

Abstract
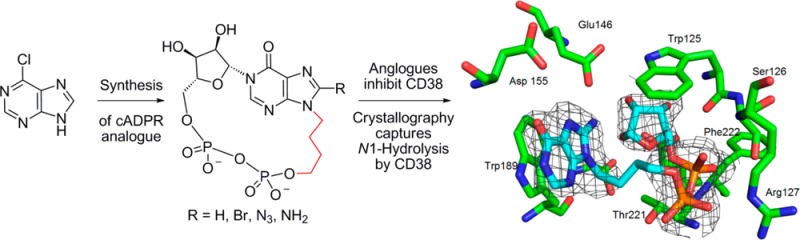
Cyclic adenosine 5′-diphosphate ribose (cADPR) analogs based on the cyclic inosine 5′-diphosphate ribose (cIDPR) template were synthesized by recently developed stereo- and regioselective N1-ribosylation. Replacing the base N9-ribose with a butyl chain generates inhibitors of cADPR hydrolysis by the human ADP-ribosyl cyclase CD38 catalytic domain (shCD38), illustrating the nonessential nature of the “southern” ribose for binding. Butyl substitution generally improves potency relative to the parent cIDPRs, and 8-amino-N9-butyl-cIDPR is comparable to the best noncovalent CD38 inhibitors to date (IC50 = 3.3 μM). Crystallographic analysis of the shCD38:8-amino-N9-butyl-cIDPR complex to a 2.05 Å resolution unexpectedly reveals an N1-hydrolyzed ligand in the active site, suggesting that it is the N6-imino form of cADPR that is hydrolyzed by CD38. While HPLC studies confirm ligand cleavage at very high protein concentrations, they indicate that hydrolysis does not occur under physiological concentrations. Taken together, these analogs confirm that the “northern” ribose is critical for CD38 activity and inhibition, provide new insight into the mechanism of cADPR hydrolysis by CD38, and may aid future inhibitor design.
Introduction
Cyclic adenosine 5′-diphosphate ribose (cADPR, 1, Figure 1)1,2 is synthesized in biological systems from nicotinamide adenine dinucleotide (NAD+) by ADP-ribosyl cyclases (ADPRCs). It acts as a second messenger, mobilizing intracellular calcium.3−6
Figure 1.
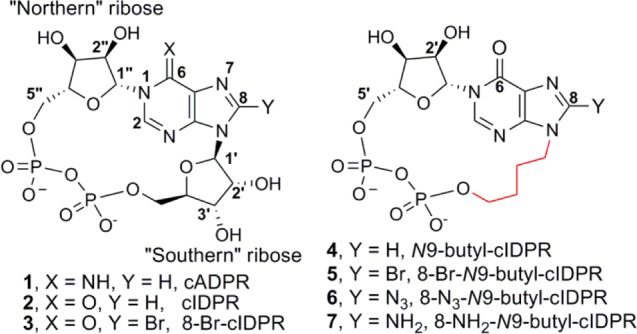
Structure of cADPR and N9-butyl analogs.
In humans, formation of cADPR is catalyzed by the multifunctional transmembrane glycoprotein ADPRC CD38.7 CD38 also acts as an NAD+ glycohydrolase (NADase) and as a cADPR hydrolase to generate adenosine 5′-diphosphate ribose (ADPR), another Ca2+-releasing second messenger.8−10 Under acidic conditions, CD38 generates the most potent Ca2+-releasing second messenger known to date, nicotinic acid adenine dinucleotide phosphate (NAADP) from nicotinamide adenine dinucleotide phosphate (NADP).11
CD38 knockout studies have revealed the importance of this pathway in a range of diseases. CD38 is a marker in AIDS progression12 and a negative prognostic marker of chronic lymphocytic leukemia.13 It acts to regulate intracellular levels of NAD+, being implicated in energy homeostasis, signal transduction, and aging,14−16 and recently has been shown to be critical for social behavior in mice.17 The emerging role of CD38 in disease states is stimulating the search for modulators of activity for chemical biological studies and to provide structural clues for drug design and potential therapeutic candidates.18 To date, CD38 inhibitors fall broadly into two categories: mechanism-based covalent inhibitors that bind to the catalytic residue, and reversible, competitive, noncovalent inhibitors. Most are derived from NAD+ and designed as inhibitors of the predominant NADase activity of CD38. Nicotinamide-based derivatives have demonstrated nanomolar inhibition of NADase activity by covalent modification of CD38 19 and low millimolar activity when designed as membrane permeable analogs.20 Competitive inhibitors are more diverse in structure, with a nonhydrolyzable NAD+ analog reported (IC50 ≈ 150 μM),21 flavonoids showing low micromolar inhibition22 and the development of a hit from commercial libraries generating the most active reported noncovalent inhibitor to date (IC50 = 4.7 μM).23
As both cADPR and ADPR are derived from a common intermediate,24 we chose to design product-like inhibitors based on the cADPR structure to exploit inhibition of CD38 cADPR hydrolase activity. cADPR itself is unattractive for inhibitor design, since it is hydrolyzed at the N1 link in both neutral aqueous solution and under physiological conditions.25−27 More stable analogs have been accessed by one of two routes: either a chemoenzymatic route modeled on the biosynthesis of cADPR from NAD+ or by total chemical synthesis. We have previously reported a chemoenzymatic route to N1-cyclic inosine 5′-diphosphate ribose (cIDPR, 2) via its 8-bromo derivative (8-Br-cIDPR, 3, Figure 1).28 Chemically and biologically stable, 2 and 3 both inhibit cADPR hydrolysis by the catalytic domain of CD38 (shCD38; IC50 of 276 and 158 μM, respectively). Furthermore, 2 acts as an agonist for Ca2+-release with almost equivalent potency to cADPR in permeabilized T-cells and 3 is a membrane permeant agonist.29,30
Until recently, total synthetic approaches have required modification of the “northern” ribose to generate a more stable N1-link by introduction of a carbocyclic “northern” ribose,31−33 replacement of the “northern” ribose, or both ribose sugars, by an alkyl or ether bridge34,35 or by attaching the “northern” ribose to the base at C-2″.36 We recently reported the use of modified Vorbrüggen conditions to effect stereo- and regiospecific introduction of an acetylated ribose at the N1-position of a protected inosine and demonstrated the utility of this method in the first total synthesis of 3.37 This paves the way for further exploration of the structure–activity relationship (SAR) of cADPR, using the cIDPR template, with the opportunity to retain an intact “northern” ribose and without the structural limitations of using enzymatic cyclization.38 We report here the synthesis of the first analogs in which the “southern” ribose is selectively replaced, the activity of these analogs as inhibitors of cADPR hydrolysis by shCD38, crystallography of the most potent inhibitor with shCD38, and HPLC studies to examine the ability of shCD38 to hydrolyze the cIDPR scaffold.
Results and Discussion
Crystallography of shCD38 has revealed the mechanism by which NAD+ is either cyclized to cADPR or hydrolyzed to ADPR.39−41 Crystal structures obtained with shCD38 and unnatural ligands 2 (PDB code 2PGJ),42 cyclic adenosine 5′-diphosphate carbocyclic ribose (cADPcR, 3UHI),43 and 8-NH2-cIDPR (3U4H) suggest a critical role for the base and “northern” ribose in the binding of cADPR analogs to CD38, as might be predicted for the locus of both cADPR formation and degradation. Glu146 hydrogen-bonds to N6 and N7 and is critical in regulating the ADPRC multifunctionality.44 Glu226 is the catalytic residue and interacts with the 2″- and 3″-OH of the “northern” ribose.45 It has recently been highlighted as crucial in orientating NAD+ for cleavage of nicotinamide.46 The “southern” ribose appears to be accommodated in a more flexible fashion across the open face of the pocket. Thus, we designed analogs in which the “southern” ribose is replaced with an N9-butyl linker (N9-butyl-cIDPRs 4–7, Figure 1) to explore whether it is required for binding to CD38. Such analogs would be predicted to have the further advantage that the acid sensitivity of the N9-ribosyl link is eliminated.47
Molecular Modeling
To predict the binding mode of analogs 4–7, which might be more flexible because of the linear N9-alkyl chain, they were first docked into the 2PGJ crystal structure of shCD38 in complex with cIDPR. The docked and minimized pose was almost identical to that of cIDPR (Figure 2A), suggesting that these analogs would mimic the critical interactions with the binding site. Minimization of the protein resulted in very little movement of the amino acid residues forming the binding site.
Figure 2.
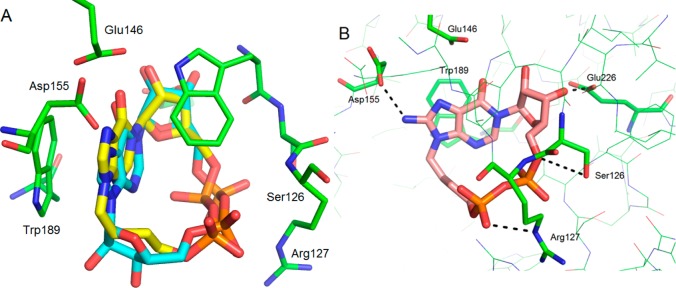
N9-Butyl-cIDPR docked into 2PGJ crystal structure. Comparison between 2PGJ crystal structure of N1-cIDPR (carbons in cyan) bound to shCD38 and (A) docked N9-butyl cIDPR 4 (carbons in yellow) showing overlap of their structures in the binding pocket; (B) interaction of 8-NH2-N9-butyl cIDPR 7 (carbons in pink) with critical residues in the binding pocket and additional predicted H-bond to Asp155.
The docked ligands display face-to-face stacking between the hypoxanthine base and Trp189. There is one predicted hydrogen bond from the ribose 3′-OH a hydroxyl group to the catalytic acid (Glu226), although the 2′-OH does not appear to be as closely located, and from the side chains of Ser126 and Arg127 to the phosphates. The 8-amino group of 7 is predicted to form an additional hydrogen bond to the side chain of Asp155. The N9-butyl chain of the docked ligand lies across the entrance of the binding pocket in an almost identical position to the “southern” ribose of cIDPR (Figure 2B).
Synthesis of N9-Butyl-cIDPR Analogs 4–7
6-Chloropurine (11, Scheme 1) was alkylated with 4-chlorobutylacetate in the presence of 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU).48 The desired major product, the N9-isomer 12, was obtained in high yield after separation from the minor N7-isomer. Treatment of 12 with methanolic ammonia at 80 °C efficiently effected both amination at the 6-position and deprotection of the N9-butyl acetyl ester to afford N9-hydroxybutyladenine 13.49 The alkyl group was then reprotected as the TBDPS ether using tert-butyldiphenylsilyl chloride (TBDPS-Cl) and imizadole to give 14. Introduction of the 8-bromo substituent to 14 is crucial at this stage to promote the subsequent N1-ribosylation.37 Attempted bromination of 14 using a solution of bromine in sodium hydrogen phosphate buffer and dioxane was not successful, and no reaction was observed. Similarly, treatment of 14 with N-bromosuccinimide or N-bromoacetamide afforded only starting material. Applying the conditions developed by Laufer et al.50 for the 8-bromination of N9-methyl chloropurines, we first deprotonated 14 using lithium diisopropylamide (LDA), followed by addition of 1,2-dibromotetrachloroethane.51 On a small scale (100 mg) the desired 8-brominated product 15 was isolated in 67% yield. However, when this reaction was scaled up (2.5 g), an inseparable mixture of two products was obtained after column chromatography. Mass spectrometry (m/z = 480.1970 and 524.1476) containing characteristic 35/37Cl and 79/81Br isotope patterns confirmed this was both the 8-bromo and 8-chloro substituted products. It was only possible to recover the starting material by treating the mixture of products with Pd/C under an atmosphere of H2. We therefore sought an alternative source of bromine that would avoid this complication and found that direct addition of Br2 to deprotonated 14 generated 15 in 66% yield, with the remainder as starting material, which could be separated by column chromatography and reused. Treatment of 15 with excess sodium nitrite converted 8-bromoadenine 15 to 8-bromohypoxanthine 16, which was a suitable substrate for N1-ribosylation. Deprotonation of 16 with DBU followed by addition of tetraacetyl-d-ribose and trimethylsilyl triflate (TMS-OTf)37 afforded stereo- and regioselective N1-coupling to generate only the desired N1-ribosylated product 17 in high yield. Deprotection of the three acetyl esters using methanolic ammonia generated triol 18.
Scheme 1. Synthesis of the N1-Ribosyl-N9-butyl-8-bromohypoxanthine Building Block.

Reagents: (i) DBU, 4-Cl-butyl-OAc, 77%; (ii) NH3, MeOH, 96%; (iii) imidazole, TBDPS-Cl, 92%; (iv) (a) diisopropylamine, n-BuLi, (b) Br2, 66%; (v) NaNO2, AcOH (aq), 84%; (vi) (a) DBU, (b) tetraacetyl-d-ribose, TMSOTf, 86%; (vii) NH3, MeOH, 98%.
The 2′,3′-diol was reprotected as an isopropylidene ketal to afford 19, which allowed selective introduction of the first phosphate ester to the 5′-OH by treatment of 19 with N,N-diisopropyldibenzylphosphoramidite and 5-phenyl-1-H-tetrazole as activator, followed by oxidation of the intermediate phosphite with hydrogen peroxide and triethylamine (Scheme 2). The silyl ether of 20 was then removed under neutral conditions to reveal the hydroxyl group. The second protected phosphate ester was introduced by treatment of 21 with cyclohexylammonium S,S-diphenylphosphorodithioate (PSS), 2,4,6-triisopropylbenzenesulfonyl chloride (TPS-Cl), and 5-phenyl-1-H-tetrazole in pyridine under strictly dry conditions to generate the fully protected precursor for cyclization, 22. Sequential deprotection of the phosphates was carried out as previously described37 using first 50% aqueous TFA to simultaneously remove the tert-butyl esters and the isopropylidene ketal, then 0.1 M sodium hydroxide in dioxane, to afford 24. The N9-butyl chain shows increased stability toward acidic conditions, compared to the natural “southern” ribose of cIDPR, which had initially suffered partial hydrolysis at the N9-glycosidic bond during deprotection of the isopropylidene acetals and tert-butyl phosphate esters.37,47 No degradation of the N9-butyl analogs was observed even over prolonged periods in 50% TFA at room temperature, since the ribose oxygen has been removed.
Scheme 2. Sequential Introduction of the Phosphate Esters.

Reagents: (i) pTsOH, H3CC(OMe)2CH3, acetone, 100%; (ii) (a) (tBuO)2PN(iPr)2, 5-Ph-1H-tetrazole, DCM; (b) H2O2, Et3N, 80%; (iii) TBAF·3H2O, AcOH, 100%; (iv) PSS, TPS-Cl, 5-Ph-1H-tetrazole, pyridine, 100%; (v) 50% TFA (aq), 100%; (vi) 0.1 M NaOH–dioxane.
Cyclization of 24 was promoted using iodine and activated 3 Å molecular sieves in pyridine,33 under very dilute conditions, to afford 5 (21% yield over two steps, Scheme 3). Because of the stability of our cIDPR-based analogs, modifications at the 8-position were possible after cyclization, which allowed us to generate three further analogs; 8-H, 8-N3, and 8-NH2. Subjecting 5 to an atmosphere of hydrogen with Pd/C catalysis generated the 8-H analog, N9-butyl-cIDPR (4). Treatment of 5 with NaN3 in an attempt to generate the 8-N3 analog 6 was not successful, despite conversion to the triethylamine salt or the free acid to improve the solubility of 5 in DMF. In each case, the DMF solution became cloudy over time as the sodium salt of the starting material was formed and became insoluble in the reaction media. However, upon treatment of the free acid form of 5 with TMSN3 in DMF, no precipitation was observed and the clear solution was stirred at 70 °C for 16 h, becoming yellow in color as the reaction proceeded. The reaction progression was followed by RP-HPLC which clearly showed the shift in wavelength as the 8-bromo starting material (λmax = 255 nm) was converted into the 8-azido product [8-N3-N9-butyl-cIDPR (6), λmax = 277 nm]. Treatment of 6 with dithiothreitol reduced the 8-azido substituent to an 8-amino group which gave 8-NH2-N9-butyl-cIDPR (7).
Scheme 3. Synthesis of N9-Butyl-cIDPR Analogs 4–7.

Reagents: (i) I2, 3 Å molecular sieves, pyridine, 21%; (ii) H2, Pd/C, NaHCO3, EtOH–H2O, 76%; (iii) TMSN3, 57%; (iv) dithiothreitol, 0.05 M TEAB, 68%.
Inhibition of CD38-Mediated cADPR Hydrolysis
The ability of the novel compounds 4–7 to inhibit shCD38-mediated hydrolysis of cADPR was assessed in a dose–response manner using a fluorimetric cycling assay.52 cIDPR (2) inhibits hydrolysis of cADPR with an IC50 of 276 μM.42 The novel N9-butyl analogs 4–7 inhibit cADPR hydrolysis in a concentration-dependent manner, with half maximal inhibition in the low micromolar range (Figure 3 and Table 1). They are all more potent than 2, showing an almost 100-fold increase in inhibition. The introduction of an 8-bromo substituent (5) gave a marginally improved inhibition (IC50 = 27 μM) compared to the parent 4 (33 μM), which was then further improved by substitution to the 8-azido (6) or 8-amino (7) group (IC50 of 6.4 and 3.3 μM, respectively). The trend of improved inhibition upon 8-H → 8-Br → 8-NH2 substitution mimics that which was observed for the cIDPR series,43 the 8-NH2 derivative benefiting from an additional binding site interaction with Asp155. This suggests that the new analogs are binding similarly in the active site (vide infra). They are of comparable potency to the recently reported non-nucleoside and flavonoid NADase inhibitors.22,23 The improved inhibition of cADPR hydrolysis suggests that the “southern” ribose is not essential for activity.
Figure 3.
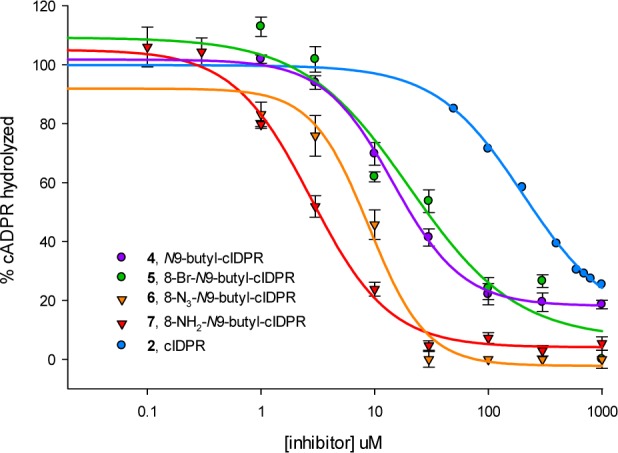
Inhibition of shCD38-mediated cADPR hydrolysis by analogs 4–7.
Table 1. Half Maximal Values for Inhibition of shCD38-Mediated cADPR Hydrolysis by Novel Analogs 4–7 and Equivalent 8-Substituted cIDPRs.
| cIDPR | IC50 (μM)43 | N9-butyl-cIDPR | IC50 (μM) | |
|---|---|---|---|---|
| 8-H | 2 | 276 | 4 | 33 |
| 8-Br | 3 | 158 | 5 | 27 |
| 8-N3 | 6 | 6.4 | ||
| 8-NH2 | 56 | 7 | 3.3 |
The significant increase in potency by substitution of the “southern” ribose with a butyl chain may be a result of the increased flexibility afforded by an alkyl chain linker, compared to a ribose sugar. This might allow the ligand to align other key residues more favorably with their binding targets, resulting in a tighter affinity for CD38. Another factor may be the hydrophobic nature of the butyl chain, which may prefer to sit further into the binding pocket, in a less polar environment away from bulk solvent.
Structure of 8-NH2-N9-butyl-cIDPR (7) Complexed with Wild-Type CD38
Preformed crystals of wild-type shCD38 were soaked with a cryoprotectant solution containing 5 mM 8-NH2-N9-butyl-cIDPR (7). X-ray diffraction data were collected to a resolution of 2.05 Å. The structure was solved by the molecular replacement method revealing two molecules of CD38 in the asymmetric unit with a ligand present in each active site (Figure 4A). Previous crystallographic work43 has generally shown occupancy of only one site by noncovalent inhibitors.
Figure 4.

Crystal structure of PDB code 4TMF. (A) Ligand (shown as sticks) occupies each active site. (B) The electron density fits that of a hydrolyzed ligand (carbons in cyan, key residues that interact with the ligand in the binding site shown as green sticks). (C) H-bonds (shown as black dotted lines) are formed between the rotated hypoxanthine base and both Glu146 and the backbone nitrogen of Asp156. Interacting residues are shown as pink sticks, and other residues are shown as green lines. The interacting binding site water is shown as a red sphere, and the other waters as red crosses.
To our surprise, the electron density within each active site did not fit the cyclic compound 7. However, hydrolysis at N1 to generate a linear compound, 7a, gave a structure with a good fit (Figure 4B). After refinement, the complex clearly showed that the catalytic residue Glu226 was still hydrogen-bonded to the ribose. For molecule A, Glu226 H-bonds to the 1′- and 3′-OH of the ribose (both 2.7 Å), which has previously been observed in hydrolyzed intermediates or products.53 In molecule B, Glu226 forms H-bonds with the 3′-OH (2.6 Å) and longer distance interactions with the 1′-OH and 2′-OH (3.3 and 3.5 Å). In both molecules, the 2′-OH interacts with an active site water molecule (Figure 4C). Addition of water to C-1′ has occurred to generate the hydrolyzed product, and the -OH group is attached to the α-face of the ribose, as has been observed previously for NAD+ hydrolysis products captured by crystallization.53,54 Such observations are at odds with both the proposed ionic or covalent hydrolysis mechanisms, which predict a predominantly or entirely β-product, respectively.55 Notably, the hydrolyzed ligand still occupies the catalytic site in the same manner as the cADPR, except that the hypoxanthine base has rotated in the binding pocket after hydrolysis. While maintaining the stacking interaction with Trp189, it has also formed new hydrogen bonds with Glu146 and Asp156 via its 8-NH2 and 6-O substituents, respectively (Figure 4C). It is unusual to capture this snapshot of the ligand directly after hydrolysis, before diffusion out of the binding site. Previously reported crystal structures of the hydrolysis products of N7-cGDPR and cADPR, GDPR and ADPR, respectively, have also captured products that were not bound in a conformation directly resulting from hydrolysis.53
Proposed Mechanism of Hydrolysis by CD38
Finding a hydrolyzed ligand in the active site of CD38 was unexpected, as the cIDPR template has previously been considered “nonhydrolyzable”.42 The absence of a partial positive charge at N1, compared to that in the N6-amino form of cADPR, was thought to contribute to this stability (Figure 5).
Figure 5.

cADPR has a partial positive charge at N1.
However, a mechanism in which the anomeric oxygen assists in breaking the N1-link could still be envisaged (Scheme 4). This would be without prejudice as to whether the role of Glu226 is to capture the resulting oxonium ion formally or interact with it as an ion pair before insertion of water to generate the final linear product. Examination of the crystal structures 2PGJ and 3U4H suggests that both binding sites contain two water molecules close to the N1-link of the cyclic ligand. One of these waters forms H-bonds to Leu123, Leu145, and the ribose 2′-OH that is consistent with other reported CD38 crystal structures.54 Notably, in the 3U4H crystal with 8-NH2-cIDPR, the remaining water molecule lies within 3 Å of the hypoxanthine carbonyl group. This water is not observed in the complex of the hydrolyzed ligand 7a (PDB code 4TMF); thus, we postulate that donation of a proton from this water molecule could be enzyme-assisted in the active site. Further examination reveals a proximal histidine residue (His133) that may potentially contribute to overall catalysis (Figure 6).
Scheme 4. Proposed Mechanism for Hydrolysis of 7 by a High Concentration of shCD38.

Figure 6.
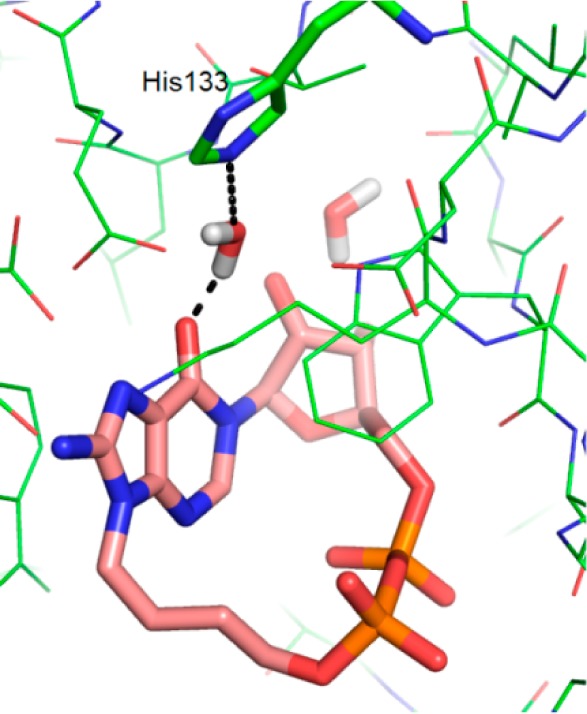
Docking of 8-NH2-N9-butyl-cIDPR (7) into 3U4H after removal of ligand (ligand carbons in pink sticks, protein carbons in green).
For the corresponding cADPR ligand to be hydrolyzed via such a mechanism would require the ligand to be present in the N6-imino form. Indeed, an earlier report indicated that cADPR exists in two forms, the N6-imino and N6-amino, the latter with a pKa of 8.3.56 Although at physiological pH the amino form will be predominant, the active form for Ca2+ release remains unresolved and crystallization of cADPR originally suggested that the C6–N6 bond displayed double bond characteristics (length 1.33 Å, cf. 1.47 Å expected for an amino group) with only one hydrogen atom bound to N6.2 This is somewhat surprising given that the molecule was crystallized as its free acid, presumably well below pH 8.3. The C6–N6 bond length in the cADPcR-shCD38 complex,43 the only cocrystal structure of a cyclic ligand with a starting C6-substituent in the amino form yet determined, measures 1.35 Å, also suggesting substantial double bond character. The authors measured the pKa of the N6-amino dissociation in this case as 8.9.26 However, cADPcR is a completely nonhydrolyzable ligand due to the absence of the ribose oxygen. Since crystallization of cADPR itself with shCD38 is not possible because of its very rapid hydrolysis, it has been impossible to capture directly the intermediates in this process. Previously, the surrogate substrate NGD+ revealed the role of Glu226 in activating and stabilizing the intermediate precursor for hydrolysis or cyclization.53 We originally showed that cIDPR, as expected, has no ionization between 6.8 and 10.9 28 and it is not likely that 7 will be any different. Thus, since it is unlikely that cIDPR or any related analog will exist in the 6-phenolic form at the pH used with shCD38 (vide infra) and the enzyme would not adopt a different mechanism to hydrolyze an analog vs cADPR itself, we can conclude that the complex of 7a with shCD38 provides the first evidence that CD38 can bind cADPR in the N6-imino form. This could have future implications for inhibitor design. We clearly cannot comment upon the ability of CD38 to bind the N6-amino form of cADPR.
Study of Ligand Hydrolysis by CD38
Following the discovery of hydrolyzed ligand 7a in the crystal complex, it was important to determine whether it is the cyclic or the hydrolyzed linear compound that inhibits hydrolysis of cADPR by shCD38. Incubation of 7 (1 mM final concentration) with 4 mg/mL shCD38 was monitored using RP-HPLC. The peak corresponding to 7 (tR = 9.1 min) reduced in intensity over time, alongside the appearance of a new peak (tR = 10.6 min), which was characteristic of an ADPR analog (see Supporting Information, Figure S1). The rate of conversion depended on the concentration of CD38 present in solution (Figure S2), and no change in the original peak was observed in a parallel control experiment containing no shCD38 (data not shown). Novel analogs 4, 5 and cIDPR (2) were also analyzed in this way (Figure 7). Surprisingly, we found that all the compounds, including cIDPR, are hydrolyzed by the enzyme at 4 mg/mL but at a slower rate than 7. The increased rate of hydrolysis of 7 could conceivably be promoted by the additional hydrogen bond generated between the 8-NH2 substituent and Asp155 promoting elongation of the N1-bond toward the transition state. This faster hydrolysis of 7 under these conditions (50% hydrolysis for 7 at T = 105 min and for cIDPR at T = 420 min), also reinforced by the extra interaction afforded by the 8-amino group after cleavage, perhaps explains why the hydrolyzed ligand 7a is captured in the 8-NH2-N9-butyl-cIDPR:CD38 complex, but a cyclic ligand is observed in the previously reported cIDPR:CD38 complex 2PGJ.42
Figure 7.
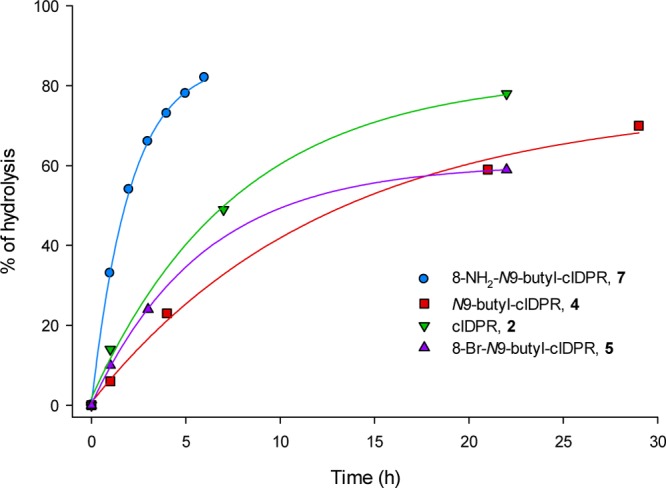
Hydrolysis of analogs 4, 5, 7, and cIDPR (2) over time with shCD38 (4 mg/mL).
These conditions are representative of crystallization concentrations of shCD38 (10 mg/mL) and are 10 000-fold more concentrated than those used in the enzyme assay (1 μg/mL). Therefore, further HPLC experiments were carried out using shCD38 concentrations of 1 μg/mL and either cIDPR or 7 to observe the effect of CD38 under conditions that reflect the enzyme assay (Supporting Information, Figure S3). Under these conditions the ligand is not hydrolyzed, confirming that the inhibition observed in the CD38 assay is a consequence of the cyclic compounds, not their hydrolyzed counterparts.
Conclusion
Two of the analogs developed in this study are potent (≤10 μM) inhibitors of cADPR hydrolysis by CD38. Analogs 4–7 illustrate that the “southern” ribose can be modified to considerably improve inhibitory activity of CD38 mediated cADPR hydrolysis. This agrees with modeling predictions based on the shCD38-ligand crystal structure complex interactions and confirms that the “northern” ribose and base are key features for the interaction of cADPR analogs with the CD38 binding pocket. The investigation of SAR in this region in isolation was not possible prior to development of a method for N1-ribosylation. This method thus opens up new avenues and the potential to introduce desirable characteristics, such as membrane permeability, into cADPR analogs by modifications in this region, without loss of activity at CD38. Crystallization of 8-NH2-N9-butyl-cIDPR with shCD38 revealed that the cIDPR scaffold can unexpectedly be hydrolyzed at high concentrations of shCD38, resulting in a cocrystal structure with linear 8-NH2-N9-butyl-IDPR (7a) in the active site. Hydrolysis of the hypoxanthine-linked scaffold offers new insight into the mechanism by which CD38 hydrolyzes cADPR to ADPR and suggests for the first time that the imino form is active in this process. However, HPLC studies suggest that hydrolysis would not occur under assay or physiologically relevant concentrations. Thus, this observation does not detract from the established use of cIDPR or its derivatives as either pharmacological tools or as nonhydrolyzable templates for future CD38 inhibitor design or indeed of 8-Br-cIDPR (3)30 as a stable membrane permeant agonist of Ca2+ release.
Experimental Section
General
All reagents and solvents were of commercial quality and were used without further purification, unless described otherwise. Unless otherwise stated, all reactions were carried out under an inert atmosphere of argon. 1H, 13C, and 31P NMR spectra were collected on a Varian Mercury 400 MHz or Bruker Avance III 500 MHz spectrometer. All 1H and 13C NMR assignments are based on gCOSY, gHMBC, gHSQC, and DEPT-135 experiments. Abbreviations for splitting patterns are as follows: b, broad; s, singlet; d, doublet; t, triplet; m, multiplet. Coupling constants are given in hertz (Hz). High resolution time-of-flight mass spectra were obtained on a Bruker Daltonics micrOTOF mass spectrometer using electrospray ionization (ESI). The purity of new tested compounds was determined to be ≥95% by analytical HPLC. Analytical HPLC analyses were carried out on a Waters 2695 Alliance module equipped with a Waters 2996 photodiode array detector (210–350 nm). The chromatographic system consisted of a Hichrom Guard column for HPLC and a Phenomenex Synergi 4 μm MAX-RP 80A column (150 mm × 4.60 mm), with elution at 1 mL/min with the following ion-pair buffer: 0.17% (m/v) cetrimide and 45% (v/v) phosphate buffer (pH 6.4) in MeOH. Synthetic phosphates were assayed and quantified by the Ames phosphate test.57
6-Chloro-9-(4-acetoxybutyl)purine (12)48
To 6-chloropurine 11 (2.50 g, 16.2 mmol) and DBU (2.91 mL, 19.4 mmol) in DMF (22 mL) was added 4-chlorobutylacetate (4.54 mL, 32.3 mmol). After the mixture was stirred at 60 °C for 14 h, the DMF was removed under reduced pressure, and the residue was purified by column chromatography on silica gel, eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (3.07 g, 71%) as a colorless oil. Rf = 0.61 (DCM/MeOH 9:1 v/v); 1H NMR (400 MHz, MeOD) δ 8.78 (s, 1H), 8.16 (s, 1H), 4.37 (t, 2H, J = 7.2, CH2), 4.15 (t, 2H, J = 6.4, CH2), 2.08–2.04 (m, 5H, CH2 and OAc), 1.74–1.67 (m, 2H, CH2) ppm.
9-(4-Hydroxybutyl)adenine (13)49
A solution of 12 (3.00 g, 11.2 mmol) in MeOH (7 mL) was cooled to 0 °C and saturated with NH3 (g), then stirred for 14 h at 80 °C. On cooling, a white solid precipitated, which was collected by filtration and air-dried to afford the title compound (2.22 g, 96%). Rf = 0.17 (DCM/MeOH 9:1 v/v); 1H NMR (400 MHz, MeOD) δ 8.23 (s, 1H), 8.16 (s, 1H), 4.30 (t, 2H, J = 7.2, CH2), 3.61 (t, 2H, J = 6.4, CH2), 2.03–1.96 (m, 2H, CH2), 1.61–1.54 (m, 2H, CH2) ppm; HRMS (ESI+) calcd for C9H14N5O1 208.1193 [(M + H)+], found 208.1195.
9-(4-tert-Butyldiphenylsilylbutyl)adenine (14)
To 13 (2.00 g, 9.65 mmol) in DMF (20 mL) were added imidazole (1.71 g, 25.09 mmol) and TBDPS-Cl (3.25 mL, 12.55 mmol). After 14 h at rt, all solvents were evaporated and the residue was purified by column chromatography on silica gel, eluting with DCM/acetone (1:0 → 0:1 v/v) to afford the title compound (3.96 g, 92%) as a white solid. Rf = 0.42 (DCM/acetone 1:3 v/v); 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.09 (s, 1H), 7.62–7.60 (m, 4H), 7.43–7.35 (m, 6H), 4.26 (t, 2H, J = 7.0, CH2), 3.71 (t, 2H, J = 6.2, CH2), 2.05–1.97 (m, 2H, CH2), 1.58–1.52 (m, 2H, CH2), 1.01 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 155.3, 153.0, 150.2, 140.5, 135.5 (4C), 133.7 (2C), 129.7 (2C), 127.7 (4C), 119.8, 63.0, 43.8, 29.5, 26.9 (3C), 26.7, 19.2 ppm; HRMS (ESI+) calcd for C25H32N5OSi 446.2371 [(M + H)+], found 446.2377.
9-(4-tert-Butyldiphenylsilylbutyl)-8-bromoadenine (15)
To diisopropylamine (4.67 mL, 33.32 mmol) in THF (20 mL) at −78 °C was added n-butyllithium (21.2 mL, 1.6 M solution, 33.97 mmol), dropwise. After 1 h, a solution of 14 (2.97 g, 6.66 mmol) in THF (25 mL) was added dropwise and stirring continued for a further 1 h. Br2 (2.04 mL, 39.96 mmol) was added dropwise and the solution allowed to warm to rt over 4 h. The reaction was quenched by addition of NH4 (aq, 2 mL), and all solvents were evaporated. The residue was taken up in DCM/H2O (1:1 v/v, 100 mL) and the organic layer separated, washed with brine, dried (Na2SO4), and evaporated to dryness. The crude material was purified by column chromatography on silica gel, eluting with DCM/acetone (1:0 → 0:1 v/v) to afford the title compound (2.18 g, 62%) as an amorphous cream solid. Rf = 0.61 (DCM/acetone 1:1 v/v); 1H NMR (400 MHz, MeOD) δ 8.31 (s, 1H, H-2), 7.64–7.62 (m, 4H), 7.40–7.33 (m, 6H), 5.85 (bs, 2H, NH2), 4.22 (t, 2H, J = 7.4, CH2), 3.70 (t, 2H, J = 6.1, CH2), 2.00–1.93 (m, 2H, CH2), 1.62–1.55 (m, 2H, CH2), 1.03 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 154.2, 153.0, 151.3, 135.5 (4C), 133.7 (2C), 129.6 (2C), 127.8 (4C), 127.3, 119.9, 63.0, 44.4, 29.4, 26.8 (3C), 26.1, 19.1 ppm; HRMS (ESI+) calcd for C25H31N5OSi79Br 524.1476 [(M + H)+], found 524.1473, calcd for C25H31N5OSi81Br 526.1455 [(M + H)+], found 526.1462.
9-(4-tert-Butyldiphenylsilylbutyl)-8-bromohypoxanthine (16)
To 15 (4.13 g, 7.87 mmol) in AcOH/H2O (20:3 v/v, 138 mL) was added NaNO2 (6.52 g, 94.48 mmol) in one portion. After 48 h, all solvents were evaporated and EtOH (100 mL) was added and evaporated to dryness. The residue was taken up in CHCl3 and washed with H2O, then NaHCO3 (aq sat.) to pH 7. The organic layer was washed with brine, dried (Na2SO4), and evaporated to dryness. The crude material was purified by column chromatography on silica gel, eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (3.49 g, 84%) as a cream foam. Rf = 0.37 (PE/EtOAc 1:3 v/v); 1H NMR (400 MHz, CDCl3) δ 13.07 (bs, 1H, NH), 8.16 (s, 1H, 2H), 7.65–7.63 (m, 4H), 7.41–7.34 (m, 6H), 4.21 (t, 2H, J = 7.3, CH2), 3.71 (t, 2H, J = 6.0, CH2), 1.96 (tt, 2H, J = 7.4, 7.3, CH2), 1.58 (tt, 2H, J = 6.5, 6.0, CH2), 1.04 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 158.0, 150.6, 145.5, 135.6 (4C), 133.8 (2C), 129.7 (2C), 127.7 (4C), 126.3, 124.8, 63.0, 44.8, 29.4, 26.9 (3C), 26.2, 19.2 ppm; HRMS (ESI+) calcd for C25H30N4O2Si79Br 525.1316 [(M + H)+], found 525.1319, calcd for C25H30N4O2Si81Br 527.1295 [(M + H)+], found 527.1301.
N1-(2′,3′,5′-Tri-O-acetyl-β-d-ribofuranosyl)-N9-[4-(tert-butyldiphenylsilyl)oxybutyl)-8-bromohypoxanthine (17)
Intermediate 16 (400 mg, 0.761 mmol) was taken up in DCM (4.0 mL), and DBU (341 μL, 2.283 mmol) was added. After 30 min, 1,2,3,5-tetra-O-acetyl-β-d-ribofuranose (266 mg, 0.837 mmol) was added and the solution cooled to −78 °C. Trimethylsilyl trifluoromethanesulfonate (551 μL, 3.044 mmol) was added dropwise and the solution stirred for a further 45 min before warming to rt. After 1 h, NaHCO3 (sat. aq.) was added and the crude material extracted into DCM (×3). The combined organic fractions were dried (Na2SO4), and solvent was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel, eluting with PE/EtOAc (1:0 → 1:0 v/v) to afford the title compound (203 mg, 90%) as a colorless glass. Rf = 0.69 (PE/EtOAc 1:3 v/v); 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H, 2H), 7.63 (dd, 4H, J = 7.8, 1.5), 7.40–7.34 (m, 6H) (10 × Ar-H), 6.39 (d, 1H, J = 4.6, H-1′), 5.47 (dd, 1H, J = 5.8, 4.6, H-2′), 5.44 (dd, 1H, J = 5.8, 4.5, H-3′), 4.42–4.38 (m, 3H, H-4′ and both H-5′), 4.17 (t, 2H, J = 7.3, CH2), 3.70 (t, 2H, J = 6.0, CH2), 2.13 (s, 3H), 2.11 (s, 3H), 2.07 (s, 3H) (3 × OAc), 1.96–1.92 (m, 2H, CH2), 1.58–1.54 (m, 2H, CH2), 1.03 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 170.2, 169.58, 169.57, 154.8, 148.7, 144.1, 135.5 (4C), 133.7 (2C), 129.7 (2C), 127.7 (4C), 126.1, 124.1, 87.3, 80.3, 74.2, 70.3, 63.0, 62.9, 44.7, 29.4, 26.9 (3C), 26.3, 20.8, 20.5, 20.4 19.2 ppm; HRMS (ESI+) calcd for C36H44N4O9Si79Br 783.2055 [(M + H)+], found 783.2046, calcd for C36H44N4O9Si81Br 785.2035 [(M + H)+], found 785.2042.
N1-(β-d-Ribofuranosyl)-N9-[4-(tert-butyldiphenylsilyl)oxybutyl)-8-bromohypoxanthine (18)
Intermediate 17 (600 mg, 0.766 mmol) was taken up in MeOH (6 mL) in a pressure tube. The solution was saturated with NH3 (g) at 0 °C, then stirred at rt for 12 h. The solvent was evaporated and the residue purified by column chromatography on silica gel, eluting with PE/EtOAc (1:0 → 1:0 v/v) to afford the title compound (492 mg, 98%) as a white amorphous solid. Rf = 0.35 (DCM/acetone 1:1 v/v); 1H NMR (500 MHz, MeOD) δ 8.79 (s, 1H, H-2), 7.62–7.60 (m, 4H), 7.42–7.35 (m, 6H) (10 × Ar-H), 6.22 (d, 1H, J = 3.1, H-1′), 4.32–4.28 (m, 2H, H-2′, H-3′), 4.23 (t, 2H, J = 7.1, CH2), 4.13 (ddd, 1H, J = 5.5, 2.9, 2.5, H-4′), 3.98 (dd, 1H, J = 12.3, 2.5, H-5a′), 3.83 (dd, 1H, J = 12.3, 2.9, H-5b′), 3.70 (t, 2H, J = 6.0, CH2), 1.98–1.92 (m, 2H, CH2), 1.55–1.49 (m, 2H, CH2), 1.02 (s, 9H) ppm; 13C NMR (125 MHz, MeOD) δ 156.9, 150.5, 147.1, 136.6 (4C), 134.8 (2C), 130.9 (2C), 128.8 (4C), 127.7, 124.6, 91.5, 86.2, 76.9, 70.6, 64.1, 61.7, 45.7, 30.4, 27.4 (3C), 27.1, 19.9 ppm; HRMS (ESI+) calcd for C30H38N4O6Si79Br 657.1739 [(M + H)+], found 657.1747, calcd for C30H38N4O6Si81Br 659.1718 [(M + H)+], found 659.1729.
N1-(2′,3′-O-Isopropylidine-β-d-ribofuranosyl)-N9-[4-(tert-butyldiphenylsilyl)oxybutyl)-8-bromohypoxanthine (19)
To 18 (430 mg, 0.634 mmol) in acetone/2,2-dimethoxypropane (5 mL, 4:1 v/v) was added p-TsOH (124 mg, 0.634 mmol). After 30 min, DCM and NaHCO3 (sat. aq) were added, the organic layer was dried (Na2SO4), and all solvents were evaporated. The residue was taken up in MeOH (2 mL), and Dowex 50WX8 H+ resin (50 mg) was added. After 30 min the resin was removed by filtration under gravity and the solvent evaporated to obtain the title compound (455 mg, 100%) as a colorless glass. Rf = 0.63 (PE/EtOAc 1:3 v/v); 1H NMR (400 MHz, CDCl3) δ 7.87 (s, 1H, H-2), 7.55 (dd, 4H, J = 5.9, 1.5), 7.35–7.26 (m, 6H) (10 × Ar-H), 5.62 (d, 1H, J = 2.8, H-1′), 5.23 (dd, 1H, J = 6.5, 2.8, H-2′), 5.08 (dd, 1H, J = 6.5, 3.6, H-3′), 4.28 (dd, 1H, J = 3.6, 2.4, H-4′), 4.09 (t, 2H, J = 7.3, CH2), 3.85 (dd, 1H, J = 12.2, 2.4, H-5a′), 3.75 (bd, 1H, J = 12.2, H-5b′), 3.61 (t, 2H, J = 6.0, CH2), 1.89–1.82 (m, 2H, CH2), 1.52 (s, 3H), 1.50–1.45 (m, 2H, CH2), 1.28 (s, 3H), 1.02 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 155.3, 149.1, 146.5, 135.5 (4C), 133.6 (2C), 129.6 (2C), 127.6 (4C), 126.5, 124.8, 114.2, 97.1, 88.1, 83.5, 80.6, 62.8, 62.8 (2C), 44.7, 29.2, 27.3, 26.8 (3C), 26.1, 25.2, 19.1 ppm; HRMS (ESI+) calcd for C33H42N4O6Si79Br 697.2052 [(M + H)+], found 697.2072, calcd for C33H42N4O6Si81Br 699.2031 [(M + H)+], found 699.2032.
N1-[2′,3′-O-Isopropylidine-5′-O-(di-tert-butyl)phosphoryl-β-d-ribofuranosyl]-N9-[4-(tert-butyldiphenylsilyl)oxybutyl)-8-bromohypoxanthine (20)
To a solution of 19 (350 mg, 0.502 mmol) in DCM (2.8 mL) was added 5-phenyl-1-H-tetrazole (147 mg, 1.003 mmol) and N,N-diisopropyldibutylphosphoramidite (238 μL, 0.753 mmol). After 2 h, the solution was cooled to 0 °C and Et3N (419 μL, 3.012 mmol) and H2O2 (138 μL, 1.255 mmol) were added. The solution was allowed to warm to rt and stirred for a further 2 h, after which DCM (20 mL) and H2O were added. The organic layer was washed with NaHCO3 (sat. aq), then brine, dried (Na2SO4), and evaporated to dryness. The residue was purified by column chromatography on silica gel, eluting with PE/EtOAc (1:0 → 1:0 v/v), where both solvents contained 0.5% v/v pyridine, to afford the title compound (356 mg, 80%) as a colorless glass. Rf = 0.43 (PE/EtOAc 1:3 v/v); 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H, H-2), 7.63–7.60 (m, 4H), 7.40–7.32 (m, 6H) (10 × Ar-H), 5.97 (d, 1H, J = 1.8, H-1′), 5.08 (dd, 1H, J = 6.4, 1.8, H-2′), 4.99 (dd, 1H, J = 6.4, 3.9, H-3′), 4.41 (dd, 1H, J = 9.6, 4.3, H-4′), 4.27–4.13 (m, 4H, both H-5′ and CH2), 3.67 (t, 2H, J = 6.0, CH2), 1.93–1.89 (m, 2H, CH2), 1.57 (s, 3H), 1.55–1.44 (m, 2H, CH2), 1.448 (s, 9H), 1.447 (s, 9H), 1.33 (s, 3H), 1.02 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 153.0, 147.2, 144.1, 133.7 (4C), 131.9 (2C), 127.8 (2C), 125.8 (4C), 124.2, 122.7, 112.5, 92.5, 85.1 (d, J = 7.9), 83.2, 80.9 (d, 2C, J = 7.2), 79.7, 64.7 (d, J = 6.2), 61.1, 42.8, 28.0 (d, 3C, J = 2.8), 27.9 (d, 3C, J = 2.9), 27.5, 25.3, 25.0 (3C), 24.4, 23.5, 17.3 ppm; 31P NMR (162 MHz, 1H decoupled, CDCl3) δ −10.2 ppm; HRMS (ESI+) calcd for C41H58N4O9PSi79BrNa 911.2786 [(M + H)+], found 911.2762, calcd for C41H58N4O9PSi81BrNa 913.2766 [(M + H)+], found 913.2763.
N1-[2′,3′-O-Isopropylidine-5′-O-(di-tert-butyl)phosphoryl-β-d-ribofuranosyl]-N9-(4-hydroxybutyl)-8-bromohypoxanthine (21)
Acetic acid (12 μL, 0.213 mmol) and TBAF·3H2O (64 mg, 0.203 mmol) were stirred in DMF (0.5 mL) for 30 min, after which the solution was cooled to 0 °C and 20 (60 mg, 0.068 mmol) in DMF (0.5 mL) added. The resulting solution was allowed to warm to rt and stirred for a further 4 h. The solution was diluted with ether, and NaHCO3 (sat. aq) and NH3Cl (sat. aq) were added. The organic layer was separated and the aqueous layer extracted with ether (×3). The combined organic layers were dried (Na2SO4), evaporated to dryness and the residue was purified by column chromatography on silica gel, eluting with DCM/acetone (1:0 → 1:0 v/v) to afford the title compound (44 mg, 100%) as a colorless glass. Rf = 0.47 (DCM/acetone 1:1 v/v); 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H, H-2), 5.98 (d, 1H, J = 1.8, H-1′), 5.05 (dd, 1H, J = 6.4, 1.8, H-2′), 4.96 (dd, 1H, J = 6.4, 3.9, H-3′), 4.39 (app. dd, 1H, J = 9.0, 4.5, H-4′), 4.25–4.11 (m, 4H, both H-5′ and CH2), 3.65 (t, 2H, J = 6.3, CH2), 2.34 (bs, 1H, OH), 1.89 (app. quintet, 2H, J = 7.2, CH2), 1.56 (app. quintet, 2H, J = 6.3, CH2), 1.55 (s, 3H), 1.44 (s, 9H), 1.43 (s, 9H), 1.31 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 154.7, 149.0, 145.9, 125.9, 124.4, 114.2, 94.1, 86.8 (d, J = 7.9), 85.1, 82.71 (d, J = 7.3), 82.69 (d, J = 7.2), 81.3, 66.4 (d, J = 6.3), 61.7, 44.5, 29.74 (d, 3C, J = 4.2), 29.72 (d, 3C, J = 4.3), 29.1, 27.1, 26.2, 25.2 ppm; 31P NMR (162 MHz, 1H decoupled, CDCl3) δ −10.4 ppm; HRMS (ESI+) calcd for C25H40N4O9P79BrNa 673.1608 [(M + H)+], found 673.1584, calcd for C25H40N4O9P81BrNa 675.1588 [(M + H)+], found 675.1570.
N1-[2′,3′-O-Isopropylidine-5′-O-(di-tert-butyl)phosphoryl-β-d-ribofuranosyl]-N9-(4-[(diphenylthio)phosphoryl]hydroxybutyl)-8-bromohypoxanthine (22)
Intermediate 21 (20 mg, 0.031 mmol) was coevaporated from pyridine (3 × 1 mL) and taken up in pyridine (0.5 mL). This solution was added to PSS (35 mg, 0.092 mmol) which had also been coevaporated from pyridine (3 × 1 mL). 5-Phenyl-1-H-tetrazole (13 mg, 0.092 mmol) and TPS-Cl (19 mg, 0.061 mmol) were added, and the solution was stirred at rt for 5 h. DCM and H2O were added. The organic layer was separated and the aqueous layer washed with DCM (×2). The combined organic layer was washed with brine, dried (Na2SO4), and evaporated to dryness. The residue was purified by column chromatography on silica gel, eluting with PE/EtOAc (1:0 → 1:0 v/v) to afford the title compound (28 mg, 100%) as a white foam. Rf = 0.73 (DCM/acetone 1:1 v/v); 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H, H-2), 7.52–7.49 (m, 4H), 7.36–7.30 (m, 6H) (10 × ArH), 5.97 (d, 1H, J = 1.8, H-1′), 5.07 (dd, 1H, J = 6.4, 1.8, H-2′), 4.98 (dd, 1H, J = 6.4, 3.9, H-3′), 4.40 (app. dd, 1H, J = 9.4, 5.2, H-4′), 4.25–4.17 (m, 4H, both H-5′ and CH2), 4.11 (t, 2H, J = 7.0, CH2), 1.82 (app. quintet, 2H, J = 7.2, CH2), 1.66 (app. quintet, 2H, J = 6.3, CH2), 1.56 (s, 3H), 1.454 (s, 9H), 1.449 (s, 9H), 1.32 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 154.7, 149.0, 146.0, 135.2 (d, 4C, J = 5.2), 129.4 (d, 2C, J = 3.2), 129.3 (d, 4C, J = 2.5), 126.3 (d, 2C, J = 6.7), 125.8, 124.5, 114.3, 94.3, 86.8 (d, J = 7.9), 85.0, 82.8 (d, 2C, J = 7.3), 81.4, 67.0 (d, J = 8.7), 66.5 (d, J = 6.2), 43.9, 29.77 (d, 3C, J = 4.2), 29.74 (d, 3C, J = 4.2), 27.1, 27.0 (d, J = 6.7), 25.6, 25.2 ppm; 31P NMR (162 MHz, 1H decoupled, CDCl3) δ 49.0, −10.3 ppm; HRMS (ESI+) calcd for C37H49N4O10P2S279BrNa 937.1441 [(M + H)+], found 937.1447, calcd for C37H49N4O10P2S281BrNa 939.1420 [(M + H)+], found 939.1440.
N1-[5′-O-Phosphoryl-β-d-ribofuranosyl]-N9-(4-[(diphenylthio)phosphoryl]hydroxybutyl)-8-bromohypoxanthine (23)
Intermediate 22 (40 mg, 0.044 mmol) was stirred in 50% TFA (2 mL) at 0 °C for 4 h. All solvents were evaporated, and the residue was coevaporated with MeOH (×4). The residue was purified by column chromatography on silica gel, eluting with EtOAc/MeOH/H2O (1:0:0 → 4:2:0 → 7:2:1 v/v/v) to afford the title compound (33 mg, 100%) as a colorless glass. Rf = 0.20 (EtOAc/MeOH/H2O 7:2:1 v/v/v); 1H NMR (500 MHz, MeOD) δ 8.59 (s, 1H, H-2), 7.43 (dd, 4H, J = 7.1, 0.7), 7.33–7.28 (m, 6H) (10 × ArH), 6.21 (d, 1H, J = 3.8, H-1′), 4.24 (d, 2H, J = 2.6, H-2′ and H-3′), 4.16–4.07 (m, 7H, H-4′, both H-5′ and 2 × CH2), 1.70 (app quintet, 2H, J = 7.1, CH2), 1.55 (app quintet, 2H, J = 5.7, CH2) ppm; 13C NMR (125 MHz, MeOD) δ 157.2, 150.6, 147.1, 136.6 (d, 4C, J = 5.3), 131.1 (d, 2C, J = 3.4), 130.7 (d, 4C, J = 2.5), 127.6, 127.0 (d, 2C, J = 6.8), 124.5, 89.8, 85.6 (d, J = 8.6), 77.1, 71.4, 69.2 (d, J = 8.9), 65.3 (d, J = 4.8), 45.2, 28.3 (d, J = 7.0), 26.8 ppm; 31P NMR (202 MHz, 1H decoupled, MeOD) δ 50.8, 0.8 ppm; HRMS (ESI–) calcd for C26H28N4O10P2S279Br 760.9911 [(M – H)−], found 760.9878, calcd for C26H28N4O10P2S281Br 762.9890 [(M – H)−], found 762.9915.
N1-(5′-O-Phosphoryl-β-d-ribofuranosyl)-N9-(4-[(diphenylthio)phosphoryl]hydroxybutyl)-8-bromohypoxanthine (24)
Intermediate 23 (20 mg, 0.026 mmol) was taken up in dioxane/H2O (1 mL, 1:1 v/v). NaOH (100 μL, 1 M) was added and the solution stirred for 30 min at rt before addition of HCl (100 μL, 1M). The solution was diluted with H2O and washed with hexane (×3) before evaporation of all solvents to give a colorless glass which was converted to the TEA salt as described below. 31P NMR (202 MHz, 1H decoupled, D2O) δ 15.6, 2.6 ppm; HRMS (ESI–) calcd for C20H24N4O11P2S79Br 668.9826 [(M – H)−], found 668.9853, calcd for C20H24N4O11P2S81Br 670.9806 [(M – H)−], found 670.9863. Conversion to TEA salt: The Na+ salt was passed through prewashed Dowex H+ resin. Acidic fractions were neutralized with TEAB (2 mL, 1 M). All solvents were evaporated, and the residue was coevaporated with H2O to remove excess buffer. The colorless glass obtained was used directly for cyclization.
N1-Cyclic-8-bromohypoxanthine-N9-hydroxybutyl Diphosphate Ribose (8-Br-N9-butyl-cIDPR, 5)
Intermediate 24 (0.026 mmol) was evaporated from pyridine (2 × 2 mL). The residue was taken up in pyridine (10 mL) and placed in a syringe. This solution was added over 15 h to a solution of iodine (70 mg, 0.30 mmol) and 3 Å molecular sieves (0.5 g) in pyridine (20 mL), in the dark. The solution was filtered through Celite, washed with H2O. After addition of TEAB (2 mL) all solvents were evaporated, and the residue was partitioned between H2O and CHCl3. The aqueous layer was washed with CHCl3 and evaporated to dryness. The residue was purified by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (3.0 mg, 21% over two steps). UV (H2O, pH 7), λmax = 256 nm (ε = 19 900); 1H NMR (500 MHz, D2O) δ 8.81 (s, 1H, H-2), 6.08 (d, 1H, J = 1.8, H-1′), 4.43 (dd, 1H, J = 4.7, 1.8, H-2′), 4.38 (dd, 1H, J = 6.9, 4.7, H-3′), 4.35–4.19 (m, 4H), 4.17 (app septet, 1H, J = 5.1, CHaH), 4.11 (dd, 1H, J = 11.0, 3.9, H-5b′), 1.92–1.83 (m, 2H, CH2), 1.41–1.38 (m, 1H, CHaH), 1.13–1.07 (m, 1H, CHbH) ppm; 13C NMR (125 MHz, D2O) δ 156.7, 150.1, 145.0, 127.7, 122.6, 91.1, 83.5 (d, J = 8.9), 75.2, 68.1, 65.2 (d, J = 6.0), 62.8 (d, J = 4.3), 42.9, 24.8 (d, J = 9.8), 23.7 ppm; 31P NMR (202 MHz, D2O, 1H-decoupled) δ −10.2 (d, J = 14.9 Hz), −11.4 (d, J = 14.9 Hz) ppm; HRMS (ESI–) calcd for C14H18N4O11P279Br 558.9636 [(M – H)−], found 558.9627; calcd for C14H18N4O11P281Br 560.9616 [(M – H)−], found 560.9612.
N1-Cyclic Hypoxanthine-N9-hydroxybutyl Diphosphate Ribose (N9-Butyl-cIDPR, 4)
Analog 5 (7.0 mg, 0.0125 mmol) was taken up in Milli-Q (3 mL), and NaHCO3 (225 mg, 2.77 mmol) was added. When all material had dissolved, EtOH (1.5 mL) and then Pd/C (6 mg) were added and the solution was placed under an atmosphere of H2. After 5 h stirring, the solution was filtered and all solvents evaporated. The residue was purified by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with MeCN/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (4.6 mg, 76%). UV (H2O, pH 7), λmax = 252 nm (ε = 11 400); 1H NMR (500 MHz, D2O) δ 8.79 (s, 1H, H-2), 8.03 (s, 1H, H-8), 6.10 (s, 1H, H-1′), 4.49 (d, 1H, J = 3.0, H-2′), 4.40 (app. t, 1H, J = 3.0, H-3′), 4.33–4.24 (m, 4H), 4.11 (dd, 1H, J = 11.8, 4.4, H-5b′), 4.05 (app septet, 1H, J = 5.2, CHaH), 3.81 (app septet, 1H, J = 5.2, CHbH), 1.96–1.84 (m, 2H, CH2), 1.41–1.35 (m, 1H, CHaH), 1.11–1.05 (m, 1H, CHbH) ppm; 13C NMR (125 MHz, D2O) δ 158.0, 148.9, 144.7, 142.3, 122.2, 91.1, 83.6 (d, J = 8.9), 75.2, 68.3, 65.1 (d, J = 5.9), 63.0 (d, J = 4.5), 42.3, 25.4, 25.0 (d, J = 9.5) ppm; 31P NMR (202 MHz, D2O, 1H-decoupled) δ −10.3 (d, J = 14.1), −11.2 (d, J = 14.2) ppm; HRMS (ESI–) calcd for C14H19N4O11P2 481.0531 [(M – H)−], found 481.0522.
N1-Cyclic-8-azidohypoxanthine-N9-hydroxybutyl Diphosphate Ribose (8-N3-N9-butyl-cIDPR, 6)
Analog 5 (4.5 mg, 8.0 μmol) was converted to the free acid by addition of Milli-Q (5 mL) and stirring with Dowex 50WX8 (H+ form) for 30 min. The resin was removed by filtration, washed with Milli-Q and the combined filtrate evaporated. The residue was evaporated from dry DMF (4 × 2 mL), taken up in DMF (1 mL), and stirred under argon. TMSN3 (50 μL, 0.43 mmol) was added and the resulting solution stirred at 70 °C in the dark for 16 h, after which ∼65% conversion of the starting material to product was observed by HPLC (λ = 255 nm → λ = 277 nm). All solvent was evaporated and the resulting residue coevaporated with Milli-Q (2 × 5 mL). The residue was then taken up in Milli-Q (5 mL), filtered through cotton wool, and purified by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/Milli-Q (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions were collected and evaporated under vacuum to give the title compound (2.4 mg, 57%). UV (H2O, pH 7), λmax = 252 nm (ε = 12 700); 1H NMR (500 MHz, D2O) δ 8.75 (s, 1H, H-2), 6.08 (d, 1H, J = 1.9, H-1′), 4.46 (dd, 1H, J = 4.8, 1.9, H-2′), 4.39 (dd, 1H, J = 6.5, 4.8, H-3′), 4.33–4.30 (m, 2H, H-4′, CHH), 4.12–4.05 (m, 3H, H-5′a, 2 × CHH), 4.00 (dt, 1H, J = 14.5, 5.2, H-5′b), 3.81 (app septet, 1H, J = 5.0, CHH), 1.81–1.76 (m, 2H, CH2), 1.40–1.36 (m, 1H, CHH), 1.14–1.09 (m, 1H, CHH) ppm; 13C NMR (125 MHz, D2O) 156.6, 149.0, 146.4, 144.2, 120.3, 91.2, 83.6 (d, J = 8.9), 75.2, 68.2, 65.1 (d, J = 6.5), 62.9 (d, J = 4.4), 40.7, 24.9 (d, J = 9.1), 23.5 ppm; 31P NMR (202 MHz, D2O) −10.4 (d, J = 14.1), −11.4 (d, J = 14.1) ppm; HRMS (ESI–) found m/z [M – H]− 522.0549; C14H18N7O11P2 requires 522.0545.
N1-Cyclic-8-aminohypoxanthine-N9-hydroxybutyl Diphosphate Ribose (8-NH2-N9-butyl-cIDPR, 7)
Analog 5 (4.5 mg, 8.0 μmol) was converted to the free acid by addition of Milli-Q (5 mL) and stirring with Dowex 50WX8 (H+ form) for 30 min. The resin was removed by filtration, washed with Milli-Q and the combined filtrate evaporated. The residue was evaporated from dry DMF (4 × 2 mL), taken up in DMF (1 mL), and stirred under argon. TMSN3 (50 μL, 0.43 mmol) was added and the resulting solution stirred at 70 °C in the dark for 16 h, after which ∼70% conversion of the starting material to product was observed by HPLC (λ = 255 nm → λ = 277 nm). All solvent was evaporated, and the resulting residue was taken up in TEAB (5 mL, 0.05 M). Dithiothreitol (12 mg, 0.08 mmol) was added and the solution stirred under an atmosphere of argon for 16 h. The crude material was directly purified by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (2.7 mg, 68% over two steps). UV (H2O, pH 7), λmax = 252 nm (ε = 14 700); 1H NMR (400 MHz, D2O) δ 8.80 (s, 1H, H-2), 6.19 (d, 1H, J = 2.1, H-1′), 4.56 (dd, 1H, J = 4.7, 2.1, H-2′), 4.50 (dd, 1H, J = 6.4, 4.7, H-3′), 4.43–4.41 (m, 2H, H-4′, CHH), 4.27–4.20 (m, 3H, H-5′a, 2 × CHH), 4.11 (ddd, 1H, J = 15.0, 5.3, 4.9, H-5′b), 3.95 (app septet, 1H, J = 5.3, CHH), 2.00–1.85 (m, 2H, CH2), 1.53–1.50 (m, 1H, CHH), 1.31–1.28 (m, 1H, CHH) ppm; 13C NMR (125 MHz, D2O) 155.3, 152.2, 148.3, 143.2, 116.7, 91.1, 83.5 (d, J = 8.9), 75.2, 68.1, 65.1 (d, J = 5.6), 62.9 (d, J = 3.1), 39.7, 24.8 (d, J = 9.6), 22.5 ppm; 31P NMR (202 MHz, D2O) −10.39 (br), −11.44 (br); HRMS (ESI–) found m/z [M – H]− 496.0637; C14H20N5O11P2 requires 496.0640.
Enzyme Assay for cADPR Hydrolysis
The inhibition of cADPR hydrolysis by various concentrations of analog (0–1 mM) was determined by incubating 1 μM cADPR with 1 μg/mL CD38 catalytic domain for 10 min at 20–24 °C in 25 mM sodium acetate, pH 4.5. The reaction was stopped by the addition of 150 mM HCl. The precipitated protein was filtered, and the pH was neutralized with Tris base. After the mixture was diluted 20-fold, the concentration of the unhydrolyzed cADPR present in the diluted reaction mixture was assayed by the fluorimetric cycling assay as described previously.52
Modeling of the Butyl Compounds
The 2PGJ crystal structure of human CD38 with cIDPR was passed through the Protein Preparation Wizard in the Schrödinger software (http://www.schrodinger.com/). Four N9-butyl compounds, where the cIDPR “southern” ribose is replaced by a four carbon linker, were built using the Schrödinger software. The four compounds differed in the substituent at the 8-position: hydrogen, bromine, azido, or an amino group. The compounds were docked into the 2PGJ structure using GOLD.58 The binding site was defined as a sphere of 5 Å radius centered on the centroid of the cIDPR ligand: the centroid of the docked ligand has to lie within this sphere. Each ligand was docked 25 times. The best ranked pose of each of the ligands was merged with the protein structure, and the resulting complex was passed through a minimization procedure using the Schrödinger software.
Crystallization, Diffraction Data Collection, and Structure Refinement
The human CD38 catalytic domain was expressed and purified as described previously.45,54 The protein was diluted to 10 mg/mL for crystallization trials. Crystals of wild type CD38 were obtained by the hanging droplet vapor diffusion method with the reservoir buffer in 0.1 M sodium acetate, pH 4.0, 15% PEG 10K, 0.2 M ammonium acetate, and 3% isopropanol. They were harvested and soaked in 0.1 M sodium acetate, pH 4.0, 15% PEG 10K, 16% ethylene glycerol, and 5 mM compound 7 for 1 h at 295 K and then flash-frozen in liquid nitrogen. The diffraction data were collected at 100 K on beamline BL17U at the Shanghai Synchrotron Radiation Facility and processed with HKL2000.59 Molecular replacement was performed using the program Phaser60 from the CCP4 suite,61 and the wild-type human CD38 (PDB code 1YH3) was used as the search model. The model was refined with Refmac62 and then cycled with manual building in Coot.63 Hydrolyzed compound 7 was built into positive difference electron-density maps of the CD38 model after a few restrained refinement runs with the stereochemical restraints generated from the program PRODRG.64 TLS refinement65 was incorporated into the later stages of the refinement process. Solvents were added automatically in Coot and then manually inspected and modified. The final model was analyzed with MolProbity,66 showing that 98% residues were in the Ramachandran favored region with only one residue (D202 in chain B) in the Ramachandran outlier region. Data collection and model refinement statistics are summarized in Supporting Information Table 1. The coordinates and structure factors are deposited in the Protein Data Bank with the code 4TMF.
HPLC Studies
The solution containing the CD38 catalytic domain was adjusted to the desired concentration using Tris-HCl buffer (20 mM, pH 8), and 50 μL therefrom was added to the inhibitor (0.05 μmol) in an Eppendorf tube at room temperature. At a given time point, a sample of 5 μL was removed and diluted with 95 μL of Milli-Q water. Then 10 μL of this sample was injected directly into the analytical HPLC system (see section General in Experimental Section), eluting at 1 mL/min with an isocratic ion-pair buffer: 0.17% (m/v) cetrimide and 45% (v/v) phosphate buffer (pH 6.4) in MeOH.
Acknowledgments
We acknowledge Grants RCGAS 201105159001 (to R.G.), HK-GRF 766911 (to Q.H.), and HK-GRF HKU 785110M (to H.Z.). BVLP is a Wellcome Trust Senior Investigator (Grant 101010).
Glossary
Abbreviations Used
- ADPR
adenosine 5′-diphosphate ribose
- ADPRC
adenosine 5′-diphosphate ribosyl cyclase
- cADPR
cyclic adenosine 5′-diphosphoribose
- cADPcR
cyclic adenosine 5′-diphosphate carbocyclic ribose
- cIDPR
cyclic inosine 5′-diphosphoribose
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NAD+
nicotinamide adenosine 5′-dinucleotide
- NADP
nicotinamide adenine dinucleotide phosphate
- N9-butyl-cIDPR
cyclic-N9-butylhypoxanthine 5′-diphosphate ribose
- shCD38
CD38 catalytic domain
Supporting Information Available
Figures S1–S3, crystallographic data collection, model refinement statistics, HPLC traces, and 1H NMR and 13C NMR spectra for all novel compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The structure of the shCD38:7a complex has been deposited in the PDB with the code 4TMF.
The authors declare no competing financial interest.
Footnotes
Note that for the N9-butyl analogs, the prime notation refers to the N1-ribose, in contrast to the natural products where the prime notation refers to the N9-ribose.
Supplementary Material
References
- Clapper D. L.; Walseth T. F.; Dargie P. J.; Lee H. C. Pyridine-nucleotide metabolites stimulate calcium release from sea-urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [PubMed] [Google Scholar]
- Lee H. C.; Aarhus R.; Levitt D. The crystal structure of cyclic ADP-ribose. Nat. Struct. Biol. 1994, 1, 143–144. [DOI] [PubMed] [Google Scholar]
- Lee H. C. Multiplicity of Ca2+ messengers and Ca2+ stores: a perspective from cyclic ADP-ribose and NAADP. Curr. Mol. Med. 2004, 4, 227–237. [DOI] [PubMed] [Google Scholar]
- Guse A. H. Biochemistry, biology, and pharmacology of cyclic adenosine diphosphoribose (cADPR). Curr. Med. Chem. 2004, 11, 847–855. [DOI] [PubMed] [Google Scholar]
- Potter B. V. L.; Walseth T. F. Medicinal chemistry and pharmacology of cyclic ADP-ribose. Curr. Mol. Med. 2004, 4303–311. [DOI] [PubMed] [Google Scholar]
- Guse A. H. Second messenger function and the structure-activity relationship of cyclic adenosine diphosphoribose (cADPR). FEBS J. 2005, 272, 4590–4597. [DOI] [PubMed] [Google Scholar]
- Zhao Y. J.; Lam C. M. C.; Lee H. C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signaling 2012, 5, ra67. [DOI] [PubMed] [Google Scholar]
- Perraud A. L.; Fleig A.; Dunn C. A.; Bagley L. A.; Launay P.; Schmitz C.; Stokes A. J.; Zhu Q. Q.; Bessman M. J.; Penner R.; Kinet J. P.; Scharenberg A. M. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [DOI] [PubMed] [Google Scholar]
- Kim H.; Jacobson E. L.; Jacobson M. K. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 1993, 261, 1330–1333. [DOI] [PubMed] [Google Scholar]
- Howard M.; Grimaldi J.; Bazan J.; Lund F.; Santos-Argumedo L.; Parkhouse R.; Walseth T.; Lee H. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [DOI] [PubMed] [Google Scholar]
- Cosker F.; Cheviron N.; Yamasaki M.; Menteyne A.; Lund F. E.; Moutin M. J.; Galione A.; Cancela J. M. The ecto-enzyme CD38 is a nicotinic acid adenine dinucleotide phosphate (NAADP) synthase that couples receptor activation to Ca2+ mobilization from lysosomes in pancreatic acinar cells. J. Biol. Chem. 2010, 285, 38251–38259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarino A.; Bensi T.; Chiocchetti A.; Bottarel F.; Mesturini R.; Ferrero E.; Calosso L.; Deaglio S.; Ortolan E.; Buttò S.; Cafaro A.; Katada T.; Ensoli B.; Malavasi F.; Dianzani U. Human CD38 interferes with HIV-1 fusion through a sequence homologous to the V3 loop of the viral envelope glycoprotein gp120. FASEB J. 2003, 17, 461–463. [DOI] [PubMed] [Google Scholar]
- Malavasi F.; Deaglio S.; Damle R.; Cutrona G.; Ferrarini M.; Chiorazzi N. CD38 and chronic lymphocytic leukemia: a decade later. Blood 2011, 118, 3470–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy P.; White T. A.; Thompson M.; Chini E. N. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [DOI] [PubMed] [Google Scholar]
- Young G. S.; Choleris E.; Lund F. E.; Kirkland J. B. Decreased cADPR and increased NAD+ in the Cd38–/– mouse. Biochem. Biophys. Res. Commun. 2006, 346, 188–192. [DOI] [PubMed] [Google Scholar]
- Chini E. N. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D.; Liu H. X.; Hirai H.; Torashima T.; Nagai T.; Lopatina O.; Shnayder N. A.; Yamada K.; Noda M.; Seike T.; Fujita K.; Takasawa S.; Yokoyama S.; Koizumi K.; Shiraishi Y.; Tanaka S.; Hashii M.; Yoshihara T.; Higashida K.; Islam M. S.; Yamada N.; Hayashi K.; Noguchi N.; Kato I.; Okamoto H.; Matsushima A.; Salmina A.; Munesue T.; Shimizu N.; Mochida S.; Asano M.; Higashida H. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 2007, 446, 41–45. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Graeff R. M.; Jin H.; Zhang L.; Zhang L. Studies on CD38 inhibitors and their application to cADPR-mediated Ca2+ signaling. Messenger 2013, 2, 19–32. [Google Scholar]
- Sauve A. A.; Schramm V. L. Mechanism-based inhibitors of CD38: a mammalian cyclic ADP-ribose synthetase. Biochemistry 2002, 41, 8455–8463. [DOI] [PubMed] [Google Scholar]
- Dong M.; Si Y. Q.; Sun S. Y.; Pu X. P.; Yang Z. J.; Zhang L. R.; Zhang L. H.; Leung F. P.; Lam C. M. C.; Kwong A. K. Y.; Yue J. B.; Zhou Y. Y.; Kriksunov I. A.; Hao Q.; Lee H. C. Design, synthesis and biological characterization of novel inhibitors of CD38. Org. Biomol. Chem. 2011, 9, 3246–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall K. A.; Klis M.; Kornet J.; Coyle D.; Amé J. C.; Jacobson M. K.; Slama J. T. Inhibition of the intrinsic NAD+ glycohydrolase activity of CD38 by carbocyclic NAD analogues. Biochem. J. 1998, 335, 631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger E.; Kuhn I.; Schuber F.; Muller-Steffner H. Flavonoids as inhibitors of human CD38. Bioorg. Med. Chem. Lett. 2011, 21, 3939–3942. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Ting K. Y.; Lam C. M. C.; Kwong A. K. Y.; Xia J.; Jin H.; Liu Z.; Zhang L.; Lee H. C.; Zhang L. Design, synthesis and biological evaluation of noncovalent inhibitors of human CD38 NADase. ChemMedChem 2012, 7, 223–228. [DOI] [PubMed] [Google Scholar]
- Sauve A. A.; Deng H. T.; Angeletti R. H.; Schramm V. L. A covalent intermediate in CD38 in responsible for ADP-ribosylation and cyclisation reactions. J. Am. Chem. Soc. 2000, 122, 7855–7859. [Google Scholar]
- Ashamu G. A.; Sethi J. K.; Galione A.; Potter B. V. L. Roles for adenosine ribose hydroxyl groups in cyclic adenosine 5′-diphosphate ribose-mediated Ca2+-release. Biochemistry 1997, 36, 9509–9517. [DOI] [PubMed] [Google Scholar]
- Guse A. H.; Cakir-Kiefer C.; Fukuoka M.; Shuto S.; Weber K.; Bailey V. C.; Matsuda A.; Mayr G. W.; Oppenheimer N.; Schuber F.; Potter B. V. L. Novel hydrolysis-resistant analogues of cyclic ADP-ribose: modification of the "northern" ribose and calcium release activity. Biochemistry 2002, 41, 6744–6751. [DOI] [PubMed] [Google Scholar]
- Bailey V. C.; Fortt S. M.; Summerhill R. J.; Galione A.; Potter B. V. L. Cyclic aristeromycin diphosphate ribose: a potent and poorly hydrolysable Ca2+-mobilising mimic of cyclic adenosine diphosphate ribose. FEBS Lett. 1996, 379, 227–230. [DOI] [PubMed] [Google Scholar]
- Wagner G. K.; Guse A. H.; Potter B. V. L. Rapid synthetic route toward structurally modified derivatives of cyclic adenosine 5′-diphosphate ribose. J. Org. Chem. 2005, 70, 4810–4819. [DOI] [PubMed] [Google Scholar]
- Wagner G. K.; Black S.; Guse A. H.; Potter B. V. L. First enzymatic synthesis of an N1-cyclised cADPR (cyclic ADP-ribose) analogue with a hypoxanthine partial structure: discovery of a membrane permeant cADPR agonist. Chem. Commun. 2003, 1944–1945. [DOI] [PubMed] [Google Scholar]
- Kirchberger T.; Moreau C.; Wagner G. K.; Fliegert R.; Siebrands C. C.; Nebel M.; Schmid F.; Harneit A.; Odoardi F.; Flugel A.; Potter B. V. L.; Guse A. H. 8-Bromo-cyclic inosine diphosphoribose: towards a selective cyclic ADP-ribose agonist. Biochem. J. 2009, 422, 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuto S.; Fukuoka M.; Manikowsky A.; Ueno Y.; Nakano T.; Kuroda R.; Kuroda H.; Matsuda A. Total synthesis of cyclic ADP-carbocyclic ribose, a stable mimic of Ca2+-mobilizing second messenger cyclic ADP-ribose. J. Am. Chem. Soc. 2001, 123, 8750–8759. [DOI] [PubMed] [Google Scholar]
- Shuto S.; Shirato M.; Sumita Y.; Ueno Y.; Matsuda A. Synthesis of cyclic IDP-carbocyclic-ribose, a stable mimic of cyclic ADP-ribose. Significant facilitation of the intramolecular condensation reaction of N1-(carbocyclic-ribosyl)inosine 5′,6″-diphosphate derivatives by an 8-bromo-substitution at the hypoxanthine moiety. J. Org. Chem. 1998, 63, 1986–1994. [Google Scholar]
- Fukuoka M.; Shuto S.; Minakawa N.; Ueno Y.; Matsuda A. An efficient synthesis of cyclic IDP and cyclic 8-bromo-IDP-carbocyclic-riboses using a modified Hata condensation method to form an intramolecular pyrophosphate linkage as a key step. An entry to a general method for the chemical synthesis of cyclic ADP-ribose analogues. J. Org. Chem. 2000, 65, 5238–5248. [DOI] [PubMed] [Google Scholar]
- Gu X.; Yang Z.; Zhang L.; Kunerth S.; Fliegert R.; Weber K.; Guse A. H. Synthesis and biological evaluation of novel membrane-permeant cyclic ADP-ribose mimics: N1-[(5″-O-phosphorylethoxy)methyl]-5′-O-phosphorylinosine-5′,5″-cyclicpyrophosphate (cIDPRE) and 8-substituted derivatives. J. Med. Chem. 2004, 47, 5674–5682. [DOI] [PubMed] [Google Scholar]
- Guse A. H.; Gu X. F.; Zhang L. R.; Weber K.; Kramer E.; Yang Z. J.; Jin H. W.; Li Q.; Carrier L.; Zhang L. H. A minimal structural analogue of cyclic ADP-ribose: synthesis and calcium release activity in mammalian cells. J. Biol. Chem. 2005, 280, 15952–15959. [DOI] [PubMed] [Google Scholar]
- Huang L. J.; Zhao Y. Y.; Yuan L.; Min J. M.; Zhang L. H. Syntheses and calcium-mobilizing evaluations of N1-glycosyl-substituted stable mimics of cyclic ADP-ribose. J. Med. Chem. 2002, 45, 5340–5352. [DOI] [PubMed] [Google Scholar]
- Swarbrick J. M.; Potter B. V. L. Total Synthesis of a cyclic adenosine 5′-diphosphate ribose receptor agonist. J. Org. Chem. 2012, 77, 4191–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeff R. M.; Walseth T. F.; Hill H. K.; Lee H. C. Fluorescent analogs of cyclic ADP-ribose: synthesis, spectral characterization, and use. Biochemistry 1996, 35, 379–386. [DOI] [PubMed] [Google Scholar]
- Graeff R.; Liu Q.; Kriksunov I. A.; Kotaka M.; Oppenheimer N.; Hao Q.; Lee H. C. Mechanism of cyclizing NAD to cyclic ADP-ribose by ADP-ribosyl cyclase and CD38. J. Biol. Chem. 2009, 284, 27629–27636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeff R.; Liu Q.; Kriksunov I. A.; Hao Q.; Lee H. C. Acidic residues at the active sites of CD38 and ADP-ribosyl cyclase determine nicotinic acid adenine dinucleotide phosphate (NAADP) synthesis and hydrolysis activities. J. Biol. Chem. 2006, 281, 28951–28957. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Kriksunov I. A.; Graeff R.; Munshi C.; Lee H. C.; Hao Q. Crystal structure of human CD38 extracellular domain. Structure 2005, 13, 1331–1339. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Kriksunov I. A.; Moreau C.; Graeff R.; Potter B. V. L.; Lee H. C.; Hao Q. Catalysis associated conformational changes revealed by human CD38 complexed with a non-hydrolysable substrate analog. J. Biol. Chem. 2007, 282, 24825–24832. [DOI] [PubMed] [Google Scholar]
- Moreau C.; Liu Q.; Graeff R.; Wagner G. K.; Thomas M. P.; Swarbrick J. M.; Shuto S.; Lee H. C.; Hao Q.; Potter B. V. L. CD38 structure-based inhibitor design using the N1-cyclic inosine 5′-diphosphate ribose template. PLoS One 2013, 8, e66247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeff R. M.; Munshi C.; Aarhus R.; Johns M.; Lee H. C. A single residue at the active site of CD38 determines its NAD cyclizing and hydrolyzing activities. J. Biol. Chem. 2001, 276, 12169–12173. [DOI] [PubMed] [Google Scholar]
- Munshi C.; Aarhus R.; Graeff R.; Walseth T. F.; Levitt D.; Lee H. C. Identification of the enzymatic active site of CD38 by site-directed mutagenesis. J. Biol. Chem. 2000, 275, 21566–21571. [DOI] [PubMed] [Google Scholar]
- Kuhn I.; Kellenberger E.; Cakir-Kiefer C.; Muller-Steffner H.; Schuber F. Probing the catalytic mechanism of bovine CD38/NAD+ glycohydrolase by site directed mutagenesis of key active site residues. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 1317–1331. [DOI] [PubMed] [Google Scholar]
- Moreau C.; Woodman T. J.; Potter B. V. L. Unusual entry to the novel 8-halo-N1-ribosyl hypoxanthine system by degradation of a cyclic adenosine 5′-diphosphate ribose analogue. Chem. Commun. 2006, 1127–1129. [DOI] [PubMed] [Google Scholar]
- Galeone A.; Mayol L.; Oliviero G.; Piccialli G.; Varra M. Synthesis of a novel N1-carbocyclic, N9 butyl analogue of cyclic ADP-ribose (cADPR). Tetrahedron 2002, 58, 363–368. [Google Scholar]
- Volpini R.; Mishra R. C.; Kachare D. D.; Ben D. D.; Lambertucci C.; Antonini I.; Vittori S.; Marucci G.; Sokolova E.; Nistri A.; Cristalli G. Adenine-based acyclic nucleotides as novel P2X3 receptor ligands. J. Med. Chem. 2009, 52, 4596–4603. [DOI] [PubMed] [Google Scholar]
- Laufer S. A.; Domeyer D. M.; Scior T. R. F.; Albrecht W.; Hauser D. R. J. Synthesis and biological testing of purine derivatives as potential ATP-competitive kinase inhibitors. J. Med. Chem. 2005, 48, 710–722. [DOI] [PubMed] [Google Scholar]
- Nolsoe J. M. J.; Gundersen L.-L.; Rise F. Synthesis of 8-halopurines by reaction of lithiated purines with appropriate halogen donors. Synth. Commun. 1998, 28, 4303–4315. [Google Scholar]
- Graeff R. M.; Lee H. C. A novel cycling assay for cellular cADP-ribose with nanomolar sensitivity. Biochem. J. 2002, 361, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Kriksunov I. A.; Graeff R.; Munshi C.; Lee H. C.; Hao Q. Structural basis for the mechanistic understanding of human CD38-controlled multiple catalysis. J. Biol. Chem. 2006, 281, 32861–32869. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Graeff R.; Lee H. C.; Hao Q. Crystal structures of human CD38 in complex with NAADP and ADPRP. Messenger 2013, 2, 44–53. [Google Scholar]
- Sauve A. A.; Munshi C.; Lee H. C.; Schramm V. L. The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry 1998, 37, 13239–13249. [DOI] [PubMed] [Google Scholar]
- Kim H.; Jacobson E. L.; Jacobson M. K. Position of cyclization in cyclic ADP-ribose. Biochem. Biophys. Res. Commun. 1993, 194, 1143–1147. [DOI] [PubMed] [Google Scholar]
- Ames B. N.; Dubin D. T. The role of polyamines in the neutralization of bacteriophage deoxyribonucleic acid. J. Biol. Chem. 1960, 235, 769–775. [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 307–325. [DOI] [PubMed] [Google Scholar]
- McCoy A.; Grosse-Kunstleve R.; Adams P.; Winn M.; Storoni L.; Read R. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Murshudov G.; Vagin A.; Dodson E. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuttelkopf A.; Van Aalten D. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Painter J.; Merritt E. TLSMD Web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 2006, 39, 109–111. [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.