

Abstract
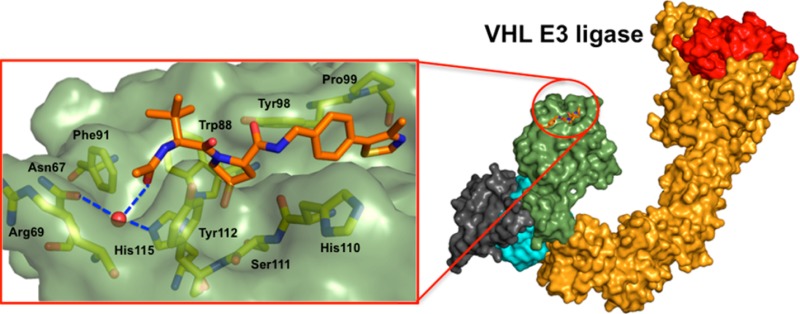
E3 ubiquitin ligases are attractive targets in the ubiquitin–proteasome system, however, the development of small-molecule ligands has been rewarded with limited success. The von Hippel–Lindau protein (pVHL) is the substrate recognition subunit of the VHL E3 ligase that targets HIF-1α for degradation. We recently reported inhibitors of the pVHL:HIF-1α interaction, however they exhibited moderate potency. Herein, we report the design and optimization, guided by X-ray crystal structures, of a ligand series with nanomolar binding affinities.
Introduction
During normal cellular homeostasis, proteins are constantly synthesized and destroyed. The most common degradation pathway for proteins is the ubiquitin–proteasome system (UPS), a highly regulated signaling cascade that is responsible for the controlled degradation of a large number of proteins upon their polyubiquitination and ultimately hydrolysis by the proteasome.1 Dysregulation of this pathway is associated with a large number of diseases, including cancer, neurodegenerative disorders, diabetes, and inflammation.2,3
The therapeutic potential of intervention in the UPS has been demonstrated by the development of proteasome inhibitors such as peptidic boronic acid bortezomid and the epoxyketone carfilzomib for the treatment of relapsed and/or refractory multiple myeloma and mantle cell lymphoma.4−6 Despite these early successes demonstrating chemical validation of the UPS as a drug target, proteasome inhibitors have several limitations including offering no selectivity for the large number of proteins being targeted.
Key to protein degradation by the UPS is the recruitment of the substrate protein by an E3 ubiquitin ligase.7 Ubiquitin ligases confer substrate specificity for ubiquitination and could provide more attractive targets for therapeutic intervention than current proteasome inhibitors, making this unconventional enzyme class most appealing for drug discovery efforts. To date, however, the development of small molecules against E3 ligases has been rewarded with limited success, in part because modulating their activity and regulation requires the targeting of protein–protein interactions (PPIs).8,9
One E3 ubiquitin ligase with important biological relevance where some success has recently been made is the von Hippel–Lindau protein (pVHL) Cullin RING ligase. The primary substrate of pVHL is the hypoxia inducible factor 1α (HIF-1α), a transcription factor that regulates over 2% of human genes,10 particularly those related to oxygen sensing and the hypoxic response.11
Under normal oxygen levels, HIF-1α is constitutively expressed and targeted for proteasomal degradation upon hydroxylation by prolyl hydroxylases domain (PHD) enzymes at Pro402 and Pro564 within its N-terminal oxygen-degradation domain (NODD) and C-terminal oxygen-degradation domain (CODD), respectively, leading to specific recruitment by pVHL and subsequent pVHL-mediated polyubiquination (Figure 1A).12 Small-molecule inhibition of this pathway would be expected to mimic the physiological response to low oxygen levels by increasing the expression of genes involved with the hypoxic response.13 To this end, small-molecule PHD inhibitors are already under examination in clinical trials for the treatment of chronic anemia associated with chronic kidney disease and cancer chemotherapy. Three PHDs isozymes are known (PHD1, PHD2, and PHD3) with different substrate specificities and cellular and tissular localization. Therefore, isoform selectivity would be desired to avoid unwanted side effects such as hematopoiesis or vasculogenesis. Unfortunately, none of the current clinical compounds show sufficient PHD isozyme selectivity.14
Figure 1.
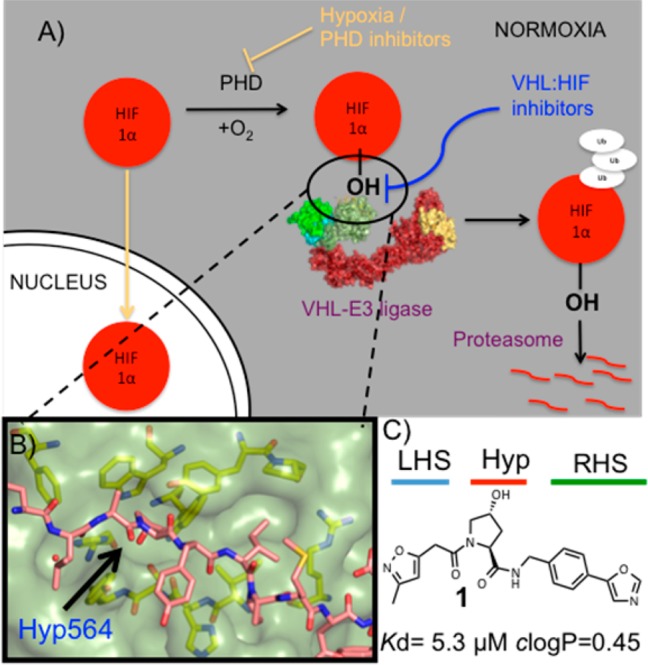
(A) Under normoxic conditions, HIF-1α is hydroxylated, recognized by VHL-E3 ligase, ubiquitinated, and degraded by the proteasome. (B) X-ray structure of VBC (pVHL–ElonginB–ElonginC complex) with the HIF-1α peptide bound (pink carbons, PDB 4AJY).17 (C) Example of first-generation pVHL ligand described (ligand 1).16
The development of a potent inhibitor of the pVHL:HIF-1α interaction provides an alternative approach to PHD inhibitors by allowing chemical intervention in the HIF pathway downstream of the PHD enzymes (Figure 1A). Such an approach may avoid the HIF-independent off-target effects observed with PHD inhibitors, thus providing a new class of lead compounds against chronic anemia as well as acute ischemic disorders where rapid revascularization driven by HIF-1α has proven to be beneficial.15
To validate the target chemically and to establish the functional and biological consequences of modulating pVHL activity, small molecules would have to bind to pVHL with sufficient potency as to be able to compete with HIF-1α inside the cell. Recently, a series of ligands were reported by us and collaborators as small-molecule inhibitors of the pVHL:HIF-1α interaction.16−19 The ligands were designed using the structure of bound HIF-1α peptide as a starting point and growing around the key hydroxyproline (Hyp) group to fill the left-hand side (LHS) and right-hand side (RHS) of the PPI surface (Figure 1B,C). These molecules bound to pVHL with moderate potency, the best only in the single-digit micromolar range, and exhibited very low lipophilicity due to the high hydrophilic nature of the Hyp core scaffold. Herein, following a metrics-, structure-, and isothermal titration calorimetry (ITC)-guided design strategy, we describe how we have optimized the pVHL:HIF-1α inhibitor ligand series by improving binding affinity and lipophilicity, both being essential considerations for designing effective and cell-active chemical probes.20
Results and Discussion
In a previous fragment-based design study using group efficiency (GE) and group lipophilicity efficiency (GLE), we observed that a t-butyl group exhibits highest GE values among the groups tested at LHS.17 Further, we showed that the LHS subpocket responded most favorably to a t-butyl group in terms of GLE values among all the other PPI subpockets.17 On the basis of these observations, and on knowledge of the importance of Hyp in the molecular recognition, we decided to select t-butyl-Hyp as a starting anchor ligand, with a balanced lipophilicity, for the ligand design. Ligand 2, bearing a 4-(oxazol-5-yl) benzyl group at the RHS as employed in 1 (Figure 1C), yielded a Kd of 22 μM by ITC (Table 1). X-ray crystal structure of the pVHL–ElonginB–ElonginC complex (VBC) with 2 bound (Figure 2) revealed that the t-butyl group points upward to make hydrophobic contacts with Phe91 and Trp88 instead of pointing toward the structural water within the LHS pocket formed by Asn67, Arg69, and His115, as does the 3-methylisoxazole fragment of 1 and earlier ligands.16,17
Table 1. Structures, Kds, and ΔH Determined by ITC, LEs, and Calculated log Ps23.
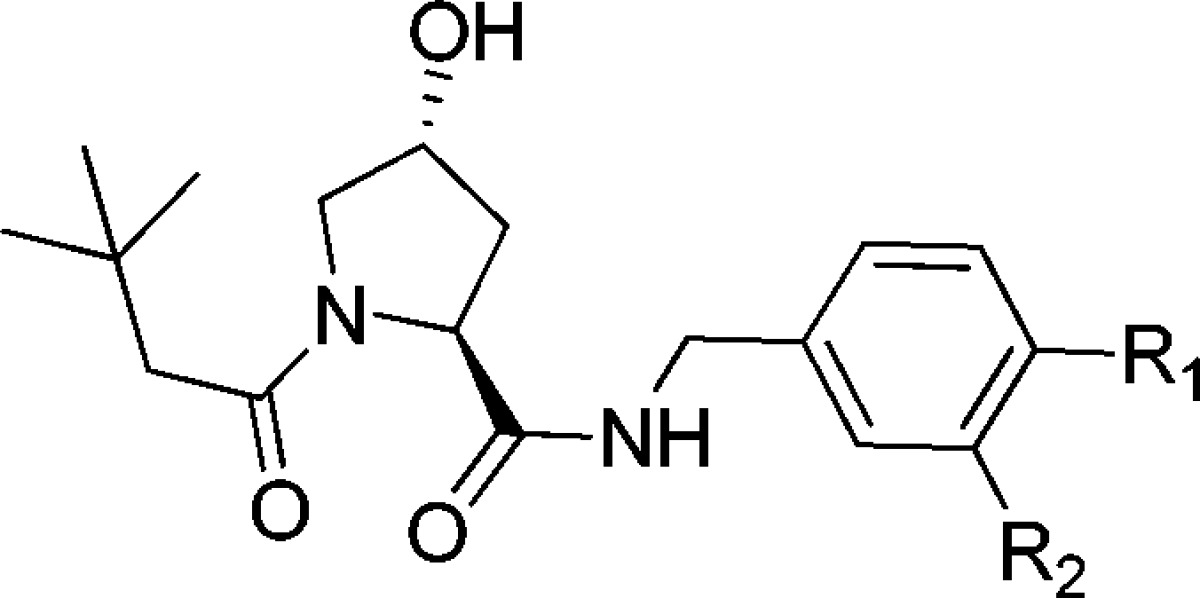
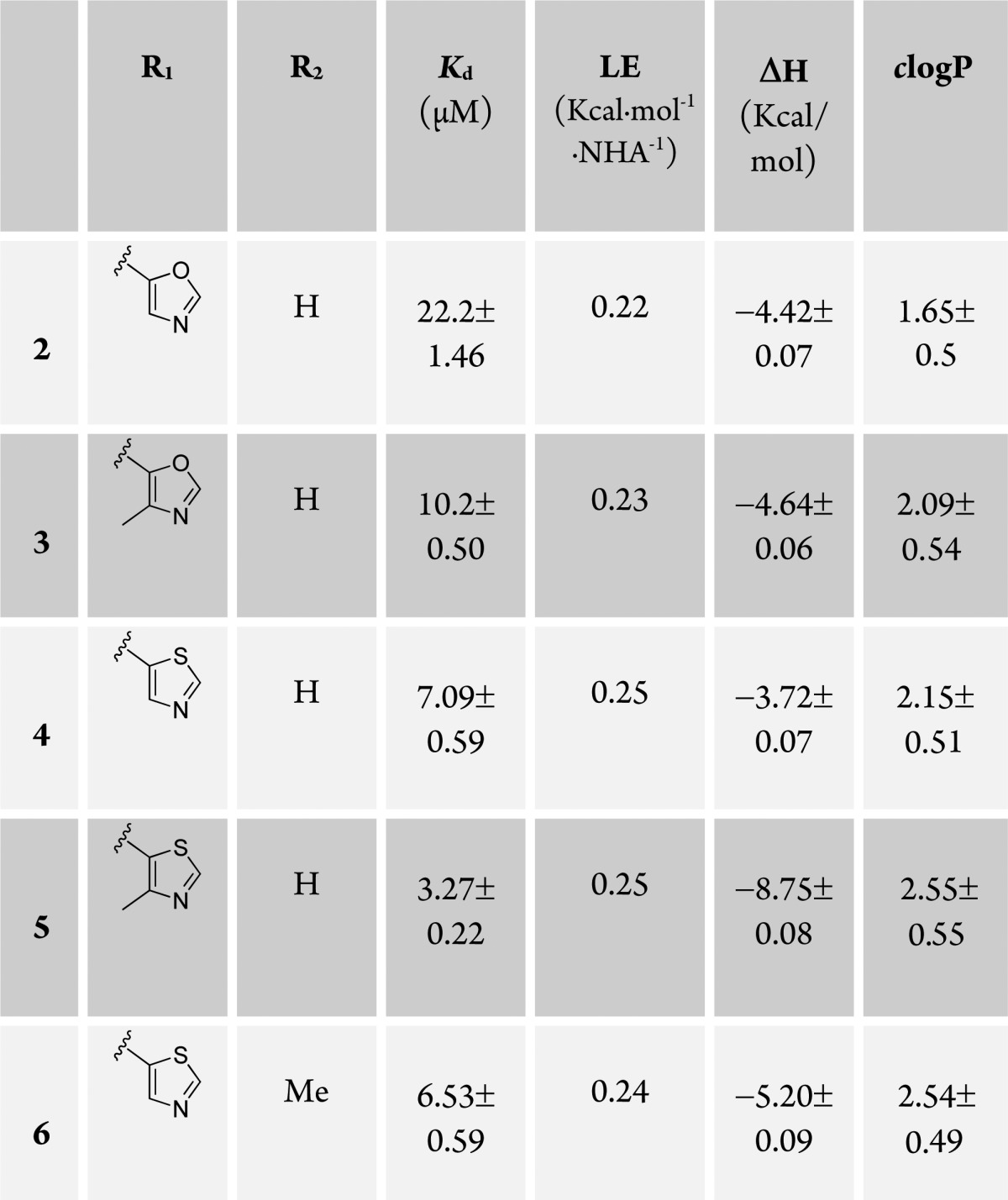
Figure 2.
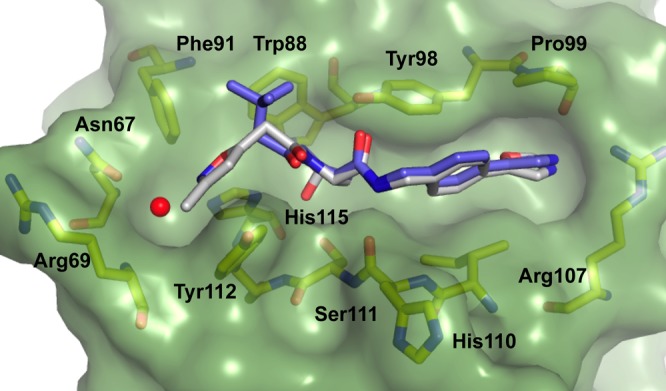
Crystal structure of VBC in complex with ligand 1 (gray carbons, PDB 3ZRC)16 and 2 (purple carbons, PDB 4W9C). pVHL is shown as a pale-green surface and the pVHL residues forming the binding pocket as yellow stick representations. The structural water at the LHS is shown as red sphere.
The unexpected binding mode of the t-butyl group was observed consistently in all four VBC protomers present in the asymmetric unit (see Supporting Information (SI) Table 1 for X-ray data processing and refinement and SI Figure 1 for ligands electron densities). The Hyp-NH-4-(oxazol-5-yl)benzyl portion of the ligand is bound as previously observed for 1, forming key hydrogen bond contacts between the Hyp-OH and His115 and Ser111 side chains, the ligand RHS amide and His110 backbone carbonyl and the side chain −OH of Tyr98, and the oxazoyl ring C-2 hydrogen and nitrogen atoms with the Pro99 backbone carbonyl and the Arg107 side chain, respectively. Notably, we observed a wide range of dihedral angles around the phenyl–oxazoyl C–C bond at the RHS of ligands 1 and 2 bound to the four liganded pVHL protomers in the crystal structures (range 24–54°). Comparison of these values with the predicted lowest-energy conformation dihedral angle calculated for this system, 0° (Macromodel/Jaguar from Schrödinger package,21SI Figure 2), suggested that the bound conformation of the biaryl at the RHS would be energetically unfavorable, so this part of the ligand warranted further optimization.
We next sought to investigate more conformationally constrained 5-membered oxazoyl and thiazoyl heteroaromatic groups and their respective 4-methyl derivatives at the para- position of the aryl ring as binding groups at the RHS (Table 1, ligands 3–5). The 4-methylthiazole substituted ligand 5, the most potent ligand of this series, yielded a Kd of 3.3 μM by ITC, with slighty better potency and ligand efficiency (LE, defined as binding energy relative to the number of heavy atoms NHA, LE = −AG/NHA = −RT ln Kd/NHA) than ligand 1, bearing the 3-methylisoxazole instead of t-butyl as the LHS group (Kd = 5.3 μM, LE = 0.24, Figure 1C).16,17 In the case of both oxazole and thiazole, methylation at position 4 of the heterocycle consistently increased the binding affinity, Kds 10.2 vs 22.2 μM and 3.3 vs 7.1 μM, respectively (Table 1).
To rationalize the observed SAR and confirm also the binding mode of the t-butyl-based ligands, X-ray crystal structures of VBC with bound ligands 3–5 were also obtained by ligand soaking and solved to 2.1–2.6 Å resolution (SI Figure 3). Remarkably, the aryl–aryl dihedral angle in the ligands (3–5) in the X-ray structures was consistently found at 38° ± 4, suggesting that this bound conformation maximizes optimal interactions at this end of the RHS pocket. In particular the methylthiazole of 5 binds to pVHL in the RHS of the pocket without the introduction of any strain in the dihedral angle (calculated, 43°; observed, 43°, SI Figure 2). Taken together, our results suggest good agreement between potency and the extent of preorganization of ligands in their bound conformation.
We next sought to obtain a ligand bearing a thiazole five-membered ring and methyl substitution at position 3 of the phenyl ring. Indeed, 6 bound in a conformation where no significant strain had to be introduced to twist the biaryl (calculated, 42°; average-observed, 43°). Although ligand 6 has a one-digit micromolar activity, it did not show a significant increase in potency compared to 4.
In all crystal structures of ligands 3–6 (SI Figure 3), the t-butyl group retains the binding mode observed for 2, reinforcing its role as a good LHS fragment. In all of the different five-membered rings, the hydrogen at position 2 of the heterocycle at the RHS forms an aromatic C–H···O=C hydrogen bond with the carbonyl group of Pro99, and the nitrogen atom of the heterocyclic ring engages the side chain of Arg107 via either a direct or a water-mediated hydrogen bond. Importantly for this series, the introduction of the lipophilic t-butyl group increases the clogP of the ligands, bringing it into a more optimal range for further optimization (Table 1).
Overlaying of the crystal structures of ligands 2–6 bound to pVHL with that of the HIF-1α peptide revealed that the −CH2 and quaternary carbons of the t-butyl group overlap very well with Cα and Cβ of the Ala563 residue of the HIF-1α peptide, respectively (SI Figure 4). This observation suggested an attractive vector to grow the compounds into the empty pocket at the LHS that is occupied by Leu562 of the peptide by mimicking the peptidic backbone structure. We therefore synthesized 7 that adds an acetamido group to ligand 5, the best binder of the previous series, by coupling a l-tert-leucine amino acid to the Hyp-RHS core and then N-terminally acetylating the resulting product. Gratifyingly, 7 showed a Kd of 185 nM as determined by ITC (LE = 0.28), an almost 20-fold increase in affinity compared to 5, affording a ligand that is even more potent than the 10-mer model HIF-1α peptide (Kd = 200 nM, LE = 0.12). The crystal structure of 7 bound to VBC (Figure 3) confirmed the expected interaction of the carbonyl group with the structural water at the LHS pocket, recapitulating well the interaction observed with the HIF peptide while the amido NH points outward toward the solvent.
Figure 3.

Crystal structure of VBC in complex with the ligand 7 (orange carbons, PDB 4W9H). Hydrogen bonds with the structural water (red sphere) are shown as blue-dashed lines. pVHL is shown as a pale-green surface and pVHL residues forming the binding pocket as yellow sticks.
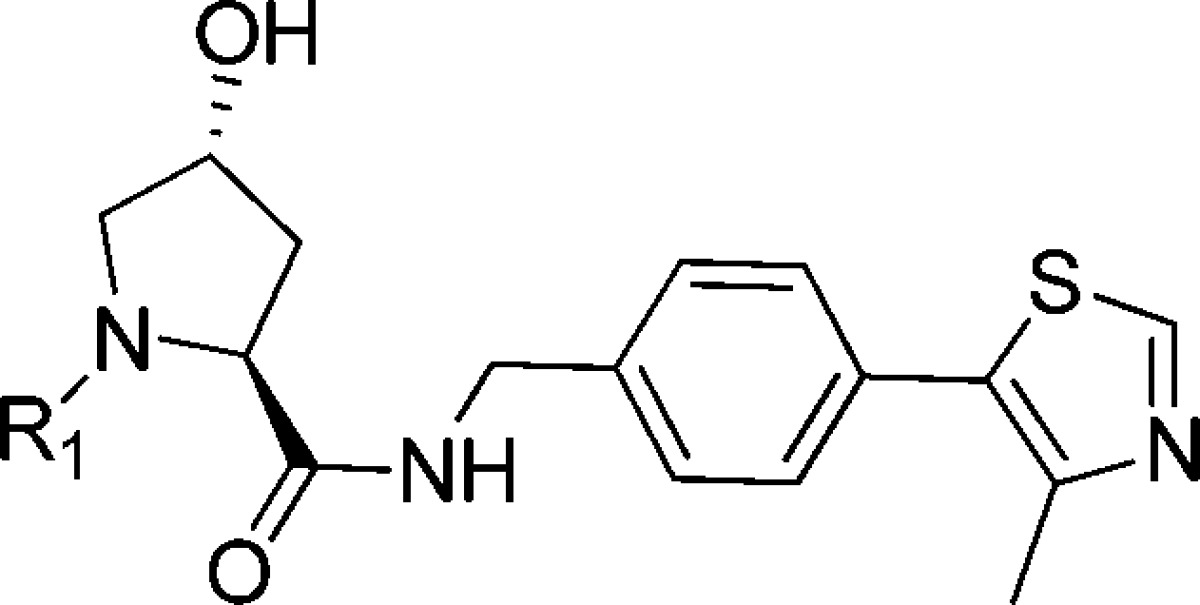
To assess the importance of the t-butyl group of 7 for the molecular recognition, we asked whether groups with different steric requirements and lipophilicities could substitute for it. We therefore synthesized analogues replacing the tert-leucine moiety of 7 with phenylglycine (8) as well as the more conformationally constrained five-membered ring proline (9) and hydroxyproline (10) and the six-membered ring pipecolic acid (11) to achieve cyclizing of the side chain with the solvent-exposed amido nitrogen. All analogues exhibited Kd values of around or below 1 μM, but weaker binding affinities when compared to 7 (Table 2), presumably the result of unfavorable steric constraints. Ligand 11 with a more relaxed six-membered ring was the best binder among this new series. The importance of steric constraints at this position was confirmed by solving the X-ray crystal structure of the hydroxyproline analogue 10 bound to VBC, which showed that the five-membered ring twists the amide bond, resulting in a less favorable interaction with the structural water (SI Figure 5). An interesting feature of this structure is the formation of water-mediated hydrogen bonds between the Hyp-OH in the LHS and the side chain of Gln96, consistent with similar interactions observed with previous ligands.18 This new interaction may be reflected in the larger ΔH contribution to binding observed for 10 with respect to other ligands in the series.
Table 2. Structures, Kds and ΔH Determined by ITC, LEs, and Calculated log Ps23.

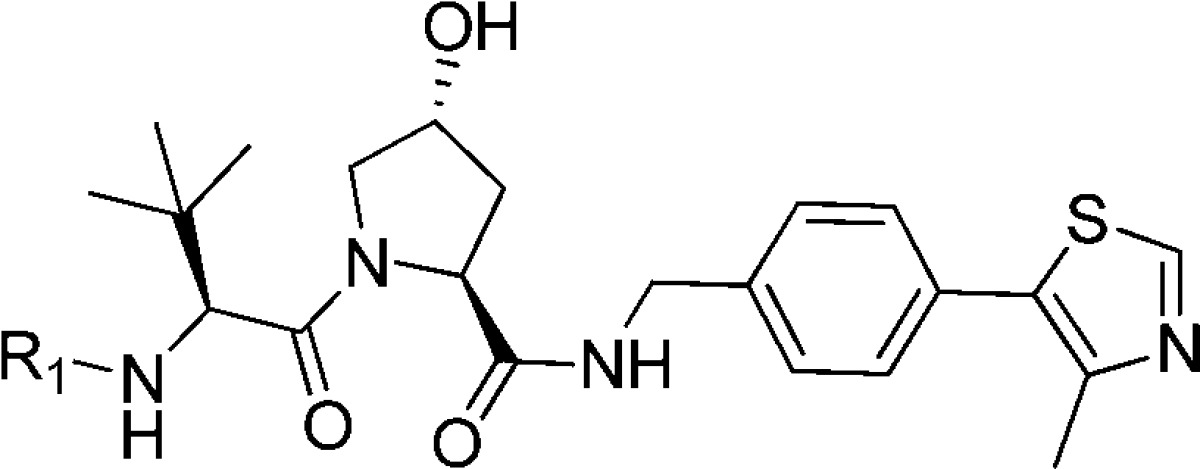
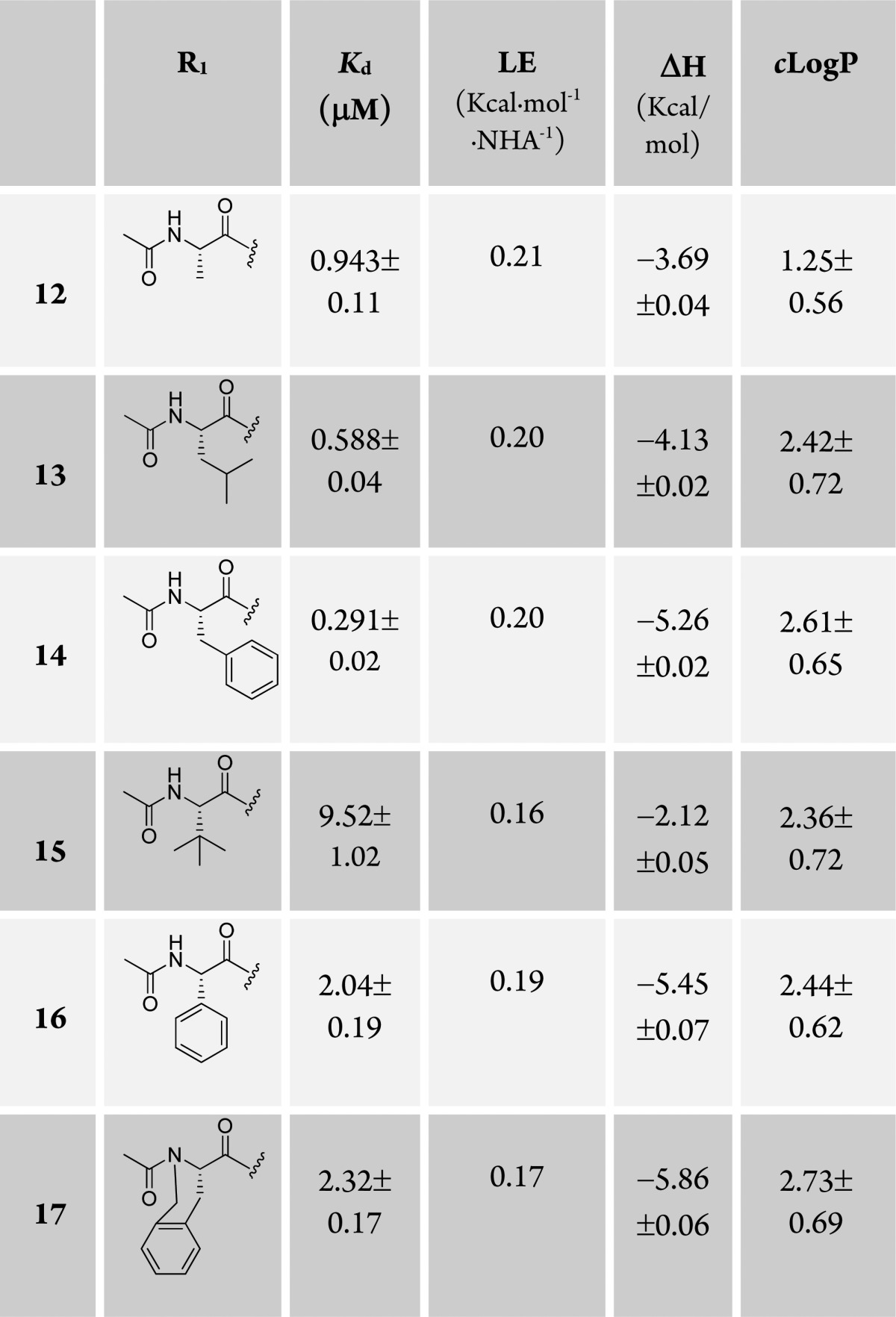
Having established the t-butyl group as the best fragment for the LHS1 pocket (from this point, we define LHS1 as the surface bound by the t-butyl group and LHS2 as the surface where the structural water remains in place, filled by Leu562 of the HIF-1α peptide), we next designed and synthesized a set of ligands growing away from the terminal acetyl group of 7, further mimicking the structure of the bound HIF peptide at the LHS2 pocket. We therefore coupled a range of N-acetylated natural and unnatural l-amino acids to the LHS1 t-butyl group, including alanine (12), leucine (13), phenylalanine (14), tert-leucine (15), phenylglycine (16), and a cyclized phenylalanine (17) (Table 3). When we attached a bulky alkyl chain directly to the backbone (15, 16), a drop in potency was observed, probably due to steric clashes with the protein. Ligand 13, mimicking the Leu562 of the HIF-1α peptide backbone, and the alanine-substituted ligand 12 yielded nanomolar affinity to pVHL. The most potent compound of this series was the phenylalanine-substituted ligand (14, Kd = 290 nM, LE = 0.20). Unfortunately, the conformationally constrained ligand 17 did not yield an increase in the affinity.
Table 3. Structures, Kds, and ΔH Determined by ITC, LEs, and Calculated log Ps23.
To gain insight into the molecular determinants that modulate the range of activities determined for the ligands of this series, crystal structures of VBC bound to 13, 14, and 15 were solved. The structures showed that the ligands maintain many of the previously highlighted interactions; introduction of these new groups at the LHS2 did not affect the binding mode of the remaining parts of the molecules. The aliphatic alkyl chains in ligands 13 (Figure 4A) and 15 (SI Figure 6) overlap well with the conformation that the Leu562 has in the natural HIF-1α peptide, however, in the case of 15, steric clashes with the Tyr112 of the protein were observed, resulting in loss of affinity. Interestingly, we observed significant conformational changes in the LHS2 of the protein to accommodate the aryl phenylalanine side chain of 14 (Figure 4B). Ligand 13 makes bidentate hydrogen bonds from the terminal acetamido with the Asn67 side chain, however, these interactions are lost in the case of the ligand 14 due to rearrangement of the terminal Ac-Phe portion of the ligand.
Figure 4.
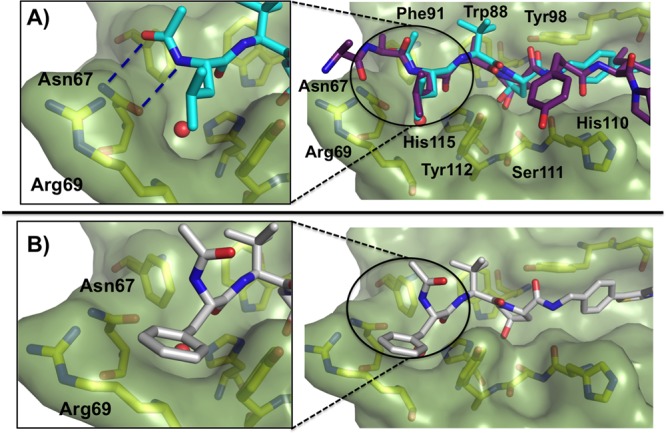
Crystal structure of the VBC in complex with (A) ligand 13 (cyan carbons, PDB 4W9J) with bound HIF-1α peptide superposed and (B) ligand 14 (gray carbons, PDB 4W9K). Hydrogen bonds are shown as blue-dashed lines. pVHL is shown as a pale-green surface and the pVHL residues forming the binding pocket as yellow sticks.
Although none of the LHS2-targeting ligands increased the affinity toward the pVHL when compared to 7, they have revealed new ligand binding modes and pocket flexibility at this part of the PPI that open up opportunities to explore further optimization of best-in-class ligand 7 within this region. Extending ligands further at LHS2 could not only improve affinity of current ligands but also add to the arsenal of linkable pVHL ligands for proteolysis-targeting chimeras (PROTACs) small-molecule approaches.22
Conclusions
Because of a historical paucity of conventional drug discovery approaches to effectively target protein–protein interactions, only a few E3 ubiquitin ligases have been successfully modulated to date and the field is still in its infancy. We describe the structure-based design, synthesis, and optimization of next-generation small molecules targeting pVHL, an important E3 ligase and a potential drug target, with binding affinities in the nanomolar range and improved lipophilicity. X-ray crystal structures and ITC studies reveal new interactions on the left-hand side of the pocket and elucidate the structural and thermodynamic determinants for the improvement in binding affinities, thus providing crucial information for future ligand optimization campaigns. Our best ligand 7, to our knowledge the most potent pVHL:HIF-1α inhibitor described to date, is more potent than a model 10-mer HIF-1α peptide, providing an excellent starting point to validate VHL as a drug target. Work is presently ongoing in our laboratory to assess the cellular activity and bioavailability of these ligands. Moreover, our group analysis has proven to be a useful tool to guide the optimization of future pVHL ligands. Our successful approach points out the importance of a parallel structure-driven and metrics-driven design to understand and modulate challenging PPIs, opening new opportunities for targeting other members of the family of E3 enzymes considered too risky for the pharmaceutical industry until this moment.
Experimental Section
Chemistry
General directions are in the SI. For each ligand, a full description of the synthetic protocols and spectroscopic analysis can be found in the SI. All of the tested ligands (2–17) were evaluated after the corresponding flash column chromatography purification, followed by preparative LC-MS purification. The purity of all compounds was analyzed by HPLC-MS (ESI) and is >95%. The synthetic procedure for the preparation of new ligands described herein is exemplified through the synthesis of 7 (Scheme 1).
Scheme 1. Synthesis of the Ligand 7.

Conditions: (i) Pd(OAc)2, KOAc, DMAc, reflux, O/N; (ii) NaBH4, CoCl2, MeOH, 0 °C, 90 min; (iii) Boc-Hyp-OH, DIPEA, HATU, DMF, rt, 30 min; (iv) TFA/DCM 1:1, rt, 30 min; (v) Boc-l-tert-leucine, DIPEA, HATU, DMF, rt, 30 min; (vi) TFA/DCM 1:1, rt, 30 min; (vii) acetic anhydride, N(Et)3, DCM, rt, 90 min.
General Method A
To a solution of amine (1 equiv) in DMF was added the appropriate acid (1 equiv), and the solution was stirred at room temperature. DIPEA (4 equiv) was added dropwise, and the mixture was stirred for 5 min at room temperature. HATU (1.1 equiv) was added, and the mixture was stirred at room temperature for another 30 min. Water was added, and the mixture was extracted with ethyl acetate (3×). The combined organic phases were washed with brine (×2), dried over MgSO4, and evaporated under reduced pressure to give the corresponding crude, which was purified by flash column chromatography purification to yield the final compound.
General Method B
The corresponding NH2 deprotected amino acid derivate (1 equiv) was dissolved in DCM, and triethylamine (3 equiv) was added to the solution. After stirring the mixture for 10 min at room temperature, acetic anhydride (1.5 equiv) was added and the reaction was stirred 90 min at room temperature. The solvents were evaporated under reduced pressure to give the corresponding crude, which was purified by flash column chromatography to yield the final compound.
(4-(4-Methylthiazol-5-yl)phenyl)methanamine (18)
To a solution of 4-bromobenzonitrile (1.5 g, 8.24 mmol, 1 equiv) and Pd(OAc)2 (2 mg, 0.08 mmol, 0.1 mol %) in DMAc (8 mL) were added KOAc (1.62 g, 16.5 mmol, 2 equiv) and 4-methylthiazole (1.63 g, 1.49 mL, 16.5 mmol, 2 equiv). The resulting mixture was heated to 150 °C and stirred overnight. The mixture was diluted with water and extracted with DCM (3×). The combined organic phases were dried over MgSO4 and evaporated under reduced pressure to give the corresponding cyano derivate as a beige solid (1.67 g, 7.99 mmol, 97%) that matched the reported spectral data.17 A solution of the cyano derivate product (270 mg, 1.3 mmol, 1 equiv) in methanol (15 mL) was cooled to 0 °C. CoCl2 (282 mg, 2.2 mmol, 1.5 equiv) was added, followed by portionwise addition of NaBH4 (274 mg, 7.2 mmol, 5 equiv). The resulting mixture was stirred for 90 min, the reaction was quenched with water and ammonium hydroxide, and the mixture was extracted with chloroform (6×). The combined organic phases were dried over MgSO4 and evaporated under reduced pressure to give a dark-brown oil which was purified by flash column chromatography to yield the product 18 as a yellow oil (76.5 mg, 0.40 mmol, 29% (isolated)) that matched the reported spectral data.18 MS (ESI): [M + 1] calculated 205.1; observed 188.1 (fragmentation).
(2S,4R)-tert-Butyl-4-hydroxy-2-((4-(4-methyl thiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carboxylate (19)
Following the general method A, from 18 (340 mg, 1.66 mmol, 1 equiv) and Boc-Hyp-OH (383 mg, 1.66 mmol, 1 equiv), ligand 19 was obtained as a yellow solid (658 mg, 1.58 mmol, 95%) that matched the reported spectral data.18 MS (ESI): [M + 1] calculated 418.2; observed 418.2.
tert-Butyl((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazole-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (20)
A solution of ligand 19 (340 mg, 0.81 mmol) in 1:1 TFA:DCM (8 mL) was stirred at room temperature for 30 min. The mixture was evaporated under reduced pressure to give the corresponding deprotected intermediate (TFA salt) as a brown oil without further purification (330 mg, 0.77 mmol, 98%) that matched the reported spectral data.18 Following the general method A, from the deprotected intermediate (330 mg, 0.77 mmol, 1 equiv) and Boc-l-tert-leucine (178 mg, 0.77 mmol, 1 equiv), ligand 20 was obtained as a yellow solid (400 mg, 0.75 mmol, 98%), which was used directly for the next step. MS (ESI): [M + 1] calculated 531.3; observed 531.3.
(2S,4R)-1-((S)-2-Acetamido-3,3-dimethyl butanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl) benzyl)pyrrolidine-2-carboxamide (7)
A solution of ligand 20 (257 mg, 0.48 mmol) in 1:1 TFA:DCM (5 mL) was stirred at room temperature for 30 min. The mixture was evaporated under reduced pressure to give the corresponding intermediate (TFA salt), which was used directly for the next step. Following the general method B, from the corresponding deprotected amine (TFA salt, 275 mg, 0.50 mmol, 1 equiv), ligand 7 was obtained as colorless oil (65.3 mg, 0.14 mmol, 27%). 1H NMR (CDCl3, 500 MHz): δ 8.67 (s, 1H), 7.35 (dd, J = 15.0, 10.0 Hz, 4H), 4.71 (t, J = 10.0 Hz, 1H), 4.56–4.48 (m, 3H), 4.33 (dd, J = 15.0, 10.0, 1H), 4.07 (d, J = 10 Hz, 1H), 3.60 (dd, J = 10.0, 5.0 Hz, 1H), 2.60 (s, 2H), 2.50 (s, 3H), 2.14–2.10 (m, 1H), 1.98 (s, 3H), 0.93 (s, 9H). 13C NMR (CDCl3, 125 MHz): δ 172.1, 170.9, 170.8, 150.4, 148.7, 138.2, 131.8, 131.2, 129.7, 128.3, 70.2, 58.6, 57.7, 56.8, 45.7, 43.2, 36.0, 35.0, 26.5, 8.6. HRMS (ESI) m/z: [M + 1] calculated for C24H33N4O4S: 473.2222; observed 473.2211.
Acknowledgments
This work was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC BB/G023123/1, David Phillips Fellowship to A.C.), the European Research Council ERC-2012-StG-311460 DrugE3CRLs (starting grant to A.C.), the EC PIEF-GA-2012-328030 (Marie-Curie Intra-European Fellowship to C.G.) and EC PIEF-GA-2010-275683 (Marie-Curie Intra-European Fellowship to I.V.M.), the Fundação para a Ciência e a Tecnologia (FCT, SFRH/BD/81735/2011 studentship to D.M.D.), and the Wellcome Trust (100476/Z/12/Z for biophysics and drug discovery and 094090/Z/10/Z for structural biology and X-ray crystallography to Dundee). S.S. was partially supported by the Italian Ministry of Education, University and Research (MIUR) grant “Messaggeri della Conoscenza” ID 497. We are thankful to Dr. Lars Sansberg for help with computational calculations, Martina Casale for help with VBC protein expression, and Dr. Paul Fyfe for support with in-house X-ray facility. We are also thankful to Diamond Light Source for beamtime (proposal mx8268) and beamline support at I03 and I04 and the European Synchrotron Radiation Facility for beamtime (proposal mx1481) and beamline support at ID14-4 and BM14.
Glossary
Abbreviations Used
- CODD
C-terminal oxygen-degradation domain
- GE
group efficiency
- GLE
group lipophilicity efficiency
- HIF-1α
hypoxia inducible factor 1 alpha
- ITC
isothermal titration calorimetry
- LE
ligand efficiency
- LHS
left-hand side
- NODD
N-terminal oxygen-degradation domain
- O/N
overnight
- PHD
prolyl hydroxylase domain
- PPI
protein–protein interaction
- RHS
right-hand side
- SAR
structure–activity relationships
- UPS
ubiquitin–proteasome system
- VBC
VHL-EloB-EloC
- pVHL
von Hippel–Lindau protein
Supporting Information Available
Supplementary figures and tables, biochemical methods, crystallographic refinement data, ITC data, computational methods, synthetic schemes, and synthesis and characterization of organic molecules. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
PDB accession codes of VBC in complex with 2, 3, 4, 5, 6, 7, 10, 13, 14, and 15 are 4W9C, 4W9D, 4W9E, 4W9F, 4W9G, 4W9H, 4W9I, 4W9J, 4W9K, and 4W9L, respectively.
Author Present Address
§ I.V.M.: Structural Biology Research Center, VIB, Structural Biology Brussels Laboratory, Vrije Universiteit Brussel and Brussels Center for Redox Biology, 1050 Brussels, Belgium.
Author Present Address
∥ S.H.: School of Chemistry, University of Leeds, Leeds, LS2 9JT, UK.
Author Contributions
C.G. and M.S.G. contributed equally. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ciechanover A. The Ubiquitin–Proteasome Proteolytic Pathway. Cell 1994, 79, 13–21. [DOI] [PubMed] [Google Scholar]
- Nalepa G.; Rolfe M.; Harper J. W. Drug Discovery in the Ubiquitin–Proteasome System. Nature Rev. Drug Discovery 2006, 5, 596–613. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Sidhu S. S. Development of Inhibitors in the Ubiquitination Cascade. FEBS Lett. 2014, 588, 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. The Proteasome: A Suitable Antineoplastic Target. Nature Rev. Cancer 2004, 4, 349–360. [DOI] [PubMed] [Google Scholar]
- Demo S. D.; Kirk C. J.; Aujay M. A.; Buchholz T. J.; Dajee M.; Ho M. N.; Jiang J.; Laidig G. J.; Lewis E. R.; Parlati F.; Shenk K. D.; Smyth M. S.; Sun C. M.; Vallone M. K.; Woo T. M.; Molineaux C. J.; Bennett M. K. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res. 2007, 67, 6383–6391. [DOI] [PubMed] [Google Scholar]
- For a review of proteasome inhibitors:Rentsch A.; Landsberg D.; Brodmann T.; Bülow L.; Girbig A.-K.; Kalesse M. Synthesis and Pharmacology of Proteasome Inhibitors. Angew. Chem., Int. Ed. 2013, 52, 5450–5488. [DOI] [PubMed] [Google Scholar]
- Cohen P.; Tcherpakov M. Will the Ubiquitin System Furnish as Many Drug Targets as Protein Kinases?. Cell 2010, 143, 686–693. [DOI] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. Reaching for High-Hanging Fruit in Drug Discovery at Protein–Protein Interfaces. Nature 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Pellecchia M. Antagonist of Protein–Protein Interactions Made Easy?. J. Med. Chem. 2013, 56, 13–14. [DOI] [PubMed] [Google Scholar]
- Manalo D. J.; Rowan A.; Lavoie T.; Natarajan T.; Kelly B. D.; Ye S. Q.; Garcia J. G.; Semenza G. L. Transcriptional Regulation of Vascular Endothelial Cell Responses to Hypoxia by HIF-1. Blood 2005, 105, 659–669. [DOI] [PubMed] [Google Scholar]
- Semenza G.L. Life with oxygen. Science 2007, 318, 62–64. [DOI] [PubMed] [Google Scholar]
- Hon W. C.; Wilson M. I.; Harlos K.; Claridge T. D.; Schofield C. J.; Pugh C. W.; Maxwell P. H.; Ratcliffe P. J.; Stuart D. I.; Jones E. Y. Structural Basis for the Recognition of Hydroxyproline in HIF-1alpha by VHL. Nature 2002, 417, 975–978. [DOI] [PubMed] [Google Scholar]
- Louie R. J.; Padró M.; Giaccia A. J.; Chan D. A.. Small Molecules Targeting the VHL/hypoxic Phenotype. In Hypoxia and Cancer: Biological Implications and Therapeutic Opportunities; Melillo G., Ed.; Springer Science+Business Media: New York, 2014: pp 253–264. [Google Scholar]
- Rabinowitz M. H. Inhibition of Hypoxia-Inducible Factor Prolyl Hydroxylase Domain Oxygen Sensors: Tricking the Body into Mounting Orchestrated Survival and Repair Responses. J. Med. Chem. 2013, 56, 9369–9402. [DOI] [PubMed] [Google Scholar]
- Muchnik E.; Kaplan J. HIF Prolyl Hydroxylase Inhibitors for Anemia. Expert Opin. Invest. Drugs 2011, 20, 545–656. [DOI] [PubMed] [Google Scholar]
- Buckley D. L.; Van Molle I.; Gareiss P. C.; Tae H. S.; Michel J.; Noblin D. J.; Jorgensen W. L.; Ciulli A.; Crews C. M. Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules to Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Molle I.; Thomann A.; Buckley D. L.; So E. C.; Lang S.; Crews C. M.; Ciulli A. Dissecting Fragment-Based Lead Discovery at the Von Hippel–Lindau Protein:Hypoxia Inducible Factor 1α Protein–Protein Interface. Chem. Biol. 2012, 19, 1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Gustafson J. L.; Van Molle I.; Roth A. G.; Tae H. S.; Gareiss P. C.; Jorgensen W. L.; Ciulli A.; Crews C. M. Small Molecules Inhibitors of the Interaction Between the E3 Ligase VHL and HIF1α. Angew. Chem., Int. Ed. 2012, 51, 11463–11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias D. M.; Van Molle I.; Baud M. G. J.; Galdeano C.; Geraldes C. F. G. C.; Ciulli A. Is NMR Fragment Screening Fine-Tuned to Assess Druggability of Protein–Protein Interactions?. ACS Med. Chem. Lett. 2013, 5, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnage M. E.; Chekler E. L. P.; Jones L. H. Target Validation Using Chemical Probes. Nature Chem. Biol. 2013, 9, 195–199. [DOI] [PubMed] [Google Scholar]
- Bochevarov A. D.; Harder E.; Hughes T. F.; Greenwood J. R.; Braden D. A.; Philipp D. M.; Rinaldo D.; Halls M. D.; Zhang J.; Friesner R. A. Jaguar: A High-Performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar]
- Buckley D. L.; Crews C. M. Small-Molecule Control of Intracellular Protein Levels Through Modulation of the Ubiquitin Proteasome System. Angew. Chem., Int. Ed. 2014, 53, 2312–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VCCLAB, Virtual Computational Chemistry Laboratory; http://www.vcclab.org, 2005. For more information: Tetko I. V.; Gasteiger J.; Todeschini R.; Mauri A.; Livingstone D.; Ertl P.; Palyulin V. A.; Radchenko E. V.; Zefirov N. S.; Makarenko A. S.; Tanchuk V. Y.; Prokopenko V. V.. Virtual Computational Chemistry Laboratory—Design and Description, J. Comput.-Aided Mol. Des. 2005, 19, 453–463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.