharmacokinetic issues related to the difficulty in reaching multiple targets or achieving peak blood concentrations of each component drug at similar times; in that sense, hybrid compounds containing multiple pharmacophores in a single molecule might present an advantage.
For some time, we have been designing potential chemotherapeutics based on the metal-drug synergism (Martinez et al. 2012; Martinez et al. 2010; Rademaker-Lakhai et al. 2004; Rajapakse et al. 2009; Strasberg et al. 2004), which may be achieved by combining a drug of known therapeutic value with a biologically relevant metal in a single molecule (Sanchez-Delgado et al. 2004). Such combinations can lead to new chemical entities with enhanced activity, decreased toxicity, and more controlled pharmacokinetic properties than each separate component, or than the mere physical combination of two drugs. Because of the high toxicity of platinum and the common occurrence of resistance, other metals are receiving increasing attention in cancer research, and ruthenium appears as a particularly interesting element in terms of anticancer activity and low toxicity. The compound NAMI-A, ImH[trans-RuCl4(DMSO)Im] (Im = imidazole), has shown very promising properties in preclinical and phase I clinical trials against lung metastases (Bergamo et al. 1999; Morbidelli et al. 2003; Rademaker-Lakhai et al. 2004; Sava et al. 2002; Sava et al. 1998) while KP1019, IndH[trans-RuCl4(Ind)2] (Ind = indazole), is effective in ovarian and colon carcinomas (Berger et al. 1989; Bratsos et al. 2007b; Garzon et al. 1987; Hartinger et al. 2008; Jakupec et al. 2008; Kapitza et al. 2005; Seelig et al. 1992). In addition, three octahedral Ru (II) complexes were found to induce apoptosis when tested on human hepatoma (BEL-7402 and HepG-2), ovarian (HeLa), and osteosarcoma (MG-63) cancer cell lines (Huang et al., 2011). Most recently, two symmetric ruthenium(II) complexes were found to exert apoptosis on the BEL-7402, HeLa, MG-63 and SKBR (breast) cancer cell lines (Xie et al., 2013). In addition, novel Ru-letrozole compounds were found to exhibit cytotoxocity against a breast cancer cell line (MCF-7) and glioblastoma (U251N) cells and one of these compounds was implicated in the induction of autophagy induced cell death (Castonguay et al., 2012). Ru-quinolone adducts were also found to have anti-cancer potential as monitored on the HeLa cancer cell line (Kljun, et al., 2013). Arene-ruthenium “semi-sandwich” compounds like [Ru(η6-arene)(X)(Y–Z)] (where Y–Z is a chelating ligand, and X is monoanionic ligand), and related derivatives incorporating the 1,3,5-triaza-7-phosphaadamantane (PTA) ligand (Aird et al. 2002; Dougan et al. 2006; Morbidelli et al. 2003; Morris et al. 2001; Scolaro et al. 2005; Wang et al. 2005) also display very interesting antitumor activities. In our search for metal-drug synergy, we synthesized a series of arene-Ru-chloroquine complexes that proved to be active against Jurkat human T lymphocyte leukemia and SUP-T1 lymphoma cell lines; we also demonstrated that induction of apoptosis is the main path for cell death in these cases (Martinez et al. 2010). The complex [Ru(η6-arene)Cl2(CQ)], also proved to be active in vitro against dedifferentiated liposarcoma (LS141), a type of tumor for which no chemotherapy is available (Rajapakse et al. 2009)
Continuing our search for new metal-based drug combinations, we have turned our attention to azole compounds like ketoconazole (KTZ) and clotrimazole (CTZ) (Figure 1), two well-known antifungal agents that also display anticancer properties. KTZ is a cytochrome P-450 inhibitor that blocks both testicular and adrenal androgen biosynthesis (Eichenberger et al. 1989a) and causes apoptosis through p53-dependent and other pathways in specific cancerous cell lines, although the molecular mechanisms are still unclear (Ho et al. 1998). Because of its ability to compete with testosterone and dihydrotestosterone in binding androgen receptors, KTZ is also used as a treatment for hormone-refractory (Rajapakse et al. 2009; Van Veldhuizen et al. 2003) and metastatic prostate cancer (Bok and Small, 1999). When KTZ is administered in combination with vinblastine or etoposide, synergistic suppression of in vitro growth on human androgen-independent prostate cancer cells was observed (Eichenberger et al. 1989a; Eichenberger et al. 1989b). The effect of KTZ has also been investigated on the glucocorticoid receptor (GR) transcriptional activity on HeLa and HepG2 (hepatic) cancer cells (Duret et al. 2006). In addition to P450 enzymes, KTZ inhibits the GR transcriptional activity and competes with dexamethasone for human GR binding (Duret et al. 2006).
Figure 1.
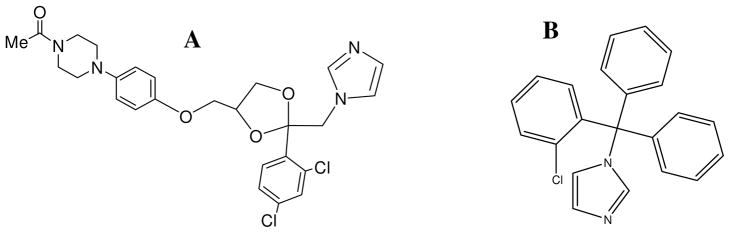
Chemical structures of (A) KTZ and (B) CTZ.
CTZ, also a cytochrome P-450 inhibitor, is in turn able to suppress tumor growth by acting as a calmodulin antagonist (Hegemann et al. 1993; Mac et al. 1993). The increase in mitocondrially bound hexokinase, process associated to the high rate of glycolysis in cancer cells (Bratsos et al. 2007a) is induced by insulin, Ca2+ and Ca2+-mobilizing hormones, and it could be prevented by calmodulin antagonists (Penso and Beitner, 1998). In addition, CTZ induces Ca2+ store-mediated inhibition of translation initiation (Cao et al. 2004) and blocks calcium-activated potassium channel (IKCa1) on human T lymphocytes (Wulff et al. 2000).
Metal complexes of KTZ and CTZ have begun to receive attention as potential drugs. Some of us reported that Ru(KTZ)2Cl2 induces cytotoxicity and apoptosis-associated caspase-3 activation in several cancer cell lines including C8161 melanoma and HT-29 colon carcinoma, irrespective of p53 status, with IC50 values around 25 μM; this complex targets the mitochondria and is more effective than KTZ, CTZ, Ru(CTZ)2Cl2, or cisplatin at inducing PARP fragmentation and proapoptotic Bak expression (Strasberg et al. 2004). Pt(CTZ)2Cl2 was found to be active against a panel of human tumor cell lines, including prostate, pancreas, breast, and colon, while the Pd analogue was inactive (Navarro et al. 2009; Navarro et al. 2006). We recently reported that the arene-Ru-CTZ and arene-Ru-KTZ compounds 1–12 (Figure 2) display high activity against Leishmania major and Trypanosoma cruzi with very low toxicity to normal mammalian cells (Iniguez et al. 2013; Martinez et al. 2012). In the present paper, we discuss the cytotoxicity of the Ru-KTZ and Ru-CTZ complexes 1–12 against a panel of tumor and non-tumor cell lines, the mechanism of cell death induced, and their ability to interact with DNA.
Figure 2.

Chemical structures of the compounds investigated. Synthesized as previously described (Iniguez et al. 2013; Martinez et al. 2012).
Materials and methods
Reagents
Calf Thymus (CT) DNA, buffers and solvents were purchased from Sigma-Aldrich. Solvents were purified by use of a PureSolv purification unit from Innovative Technology, Inc.; all other chemicals were used as received. Thermal denaturation experiments were performed on an Agilent 8453 diode-array spectrophotometer equipped with a HP 89090 Peltier temperature control accessory. CD spectra were taken in a Chirascan CD Spectrometer also equipped with a thermostated cuvette holder. Complexes 1–12 were synthesized as previously described by us (Iniguez et al. 2013; Martinez et al. 2012; Penso and Beitner, 1998).
Cell culture
All cell lines were obtained from American Tissue Culture Collection (ATCC; Manassas, VA, USA). Cells were cultured at 37°C in a 5% CO2 humidified atmosphere, by incubating them in a regular water-jacketed incubator or in a temperature controlled chamber within the bioimaging system. Only cell cultures demonstrating cell viabilities of 90% or higher were used for the cytotoxicity experiments (Lema et al. 2011). Human non-transformed keratinocyte HaCaT cells (Boukamp et al. 1988), mammary epithelial MCF-10A cells (Soule and McGrath, 1986) and human cervical HeLa cancer cells were cultured in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% heat-inactivated newborn calf serum (Hyclone, Logan, UT, USA) and antibiotics; 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 0.25 μg/ml of amphotericin B (Invitrogen, Carlsbad, CA, USA). Wild type mouse embryonic fibroblasts (MEFs) were grown as above with the addition of 2 mM L-glutamine (Glutamax; Life Technologies) as previously described (Song et al., 2006). DU145 human prostate carcinoma cells (Stone et al. 1978) were grown in the previously described medium in which newborn calf serum was substituted with fetal bovine serum. Human prostate cancer lines 22RV1 (Sramkoski et al. 1999) and LNCaP (Horoszewicz et al. 1983) and the YT NK-like lymphoblastic lymphoma (Yodoi et al. 1985) cell lines were cultivated in Rockwell Park Memorial Institute 1640 (RPMI-1640) culture medium, supplemented with 10% heat inactivated fetal bovine serum (FBS; Hyclone) and same three antibiotics as described above.
Drug screening assays
The cytotoxic effect of KTZ, CTZ, and complexes 1–12 was measured by using the differential nuclear staining (DNS) assay essentially as previously described (Lema et al. 2011). Cell lines plated on 96-well microplates were seeded at a density of 10,000 cells/200 μl media per well and incubated overnight to promote cell attachment. Subsequently, cells were incubated for a total of 20 h in the presence of the compounds. One hour prior to image capture in a live-cell mode, a mixture of Hoechst 33342 (Invitrogen, Eugene, OR) and Propidium iodide (PI; MP Biomedicals, Solon, OH) was added to a final concentration of 1 μg/ml each fluorophore. In this assay, Hoechst was utilized to label the total number of cells, while PI was used to obtain the number of dead cells. Dimethyl sulfoxide (DMSO), which was the compound diluent, was used as the solvent control at a final concentration of 0.5% v/v. Hydrogen peroxide (H2O2; 300 μM) was used as the positive control for cytotoxicity while untreated cells were included to estimate the background of dead cells generated by the plating process. The fluorescent signal of stained cells was captured in a series of microscopic images utilizing a Pathway 855 Bioimager system (BD Biosciences Rockville, MD) and data analysis was conducted as previously described (Lema et al. 2011). To obtain adequate numbers of regions of interest (ROIs; cells), montages (3×3) from nine contiguous image fields were captured per well utilizing a 20X objective. Every experimental point, as well as all controls, was evaluated in triplicate. The 50% cytotoxic concentration (CC50) values were determined as previously described (Varela-Ramirez et al. 2011). Briefly, the average percentage from triplicates of the two compound concentrations closest to the 50% cytotoxicity value was plotted against the compound concentration in a xy (scatter) chart function (Microsoft Excel). The best-fit regression line and its equation was used to calculate the concentration of chemical compound required to damage the plasma membrane by 50% of the cell population, as compared to solvent treated cells (DMSO). Standard deviations of the mean are not shown since the CC50 values were determined by linear extrapolation.
Apoptosis assays
After treatment of cells with the novel KTZ- and CTZ-ruthenium complexes, all subsequent operations were carried out on ice to slow down or arrest cell deterioration. Cells from each individual well were then collected and processed as previously described (Varela-Ramirez et al. 2011). Briefly, cells were simultaneously stained with annexin V-FITC and PI following the manufacture’s specification and immediately analyzed via flow cytometry using a Cytomics FC500 flow cytometer (Beckman Coulter, Miami, FL). Approximately, 10,000 events were acquired for each sample and the recorded data analyzed using CXP software (Beckman Coulter). In each experiment, the same controls used in the DNS assays were used which included apoptosis, solvent, and untreated controls. The total percentage of apoptosis was calculated by summing the total annexin V-FITC positive signal that included early and late apoptosis (Shaik et al. 2009).
Circular dichroism (CD) and thermal denaturation (Tm) experiments
For CD experiments, stock solutions (1.5 mM) of each complex were freshly prepared in water prior to use. An appropriate volume of those solutions was added to 3 ml samples of a freshly prepared solution of CT DNA (60 μM) in Tris/HCl buffer (5 mM Tris/HCl, 50 mM NaClO4, pH=7.39) to achieve molar ratios ranging from 0.5 to 2.0 drug/DNA. The samples were incubated at 37 °C for 20 h. All CD spectra of DNA and DNA-drug adducts were recorded at 25 °C over a 220–420 nm range and finally corrected with blank and noise reduction. The final data is expressed in molar ellipticity (millidegrees). Melting curves were recorded in 5 mM Tris/HCl, 50 mM NaClO4, pH 7.39. The absorbance at 260 nm was monitored for solutions of CT DNA (35 μM) before and after incubation with a solution of the drug under study (17.5 μM in Tris/HCl buffer) for periods of 3 and 20 h at room temperature. The temperature was increased by 0.5°C/min in the range 65–82 °C and by 3 °C/min in the ranges 25–65 °C and 82–97 °C.
Results and discussion
Ru-complexes are cytotoxic to cancer cell lines
The cytotoxicity of compounds 1–12 (chemical structures shown in Figure 2) was measured by differential nuclear staining (DNS) against three prostate tumor cell lines (DU145, 22Rv1, LNCaP), a cervical cancer (HeLa), a lymphoblastic lymphoma (YT NK, Natural Killer-like) and non-tumorigenic human cell lines (immortalized keratinocyte, HaCaT, breast epithelial MCF-10A, and MEFs). In order to ascertain the effectiveness of the metal-drug synergy, the activity of KTZ and CTZ, and of metal compounds with molecular structures similar to complexes 1–12 but lacking the organic drug, viz. Na[trans-RuCl4(DMSO)2] (C1), cis-[RuCl2(DMSO)4] (C2) and [Ru(p-cymene)Cl2]2 (C3) was also measured. The results of these assays are shown in Table 1. We note that both KTZ and CTZ display moderate cytotoxicity against all the cancer cells lines studied, while the control metal compounds C1–C3 not containing KTZ or CTZ were essentially inactive. The Ru-KTZ and Ru-CTZ complexes, in turn, display activity against all cancer cell lines, in some cases considerably higher than the uncomplexed KTZ or CTZ, validating our synergistic approach. Importantly, the new complexes display low to very low toxicity to non-cancerous HaCaT cells. In addition to the HaCaT cell line, the experimental compounds were also tested on the MCF-10A human breast epithelium cell line and mouse embryonic fibroblasts (MEFs). As shown on Table 1, half of the compounds tested (1, 3, 4, CTZ, 10–12) exhibited similar toxicity levels when tested on HaCaT and the MCF-10A cell line while only two compounds (1 and 12) had similar effects on the three control cell lines. Control compounds C1–C3, were not evaluated on the MCF-10A and MEF cell lines as these proved to have little if any inherent cytotoxicity
Table 1.
CC50 values, Relative Activity (R.A.) and Selectivity Index (S.I.) of Ru-KTZ and Ru-CTZ complexes.
| Cell Line | DU145 | 22Rv1 | LNCaP | YT | HeLa | HaCaT | MCF-10A | MEFs |
|---|---|---|---|---|---|---|---|---|
| C1 | † | 113.7* | 46.5 | † | † | † | N.D. | N.D. |
| C2 | † | † | 232.4 | 438.6 | † | † | N.D. | N.D. |
| C3 | 217.6 | 140.9 | 29 | 164.2 | † | † | N.D. | N.D. |
|
KTZ R.A./S.I.** |
28.6 1/>11 |
24.2 1/>13 |
27.9 1/>11 |
4.9 1/>64 |
66.7 1/>4 |
† | 37 | 31.3 |
|
1 R.A./S.I. |
† | 49.4 0.5/1.6 |
55.1 0.5/1.4 |
56.3 0.1/1.4 |
† | 76.8 | 62* | 55.8 |
|
2 R.A./S.I. |
8.1 3.5/>39 |
21.4 1.1/>15 |
4.3 6.6/>74 |
32.5 0.2/>10 |
47.8 1.4/>7 |
† | 6.7 | 5.2 |
|
3 R.A./S.I. |
74.4 0.4/0.8 |
7.4 3.3/8.3 |
36.4 0.8/1.7 |
8.1 0.6/7.7 |
61.4 1.1/1 |
61.6 | 66.4 | 25.3 |
|
4 R.A./S.I. |
† | 132.3 0.2/>2.4 |
7.4 3.8/>43 |
<4‡ >1.2/>79 |
† | † | † | 212.5 |
|
5 R.A./S.I. |
34.3 0.8/9.1 |
14.7 1.6/21.3 |
6.6 >3/>31 |
30.0 0.2/10.4 |
254.1 0.3/1.2 |
312.6†† | 171 | 39.2 |
|
6 R.A.-S.I |
94.8 0.3/1.2 |
128 0.2/0.9 |
6.8 >2.8/>11 |
30.6 0.2/3.7 |
33.3 2.0/3.4 |
113.1 | 12.3 | 37.3 |
|
CTZ R.A./S.I. |
43.6 1/2.2 |
19.2 1/5 |
15.4 1/6.2 |
16.7 1/5.7 |
94.3 1/1 |
95.3 | 79.4 | 35.3 |
|
7 R.A./S.I. |
† | † | 23.2 0.7/>14 |
12.7 1.3/>25 |
† | † | 31.6 | 31.8 |
|
8 R.A./S.I. |
† | 3.6 5.4/>87 |
2.4 6.3/>128 |
3.6 4.7/>87 |
38.2 2.5/>8 |
† | 19.5 | 4.8 |
|
9 R.A./S.I. |
† | 24.2 0.8/>13 |
5.0 3.1/>63 |
4.3 3.9/>72 |
† | † | 33.5 | 4.5 |
|
10 R.A./S.I. |
67.1 0.7/0.4 |
20.4 0.9/1.3 |
5.3 2.9/5 |
5.2 2.9/5 |
36.9 2.6/0.7 |
26.1 | 42.9 | 5.6 |
|
11 R.A./S.I. |
† | 36.1 0.5/>9 |
14.7 1.1/>21 |
11.2 1.5/>28 |
126.7 0.7/>2 |
† | † | 12.8 |
|
12 R.A./S.I. |
546.97 0.1/0.2 |
† | 25.38 0.6/3.8 |
42.18 0.4/2.3 |
359.3 0.3/0.3 |
97.1 | 79.1 | 64.7 |
Concentration of chemical compounds is in μM.
Experimental chemical compound with cytotoxicity below 25% at the maximum compound concentration tested.
N.D. Not determined
When this compound was tested at 4 μM the percentage of dead cells observed was 82.48
R.A.= CC50 of compound/CC50 of KTZ or CTZ; S.I.= CC50 in HaCat cells/CC50 in tumor cells
For those complexes where the toxicity on HaCaT could not be established, the selectivity indexes were calculated using the highest value reported for HaCaT toxicity (312.58 μM), corresponding to compound 5.
It is interesting to highlight some observations concerning the three prostate cancer cell lines employed in this study: First, our Ru-KTZ and Ru-CTZ complexes are more active on 22Rv1 and LNCaP than on DU145. DU145 cells are androgen receptor negative (Alimirah et al. 2006), while 22Rv1 cells are androgen receptor positive and weakly stimulated by dihydroxytestosterone (DHT), and LNCaP cells express high levels of androgen receptors and are highly sensitive to DHT (Horoszewicz et al. 1983). KTZ has been used as treatment for androgen-dependent prostate cancer (De et al. 1996) on the basis that its cytochrome 450 inhibition properties may result in inhibition of testicular and adrenal biosynthesis of androgen by competing with testosterone and dihydrotestosterone (DHT) in the binding of androgen receptors (Borlak and Thum, 2001; Thum and Borlak, 2000). The ability of CTZ to inhibit cytochrome P450, on the other hand, has been linked to the modulation of Ca2+ channels and consequently, to its cytostatic antitumor properties (Xiao et al. 1998).
The higher cytotoxicity observed for the Ru-KTZ and Ru-CTZ complexes to the androgen receptor positive lines might thus be related to: (i) the ability of Ru-KTZ compounds to act as androgen receptor antagonists, and/or (ii) changes in Ca2+ levels induced by the Ru-CTZ drugs, as reported in the case of apoptotic prostatic cell death of LNCaP (Gong et al. 1995). Interestingly, the same trend is not observed for the parent compounds KTZ or CTZ, indicating that new mechanisms of action are triggered when the azole molecules are linked to the metal fragment. As a result, some of the Ru compounds exhibit CC50 values in the low micromolar range, an enhanced activity relative to KTZ or CTZ, together with good selectivity in relation to non-cancerous HaCaT cells. For instance, compounds 2 and 8 are particularly active against the LNCaP line, over 6 times more active than the parental drugs KTZ or CTZ, in line with our metal-drug synergy hypothesis. They also display the highest selectivity indexes, more than 74 and 128 times less toxic to HaCaT than to LNCaP cells.
In the case of lymphoblastic lymphoma YT NK, we note that the Ru-KTZ complexes are less active than KTZ alone, while Ru-CTZ compounds show significantly higher activity than CTZ, with CC50 values in the low micromolar range. These results suggest that Ca2+ homeostasis could be related to the apoptotic process in some lymphomas, as previously described (Ito et al. 2002). For HeLa cells, complex 2 is again the most active of the Ru-KTZ series, and complex 8 is the best of the Ru-CTZ series in terms of relative activity and selectivity index. Other Ru complexes, including KP1019 analogues with a chemical structure similar to 8 have been reported to be active against HeLa cells with IC50 values in the micromolar range, comparable to our present data (Frasca et al. 1996; Qian et al. 2013)
Ru-complexes induce cytotoxicity via apoptosis
After a cytotoxic treatment, cells can undergo two main death pathways: necrosis or programmed cell death (apoptosis). Necrosis is typically initiated at the cell surface and is the consequence of a direct injury; it does not require metabolic energy and evolves through a traumatic cellular lysis. On the other hand, apoptosis is a much slower metabolic energy-dependent structured program that requires RNA and protein synthesis, leading to a series of concerted biochemical events that culminate in cellular suicide (Fink and Cookson, 2005; Kanduc et al. 2002). Both processes have different biochemical implications and, consequently, discerning the mode of death mediated by any drug is of evident relevance. In line with this, we have quantified the percentage of apoptotic cells after treatment with our Ru complexes by flow cytometry. The results obtained for KTZ, CTZ and the Ru complexes 1–12 on YT NK-like cells after 18 h exposure are shown in Figure 3. One of the significant biochemical features in the early stages of apoptosis is loss of plasma membrane phospholipid asymmetry because of translocation of phosphatidylserine (PS) from the cytoplasmic to the extracellular side. This characteristic allows detection of externalized PS by the specific binding of annexin V (FITC conjugated). Initiation of cell death will eventually result in the permeabilization of the cell membrane, allowing Propidium iodide (PI) to stain DNA within the nucleus. Cells identified as annexin V-FITC positive were considered as early apoptotic. Both annexin V-FITC and PI staining is typical at late stages of apoptosis, whereas cells that were stained only with PI due to the loss of plasma membrane integrity were considered necrotic cells (black bars in Figure 3). The total percentage of apoptotic cell populations is expressed as the sum of percentages of both early and late stages of apoptosis (white bars in Figure 3).
Figure 3.

Evaluation of cell death via flow cytometry. YT cells were exposed to the various Ru-based compounds for 18 hrs and stained with annexin V-FITC and PI. Cells treated with Ru-KTZ (A) or Ru-CTZ (B) complexes were compared to cells treated with uncomplexed KTZ and CTZ. Cells treated with the solvent control (0.3% v/v DMSO) and cells exposed to 600 μM Hydrogen peroxide (H2O2), were included as negative and positive cytotoxicity controls, respectively. Untreated (Unt) cells were also included to determine the viability of cells during the course of the experiments. Cells identified as annexin V-FITC positive were considered as apoptotic and the percentage of these cells is expressed as the sum of percentages of both early and late stages of apoptosis (white bars). Cells that were only stained with PI (but not with annexin V-FITC) due to the loss of plasma membrane integrity are considered necrotic cells (grey bars). The concentration (in μM), as well as the name of each experimental compound is shown below of the x-axis. Each bar represents the mean of three independent measurements with their respective standard deviations.
Our results show different trends when YT NK-like (YT) cells are treated with Ru-KTZ, Ru-CTZ complexes and their parent compounds, KTZ and CTZ. On one hand, cells treated with 15 and 10 μM concentrations of KTZ and CTZ, respectively, undergo death preferentially through apoptosis. This behavior agrees with previously reported results in which the treatment of primary bone marrow cell cultures from patients suffering of acute lymphoblastic leukemia with 10 μM clotrimazole depleted intracellular Ca2+ stores, provoking cytotoxicity via apoptosis, without affecting mature T lymphocytes and bone marrow stromal cells (Ito et al. 2002). Regarding the Ru-KTZ metal compounds, the presence of increasing concentrations (ranging 20–60 μM) of complexes 3, 4, and 5 induce YT cells to undergo cell death primarily by apoptosis, especially in the case of 4 (~52% apoptosis vs. ~8% necrosis) and 5 (~41% apoptosis vs. ~4% necrosis), in agreement with what was observed for other Ru compounds (Martinez et al. 2010; Strasberg et al. 2004). Complex 4 was significantly more active against YT (CC50 <4 μM) and the prostate cancer cell line LNCaP (CC50=7.37 μM) while 5 had similar activity to LNCaP (CC50=6.64 μM) and was more cytotoxic to the other prostate cancer cell lines used (CC50 between 16 and 34 μM; see Table 1). Interestingly, only two of the Ru-CTZ Complexes 8 and 11 elicited slightly (no more than two-fold) more apoptosis than necrosis while the rest of the CTZ-based compounds were more necrotic. It is important to note that CTZ-alone induced more (≥3 fold) apoptosis than any of the Ru-CTZ complexes. Taken together, our results indicate that Ru-KTZ complexes are more cytotoxic than the Ru-CTZ ones and that KTZ-based complexes act via distinct cytotoxic mechanisms (compare 4 and 6 for example, Fig. 3A).
Interaction of Ru-complexes with DNA
The cytotoxicity of cisplatin, a widely used heavy metal-based anti-tumor agent, results mainly from the formation of bifunctional DNA cross-links, especially between consecutive guanines (Jamieson and Lippard, 1999). This interaction induces important DNA structure modifications that trigger a cascade of biological processes, ultimately leading to inhibition of both DNA synthesis and replication, and resulting in death of cancer cells, primarily through apoptosis. It is also known that the anti-tumor activity of some ruthenium compounds like KP1019 (Kapitza et al. 2005) and [Ru(η6-arene)(X)(Y–Z)] (Aird et al. 2002; Dougan et al. 2006; Morris et al. 2001; Wang et al. 2005) is related to their interactions with DNA. In contrast, the anti-metastasic NAMI and the arene-Ru-CQ complexes reported by us show some ability to interact with DNA but their cytotoxic behavior appears to follow mechanisms different to DNA interactions. In view of the cytotoxic properties displayed by complexes 1–12, it was significant to determine whether they interact with DNA, and if so, whether that interaction contributes to the observed activity to a significant extent.
For this purpose, circular dichroism (CD) and thermal denaturation (Tm) experiments were carried out on selected complexes (Figure 4 and Table 2). The CD spectral technique is very sensitive for diagnosing changes in secondary structure of DNA resulting from drug-DNA interactions. A typical CD spectrum of Calf Thymus (CT) DNA shows a maximum at 275 nm due to the base stacking, and a minimum at 248 nm attributed to the right-handed helicity, characteristic of the B conformation (Figure 4) (Cutts et al. 1997). Thus, changes in CD signals can be assigned to corresponding changes in DNA structure. It is known that simple groove binding or electrostatic interaction of small molecules cause little or no alteration in any of the characteristic bands, whereas covalent interaction or intercalation between the base pairs will induce major perturbations in the CD spectrum. On the other hand, the thermal denaturation technique is an essential tool to detect the nature of drug-DNA interactions; destabilizing interactions (typically covalent) tend to decrease the thermal denaturation temperature of DNA, whereas a stabilizing interaction (electrostatic or intercalation) will increase it (Novakova et al. 2003)
Figure 4.

CD spectra of CT DNA and CT DNA incubated with 1.0, 1.5 and 2.0 equivalents of A) KTZ, B) CTZ and complexes C) 1, D) 5, E) 7 and F) 8. See Materials and Methods for assay details.
Table 2.
Change in the melting temperature (Tm) of DNA incubated with compounds†.
| Compound | ΔTm DNA (°C) |
|---|---|
| KTZ | NC |
| CTZ | NC |
| 1 | −1.1 |
| 2 | * |
| 3 | NC |
| 4 | ** |
| 5 | NM |
| 6 | NM |
| 7 | −2.3 |
| 8 | NC |
| 9 | NC |
| 10 | NC |
| 11 | −0.3 |
| 12 | * |
DNA was incubated with 0.5 equivalents of every compound for 3–24 hours in 5 mM Tris/HCl buffer at pH=7.39
Complex decomposes at high temperature
Tm cannot be measured because of band overlap
NC: no change in Tm beyond error is observed
NM: Tm non measured
CD spectra of CT DNA incubated for 20 h at 37 °C and pH 7.39 in Tris/HCl buffer with CTZ and KTZ up to ri 2.0 (molar ratio drug/DNA) show no modifications with respect to untreated DNA (Figure 4A and 4B), suggesting that none of the parent compounds interacts with CT DNA to a significant extent, or that the interaction does not induce any observable perturbation in the secondary structure. This is further supported by no changes being observed in the melting temperature (Tm) of CT DNA upon incubation with the azole molecules (Table 2). In contrast, the Ru-KTZ and Ru-CTZ complexes studied are able to modify the CD spectrum of CT DNA to a different extent, depending on the compound and the concentration. Complexes 1, 5 and 7 induce the largest alteration on the CT DNA secondary structure. A significant decrease in the intensity of the positive and/or the negative bands was observed, indicating a perturbation of the nucleobase pi-stacking and depletion of the B conformation, similar to the effect induced by cisplatin (Brabec et al. 1990). Furthermore, complexes 1 and 7 decrease Tm of CT DNA by 1.1 and 2.3 °C, respectively (Table 2), suggesting that the strong interaction observed by CD is destabilizing DNA.
The amount of Ru covalently bound to DNA, determined by the Burton assay after 48 h of incubation with complex 7 was 585 nmol/mg DNA, which confirms that the changes observed in the CD spectra and Tm experiments are the result of a strong covalent interaction of the metal with the nucleobases of DNA (Anzelloti, 2004). It is reasonable to hypothesize that the similar effect observed for complex 1 in CD spectral and Tm experiments is a consequence of the same type of interaction, given the structural analogy between both complexes. In the case of complex 5, the presence of ethylenediamine ligand and the overall positive charge likely allow this compound to form hydrogen bonds with the bases of DNA and establish electrostatic interactions with the phosphate backbone. The weak interaction observed for the other compounds (data not shown), reminiscent of that described for NAMI (Gallori et al. 2000), is likely due to electrostatic attraction/repulsion or minor groove binding, as previously reported for Pt-CTZ complexes (Navarro et al. 2009). This is further supported by results of the Burton Assay on complexes 8 and 9 (Anzelloti, 2004), revealing 87 and 15 nmol/mg DNA of drug covalently bound to DNA after 48 h of incubation, respectively, much lower than was measured for complex 7. Importantly, our results show no correlation between the ability to interact with CT DNA and anti-tumor activity; complex 8 shows the best cytotoxic profile on 22Rv1, LNCaP and YT NK, but induces only slight changes in both the CD spectra (Figure 4F) and the Tm of CT DNA, whereas complexes 1, 5, and 7 display a remarkably poor anti-tumor activity (Table 1) and strong interactions with DNA. This suggests that the enhancement in the anti-tumor properties of KTZ and CTZ when they are attached to ruthenium in a molecular complex is due to mechanisms/factors other than DNA interactions, as has been previously observed for other ruthenium complexes (Sanna et al. 2002). Importantly, the toxicity to non-tumorigenic cells (HaCaT) is not related to the ability of the complexes to interact with DNA, which could have important implications in drug safety.
Conclusions
We report the activity of a series of twelve Ru-KTZ and Ru-CTZ compounds against three prostate tumor cell lines with different androgen sensitivity, as well as cervical cancer and lymphoblastic lymphoma cell lines. A human epidermal cell line was used to evaluate the toxicity against non-transformed cells and to establish selectivity indexes. Our results show that the combination of ruthenium and KTZ/CTZ in a single molecule resulted in some of the complexes exhibiting more cytotoxic than any of the individual components separately. Additionally, some of the compounds are more cytotoxic against cancer cell lines with lower activity against non-transformed cell lines. Studies of DNA interactions reveal that neither the antitumor properties, nor the toxicity against non-tumorigenic human cells for any of the metal compounds are related to their ability to interact with DNA. Some of the complexes display promising cytotoxic profiles among the different cell lines tested, including high activity accompanied by high selectivity indexes and cell death preferentially induced through apoptosis.
Acknowledgments
We thank to Dr. Chuanshu Huang at New York University School of Medicine for gift of MEFs. We also thank the Biomolecule Analysis and the Cytometry, Screening and Imaging Core Facilities at the University of Texas at El Paso, supported by RCMI program Grant No. 8G12MD007592, to the Border Biomedical Research Center, from the NIMHD-NIH. We acknowledge support from the NIGMS SCORE Grants 1SC3GM103713-01 to RJA and 1SC1GM089558 to RAS-D. ERE was supported by the RISE Scholars Program at UTEP through NIGMS Grant No. R25GM069621-10.
References
- Aird RE, Cummings J, Ritchie AA, Muir M, Morris RE, Chen H, et al. In vitro and in vivo activity and cross resistance profiles of novel ruthenium (II) organometallic arene complexes in human ovarian cancer. Br J Cancer. 2002;86(10):1652–7. doi: 10.1038/sj.bjc.6600290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzelloti A. M Sc Thesis. Venezuelan Institute for Scientific Research (IVIC); Caracas, Venezuela: 2004. Study of Possible Mechanisms of Action of Ruthenium-azole Complexes against Trypanosoma cruzi. [Google Scholar]
- Bergamo A, Gagliardi R, Scarcia V, Furlani A, Alessio E, Mestroni G, et al. In vitro cell cycle arrest, in vivo action on solid metastasizing tumors, and host toxicity of the antimetastatic drug NAMI-A and cisplatin. J Pharmacol Exp Ther. 1999;289(1):559–64. [PubMed] [Google Scholar]
- Berger MR, Garzon FT, Keppler BK, Schmahl D. Efficacy of new ruthenium complexes against chemically induced autochthonous colorectal carcinoma in rats. Anticancer Res. 1989;9(3):761–5. [PubMed] [Google Scholar]
- Bok RA, Small EJ. The treatment of advanced prostate cancer with ketoconazole: safety issues. Drug Saf. 1999;20(5):451–8. doi: 10.2165/00002018-199920050-00005. [DOI] [PubMed] [Google Scholar]
- Borlak J, Thum T. Induction of nuclear transcription factors, cytochrome P450 monooxygenases, and glutathione S-transferase alpha gene expression in Aroclor 1254-treated rat hepatocyte cultures. Biochem Pharmacol. 2001;61(2):145–53. doi: 10.1016/s0006-2952(00)00537-2. [DOI] [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988:761–71. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabec V, Kleinwachter V, Butour JL, Johnson NP. Biophysical studies of the modification of DNA by antitumour platinum coordination complexes. Biophys Chem. 1990;35(2–3):129–41. doi: 10.1016/0301-4622(90)80003-p. [DOI] [PubMed] [Google Scholar]
- Bratsos I, Jedner S, Gianferrara T, Alessio E. Ruthenium Anticancer Compounds:Challenges and Expectations. Chimia. 2007;61:692–7. [Google Scholar]
- Bratsos I, Serli B, Zangrando E, Katsaros N, Alessio E. Replacement of chlorides with dicarboxylate ligands in anticancer active Ru(II)-DMSO compounds: a new strategy that might lead to improved activity. Inorg Chem. 2007;46(3):975–92. doi: 10.1021/ic0613964. [DOI] [PubMed] [Google Scholar]
- Cao MY, Lee Y, Feng NP, Al-Qawasmeh RA, Viau S, Gu XP, et al. NC381, a novel anticancer agent, arrests the cell cycle in G0–G1 and inhibits lung tumor cell growth in vitro and in vivo. J Pharmacol Exp Ther. 2004;308(2):538–46. doi: 10.1124/jpet.103.059618. [DOI] [PubMed] [Google Scholar]
- Castonguay A, Doucet C, Juhas M, Maysinger D. New ruthenium(II)-letrozole complexes as anticancer therapeutics. J Med Chem. 2012;55(20):8799–806. doi: 10.1021/jm301103y. [DOI] [PubMed] [Google Scholar]
- Cutts SM, Masta A, Panousis C, Parsons PG, Sturm RA, Phillips DR. A gel mobility shift assay for probing the effect of drug-DNA adducts on DNA-binding proteins. Methods Mol Biol. 1997:95–106. doi: 10.1385/0-89603-447-X:95. [DOI] [PubMed] [Google Scholar]
- De CR, Wouters W, Bruynseels J. P450-dependent enzymes as targets for prostate cancer therapy. J Steroid Biochem Mol Biol. 1996;56:133–43. doi: 10.1016/0960-0760(95)00230-8. [DOI] [PubMed] [Google Scholar]
- Dougan SJ, Melchart M, Habtemariam A, Parsons S, Sadler PJ. Phenylazo-pyridine and phenylazo-pyrazole chlorido ruthenium(II) arene complexes: arene loss, aquation, and cancer cell cytotoxicity. Inorg Chem. 2006;45 (26):10882–94. doi: 10.1021/ic061460h. [DOI] [PubMed] [Google Scholar]
- Duret C, Daujat-Chavanieu M, Pascussi JM, Pichard-Garcia L, Balaguer P, Fabre JM, et al. Ketoconazole and miconazole are antagonists of the human glucocorticoid receptor: consequences on the expression and function of the constitutive androstane receptor and the pregnane X receptor. Mol Pharmacol. 2006;70(1):329–39. doi: 10.1124/mol.105.022046. [DOI] [PubMed] [Google Scholar]
- Eichenberger T, Trachtenberg J, Chronis P, Keating A. Synergistic effect of ketoconazole and antineoplastic agents on hormone-independent prostatic cancer cells. Clin Invest Med. 1989:363–6. [PubMed] [Google Scholar]
- Eichenberger T, Trachtenberg J, Toor P, Keating A. Ketoconazole: a possible direct cytotoxic effect on prostate carcinoma cells. J Urol. 1989:190–1. doi: 10.1016/s0022-5347(17)40639-2. [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasca D, Ciampa J, Emerson J, Umans RS, Clarke MJ. Effects of hypoxia and transferrin on toxicity and DNA binding of ruthenium antitumor agents in hela cells. Met Based Drugs. 1996;3(4):197–209. doi: 10.1155/MBD.1996.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallori E, Vettori C, Alessio E, Vilchez FG, Vilaplana R, Orioli P, et al. DNA as a possible target for antitumor ruthenium(III) complexes. Arch Biochem Biophys. 2000;376(1):156–62. doi: 10.1006/abbi.1999.1654. [DOI] [PubMed] [Google Scholar]
- Garzon FT, Berger MR, Keppler BK, Schmahl D. Comparative antitumor activity of ruthenium derivatives with 5′-deoxy-5-fluorouridine in chemically induced colorectal tumors in SD rats. Cancer Chemother Pharmacol. 1987;19(4):347–9. doi: 10.1007/BF00261487. [DOI] [PubMed] [Google Scholar]
- Gong Y, Blok LJ, Perry JE, Lindzey JK, Tindall DJ. Calcium regulation of androgen receptor expression in the human prostate cancer cell line LNCaP. Endocrinology. 1995;136(5):2172–8. doi: 10.1210/endo.136.5.7720667. [DOI] [PubMed] [Google Scholar]
- Hartinger CG, Jakupec MA, Zorbas-Seifried S, Groessl M, Egger A, Berger W, et al. KP1019, a new redox-active anticancer agent--preclinical development and results of a clinical phase I study in tumor patients. Chem Biodivers. 2008;5(10):2140–55. doi: 10.1002/cbdv.200890195. [DOI] [PubMed] [Google Scholar]
- Hegemann L, Toso SM, Lahijani KI, Webster GF, Uitto J. Direct interaction of antifungal azole-derivatives with calmodulin: a possible mechanism for their therapeutic activity. J Invest Dermatol. 1993;100(3):343–6. doi: 10.1111/1523-1747.ep12470043. [DOI] [PubMed] [Google Scholar]
- Ho YS, Tsai PW, Yu CF, Liu HL, Chen RJ, Lin JK. Ketoconazole-induced apoptosis through P53-dependent pathway in human colorectal and hepatocellular carcinoma cell lines. Toxicol Appl Pharmacol. 1998:39–47. doi: 10.1006/taap.1998.8467. [DOI] [PubMed] [Google Scholar]
- Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, et al. LNCaP model of human prostatic carcinoma. Cancer Res. 1983:1809–18. [PubMed] [Google Scholar]
- Huang HL, Li ZZ, Liang ZH, Liu YJ. Cell Cycle Arrest, Cytotoxicity, Apoptosis, DNA-Binding, Photocleavage, and Antioxidant Activity of Octahedral Ruthenium(II) Complexes. Eur J Inorg Chem. 2011:5538–5547. [Google Scholar]
- Iniguez E, Sanchez A, Vasquez M, Martinez A, Olivas J, Sattler A, et al. Metal-drug synergy: new ruthenium(II) complexes of ketoconazole are highly active against Leishmania major and Trypanosoma cruzi and nontoxic to human or murine normal cells. J Biol Inorg Chem. 2013;18(7):779–90. doi: 10.1007/s00775-013-1024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito C, Tecchio C, Coustan-Smith E, Suzuki T, Behm FG, Raimondi SC, et al. The antifungal antibiotic clotrimazole alters calcium homeostasis of leukemic lymphoblasts and induces apoptosis. Leukemia. 2002:1344–52. doi: 10.1038/sj.leu.2402510. [DOI] [PubMed] [Google Scholar]
- Jakupec MA, Galanski M, Arion VB, Hartinger CG, Keppler BK. Antitumour metal compounds: more than theme and variations. Dalton Trans. 2008;(2):183–94. doi: 10.1039/b712656p. [DOI] [PubMed] [Google Scholar]
- Jamieson ER, Lippard SJ. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem Rev. 1999;99(9):2467–98. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- Kanduc D, Mittelman A, Serpico R, Sinigaglia E, Sinha AA, Natale C, et al. Cell death: apoptosis versus necrosis (review) Int J Oncol. 2002;21(1):165–70. [PubMed] [Google Scholar]
- Kapitza S, Pongratz M, Jakupec MA, Heffeter P, Berger W, Lackinger L, et al. Heterocyclic complexes of ruthenium(III) induce apoptosis in colorectal carcinoma cells. J Cancer Res Clin Oncol. 2005;131(2):101–10. doi: 10.1007/s00432-004-0617-0. [DOI] [PubMed] [Google Scholar]
- Kljun J, Bratsos I, Alessio E, Psomas G, Repnik U, Butinar M, Turk B, Turel I. New uses for old drugs: attempts to convert quinolone antibacterials into potential anticancer agents containing ruthenium. Inorg Chem. 2013;52(15):9039–52. doi: 10.1021/ic401220x. [DOI] [PubMed] [Google Scholar]
- Lema C, Varela-Ramirez A, Aguilera RJ. Differential nuclear staining assay for high-throughput screening to identify cytotoxic compounds. J Current Cellular Biochemistry. 2011;1(1):1–14. [PMC free article] [PubMed] [Google Scholar]
- Lippert BE. Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug. Verlag Helvetica Chimica Acta. 2006 [Google Scholar]
- Lorch JH, Goloubeva O, Haddad RI, Cullen K, Sarlis N, Tishler R, et al. Induction chemotherapy with cisplatin and fluorouracil alone or in combination with docetaxel in locally advanced squamous-cell cancer of the head and neck: long-term results of the TAX 324 randomised phase 3 trial. Lancet Oncol. 2011:153–9. doi: 10.1016/S1470-2045(10)70279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac NS, Dawson RA, Crocker G, Tucker WF, Bittiner B, Singleton JG, et al. Antiproliferative effects on keratinocytes of a range of clinically used drugs with calmodulin antagonist activity. Br J Dermatol. 1993:143–50. doi: 10.1111/j.1365-2133.1993.tb15143.x. [DOI] [PubMed] [Google Scholar]
- Martinez A, Carreon T, Iniguez E, Anzellotti A, Sanchez A, Tyan M, et al. Searching for new chemotherapies for tropical diseases: ruthenium-clotrimazole complexes display high in vitro activity against Leishmania major and Trypanosoma cruzi and low toxicity toward normal mammalian cells. J Med Chem. 2012:3867–77. doi: 10.1021/jm300070h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Rajapakse CS, Sanchez-Delgado RA, Varela-Ramirez A, Lema C, Aguilera RJ. Arene-Ru(II)-chloroquine complexes interact with DNA, induce apoptosis on human lymphoid cell lines and display low toxicity to normal mammalian cells. J Inorg Biochem. 2010;104(9):967–77. doi: 10.1016/j.jinorgbio.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morbidelli L, Donnini S, Filippi S, Messori L, Piccioli F, Orioli P, et al. Antiangiogenic properties of selected ruthenium(III) complexes that are nitric oxide scavengers. Br J Cancer. 2003;88(9):1484–91. doi: 10.1038/sj.bjc.6600906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RE, Aird RE, Murdoch PS, Chen H, Cummings J, Hughes ND, et al. Inhibition of cancer cell growth by ruthenium(II) arene complexes. J Med Chem. 2001;44(22):3616–21. doi: 10.1021/jm010051m. [DOI] [PubMed] [Google Scholar]
- Navarro M, Highera-Padilla AR, Arsenak M, Taylor P. Synthesis, characterization, DNA interaction studies and anticancer activity of platinum–clotrimazole complexes. Trans Met Chem. 2009;34:869–875. [Google Scholar]
- Navarro M, Pena NP, Colmenares I, Gonzalez T, Arsenak M, Taylor P. Synthesis and characterization of new palladium-clotrimazole and palladium-chloroquine complexes showing cytotoxicity for tumor cell lines in vitro. J Inorg Biochem. 2006;100(1):152–7. doi: 10.1016/j.jinorgbio.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Novakova O, Chen H, Vrana O, Rodger A, Sadler PJ, Brabec V. DNA interactions of monofunctional organometallic ruthenium(II) antitumor complexes in cell-free media. Biochemistry. 2003;42(39):11544–54. doi: 10.1021/bi034933u. [DOI] [PubMed] [Google Scholar]
- Penso J, Beitner R. Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells. Eur J Pharmacol. 1998;342(1):113–7. doi: 10.1016/s0014-2999(97)01507-0. [DOI] [PubMed] [Google Scholar]
- Posner MR, Hershock DM, Blajman CR, Mickiewicz E, Winquist E, Gorbounova V, et al. Cisplatin and fluorouracil alone or with docetaxel in head and neck cancer. N Engl J Med. 2007:1705–15. doi: 10.1056/NEJMoa070956. [DOI] [PubMed] [Google Scholar]
- Qian C, Wang JQ, Song CL, Wang LL, Ji LN, Chao H. The induction of mitochondria-mediated apoptosis in cancer cells by ruthenium(ii) asymmetric complexes. Metallomics. 2013 Jun 27;:844–54. doi: 10.1039/c3mt20270d. [DOI] [PubMed] [Google Scholar]
- Rademaker-Lakhai JM, van den Bongard D, Pluim D, Beijnen JH, Schellens JH. A Phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin Cancer Res. 2004;10(11):3717–27. doi: 10.1158/1078-0432.CCR-03-0746. [DOI] [PubMed] [Google Scholar]
- Rajapakse CS, Martinez A, Naoulou B, Jarzecki AA, Suarez L, Deregnaucourt C, et al. Synthesis, characterization, and in vitro antimalarial and antitumor activity of new ruthenium(II) complexes of chloroquine. Inorg Chem. 2009;48(3):1122–31. doi: 10.1021/ic802220w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Delgado RA, Anzelloti A, Suarez L. Metal ions and their complexes in medication. In: Siegel H, Siegel A, editors. Metal Ions in Biological Systems. Marcel Dekker; New York: 2004. p. 379. [PubMed] [Google Scholar]
- Sanna B, Debidda M, Pintus G, Tadolini B, Posadino AM, Bennardini F, et al. The anti-metastatic agent imidazolium trans-imidazoledimethylsulfoxide-tetrachlororuthenate induces endothelial cell apoptosis by inhibiting the mitogen-activated protein kinase/extracellular signal-regulated kinase signaling pathway. Arch Biochem Biophys. 2002;403(2):209–18. doi: 10.1016/s0003-9861(02)00218-7. [DOI] [PubMed] [Google Scholar]
- Sava G, Bergamo A, Zorzet S, Gava B, Casarsa C, Cocchietto M, et al. Influence of chemical stability on the activity of the antimetastasis ruthenium compound NAMI-A. Eur J Cancer. 2002;38(3):427–35. doi: 10.1016/s0959-8049(01)00389-6. [DOI] [PubMed] [Google Scholar]
- Sava G, Capozzi I, Clerici K, Gagliardi G, Alessio E, Mestroni G. Pharmacological control of lung metastases of solid tumours by a novel ruthenium complex. Clin Exp Metastasis. 1998;16(4):371–9. doi: 10.1023/a:1006521715400. [DOI] [PubMed] [Google Scholar]
- Scolaro C, Bergamo A, Brescacin L, Delfino R, Cocchietto M, Laurenczy G, et al. In vitro and in vivo evaluation of ruthenium(II)-arene PTA complexes. J Med Chem. 2005;48(12):4161–71. doi: 10.1021/jm050015d. [DOI] [PubMed] [Google Scholar]
- Seelig MH, Berger MR, Keppler BK. Antineoplastic activity of three ruthenium derivatives against chemically induced colorectal carcinoma in rats. J Cancer Res Clin Oncol. 1992;118(3):195–200. doi: 10.1007/BF01410134. [DOI] [PubMed] [Google Scholar]
- Shaik N, Martinez A, Augustin I, Giovinazzo H, Varela-Ramirez A, Sanau M, et al. Synthesis of apoptosis-inducing iminophosphorane organogold(III) complexes and study of their interactions with biomolecular targets. Inorg Chem. 2009;48(4):1577–87. doi: 10.1021/ic801925k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Li J, Zhang D, Liu ZG, Ye J, Zhan Q, Shen HM, Whiteman M, Huang C. IKKbeta programs to turn on the GADD45alpha-MKK4-JNK apoptotic cascade specifically via p50 NF-kappaB in arsenite response. J Cell Biol. 2006;175:607–17. doi: 10.1083/jcb.200602149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soule HD, McGrath CM. A simplified method for passage and long-term growth of human mammary epithelial cells. In Vitro Cell Dev Biol. 1986;22:6–12. doi: 10.1007/BF02623435. [DOI] [PubMed] [Google Scholar]
- Sramkoski RM, Pretlow TG, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, et al. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev Biol Anim. 1999:403–9. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- Stone KR, Mickey DD, Wunderli H, Mickey GH, Paulson DF. Isolation of a human prostate carcinoma cell line (DU 145) Int J Cancer. 1978:274–81. doi: 10.1002/ijc.2910210305. [DOI] [PubMed] [Google Scholar]
- Strasberg RM, Anzellotti A, Sanchez-Delgado RA, Rieber M. Tumor apoptosis induced by ruthenium(II)-ketoconazole is enhanced in nonsusceptible carcinoma by monoclonal antibody to EGF receptor. Int J Cancer. 2004;112(3):376–84. doi: 10.1002/ijc.20415. [DOI] [PubMed] [Google Scholar]
- Sweetman SCEd. Martindale: The Complete Drug Reference. 37. Pharmaceutical Press; London: 2010. [Google Scholar]
- Thum T, Borlak J. Cytochrome P450 mono-oxygenase gene expression and protein activity in cultures of adult cardiomyocytes of the rat. Br J Pharmacol. 2000;130(8):1745–52. doi: 10.1038/sj.bjp.0703465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Veldhuizen PJ, Reed G, Aggarwal A, Baranda J, Zulfiqar M, Williamson S. Docetaxel and ketoconazole in advanced hormone-refractory prostate carcinoma: a phase I and pharmacokinetic study. Cancer. 2003;98(9):1855–62. doi: 10.1002/cncr.11733. [DOI] [PubMed] [Google Scholar]
- Van CE, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol. 2006:4991–7. doi: 10.1200/JCO.2006.06.8429. [DOI] [PubMed] [Google Scholar]
- Varela-Ramirez A, Costanzo M, Carrasco YP, Pannell KH, Aguilera RJ. Cytotoxic effects of two organotin compounds and their mode of inflicting cell death on four mammalian cancer cells. Cell Biol Toxicol. 2011;27(3):159–68. doi: 10.1007/s10565-010-9178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Bella J, Parkinson JA, Sadler PJ. Competitive reactions of a ruthenium arene anticancer complex with histidine, cytochrome c and an oligonucleotide. J Biol Inorg Chem. 2005;10(2):147–55. doi: 10.1007/s00775-004-0621-5. [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci U S A. 2000:8151–6. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao YF, Huang L, Morgan JP. Cytochrome P450: a novel system modulating Ca2+ channels and contraction in mammalian heart cells. J Physiol. 1998:777–92. doi: 10.1111/j.1469-7793.1998.777bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie YY, Huang HL, Yao JH, Lin GJ, Jiang GB, Liu YJ. DNA-binding, photocleavage, cytotoxicity in vitro, apoptosis and cell cycle arrest studies of symmetric ruthenium(II) complexes. Eur J Med Chem. 2013;63:603–610. doi: 10.1016/j.ejmech.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Yodoi J, Teshigawara K, Nikaido T, Fukui K, Noma T, Honjo T, et al. TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells) J Immunol. 1985:1623–30. [PubMed] [Google Scholar]