

Abstract
Epidermal growth factor receptor (EGFR) was one of the first oncogenes identified in glioblastoma (GBM) and remains one of the most attractive therapeutic targets. Genomic alterations in EGFR are present in 57% of patients and are strikingly diverse, including gene amplification, rearrangements, and point mutations. Each aberration class has important clinical implications for diagnosis, prognosis, or therapeutic investigation of EGFR in clinical trials. Somatic copy number alterations (SCNAs) are the most common abnormalities in EGFR, with gene amplification present in >43% of patients. The presence of EGFR amplification is often used now to support the diagnosis of GBM and discriminate GBM from other gliomas. It is currently detected in clinical labs using fluorescence in situ hybridization, colorimetric in situ hybridization or, more recently multiplex genomic technologies such as array CGH or targeted next-generation sequencing approaches. Rearrangements of EGFR are most commonly internal deletions leading to activation of the receptor including EGFRvIII and, less commonly, EGFRvII and other variants, which are collectively seen in 25% of GBM patients. EGFRvIII is readily detected via mutation-specific antibodies, but heterogeneity of this and other deletion variants has hindered reliable detection of these aberrations using genomic DNA-based methods. RNA expression profiling (Nanostring and anchored multiplex PCR) has additional potential as a rapid and reliable strategy for detecting EGFR rearrangements with high sensitivity. Single nucleotide variants in EGFR are relatively rare and diverse but are efficiently detected using the targeted or exome-sequencing assays that are now entering clinical pathology practice. The advent of multiplex technologies has revealed the fact that multiple aberrations of EGFR are present in at least 30% of patients with EGFR disruption, a fact recently highlighted by more quantitative sequencing techniques and single cell analysis of GBM. Diagnostic assays used to evaluate EGFR and other receptor tyrosine kinases will therefore be increasingly used to measure and resolve this heterogeneity in order to better understand their mechanisms of resistance. In summary, the diagnostic approaches for identifying clinically relevant EGFR aberrations have rapidly advanced and are providing insights into more effective inhibition of this familiar oncogene in GBM and other cancers.
Keywords: diagnostics, EGFR, GBM, pathology, sequencing
Epidermal Growth Factor Receptor Biology
The epidermal growth factor receptor (EGFR) erbB1 is a receptor tyrosine kinase (RTK) located on chromosome 7p12 and is responsive to extracellular ligands such as EGF and TGF-α. EGFR was one of the first proto-oncogenes recognized to play a potential role in glioblastoma (GBM) pathogenesis because of its high level of expression in the majority of patients (up to 90%) and frequent genomic amplification. Receptor signaling occurs through multiple pathways, but the most studied are recruitment and activation of the phosphatidylinositol-3-kinase (PI3K) signaling network with ultimate downstream activation of AKT and mTOR proteins that sustain tumor growth. Evidence suggests that PTEN is a negative regulator of the pathway, with specific evidence suggesting that these 2 pathways may interact in the context of GBM cells in a complex manner.1 Genomic alterations in EGFR generally lead to receptor activation and increased signaling, but the oncogenic contributions of wild-type receptor signaling, compared with mutant receptor signaling in actual patient tumors, has not been well established.
As the scientific understanding of EGFR has advanced, clinical applications have rapidly emerged around attempts to therapeutically inhibit EGFR function in multiple cancers. Targeted therapies have shown the most promise for inhibition of receptor function, leading to tumor responses in lung and other cancers, and still being evaluated in GBM. Results in GBM to date have used early inhibitors that were not specifically designed to inhibit the aberrations most common to GBM. As such, the results of these EGFR inhibitor studies in GBM have been inconsistent, and further study is generally needed. Current trends involve a shift toward design of more appropriate inhibitors in combination with specific selection or stratification of patients for treatment based on the specific alterations of EGFR in their tumors.1 Given the intense level of interest in measuring EGFR alterations and its biology in both the clinical and research settings, numerous assays capable of measuring EGFR have evolved rapidly and offer current and future clinical value (Fig. 1).
Fig. 1.
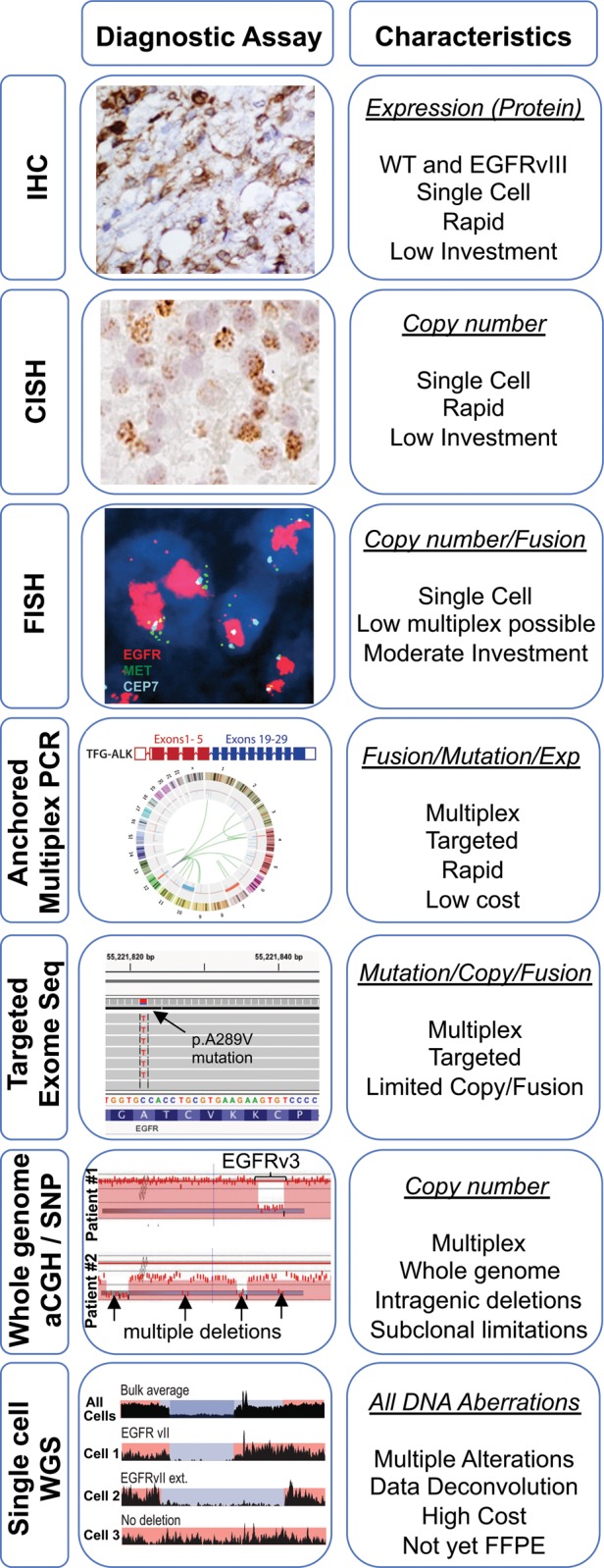
Summary of assays and their utility for epidermal growth factor receptor assessment in glioblastoma.
Wild-type EGFR Expression
Wild-type EGFR (EGFRwt) is highly expressed in the vast majority of GBMs and in lower-grade astrocytomas as well.2 Expression is generally uniform geographically throughout the tumor, and nearly all cells within the tumor express high levels of EGFR protein (>90%) by immunohistochemistry (IHC). The high levels of expression may parallel the developmental expression pattern of EGFR, a central regulator of neural stem and progenitor cells from which some GBM tumors are felt to derive. Studies of EGFR expression in adult neural stem cell niches, in particular, correlate with expression in type C transit-amplifying cells3 and have shown that EGF stimulation of these regions causes glioma-like growths in the subventricular and subependymal zones of mice.4–6
While biologically important, both the practical and diagnostic utility of EGFRwt expression in the clinical setting are still under investigation. Review of the literature suggests that high expression of wild-type receptor in GBM versus other gliomas have been proposed, but has not led to common implementation by pathologists.7 The levels of RNA and protein expression can vary widely among patients8 and clearly correlate with EGFR amplification, but formal studies have shown that such correlations are not specific enough for reliable identification of EGFR-amplified tumors by IHC. Given these limitations, there is currently no consensus standard-of-care role for testing EGFR expression in the clinical diagnostic and prognostic setting, even though there is clearly a critical pathway.
EGFR protein expression has been used as a diagnostic in the setting of clinical trials involving EGFR inhibitors. Several trials in adult and pediatric gliomas have used these markers as prospective markers for patient selection and enrichment of target expression prior to treatment. EGFR protein expression has also been routinely used in clinical trials for retrospective marker analysis of the results from EGFR inhibitors. While the lack of response and prevalence of the target have not led to a clear indication for testing, one interesting area for future exploration is that quantification of total levels of EGFRwt protein may be needed to determine whether response to treatment with EGFR inhibitors and RTK dosage may in fact be relevant for predicting response.
Epidermal Growth Factor Receptor Copy Number Alterations
The most common aberration of EGFR in adult GBM is an increased number of copies of the gene by focal gene amplification and/or broad genomic gains that include the EGFR locus. Gene amplification is a genetic term indicating focally increased gene copy number arising through specific mechanisms, including formation of extrachromosomal double minutes (dmin) or heterogeneously staining regions (HSR) occurring at a specific gene locus or chromosomal region. As a result, large increases in copy number (eg, >5 and even hundreds of copies in some tumors) are observed. The term “gain” is used to indicate a lower level increase of gene number arising through distinct mechanisms (ie, tandem gene duplication, polysomy, etc) that lead to more modest copy number increases (eg, 3–5 copies of a gene instead of the normal 2 copies). Such terms should be differentiated from the more general term “amplification/amplified,” which is increasingly used in cancer genetics communities in reference to any level of increase in copy number without implication as to the mechanism.
Polysomy of the entire chromosome 7 (1–2 extra copies of the entire chromosome) is the most common EGFR copy number alteration in all gliomas. Additional alterations leading to a gain of 7q containing the EGFR genes are also seen in a significant percent of glioma patients. While these large chromosomal gains are frequent in gliomas, they are not specific to GBM. Furthermore, the broad nature of events containing many different genes does not suggest that EGFR function is altered or is supporting the tumorigenic phenotype in any specific manner when these occur. In contrast, more than 40% of GBMs have focal EGFR gene amplification with amplicons containing >50–100 copies of the gene.9,10 Such amplifications are present in a high number of tumor cells in most patients but may also occur diagnostically in a subset of tumor cells, particularly in association with coamplification of other RTK genes (PDGFRA, MET).11 In contrast to polysomy 7, evidence that focal amplifications of EGFRwt receptor are driver events is supported by laboratory investigations; it is therefore important to identify these diagnostically and target them therapeutically.
Clinically, the detection of EGFR copy number alterations is most commonly used for diagnosis and classification of GBM versus other gliomas with similar histological appearance. The presence of amplification is almost exclusively seen in GBM. Fewer than 3%12 of other diffuse gliomas of any class or grade are amplified based on recent studies of carefully evaluated patient cohorts. EGFR amplification in the presence of lower-grade histology on biopsy frequently indicates surgical undersampling of a histologically heterogeneous tumor, in which the features required to diagnose GBM (vascular proliferation and necrosis) are not present in the samples. While initial studies had suggested that the presence of EGFR amplification was prognostically unfavorable in GBM, these findings have not been reproduced in later well-characterized cohorts.13 Such prognostic value was likely to be conferred by including histological variants of anaplastic oligodendroglioma, which overlap GBM appearance but invariably lack EGFR amplification and have an inherently more favorable prognosis. EGFR amplification as a predictive marker has been analyzed in several clinical trials of EGFR inhibitors but has not consistently shown strong power as an independent biomarker.1,14
Currently, the most common diagnostic tools used to clinically evaluate EGFR copy number are fluorescence in situ hybridization (FISH) or colorimetric in situ hybridization (CISH). These techniques allow careful quantification of individual copies that have been noted in single cells/nuclei within the tumor. CISH has the advantage of using standard brightfield microscopy for looking at tumor morphology.15 Interpretation can be readily performed for qualitative presence/absence of amplification (>10 copies per nucleus), abnormal or consistent with polysomy (3–10 copies) or normal (2 copies) by neuropathologists in a manner similar to IHC. FISH performance is technically more consistent and robust and facilitates accurate quantification of individual nuclei and colocalization with other RTKs. The disadvantages of FISH are the need for specific equipment and training to read darkfield microscopy.
Given the common occurrence of multiple copy number changes in individual GBM tumors (eg, EGFR, PDGFRA, MET, CDKN2A, PTEN) multiplex assays based on DNA hybridization arrays (array CGH or SNP arrays) are already essential in the research arena and are now emerging clinically as routine assays for patient care. Until recently, these assays were limited in clinical potential because of poor performance in FFPE tumor tissues. However, development of novel methods has allowed highly reproducible and effective performance using FFPE samples of GBM and other cancers.16,17 The most common platforms currently used are CGH or SNP arrays.12,18 Alternate methods based on molecular inversion probe technology have also emerged as effective for analyzing FFPE samples with lower DNA input requirements than arrays, but their resolution is generally lower.19 These dedicated copy number assays have become valuable in clinical care of GBM and clinical trials, given the ever-increasing number and decreasing size of clinically relevant aberrations that are not readily detected by targeted next-generation sequencing strategies or other sequence-based methods.20
Several limitations of these multiplex technologies are now apparent. One limitation is their longer turnaround time compared with the conventional FISH/CISH assays used previously, and another is their requirement for significantly more tissue (generally 10 unstained slides of 4 μm thickness). In addition, since they are based on average hybridization kinetics or analysis of bulk populations, interpretation and analysis of the results identify potential subclonal events but do not readily distinguish these from lower level copy number events using current algorithms. This issue usually arises as a practical consideration in GBMs with EGRR amplification and concurrent low level gains in PDGFRA or MET. FISH analysis of most of these cases shows PDGFRA and MET are highly amplified in distinct cell subclonal cell populations and not low level gains at these loci in all the cells.9,11 For treatment decisions, laboratories could formally test whether screening patients first with these whole genome or multiplex assays, followed by reflex testing via FISH or other methods, adds practical clinical value beyond the use of “gain” as a biomarker.
Epidermal Growth Factor Receptor Rearrangements
EGFRvIII
The most frequently recurring rearrangements in EGFR are intragenic deletions, the most common of which is the variant EGFRvIII that can be detected in 19% of GBMs.10 This variant involves intragenic in-frame deletion of exons 2–7 and occurs only in tumors with EGFR amplification. The biology of EGFRvIII has been studied extensively and has been shown to be a strong oncogenic driver that can transform cells and produce a more aggressive phenotype in GBM. While its functional significance has been well established in model systems, its clinical relevance for patients is still being explored, and no independent prognostic value has been established in early studies or by The Cancer Genome Atlas (TCGA).21 Diagnostically, the presence of EGFRvIII is useful as a tumor-specific marker that is fairly specific to GBM, as it has only rarely been identified in other cancers (eg, lung).
Clinical detection of EGFRvIII, while not standard-of-care, is often performed in CLIA labs as a clinical research assay to identify candidates for clinical trials that are specifically directed against the antigen (eg, EGFRvIII vaccine or antibody trials). Several trials seek to enroll patients with EGFRvIII-positive tumors, most often by IHC using tumor-specific EGFRvIII antibodies21 or by reverse transcription PCR for detecting expressed RNA transcripts in the rearranged locus. Studies using IHC have highlighted the heterogeneous geographic expression of EGFRvIII within tumors, which is also supported by patterns seen in genomic assays22 that represent a significant concept in potential resistance by nonexpressing cells. A disadvantage of EGFRvIII IHC is that specific EGFRvIII antibodies suitable for routine clinical IHC are not available commercially and are instead accessed only through association with clinical trials. Use of array CGH or SNP platforms is advantageous in these scenarios because one can identify the coassociated amplification of the locus, and the signal for EGFR intragenic deletion is highly reliable when present. However, such assays, by their nature of averaging results from numerous tumor cells, are less sensitive given the known heterogeneity of EGFRvIII in tissues and cells. Single-cell analysis by oligoFISH-based methods (eg, Agilent SureFISH) allows visual detection of missing signals in nuclei with vIII-deleted regions, but testing for their efficacy in clinical practice just started recently.9
Other Deletion Variants
Multiple additional intragenic deletion variants of EGFR were described early in the study of GBM, but they remained relatively obscure for years after their initial discovery, until the recent advent of improved sequencing technology.23 Recent studies have established that intragenic deletions are detectable in 71% of EGFR-amplified tumors. Furthermore, there is evidence that EGFRvII is transforming similar to EGFRvIII, suggesting that assays to measure such variants are likely to become clinically relevant for GBM and its pathogenesis.9 Other notable EGFR rearrangements are those involving the carboxy-terminal intracellular domain.24,25 Exon array analysis (SNP data from TCGA; validated by Sanger PCR) has identified multiple deletions (8/469) involving exons 25–27, exons 27–28, and exons 25–28, all within the carboxyl-terminus domain.26 These deletions are able to induce cell transformation without ligand and are highly sensitive to the EGFR inhibitor cetuximab in a xenograft model.26
Diagnostically, the diversity of EGFR rearrangements presents a challenge for capture with a single assay or approach because the boundaries and breakpoints for EGFR rearrangements can vary between cells within a tumor and also across patients. Therefore, it will be increasingly important for clinical labs to identify the entire spectrum of EGFR genomic rearrangements with appropriate technologies. While targeted exome-sequencing platforms can detect these rearrangements in theory, such events are more likely to be optimally identified by other targeted (eg, Nanostring RNA)8 or untargeted (eg, whole genome array CGH for DNA) approaches in practice to obtain more complete genotyping of EGFR. Recent data from the Nanostring assay have carefully demonstrated its improved quantitative and sensitive detection of EGFRvIII and other events in GBM compared with array-based methods; however, the presence of low levels of transcripts in a high number of tumors raises questions about which levels are ultimately relevant clinically for future clinical reporting.
EGFR Fusions
Recent examination of a large RNA-sequencing dataset of glioblastoma from the TCGA,27 as well as the Swedish Hospital Ivy Center cohort,28 revealed several in-frame gene fusions and complex rearrangements. EGFR emerged as the most frequent gene fusion in GBM and was present in 4% of GBM patients. EGFR is most commonly fused to intron 9 of SEPT14 or to the gene PSPH (2.2%), and both events were noted to leave the tyrosine kinase domain intact.27 Although the majority of gene fusions in GBM are associated with unbalanced genomic rearrangements, EGFR fusions can occur without accompanying copy number changes. Interestingly, most of the EGFR-SEPT14 or PSPH fusions lacked expression of EGFRvIII (exon2–7 deletion). It is therefore possible that the C-terminal deletion of EGFR through gene fusion acts as an alternative mechanism of EGFR activation. Indeed, Frattini et al reported that EGFR-SEPT14 was able to confer mitogen independence and sensitivity to EGFR inhibitors such as erlotinib and lapatinib in a manner similar to EGFRvIII. The clinical relevance of fusions has not yet been established within the diagnostic or clinical trial arena. However, the diagnostic identification of such events is increasingly likely and efficient with the advent of RNA/cDNA sequencing techniques, such as anchored multiplex PCR (AMP)29 or Nanostring assays, designed to capture rearrangements that might be known as clinically relevant. Eventual progress towards whole genome sequencing and RNA sequencing offers even more potential for diagnostic convergence to aid identification of these events, as this technology does currently in the research arena.
EGFR Mutations
While GBM appears to be predominantly a copy number disease, results of large scale sequencing of GBM patients have demonstrated that up 20% of GBMs harbor EGFR point mutations, which are frequently combined with rearrangements or copy alterations (44% of amplified patients).10 Clinical trials using EGFR inhibitors, such as erlotinib, on unselected GBM patients have not shown a clear clinical benefit; however, 15%–20% of treated patient in some studies experienced significant tumor reduction in response to small molecule EGFR inhibitors.1,30 In order to identify more genomic alteration in EGFR, a significant effort has been made to analyze large cohorts of GBM patients, originally by using PCR sequencing and mass-spectrometric genotyping24,31,32 and more recently by using whole-exome and RNA sequencing.10 EGFR point mutations in GBM are more frequently located in the extracellular domain (13% of patients) unlike lung cancer, in which most mutations affect the kinase domain.32–34 The selectivity of drugs for kinase-domain inhibition in current trials has been proposed as an explanation for GBM resistance to first-generation EGFR inhibitors targeting the kinase domain. Identification of such mutations, however, might indicate possible sensitivity to ATP-site competitive EGFR kinase inhibitors, such as lapatinib.33 Point mutations in the extracellular domain may be enriched in EGFR-amplified tumors but can be observed without amplification.10,35
Given these findings, clinical detection of point mutations has a direct implication for treatment with EGFR inhibitors and interpretation of clinical trials. While some recurrent EGFR mutational events are common (eg, A289 and R108), the diversity of mutations suggests that multiplexed targeted genotyping36,37 or targeted exome-sequencing approaches are necessary for identifying a broader set of events. Such platforms are now in place at several of the larger tertiary care centers, and private laboratories are increasingly offering access to services such as targeted exome sequencing for patients. Several GBM nonprofit foundations (eg, ABC2, Sontag Foundation) have also prioritized facilitating patient access to clinical sequencing programs as a means to accelerating integration of genomics with clinical trials and eventual improved patient outcomes. These efforts should greatly increase the knowledge about specific mutations and their natural history of disease progression and response to EGFR inhibitors.
Conclusions
Recent in-depth analyses of genomic alterations in EGFR have revealed an unprecedented level of complexity due to diverse deletions, fusions, and point mutations that all occur frequently in a highly amplified locus. The unique combination of these multiple hits and the mutations in other genes produces a variety of functionally distinct EGFR-altered tumors. Past studies and trials had focused on investigating only one type of alteration (eg, EGFRvIII deletion), but the focus on single alterations might not be enough to decipher results in the background of the multiplex heterogeneity of GBM.1,38 The advent of single-cell sequencing and quantitative sequencing methods offers the opportunity to perform precise deconvolution of the EGFR state in tumor cells and demonstrates that multiple clones in the same tumor may each behave differently once mutations are combined.9 Such attention to subclonal populations has implications not only for EGFR but also for other amplified RTKs such as PDGFRA and MET, which are common in other cancers. Given the resistance of GBM to EGFR inhibition to date, the use of novel diagnostic approaches for assessing EGFR could reveal new pathways to more effective inhibition.
Funding
K.L.L. and C.M. are supported by funding from the NIH (R01CA170592, P50CA165962, and P01CA14256), the MIT/DFHCC Bridge Project for Cancer Research, and the Pediatric Low Grade Astrocytoma Foundation. K.L.L. has also received support from the Sontag Foundation.
Conflict of interest statement. K.L.L. participates in clinical research at the DFCI that receives sponsored research support from the following sources: Novartis, GSK, and EMD Serono. K.L.L. serves as a paid consultant to EMD Serono and Midatech LLC. K.L.L. is an inventor on a patent filed by DFCI related to cancer genomics testing.
References
- 1.Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 2.Biernat W, Huang H, Yokoo H, et al. Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol. 2004;14(2):131–136. doi: 10.1111/j.1750-3639.2004.tb00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doetsch F, Petreanu L, Caille I, et al. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002;36(6):1021–1034. doi: 10.1016/s0896-6273(02)01133-9. [DOI] [PubMed] [Google Scholar]
- 4.Bachoo RM, Maher EA, Ligon KL, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1(3):269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 5.Tavazoie M, Van der Veken L, Silva-Vargas V, et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3(3):279–288. doi: 10.1016/j.stem.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aguirre A, Dupree JL, Mangin JM, et al. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10(8):990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- 7.Zhou YH, Hess KR, Raj VR, et al. Establishment of prognostic models for astrocytic and oligodendroglial brain tumors with standardized quantification of marker gene expression and clinical variables. Biomark Insights. 2010;5:153–168. doi: 10.4137/BMI.S6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kastenhuber ER, Huse JT, Berman SH, et al. Quantitative assessment of intragenic receptor tyrosine kinase deletions in primary glioblastomas: their prevalence and molecular correlates. Acta Neuropathol. 2014;127(5):747–759. doi: 10.1007/s00401-013-1217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francis JM, Zhang CZ, Maire CL, et al. EGFR Variant Heterogeneity in Glioblastoma Resolved through Single-Nucleus Sequencing. Cancer Discov. 2014;4(8):956–971. doi: 10.1158/2159-8290.CD-13-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snuderl M, Fazlollahi L, Le LP, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Cryan JB, Haidar S, Ramkissoon LA, et al. Clinical multiplexed exome sequencing distinguishes adult oligodendroglial neoplasms from astrocytic and mixed lineage gliomas. Oncotarget. 2014 doi: 10.18632/oncotarget.2342. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64(19):6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 14.Wen PY, Chang SM, Lamborn KR, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04–02. Neuro Oncol. 2014;16(4):567–578. doi: 10.1093/neuonc/not247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer I, de la Cruz C, Rivera AL, et al. Utility of chromogenic in situ hybridization (CISH) for detection of EGFR amplification in glioblastoma: comparison with fluorescence in situ hybridization (FISH) Diagn Mol Pathol. 2008;17(4):227–230. doi: 10.1097/PDM.0b013e3181642230. [DOI] [PubMed] [Google Scholar]
- 16.Craig JM, Vena N, Ramkissoon S, et al. DNA fragmentation simulation method (FSM) and fragment size matching improve aCGH performance of FFPE tissues. PloS One. 2012;7(6):e38881. doi: 10.1371/journal.pone.0038881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohapatra G, Engler DA, Starbuck KD, et al. Genome-wide comparison of paired fresh frozen and formalin-fixed paraffin-embedded gliomas by custom BAC and oligonucleotide array comparative genomic hybridization: facilitating analysis of archival gliomas. Acta Neuropathol. 2011;121(4):529–543. doi: 10.1007/s00401-010-0773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dougherty MJ, Tooke LS, Sullivan LM, et al. Clinical utilization of high-resolution single nucleotide polymorphism based oligonucleotide arrays in diagnostic studies of pediatric patients with solid tumors. Cancer Gen. 2012;205(1-2):42–54. doi: 10.1016/j.cancergen.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 19.Gessi M, Zur Muhlen A, Hammes J, et al. Genome-wide DNA copy number analysis of desmoplastic infantile astrocytomas and desmoplastic infantile gangliogliomas. J Neuropathol Exp Neurol. 2013;72(9):807–815. doi: 10.1097/NEN.0b013e3182a033a0. [DOI] [PubMed] [Google Scholar]
- 20.Ducray F, de Reynies A, Chinot O, et al. An ANOCEF genomic and transcriptomic microarray study of the response to radiotherapy or to alkylating first-line chemotherapy in glioblastoma patients. Mol Cancer. 2010;9:234. doi: 10.1186/1476-4598-9-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aldape KD, Ballman K, Furth A, et al. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol. 2004;63(7):700–707. doi: 10.1093/jnen/63.7.700. [DOI] [PubMed] [Google Scholar]
- 22.Nishikawa R, Sugiyama T, Narita Y, et al. Immunohistochemical analysis of the mutant epidermal growth factor, deltaEGFR, in glioblastoma. Brain Tumor Pathol. 2004;21(2):53–56. doi: 10.1007/BF02484510. [DOI] [PubMed] [Google Scholar]
- 23.Callaghan T, Antczak M, Flickinger T, et al. A complete description of the EGF-receptor exon structure: implication in oncogenic activation and domain evolution. Oncogene. 1993;8(11):2939–2948. [PubMed] [Google Scholar]
- 24.Frederick L, Wang XY, Eley G, et al. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60(5):1383–1387. [PubMed] [Google Scholar]
- 25.Ekstrand AJ, Sugawa N, James CD, et al. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci USA. 1992;89(10):4309–4313. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho J, Pastorino S, Zeng Q, et al. Glioblastoma-derived epidermal growth factor receptor carboxyl-terminal deletion mutants are transforming and are sensitive to EGFR-directed therapies. Cancer Res. 2011;71(24):7587–7596. doi: 10.1158/0008-5472.CAN-11-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frattini V, Trifonov V, Chan JM, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nature Gen. 2013;45(10):1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah N, Lankerovich M, Lee H, et al. Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genomics. 2013;14:818. doi: 10.1186/1471-2164-14-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McFadden DG, Dias-Santagata D, Sadow PM, et al. Identification of oncogenic mutations and gene fusions in the follicular variant of papillary thyroid carcinoma. J Clin Endocrinol Metab. 2014:jc20142611. doi: 10.1210/jc.2014-2611. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haas-Kogan DA, Prados MD, Tihan T, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97(12):880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 31.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JC, Vivanco I, Beroukhim R, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006;3(12):e485. doi: 10.1371/journal.pmed.0030485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vivanco I, Robins HI, Rohle D, et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2(5):458–471. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Idbaih A, Aimard J, Boisselier B, et al. Epidermal growth factor receptor extracellular domain mutations in primary glioblastoma. Neuropathol Appl Neurobiol. 2009;35(2):208–213. doi: 10.1111/j.1365-2990.2008.00977.x. [DOI] [PubMed] [Google Scholar]
- 35.Ekstrand AJ, James CD, Cavenee WK, et al. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991;51(8):2164–2172. [PubMed] [Google Scholar]
- 36.Chi AS, Batchelor TT, Dias-Santagata D, et al. Prospective, high-throughput molecular profiling of human gliomas. J Neuroncol. 2012;110(1):89–98. doi: 10.1007/s11060-012-0938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacConaill LE, Garcia E, Shivdasani P, et al. Prospective Enterprise-Level Molecular Genotyping of a Cohort of Cancer Patients. J Mol Diagn. 2014 doi: 10.1016/j.jmoldx.2014.06.004. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Del Vecchio CA, Giacomini CP, Vogel H, et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene. 2013;32(21):2670–2681. doi: 10.1038/onc.2012.280. [DOI] [PubMed] [Google Scholar]