

Abstract
Objective
There is limited evidence for efficacy of analgesics as monotherapy for neuropathic pain associated with HIV-associated polyneuropathies, in spite of demonstrated efficacy in other neuropathic pain conditions. We evaluated the tolerability and analgesic efficacy of duloxetine, methadone, and the combination of duloxetine-methadone compared to placebo.
Design
This study was a phase II, randomized, double blind, placebo-controlled, four-period crossover multi-center study of analgesic therapy for patients with at least moderate neuropathic pain due to HIV-associated polyneuropathy. Duloxetine, methadone, combination duloxetine-methadone, and placebo were administered in four different possible sequences. The primary outcome measure was mean pain intensity (MPI) measured daily in a study-supplied pain diary.
Results
A total of 15 patients were enrolled from 8 study sites and 8 patients completed the entire trial. Study treatments failed to show statistically significant change in MPI compared to placebo. Adverse events were frequent and associated with high rates of drug discontinuation and study drop-out.
Conclusions
Challenges with participant recruitment and poor retention precluded trial completion to its planned targets, limiting our evaluation of the analgesic efficacy of the study treatments. Challenges to successful completion of this study and lessons learned are discussed.
Introduction
Polyneuropathy (PN) is considered the most common neurological complication of HIV infection, generally associated with moderate to severe immunodeficiency (HIV distal sensory polyneuropathy) or as a treatment-related toxicity related to certain – particularly dideoxynucleotide -- antiretroviral drugs (i.e., antiretroviral toxic neuropathy). [1-6] While the mechanism for PN associated with HIV infection is likely immune activation, PN related to antiretroviral exposure is likely due to mitochondrial toxicity. In spite of these mechanistic differences, the clinical presentation of these entities is sufficiently similar that they are frequently considered together as HIV-associated polyneuropathy (HIV-PN). Overall, it has been estimated that up to one-third of HIV-infected patients suffer from symptomatic HIV-PN.[1, 2, 7, 8] HIV- PN is clinically important given neuropathic pain and its adverse impact on quality of life, functional status, and disability.[8-10]
Neuropathic pain is challenging condition to treat as only about half of treated patients report at least moderate relief in response to common analgesic monotherapy regimens.[11] Evidence-based analgesic options for the symptomatic treatment of painful HIV-PN are limited: multiple agents from diverse drug classes - including tricyclic antidepressants, sodium channel antagonists, and gabapentinoids - have failed to demonstrate significant pain relief compared to placebo.[12-17] Although efficacy has been reported with some agents currently available on the market, including lamotrigine, high-dose topical capsaicin, and smoked marijuana, for variable reasons these drugs do not generally benefit from widespread use.[18-21]
Acknowledging the limitations of available analgesic options for painful HIV-PN, combination therapy offers a promising alternative to monotherapy. As the underlying pathophysiology of chronic neuropathic pain likely stems from multiple processes affecting both the peripheral and central nervous system, different medications characterized by unique mechanisms of action may provide relief for different aspects of neuropathic pain. The potential exists for a combination regimen to act in a synergistic, or greater than additive, fashion. In clinical trials of diabetic neuropathy and post-herpetic neuralgia, combination pharmacotherapy has been shown to afford greater reductions in pain intensity than that afforded by monotherapy.[22, 23] Combination regimens may additionally afford fewer side effects, which may be a consequence of lower mean drug doses.[24]
We sought to evaluate the efficacy of duloxetine, methadone, and the combination of duloxetine -methadone compared to placebo for the symptomatic treatment of painful HIV- PN. Duloxetine is a balanced serotonin-norepinephrine reuptake inhibitor which provides a significant reduction in mean 24-hour pain intensity at a dose of 60mg per day in patients with painful diabetic PN.[25-27] Formal evaluation of duloxetine's efficacy was felt to be a pressing need as neuropathic pain and depression frequently co-exist and duloxetine was increasingly utilized off-label in our clinics. Methadone is a generic synthetic opioid approved by the FDA for moderate to severe pain not responsive to non-opioid analgesics, with analgesic effects mostly stemming from activity at μ opioid receptors. While short term analgesic efficacy has been demonstrated with other opioid compounds,[28] limited data suggests that methadone might be effective at a relatively low dose.[29, 30] Methadone was considered an attractive study drug as it is an affordable and a realistic option for resource-limited settings, and we recognized a trend towards increasing use of chronic opioid therapy for neuropathic pain in HIV clinics in spite of a complete absence of clinical efficacy data. Human trials have suggested differential effects of these drug classes,[31] and animal data suggest the possibility of analgesic synergy between these drug classes.[32, 33]
In the pursuit of this study, we encountered serious challenges to accrual and study drug selection, as well as unanticipated adverse events and high dropout. We believe our experience is instructive for future neuropathic pain trials, specifically those targeting the HIV population, as it brings attention to important issues related to studying analgesic therapies for painful HIV-PN.
Materials and Methods
Recruitment/enrollment
Eligible patients included those ≥18 years of age with at least moderately painful HIV-PN. HIV-PN was defined by the presence of symmetrical pain, burning, or dysesthesias in a stocking distribution for at least 6 months with abnormal ankle reflexes or at least one abnormal sensory sign (elevated vibratory thresholds, stocking loss of pinprick or temperature, or cutaneous allodynia). [34] Moderately severe neuropathy pain was defined by a baseline mean pain intensity (MPI) of ≥4 measured on an 11-point Likert scale ranging from 0 = “No pain” to 10 = “Pain as bad as you can imagine” in a study-supplied diary.
Mean baseline pain intensity was measured daily over a 1-week period, during which time subjects were required to complete a minimum of 6 of 7 daily diary entries. Other inclusion criteria included the following: Karnofsky performance scale ≥60 (requires occasional assistance, but is able to care for most personal needs), stable use or non-use of antiretrovirals for 30 days prior to entry, as well as QTc interval ≤0.45 seconds within 90 days of entry.
Patients were ineligible for the following conditions due to their impact on HIV-PN diagnosis and impact on PN stability over time: (1) current B12 level <200 pg/ml or recent diagnosis of B12 deficiency and treated within 45 days of entry, (2) poorly controlled diabetes, defined by HgbA1c >7, diagnosed at any time prior to the diagnosis of HIV-PN; (3) discontinuation of neurotoxic ARV therapy within 16 weeks of entry; (4) treatment with any neurotoxic drug within 120 days of entry; and (4) hereditary-PN, lumbosacral radiculopathy, lumbosacral stenosis, or HIV-associated myelopathy that would interfere with ability to evaluate neuropathy pain.
Medical co-morbidities prompting exclusion included end stage renal disease requiring hemodialysis, hepatic cirrhosis, at high risk of opioid-induced respiratory compromise (acute bronchial asthma, body mass index >30, sleep apnea not on CPAP), seizure disorder or seizure within 90 days of entry, or history acute angle closure glaucoma. Participants were excluded if they were pregnant, breastfeeding, or were unwilling to use a reliable form of contraception. Additional laboratory abnormities which prompted exclusion: (1) absolute neutrophil count (ANC) ≤500mm3;, (2) hemoglobin ≤8 g/dL for males and ≤7.5g/dL for females; (3) direct bilirubin greater than 1.5 times the upper limit of normal; (4) serum alanine aminotransferase or aspartate aminotransferase greater than 3 times the upper limit of normal; and (5) creatinine clearance <30mL/min.
Finally, patients were deemed ineligible for the following mental health conditions: (1) significant premorbid depression on antidepressant therapy precluding withdrawal from antidepressants for participation in the study; and (2) active substance abuse or dependence that poses risk for the participant, significant alcohol-related complications in the 6 months prior to entry. Patients on baseline opioid therapy were allowed to participate in the study provided that the total daily dose of opioids did not exceed the equivalent of 60 mg morphine daily, as such a dose was deemed reasonable to taper in a relatively short time period.
Study Design
This was a phase II, randomized, double blind, placebo-controlled, four-period crossover multicenter study designed to evaluate the efficacy and safety of duloxetine, methadone, and the combination of duloxetine and methadone for the treatment of painful HIV-PN. After a screening visit, subjects on selected analgesic pain regimens were required to taper and wash out the following prohibited medications during a 1-2 week period prior to baseline assessment of pain intensity: tricyclic antidepressants, selective serotonin uptake inhibitors, monoamine reuptake inhibitors, bupropion, serotonin-norepinephrine reuptake inhibitors, tramadol, and opioid regimens approximating 60 mg oral morphine daily. Continued use of gabapentin, pregabalin, lamotrigine, topical capsaicin, topical lidocaine, and opioid therapy < 60 mg daily morphine equivalent were permitted if maintained at stable doses during study participation if used at a stable dose for at least 8 weeks prior to study entry. Washout duration was at the discretion of the site investigator and, for those not taking prohibited medications at screening, the 1-2 week washout period was waived. After washout subjects documented their daily mean pain intensity (MPI) each day in a study-supplied diary each morning over baseline period of one week. Patients with a MPI of ≥ 4 on an 11-point Likert scale were assigned in a randomized, double-blind fashion via a balanced Latin-square crossover design to one of four treatment sequences: (1) duloxetine, methadone, duloxetine -methadone, and placebo; (2) methadone, placebo, duloxetine, and duloxetine-methadone; (3) duloxetine-methadone, duloxetine, placebo, and methadone; or (4) placebo, duloxetine -methadone, methadone, and duloxetine. Computer-generated randomized permuted blocks using a block size of 8 was utilized to create sequence assignments. The total duration of the double blind phase was 20 weeks, comprised of four treatment periods lasting four weeks apiece punctuated by 1-week combination drug taper and discontinuation. Both patients and investigators completed blinding questionnaires at the end of each treatment period to gauge the quality of blinding and allocation concealment.
Study Medications
Patients received oral duloxetine (Cymbalta®, Eli Lilly, USA) monotherapy, oral methadone (Mallinckrodt, USA) monotherapy, the combination of duloxetine-methadone, or placebo for 28 days. Study drugs were color-coded with color-matched placebo according to a double-dummy design. Duloxetine and matching placebo were initiated at 30mg daily and titrated to 60mg for Days 6-28. Methadone and matching placebo were initiated at 5mg twice daily (b.i.d.), titrated to 5 mg three times daily (t.i.d.) on Day 6, and thereafter increased to 10mg t.i.d. for Days 11-28. Each treatment period lasted 4 weeks and was followed by a 1-week crossover period which combined drug taper and discontinuation. During the one week crossover period duloxetine was reduced to one capsule daily for 3 days (Days 29-31) and discontinued on Day 32. Methadone was similarly decreased every three days as follows (assuming MTD of 10mg t.i.d., the taper could be modified as deemed proper by the site investigator): : Days 29-31 5 mg t.i.d., Days 32-34 5mg b.i.d., and discontinued on Day 35.
Flexible dosing allowed participants to receive either the target ceiling dose or the maximum tolerated dose (MTD) of study treatments. Subjects were contacted by study personnel weekly for the combined purpose of facilitating drug titration and assisting with dose modifications as needed for treatment-emergent drug toxicities. Drug titration was guided by the presence or absence of dose-limiting side effects, with the goal of reaching target ceiling doses and thus maximizing treatment efficacy.
Acetaminophen 500mg capsules were provided as rescue pain mediation throughout study participation, with explicit instructions to use only when needed for neuropathic pain at a ceiling dose of 3g per day.
Outcome measures
The primary outcome measure was mean 24 hour pain intensity measured on an 11-point Likert numerical rating scale ranging from 0 = “No pain” to 10 = “Pain as bad as you can imagine” documented in a study-supplied pain diary. Patients recorded pain intensity in the morning to describe average neuropathy pain over the previous 24 hours. A secondary outcome measure was nighttime pain intensity, which was documented in the morning and described average pain over the preceding night.
The primary analysis proposed 5 pair-wise comparisons of the daily mean pain intensity (MPI) measured over the fourth week of each period between duloxetine, methadone, duloxetine-methadone, and placebo as well as between each monotherapy and combination therapy. We calculated that 120 patients would be required to provide the study with 90 percent power to detect a 1.5 point difference in MPI amongst the study treatments at an alpha of 0.01.
Results
Patient Disposition
The protocol was reviewed and approved by all relevant committees of the AIDS Clinical Trials Group (ACTG) as well as by the Institutional Review Boards at all participating study sites. Each patient gave written informed consent before entering the study. A total of 24 patients were screened with 15 patients with moderate-to-severe HIV- PN were randomized between August 2009 and October 2010 to receive treatment (Figure 1). Subjects were enrolled across 8 of 15 study sites in the ACTG system. Overall, only 53% (8/15) of the randomized patients completed the 20 week study on treatment, while another 3 subjects completed the study off treatment (completed all study visits though discontinued study drugs).
Figure 1. Consort Chart.
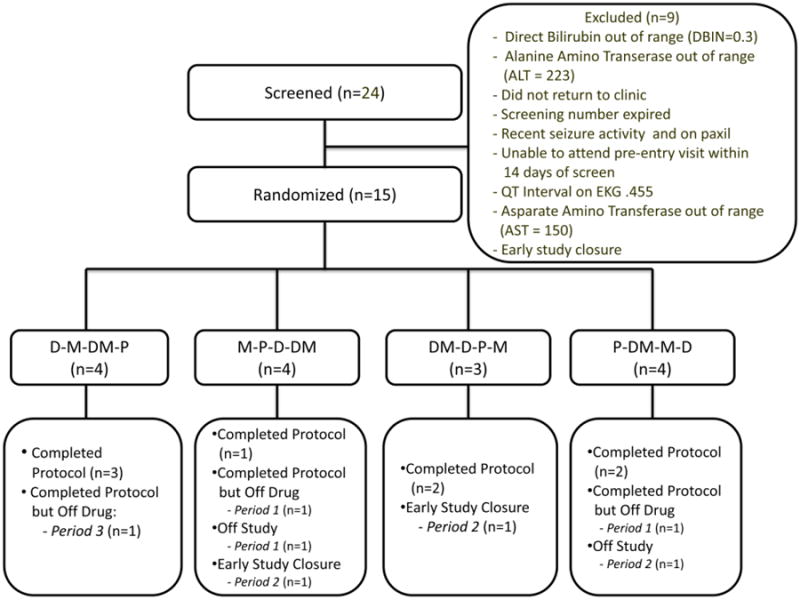
Patients who did not complete the 20 week protocol included 4 who prematurely discontinued study drugs in period 1 (one of whom dropped out after period 2) and 3 who discontinued both study treatment and participation in period 2. Reasons for subjects discontinuing study drugs in period 1 are as follows: intolerable fatigue (placebo), intolerable nausea and dizziness (methadone) followed by withdrawal symptoms, aberrant drug behavior (methadone), and urinary symptoms (duloxetine). The only patient who discontinued study drugs in period 2 reported intolerable tinnitus (duloxetine-methadone). The other two patients who discontinued study medications prior to completing the protocol did so due to premature cessation of the study.
All subjects who successfully completed the second treatment period finished the entire 20 week protocol.
Baseline Characteristics
The majority of participants were male (87%), older than 50 years old (73%), and non-white (53%). Median CD4 was 499 (IQR, 182-749), with an undetectable plasma viral load observed in 67% (10/15). Median CES-D score was 8 (IQR 4-19), indicating that depression symptoms were mild. Baseline daily and nighttime mean MPI score was 7 (IQR, 6-8), indicating moderate-severe neuropathic pain.
Efficacy
Refer to supplemental material.
Safety
Descriptive statistics were utilized for evaluation of tolerability and safety. No deaths, life-threatening adverse events (AEs), or severe laboratory abnormalities were reported during this study. Overall, four patients reported five Grade 2-3 AEs (Grade 2 moderate, Grade 3 severe) while on duloxetine, six participants reported 17 grade 2 AEs while on methadone, five patients reported 17 Grade 2 AEs while on combination therapy, and five participants reported 6 Grade 2-3 AEs while on placebo. Severe (Grade 3) adverse events on duloxetine included nausea (n=1), vomiting (n=1), renal dysfunction (n=1). Severe adverse events on placebo included pain (n=1) and fatigue (n=1). No severe adverse events were noted on either methadone or combination duloxetine-methadone.
A single severe laboratory abnormality, elevated creatinine, was noted with duloxetine monotherapy but was not deemed to be related to treatment; no other laboratory abnormalities were noted with duloxetine therapy. Other laboratory abnormalities observed during the study included: elevated SGOT (n=1) and alkaline phosphate (n=1) on methadone; elevated total bilirubin (n=2) with duloxetine-methadone; and elevated total bilirubin (n=1) with placebo.
Discussion
This study was designed to evaluate the efficacy, safety, and tolerability of duloxetine, methadone, and the combination of duloxetine-methadone for symptomatic treatment of painful HIV-PN. The primary efficacy endpoint measure, MPI, failed to demonstrate statistically significant differences across treatments. After the trial had been accruing for approximately eighteen months, and only one year remaining for the shelf-life of methadone study drug, it was deemed that the sample size achieved by continued accrual over a 12-month period would not be sufficiently powered to conduct the primary analysis (25% power to detect an MPI effect size of 1.5, data not shown). While we believe combination therapy pain treatment studies remain highly important to the field, our experience conducting this trial illustrates the formidable challenges of such studies. In the ensuing discussion, we detail many key issues our team identified as contributory factors to the premature closure of the study. We sought to explore these factors in context of a root-cause analysis (Figure 2). We feel that our experience will be valuable for the planning of future analgesic trials in this clinical population, as a recent meta-analysis of symptomatic treatments for painful HIV-PN specifically identified duloxetine and opioids as viable treatment options whose efficacy require formal investigation.[35]
Figure 2.

Poor accrual was a primary reason for premature study termination. In spite of excellent support and an appropriate research base, this study accrued too poorly to provide power to support the efficacy of the treatment regimens tested. Seven sites failed to enroll a single subject. As all 15 study sites were experienced clinical trial units participating in the AIDS Clinical Trials Group (ACTG), site inexperience was not a significant contributor to the accrual rates. The trial had combined support of the ACTG system and the Neurologic AIDS Research Consortium (NARC), with substantial support based on productivity, so that resources were available to all sites to support the effort of recruitment and the study. No other analgesic trials competing for the targeted study population were identified. The protocol team orchestrated a start-up teleconference with investigators and study coordinators, an annual face-to-face meeting with sites, monthly conference calls, and direct contact with site investigators. One explanation for slow enrollment is changing trends in HIV-associated morbidities in the face of evolving clinical care. While most epidemiological studies in the antiretroviral era describe symptomatic HIV-PN rates up to 30%, recent data from the ACTG consortium suggest that symptomatic PN may impact a smaller percentage (10%) of the overall HIV-infected population.[7] Trends in earlier HIV diagnosis, earlier initiation of effective antiretroviral therapy, and non-neurotoxic antiretroviral treatment options all contribute to this observation. With this in mind one might posit that the overall population of HIV patients with moderately severe ongoing neuropathic pain in spite of available therapies likely comprises a decreasing proportion of the overall HIV-infected population, particularly in resource-rich settings.[7] Other studies in the current treatment era describe higher HIV-PN rates that are consistent with prior studies.[8] It may be that patients in the ACTG system differ in important HIV-PN risk factors. The study team had conducted a site survey amongst all ACTG sites during the protocol development phase for this study, unfortunately not all sites professing interested opened the study at their sites and, furthermore, individual estimates of accrual projects were not met at any site.
A second factor was a delay in opening screens at study sites. The primary reason for this delay was in clearing the local pharmacies for study initiation. Opioid-related regulatory hurdles were significant contributors to delays in opening screening at many sites, as we faced diverse challenges posed by state- and site-specific schedule II opioid regulations regarding delivery, handling, and disposal. In consequence, many sites opened as late as six months after the first site screened its first patient. This delay had significant implications for drug supply given the shelf life for the methadone study drug was less than two years at the time of initial receipt. Aside from investigator licensure, study sites are required to have active licenses for the receipt and distribution of schedule II opioids and the Drug Enforcement Agency (DEA) requires completion of the DEA Forms 222 and 223 as well as transfer of unused and expired opioids to a DEA-approved Pharmaceutical Returns Processor. Sites in California faced the additional step of submitting the protocol to the California Research Advisory Panel, which reviews all protocols which utilize a schedule I or II controlled substance as a study drug. We found that defining the proper method of unused and expired drug disposal was particularly problematic, as there were site and institution-dependent factors, and in some sites FDA's recommendation was, surprisingly, to flush unused study drug in the toilet rather than return it to the pharmacy. In the ACTG system, all sites are eligible to enroll in the available study protocols, which made it difficult to proactively plan for the pharmacy needs at each site. Early investigation and identification of the specific regulations at each study site are necessary to avoid this complication.
An additional consideration for slow accrual is that the protocol's inclusion and exclusion criteria were rigorous, potentially excluding a large number of HIV-PN patients. We carefully considered inclusion and exclusion criteria and employed to precisely define a clinically stable painful HIV-PN population while ensuring their safety. Safety was paramount given that we were investigating two study treatments, one of which a controlled substance, in a medically complicated population. While many exclusion criteria related to the methadone, we were most concerned about the impact of duloxetine-related exclusion criteria. Given duloxetine's serotonergic activity most antidepressants were contraindicated, which was important when recognizing the high depression rates and frequent use of psychiatric medications in the HIV population, particularly in patients with painful HIV-PN[8, 36]. Rates of major depression approach 9%, approximately twice that of non-HIV infected populations, with depressive symptoms reported by up to one third of patients with use of psychotherapeutic medications in up to 30%[37-39]. We are, unfortunately, unable to determine the impact of this factor on accrual as HIPPA waivers allow staff to ‘pre-screen’ charts prior to an official screening visit. Once patients completed the screening visit enrollment rate was fairly high (24 screened, 15 randomized), though the low depression scores imply that those with even mild-moderate depressive symptoms did not enter screening. This potential barrier to recruitment could affect the future neuropathic pain study of any serotonergic medication in the HIV population.
Eligible patients may have been reluctant to enroll in the study because of methadone. As the protocol development period lasted approximately 2 years due to multiple scientific and regulatory reviews, we witnessed significant changes in public attitudes and perceptions regarding opioid therapy as the medical community and the general public faced an increasing epidemic of opioid-related abuse and overdose deaths.[40, 41] Given longstanding experience with the use of methadone to prevent heroin withdrawal, in many circles methadone is considered as a drug of ‘addicts’ rather than an analgesic with pain-relieving potential. Indeed, in a survey of members from the ACTG Community Advisory Board, 10/29 (38%) stated that, if eligible, they would not consider participation in the protocol due to fears of addiction. Such stigma may be in some ways particularly pervasive in the HIV-infected population, which in the early years was driven significantly by IV drug abuse. We sought to allay those concerns through development of a patient information sheet which was supplied to sites with a frank discussion of risks and benefits of methadone for pain management. The consent form and placebo patient information sheet are available as supplemental material.
Finally, we may have underestimated the willingness of patients with painful HIV-PN to endure placebo. We believed placebo was necessary for the scientific rigor of the study and sought to minimize its potential impact by allowing ongoing co-administration of multiple baseline neuropathic pain medications. Co-administered analgesics were considered to potentially mollify not only the placebo treatment period but also the effect of multiple washouts, though at the potential cost of muting differences between study treatments and placebo. Our study design furthermore offered each subject exposure all study treatments and thus active drug in 75% of the treatment periods. Placebo-controlled trials of marketed drugs have distinct challenges given that physicians can offer the same treatment in clinic in an unblinded fashion as part of routine clinical care, and in many cases such therapies may have already been tried and potentially rejected. Unapproved ‘novel’ drugs, only available through clinical trials, have a significant advantage since no prior experience or alternative source is available. It is not known whether our study may have been hampered by the current availability of these drugs off label, as it is not known how frequently these medications are utilized in our clinical population. Prior use is likely to have substantially reduced the eligible population willing to participate in the controlled trial we designed.
Accrual challenges were further underscored by significant premature discontinuation. Had accrual been more robust, successful completion of the protocol would have remained challenging given high dropout. Dropouts are particularly costly in a crossover study design as each patient serves as his or her own control. Although dropout may be seen due to lack of efficacy in placebo groups, we observed high rates of toxicity reflected in AEs and particularly noted with methadone-containing treatment regimens. Although we had estimated a 25% dropout rate based on prior crossover studies we observed high adverse event rates which were associated with frequent drug discontinuation and dropout.
One explanation for the observed AE rates is that methadone, even at low doses may be too toxic for opioid-naïve patients. There exist no data to suggest that study drugs affected each other's pharmacokinetics, and of the varied antiretroviral regimens the vast majority of recognized interactions would serve to increase methadone metabolism. Although we sought to propose a conservative titration regimen, it is possible that methadone initiated at a lower dose, such as 5mg daily, would have been more appropriate for opioid-naïve patients. Another possibility is that the study drugs had overlapping side effect profiles and that the combination regimen thus amplified side effect frequency and/or severity. This factor may have played a role given that we titrated study drugs in tandem to either target ceiling dose or MTD. In anticipation of higher adverse event rates particularly in the combination treatment period we proposed toxicity management algorithms for the more common anticipated toxicity syndromes, including constipation and nausea. A complicated protocol with multiple periods of tandem drug titration and taper is challenging to employ, particularly in a multi-center model. Presentations on study coordinator calls, a team meeting at the annual consortium conference, and monthly study team calls were orchestrated to streamline the learning curve. It is also possible that, had more subjects accrued, dropout from toxicities would have decreased as sites became more experienced with the symptom management. To our knowledge this study design had been previously employed successfully only in the context of a single center study.
A potential contributor to dropout was the rapid washout in between treatment periods. Decision regarding washout duration was balanced between risk of opioid withdrawal and/or serotonergic discontinuation symptoms against the overall duration of the study and time off study drugs. It was recognized that there were likely carry over effects at the beginning of each treatment period, but endpoints were measured only over the fourth week of each treatment period and we believed that sufficient time had transpired to minimize effects from the prior study treatment.
In summary, we sought to evaluate the efficacy of moderate dose duloxetine and methadone, both alone and in combination, for the treatment of moderately severe neuropathic pain due to HIV-PN. While this trial design was theoretically attractive and efficient, it proved impossible to complete. In the process of pursing this study, we identified many factors that precluded the successful completion of this trial that will need to be addressed in future neuropathic pain trials. We strongly believe that it is imperative for research to continue to address the unmet need of patients with unsuccessfully treated neuropathic pain.
Statistical Methods
Given the small number of participants, descriptive statistics and Wilcoxon Signed Rank tests were performed. Daily MPI scores were examined in two ways: (1) fourth week MPI scores and (2) four-week change in MPI scores. Secondary analyses examined the nighttime MPI scores, which were analyzed in a similar fashion. The analyses were as-treated and the missing outcomes were not imputed. Type I error was not adjusted for multiple testing.
Efficacy
No differences in the daily MPI scores were detected between any of the active treatments and placebo, or between combination duloxetine-methadone and duloxetine or methadone monotherapy (Supplemental Table1). Scatterplots for first and fourth week MPI scores are presented in Supplemental Figure 1. When evaluating pain improvement from baseline, MPI scores during combination duloxetine-methadone showed significant improvement from baseline (median change -1.0; Q1 and Q3 -2.0, 1.0; p=0.008); MPI score during placebo, however, also resulted in significant improvement (median change -1.0; Q1 and Q3 2.0, 0.0; p=0.016) and the observed effect of combination therapy was not statistically different from that observed with the other study treatments or placebo (data not shown).
Significant pair-wise differences were not detected in nighttime MPI scores between treatments. Nighttime MPI scores in the combination and monotherapy treatments resulted in significant improvement from baseline (p=0.004 for DM, p=0.016 for both duloxetine and methadone).
All patients on duloxetine took 60 mg as the MTD. The mean MTD for methadone was 24.6 mg. Pill counts corresponded with study regimen 79% (58/74) with duloxetine and 72% (53/74) with methadone.
According to blinding questionnaire responses, correct guesses by patients with respect to treatment arm was observed with 4 (40%) patients on duloxetine, 4 (40%) patients on methadone, and 5 (50%) patients on duloxetine-methadone. Correct guesses by investigators with respect to treatment arm was noted in 6 (60%) on duloxetine, 3 (30%) on methadone, and 5 (50%) on duloxetine-methadone. Given the small sample size, these data do not make it possible to determine whether unblinding from the side effects or other factors may have occurred in the trial.
Supplementary Material
Supplemental Figure 1: Scatter plots of baseline and 4th week MPI scores by treatment
Supplemental Table 1: Fourth week daily mean pain intensity scores between treatment groups
Acknowledgments
Funding and other support: The project described was supported by Award Number U01AI068636 from the National Institute of Allergy and Infectious Diseases and supported by National Institute of Mental Health (NIMH), National Institute of Dental and Craniofacial Research (NIDCR). The project was also supported by the Neurological AIDS Research Consortium, NS32228, and the National Institute of Neurological Diseases and Stroke (NINDS). Pharmaceutical support (duloxetine study drug and matching placebo) was provided by Eli Lilly. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. Supported in part by grants funded by the National Center for Research Resources.
The protocol team is grateful to the individuals who volunteered to participate in this study. The authors would like to acknowledge the invaluable support provided by other members of the A5252 study team: Jane Baum, Travis Behm, Laura Blair, William (Royce) Hardin, Mallory Harvey, Evan Kharasch, Justin McArthur, David Moody, Kimberly Scarsi, Holly Shaw, Katherine Shin, David Simpson, Elizabeth Smith, and Tzu-min Yeh.
ACTG site staff (in order of accrual by site): Eric S Daar, MD and Edward Lozano, MD [Harbor-UCLA Med. Ctr. CRS (Site 603) CTU Grant # A1069424], Teresa Spitz BSN and Judy Frain MSN [Washington U CRS (Site 2101) CTU Grant # AI0694595], Nagagopal Venna, MD and Tracey Cho, MD [Massachusetts General Hospital ACTG CRS (Site 101) CTU Grant # AI069472], Kathleen Nuffer, NP and Susan Cahill, RN [UCSD, AVRC CRS (Site 701) CTU Grant # AI069432], Robert Kalayjian, MD and Ann Marie Anderson, RN [MetroHealth CRS (Site 2503) CTU Grant # AI069501], Nina Lambert, RN and Bruce Cohen, MD [Northwestern University CRS (Site 2701) CTU Grant # AI069471], Beverly Putnam, RN, ANP and Steven Johnson, MD [University of Colorado Hospital CRS (Site 6101) CTU Grant # AI69450; RR025780], Tanvir K. Bell, MD and Maria Laura Martinez, BS [Houston AIDS Research Team CRS (Site 31473) CTU Grant # AI069503].
A5252 is registered with the ClinicalTrials.gov (NCT00863057).
Appendix
Eric S Daar, M.D. and Edward Lozano, M.D., - Harbor-UCLA Med. Ctr. CRS (Site 603) CTU Grant # A1069424
Teresa Spitz BSN, and Judy Frain MSN, - Washington U CRS (Site 2101) CTU Grant # AI0694595
Nagagopal Venna, M.D. and Tracey Cho, M.D. - Massachusetts General Hospital ACTG CRS (Site 101) CTU Grant # AI069472
Kathleen Nuffer, NP and Susan Cahill, R.N. - UCSD, AVRC CRS (Site 701) CTU Grant # AI069432
Robert Kalayjian, MD., and Ann Marie Anderson, RN., - MetroHealth CRS (Site 2503) CTU Grant # AI069501
Nina Lambert, RN, and Bruce Cohen, M.D., - Northwestern University CRS (Site 2701) CTU Grant # AI069471
Beverly Putnam, R.N., A.N.P. and Steven Johnson., M.D., - University of Colorado Hospital CRS (Site 6101) CTU Grant # AI69450; RR025780
Tanvir K. Bell, M.D., and Maria Laura Martinez, B.S., - Houston AIDS Research Team CRS (Site 31473) CTU Grant # AI069503
Footnotes
Conflict of Interest/Disclosure: Taylor Harrison: Funding support from the NINDS/NIH.
Sachiko Miyahara: Funding support from NINDS/NIH and NIAID/NIH.
Anthony Lee: Funding support from NIAID/NIH
Scott Evans: Funding support from NIAID/NIH
Barbara Bastow: No funding support for this project.
David Simpson: Funding support from NINDS/NIH.
Ian Gilron: No funding support for this project.
Robert Dworkin: No funding support for this project.
Eric Daar: Funding support from the NIAID and NIMH (NIH)
Linda Wieclaw: Funding support from the NIAID (NIH).
David Clifford: Funding support from NINDS and NIAID (NIH).
References
- 1.Schifitto G, et al. Incidence of and risk factors for HIV-associated distal sensory polyneuropathy. Neurology. 2002;58(12):1764–8. doi: 10.1212/wnl.58.12.1764. [DOI] [PubMed] [Google Scholar]
- 2.Morgello S, et al. HIV-associated distal sensory polyneuropathy in the era of highly active antiretroviral therapy: the Manhattan HIV Brain Bank. Arch Neurol. 2004;61(4):546–51. doi: 10.1001/archneur.61.4.546. [DOI] [PubMed] [Google Scholar]
- 3.Dalakas MC, Semino-Mora C, Leon-Monzon M. Mitochondrial alterations with mitochondrial DNA depletion in the nerves of AIDS patients with peripheral neuropathy induced by 2′3′-dideoxycytidine (ddC) Lab Invest. 2001;81(11):1537–44. doi: 10.1038/labinvest.3780367. [DOI] [PubMed] [Google Scholar]
- 4.Blum AS, et al. Low-dose zalcitabine-related toxic neuropathy: frequency, natural history, and risk factors. Neurology. 1996;46(4):999–1003. doi: 10.1212/wnl.46.4.999. [DOI] [PubMed] [Google Scholar]
- 5.Abrams DI, et al. A comparative trial of didanosine or zalcitabine after treatment with zidovudine in patients with human immunodeficiency virus infection. The Terry Beirn Community Programs for Clinical Research on AIDS. N Engl J Med. 1994;330(10):657–62. doi: 10.1056/NEJM199403103301001. [DOI] [PubMed] [Google Scholar]
- 6.Berger AR, et al. 2′,3′-dideoxycytidine (ddC) toxic neuropathy: a study of 52 patients. Neurology. 1993;43(2):358–62. doi: 10.1212/wnl.43.2.358. [DOI] [PubMed] [Google Scholar]
- 7.Evans SR, et al. Peripheral neuropathy in HIV: prevalence and risk factors. AIDS. 2011;25(7):919–28. doi: 10.1097/QAD.0b013e328345889d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellis RJ, et al. Continued high prevalence and adverse clinical impact of human immunodeficiency virus-associated sensory neuropathy in the era of combination antiretroviral therapy: the CHARTER Study. Arch Neurol. 2010;67(5):552–8. doi: 10.1001/archneurol.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pandya R, et al. HIV-related neurological syndromes reduce health-related quality of life. Can J Neurol Sci. 2005;32(2):201–4. doi: 10.1017/s0317167100003978. [DOI] [PubMed] [Google Scholar]
- 10.Pettersen JA, et al. Sensory neuropathy in human immunodeficiency virus/acquired immunodeficiency syndrome patients: protease inhibitor-mediated neurotoxicity. Ann Neurol. 2006;59(5):816–24. doi: 10.1002/ana.20816. [DOI] [PubMed] [Google Scholar]
- 11.Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010;150(3):573–81. doi: 10.1016/j.pain.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 12.Kieburtz K, et al. A randomized trial of amitriptyline and mexiletine for painful neuropathy in HIV infection. AIDS Clinical Trial Group 242 Protocol Team. Neurology. 1998;51(6):1682–8. doi: 10.1212/wnl.51.6.1682. [DOI] [PubMed] [Google Scholar]
- 13.Shlay JC, et al. Acupuncture and amitriptyline for pain due to HIV-related peripheral neuropathy: a randomized controlled trial. Terry Beirn Community Programs for Clinical Research on AIDS. Jama. 1998;280(18):1590–5. doi: 10.1001/jama.280.18.1590. [DOI] [PubMed] [Google Scholar]
- 14.Kemper CA, et al. Mexiletine for HIV-infected patients with painful peripheral neuropathy: a double-blind, placebo-controlled, crossover treatment trial. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;19(4):367–72. doi: 10.1097/00042560-199812010-00007. [DOI] [PubMed] [Google Scholar]
- 15.Estanislao L, et al. A randomized controlled trial of 5% lidocaine gel for HIV-associated distal symmetric polyneuropathy. J Acquir Immune Defic Syndr. 2004;37(5):1584–6. doi: 10.1097/00126334-200412150-00010. [DOI] [PubMed] [Google Scholar]
- 16.Hahn K, et al. A placebo-controlled trial of gabapentin for painful HIV-associated sensory neuropathies. J Neurol. 2004;251(10):1260–6. doi: 10.1007/s00415-004-0529-6. [DOI] [PubMed] [Google Scholar]
- 17.Simpson DM, et al. Pregabalin for painful HIV neuropathy: a randomized, double-blind, placebo-controlled trial. Neurology. 2010;74(5):413–20. doi: 10.1212/WNL.0b013e3181ccc6ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson DM, et al. Lamotrigine for HIV-associated painful sensory neuropathies: a placebo-controlled trial. Neurology. 2003;60(9):1508–14. doi: 10.1212/01.wnl.0000063304.88470.d9. [DOI] [PubMed] [Google Scholar]
- 19.Simpson DM, Brown S, Tobias J. Controlled trial of high-concentration capsaicin patch for treatment of painful HIV neuropathy. Neurology. 2008;70(24):2305–13. doi: 10.1212/01.wnl.0000314647.35825.9c. [DOI] [PubMed] [Google Scholar]
- 20.Ellis RJ, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology. 2009;34(3):672–80. doi: 10.1038/npp.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abrams DI, et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68(7):515–21. doi: 10.1212/01.wnl.0000253187.66183.9c. [DOI] [PubMed] [Google Scholar]
- 22.Gilron I, et al. Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med. 2005;352(13):1324–34. doi: 10.1056/NEJMoa042580. [DOI] [PubMed] [Google Scholar]
- 23.Gilron I, et al. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet. 2009;374(9697):1252–61. doi: 10.1016/S0140-6736(09)61081-3. [DOI] [PubMed] [Google Scholar]
- 24.Gatti A, et al. Controlled-release oxycodone and pregabalin in the treatment of neuropathic pain: results of a multicenter Italian study. Eur Neurol. 2009;61(3):129–37. doi: 10.1159/000186502. [DOI] [PubMed] [Google Scholar]
- 25.Raskin J, et al. A double-blind, randomized multicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic pain. Pain Med. 2005;6(5):346–56. doi: 10.1111/j.1526-4637.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- 26.Wernicke JF, et al. A randomized controlled trial of duloxetine in diabetic peripheral neuropathic pain. Neurology. 2006;67(8):1411–20. doi: 10.1212/01.wnl.0000240225.04000.1a. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein DJ, et al. Duloxetine vs. placebo in patients with painful diabetic neuropathy. Pain. 2005;116(1-2):109–18. doi: 10.1016/j.pain.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 28.Eisenberg E, McNicol ED, Carr DB. Efficacy and safety of opioid agonists in the treatment of neuropathic pain of nonmalignant origin: systematic review and meta-analysis of randomized controlled trials. Jama. 2005;293(24):3043–52. doi: 10.1001/jama.293.24.3043. [DOI] [PubMed] [Google Scholar]
- 29.Gagnon B, Almahrezi A, Schreier G. Methadone in the treatment of neuropathic pain. Pain Res Manag. 2003;8(3):149–54. doi: 10.1155/2003/236718. [DOI] [PubMed] [Google Scholar]
- 30.Morley JS, et al. Low-dose methadone has an analgesic effect in neuropathic pain: a double-blind randomized controlled crossover trial. Palliat Med. 2003;17(7):576–87. doi: 10.1191/0269216303pm815oa. [DOI] [PubMed] [Google Scholar]
- 31.Enggaard TP, et al. The analgesic effect of codeine as compared to imipramine in different human experimental pain models. Pain. 2001;92(1-2):277–82. doi: 10.1016/s0304-3959(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 32.Luccarini P, et al. Synergistic antinociceptive effect of amitriptyline and morphine in the rat orofacial formalin test. Anesthesiology. 2004;100(3):690–6. doi: 10.1097/00000542-200403000-00033. [DOI] [PubMed] [Google Scholar]
- 33.Taiwo YO, et al. Potentiation of morphine antinociception by monoamine reuptake inhibitors in the rat spinal cord. Pain. 1985;21(4):329–37. doi: 10.1016/0304-3959(85)90162-9. [DOI] [PubMed] [Google Scholar]
- 34.Ellis RJ, et al. Clinical validation of the NeuroScreen. J Neurovirol. 2005;11(6):503–11. doi: 10.1080/13550280500384966. [DOI] [PubMed] [Google Scholar]
- 35.Phillips TJ, et al. Pharmacological treatment of painful HIV-associated sensory neuropathy: a systematic review and meta-analysis of randomised controlled trials. PLoS One. 2010;5(12):e14433. doi: 10.1371/journal.pone.0014433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson-Papp J, et al. Association of self-reported painful symptoms with clinical and neurophysiologic signs in HIV-associated sensory neuropathy. Pain. 2010;151(3):732–6. doi: 10.1016/j.pain.2010.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katz MH, et al. Depression and use of mental health services among HIV-infected men. AIDS Care. 1996;8(4):433–42. doi: 10.1080/09540129650125623. [DOI] [PubMed] [Google Scholar]
- 38.Burnam MA, et al. Use of mental health and substance abuse treatment services among adults with HIV in the United States. Arch Gen Psychiatry. 2001;58(8):729–36. doi: 10.1001/archpsyc.58.8.729. [DOI] [PubMed] [Google Scholar]
- 39.Ciesla JA, Roberts JE. Meta-analysis of the relationship between HIV infection and risk for depressive disorders. Am J Psychiatry. 2001;158(5):725–30. doi: 10.1176/appi.ajp.158.5.725. [DOI] [PubMed] [Google Scholar]
- 40.Kuehn BM. Opioid prescriptions soar: increase in legitimate use as well as abuse. Jama. 2007;297(3):249–51. doi: 10.1001/jama.297.3.249. [DOI] [PubMed] [Google Scholar]
- 41.Okie S. A flood of opioids, a rising tide of deaths. N Engl J Med. 2010;363(21):1981–5. doi: 10.1056/NEJMp1011512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Scatter plots of baseline and 4th week MPI scores by treatment
Supplemental Table 1: Fourth week daily mean pain intensity scores between treatment groups