

Abstract
Pathologic obliterative bronchiolitis (OB)/Bronchiolitis obliterans syndrome (pathologic OB/BOS) is the major obstacle to long-term survival post-lung transplantation (LT). Our group has demonstrated that pulmonary hypertension (PH) complicates the course of chronic inflammatory lung diseases that have similarities to pathologic OB/BOS and that vascular remodeling of the bronchial circulation occurs during BOS. Consequently, we hypothesized that PH is associated with pathologic OB/BOS and may result from a vasculopathy of the allograft pulmonary circulation.
We conducted a single-center, retrospective study and examined the presence of PH and vasculopathy in patients with pathologic OB/BOS. Fifty-two pathologic specimens post-LT were recovered from January 10, 1997 to January 5, 2007 and divided into two groups, those with and without pathologic OB/BOS. PH was defined as a mean pulmonary artery pressure (mPAP) > 25 mmHg by right heart catheterization (RHC) or right ventricular systolic pressure (RVSP) ≥45 mmHg by transthoracic echocardiogram (TTE).
PH was more prevalent in those LT recipients with pathologic OB/BOS (72% vs. 0%, p = 0.003). Furthermore, pulmonary arteriopathy and venopathy were more prevalent in patients with pathologic OB/BOS (84% vs. 4%, p < 0.0001, and 77% vs. 35%, p = 0.004, respectively).
PH is common in LT recipients with pathologic OB/BOS and is associated with a vasculopathy of the allograft pulmonary circulation.
Keywords: Arteriopathy, bronchiolitis obliterans, chronic rejection, lung transplantation, pulmonary hypertension, vasculopathy, venopathy
Introduction
Pulmonary hypertension (PH) is a known complication of inflammatory lung diseases and is an independent predictor of worse survival (1–3). Our group has recently demonstrated noninvasive determinants of PH (4,5) and promising therapeutic options targeted at the PH complicating interstitial lung diseases (ILD) (6). Another chronic inflammatory disease of the lung that is accompanied by vascular remodeling is bronchiolitis obliterans syndrome (BOS). BOS is a form of chronic lung allograft rejection that pathologically is characterized by periairway inflammation that eventually gives way to luminal fibrosis in the form of obliterative bronchiolitis (OB) (7).
BOS is the foremost cause of death post-lung transplantation (LT) (8). The number and magnitude of acute rejection episodes are the most important risk factors for the development of BOS (7). We have recently demonstrated that the perivascular mononuclear cell infiltration that occurs during rejection typically results in allograft parenchymal and airway damage as well as vessel injury that progresses to cause vascular remodeling of the allograft airway circulation (9–12). Based on the above studies, we hypothesized that PH occurs in LT recipients that develop pathologic OB in the context of clinical BOS (pathologic OB/BOS) is, in part, a result of a vasculopathy affecting the lung allograft circulation.
The purpose of this study is to retrospectively determine if PH is associated with pathologic OB/BOS after LT and if this exists, in part, as a consequence of an underlying vascular remodeling of the allograft pulmonary circulation.
Materials and Methods
Selection of pathologic specimens
This is a single-center retrospective study evaluating all retrievable autopsy, surgical lung biopsy and explant lung material collected from January 10, 1997 until January, 5 2007. This start date was chosen as it marked the arrival of a different lung transplant team and operative approach. The local Institutional Review Board approved the study.
There were a total of 287 LT performed at our institution from January 10, 1997 to January, 5 2007. During this time period 52 pathologic specimens were collected post-LT that included 21 autopsies, 10 explants obtained as a result of retransplantation for BOS, 19 surgical wedge biopsies and 2 pneumonectomies.
Review of pathologic specimens
All cases were reviewed by a panel of three experienced lung transplant pathologists (MCF, WDW, CKL). The pathologic findings represent a consensus of this core group that was blinded to any clinical patient information. The specimens were evaluated for lung allograft rejection as well as other pathologic diagnoses as described by the ISHLT working formulation for the grading of pulmonary allograft rejection (13). The pathologic specimens were sectioned and stained with hematoxylin and eosin (H&E) as well as the combined Masson’s trichrome/elastic Verhoeff van Gieson stain (MT/EVG); the latter identifies vascular hyperplasia and fibrosis, with or without accompanying elastosis. In addition, if there was a suspicion of capillary proliferation by H&E staining, a reticulin stain was also performed to highlight capillary duplication. ISHLT criteria for arteriopathy and venopathy were used and defined as intimal fibrosis with a variable infiltration of mononuclear cells, with or without medial fibrosis (14). Capillary proliferation was defined as alveolar wall capillary duplication, as seen in the setting of pulmonary capillary hemangiomatosis (PCH) (15) and pulmonary veno-occlusive disease (PVOD) (16).
Methods used to evaluate PH
Right heart catheterization (RHC) was performed at rest in the supine position by means of a Swan-Ganz catheter (Edwards Lifesciences LLC, Irvine, CA). Oxygen was given when necessary to maintain peripheral pulse oximetry greater than 92% at all times. All hemodynamic measurements were made at end expiration and all cardiac output measurements were determined as an average of triplicate readings by thermodilution.
Transthoracic echocardiogram (TTE) was performed using conventional echocardiographic equipment (Hewlett-Packard 5500 model; Hewlett-Packard, Palo Alto, CA) with either 2.5 or 3.5 MHz transducers. Echocardiographic variables pertinent to PH were evaluated, including left ventricular diastolic dysfunction (LVDD) and systolic dysfunction (LVSD), estimated right ventricular systolic pressure (RVSP) based on the modified Bernoulli equation (assumed no obstruction to right ventricular outflow in estimating the systolic pulmonary artery pressure), qualitative right ventricular dysfunction (RVD) (right ventricular enlargement or hypertrophy and/or decreased right ventricular ejection fraction <40%) as previously described (17), and the acceleration time (i.e. time to peak velocity in the right ventricular outflow tract) as previously described (18). The mean right atrial pressure was conservatively estimated at 5 mmHg for all cases. LVSD was considered clinically meaningful if the ejection fraction was less than 50%.
Diagnosis of PH and secondary pulmonary arterial hypertension
For purposes of this study, PH was defined as a mean pulmonary artery pressure (mPAP) >25 mmHg by RHC or RVSP ≥45 mmHg by TTE. We have chosen this value based on the criteria established by the World Health Organization Symposium on Pulmonary Arterial Hypertension, which defines PH as a systolic PAP of 40–50 mmHg. Importantly, the value of 45 mmHg is clinically relevant as patients with emphysema and systolic PAP ≥45 mmHg were excluded from lung volume reduction surgery in the National Emphysema Treatment Trial (19). At our institution, a RHC was only performed in the context of considering a repeat LT in the setting of clinical BOS. Secondary pulmonary arterial hypertension (SPAH) was defined as PH in association with a left ventricular end diastolic pressure (LVEDP) < 15 mmHg (if LVEDP not available, a pulmonary artery wedge pressure (PAWP) < 15 mmHg), and a pulmonary vascular resistance (PVR) > 3 Wood Units (WU). For purposes of this study, a RHC or TTE was only included in the analysis if performed within 3 months of recovering pathologic lung allograft material.
Evaluation for clinical BOS
The diagnosis and severity of clinical BOS were determined based on existing standard criteria as previously described (7). Pulmonary function tests were performed according to American Thoracic Society standards with appropriate spirometric reference values. A diagnosis of clinical BOS was made only after exclusion of clinically significant acute rejection, infection, native disease progression or recurrence, increased body mass index, and/or anastomotic complications.
Statistical analysis
Characteristics of the study population are expressed as means ± SD or proportions where appropriate, unless otherwise noted. The prevalence of PH, right ventricular dysfunction, arteriopathy and venopathy seen on pathological specimens from LT recipients with and without pathologic OB/BOS were compared using the two-tailed Fisher’s exact test. RVSP measurements from LT recipients with and without pathologic OB/BOS were compared using the Student’s t test and data are presented as means ± SD. Univariate logistic regression models were constructed to further assess the effect of pathologic OB/BOS, as well as time post-LT, on the development of allograft arteriopathy and venopathy. We also performed multivariate logistic regression to assess the impact of pathologic OB/BOS on the development of allograft arteriopathy and venopathy, independent of the effects of time posttransplant. Data are displayed using bar graphs where appropriate. All analyses were carried out using JMP IN version 5.1 (SAS Institute, Cary, NC).
Results
Clinical-pathological diagnoses from LT recipients with and without OB
All 52 available pathologic specimens were divided into two groups based on the presence or absence of pathologic OB. LT recipient demographics with the type of lung specimen obtained are included in Table 1. We found that 29 of the 52 pathologic specimens had pathologic OB and 23 of the 52 did not have any pathologic OB (Table 1).
Table 1.
Lung transplant recipient demographics
| Patient characteristic | OB(+) n = 29 |
OB(−) n = 23 |
|---|---|---|
| Age, y (SD) | ||
| Age at transplant | 49 ± 15 | 57 ± 11 |
| Sex, n (%) | ||
| Female | 12 (41) | 9 (39) |
| Male | 17 (59) | 14 (61) |
| Transplant indication, n (%) | ||
| COPD-smoking | 4 (14) | 3 (13) |
| COPD-alpha 1 anti-trypsin | 1 (3) | 1 (4) |
| Idiopathic pulmonary arterial hypertension | 2 (7) | 0 (0) |
| Re-lung transplant BOS | 12 (41) | 0 (0) |
| Sarcoidosis | 1 (3) | 1 (4) |
| Idiopathic pulmonary fibrosis | 5 (18) | 12 (53) |
| Congenital heart disease | 0 (0) | 3 (13) |
| Hypersensitivity pneumonitis-interstitial lung disease | 2 (7) | 0 (0) |
| Other | 2 (7) | 3 (13) |
| Type of transplant, n(%) | ||
| Single lung | 12 (41) | 6 (26) |
| Bilateral lung | 16 (55) | 14 (61) |
| Heart-lung | 1 (4) | 3 (13) |
| Pathology source, n(%) | ||
| Autopsy | 14 (48) | 7 (30) |
| Re-lung transplant BOS explants | 10 (34) | 0 (0) |
| Surgical specimens | 5 (18) | 16 (70) |
| Days posttransplant for pathologic sampling | ||
| Median | 518 | 20 |
| Range | 38–5851 | 2–2225 |
Since community acquired respiratory viral infections and primary graft dysfunction (PGD) among other immune and nonimmune injuries can propagate allograft changes consistent with pathologic OB, we determined the clinical-pathologic diagnosis for each LT recipient at the time of obtaining pathologic lung tissue. The clinical-pathologic diagnoses from the 29 specimens with pathologic OB were: BOS (n = 25) [Stage 0p (n = 1), Stage I (n = 4), Stage II (n = 3), Stage III (n = 17)], PGD 3 (n = 3) and adenovirus pneumonia (n = 1) (Table 2). The clinical-pathologic diagnoses for the 23 specimens without pathologic OB are described in Table 2. Based on these clinical-pathologic diagnoses, we divided our LT recipients into two pure populations, those with pathologic OB and clinical BOS (pathologic OB/BOS) (n = 25), and those without pathologic OB/BOS (nonpathologic OB/non-BOS) (n = 23) (Figure 1).
Table 2.
Clinical and clinical-pathologic diagnoses of the LT recipient cohorts
| Clinical diagnosis | Pathologic diagnosis | Clinical-pathologic diagnosis |
|---|---|---|
| Obliterative bronchiolitis (OB+) Group (n = 29) | ||
| Bronchiolitis obliterans syndrome | ||
| (BOS) (n = 25) | OB | Pathologic OB/BOS (n = 25) |
| Stage 0p | OB | Pathologic OB/BOS (n = 1) |
| Stage I | OB | Pathologic OB/BOS (n = 4) |
| Stage II | OB | Pathologic OB/BOS (n = 3) |
| Stage III | OB | Pathologic OB/BOS (n = 17) |
| PGD | Areas of OB with diffuse alveolar damage (DAD) | PGD (n = 3) |
| Adenovirus pneumonia | Areas of OB with hemorrhagic adenovirus pneumonia | Hemorrhagic adenovirus pneumonia (n = 1) |
| Obliterative bronchiolitis negative (OB−) Group (n = 23) | ||
| Hemothorax | Normal lung allograft | Hemothorax/normal lung allograft (n = 3) |
| Hemothorax | DAD | Hemothorax/PGD (n = 1) |
| Hemothorax/hyperammonemia | Acute rejection (AR)/lymphocytic bronchiolitis (LB) | Hemothorax/hyperammonemia/AR/LB (n = 1) |
| Hemothorax | AR/pulmonary embolism (PE) | Hemothorax/AR/PE (n = 1) |
| Hemothorax | Respiratory bronchiolitis/PE | Hemothorax/respiratory Bronchiolitis/PE (n = 1) |
| Hemothorax | Diffuse organizing pneumonia/LB | Hemothorax/atypical AR (n = 1) |
| Progressive respiratory failure/acute respiratory distress syndrome | DAD | Acute respiratory distress syndrome (n = 1) |
| Acute respiratory distress syndrome/multiorgan failure | DAD/PTLD/PE | Acute respiratory distress syndrome//multiorgan failure/PTLD/PE (n = 1) |
| Progressive respiratory failure/acute respiratory distress syndrome | DAD with capillaritis/LB | Antibody-mediated rejection (n = 1) |
| PGD | CMV pneumonitis/DAD | PGD with CMV pneumonitis (n = 2) |
| Primary graft dysfunction/ECMO | DAD | PGD (n = 1) |
| PGD/ECMO | DAD/PE | PGD/PE (n = 1) |
| Sternal wound dehiscence (SWD) | DAD | SWD with PGD (n = 1) |
| Massive unilateral PE | Angioinvasive aspergillosis/PE | Angioinvasive aspergillosis/PE (n = 1) |
| PGD | Necrotizing pneumonia/PE/infarction | Nosocomial necrotizing pneumonia/PE with infarction (n = 1) |
| Trapped lung/chylothorax | Inflamed visceral pleura/normal lung allograft | Trapped lung/chylothorax with normal lung allograft (n = 1) |
| Hyperammonemia | Normal lung allograft | Hyperammonemia/normal lung allograft (n = 1) |
| Enterococcal sepsis | Valvular vegetations with septic pulmonary embolism | Spontaneous bacterial endocarditis with septic pulmonary embolism (n = 1) |
| Sudden death | Acute PE | Acute PE (n = 1) |
| PGD/massive coagulopathy | Diffuse bilateral pulmonary hemorrhage/PE | Bilateral pulmonary hemorrhage/PE (n = 1) |
PTLD = posttransplant lymphoproliferative disease; CMV = cytomegalovirus; ECMO = extracorporeal membrane oxygenation.
Figure 1. Algorithm for subject selection in this retrospective study.
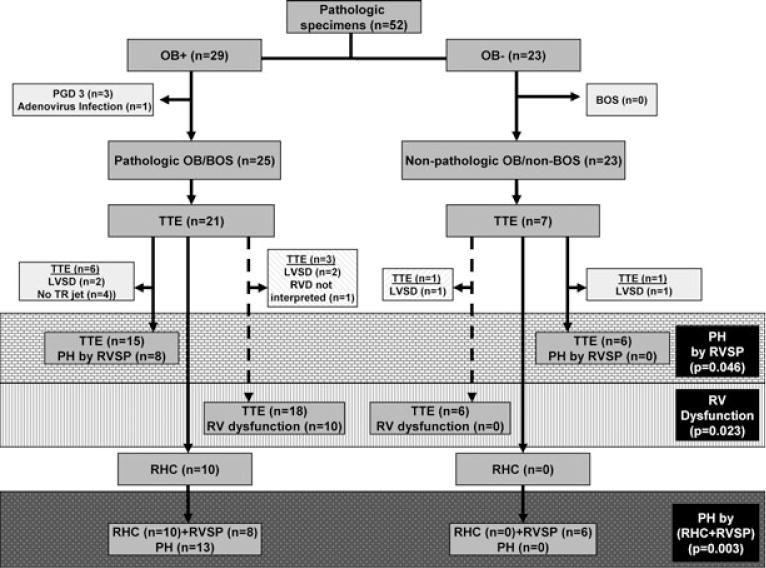
There were 287 lung transplants performed from January 10, 1997 to January, 5 2007 at our medical center. During this time period, 52 pathologic specimens were collected post-LT. These 52 specimens were blindly evaluated and categorized into those with pathologic OB (n = 29) and those without pathologic OB (n = 23). We then divided our LT recipients into two populations: those with pathologic OB in the context of clinical BOS (n = 25) and those without pathologic OB or clinical BOS (n = 23). All available TTEs performed within 3 months of obtaining the pathologic lung specimen were used to evaluate RVSP and RVD. The gray brick zone represents the prevalence of PH by RVSP between groups; the gray vertical hashed zone represents the prevalence of RVD between groups; the gray dotted zone represents the prevalence of PH by RHC when available in place of RVSP.
PH occurs in the context of pathologic OB/BOS
We assessed whether there was an association of PH with pathologic OB/BOS by evaluating all available RVSP measurements obtained by TTE from our LT recipients with and without pathologic OB/BOS. LT recipients with pathologic OB/BOS (n = 25) had a TTE performed (n = 21), of which six could not be included in our analysis due to either LVSD (n = 2) or no tricuspid regurgitant (TR) jet available to evaluate RVSP (n = 4) (Figure 1). LT recipients without pathologic OB/BOS (n = 23) had a TTE performed (n = 7), of which one could not be included in our analysis due to LVSD (n = 1) (Figure 1). The clinical-pathologic diagnoses for these patients without pathologic OB/BOS with an available TTE (n = 6) were nosocomial necrotizing pneumonia with PE and infarction (n = 1), bilateral pulmonary hemorrhage/PE (n = 1), spontaneous bacterial endocarditis with septic PE (n = 1), hyperammonemia with normal lung allograft (n = 1), PGD with CMV pneumonitis (n = 1) and trapped lung/chylothorax with normal lung allograft (n = 1). PH by RVSP was more prevalent in LT recipients with, as compared to those without, pathologic OB/BOS (53% vs. 0%; respectively, p = 0.046) (Figure 1; gray brick zone). Additionally, the absolute mean RVSP from our LT recipients with, as compared to those without, pathologic OB/BOS was significantly higher (53.0 ± 25.7 mmHg vs.33.1 ± 13.1 mmHg; respectively, p = 0.007). Previous studies evaluating PH associated with advanced lung disease have demonstrated that RV dysfunction is a better predictor of PH than RVSP (17). Importantly, since the evaluation for RV dysfunction does not require a TR jet, this increased the number of TTEs available for our analysis. Thus, we evaluated RV dysfunction in LT recipients with and without pathologic OB/BOS. LT recipients with pathologic OB/BOS had a TTE performed (n = 21) of which three could not be included in our analysis due to either LVSD (n = 2) or no interpretation of RVD (n = 1) (Figure 1; gray angled hashed zone). The LT recipients without pathologic OB/BOS had a TTE performed (n = 7) of which one could not be included due to LVSD (Figure 1; gray angled hashed zone). RV dysfunction was significantly more prevalent in LT recipients with, as compared to those without, pathologic OB/BOS (56% vs. 0%; respectively, p = 0.023) (Figure 1; gray vertical hashed zone).
The most accurate method of diagnosing PH is by RHC (20). LT recipients with pathologic OB/BOS had a TTE available (n = 21); 10 of these 21 LT recipients also underwent RHC (Figure 1). LT recipients without pathologic OB/BOS had a TTE available (n = 7) of which none had a RHC (Figure 1). Using RHC data when available in place of RVSP, we found an increased prevalence of PH in LT recipients with pathologic OB/BOS (RHC (n = 10) + RVSP (n = 8)) as compared to those without pathologic OB/BOS (RHC (n = 0) + RVSP (n = 6) (72% vs. 0% respectively, p = 0.003) (Figure 1; gray dotted zone). Importantly, of the LT recipients with pathologic OB/BOS and a RHC (n = 10), all had PH except for two LT recipients with mPAPs of 18 mmHg and 23 mmHg (Table 3).
Table 3.
Hemodynamics from right heart catheterizations performed on lung transplant recipients undergoing repeat LT for pathologic OB/BOS
| Type of PH | ||||||||||
| Echocardiographic PH | No | – | No | – | Yes | No | – | No | Yes | Yes |
| RHC PH | Yes | Yes | No | Yes | Yes | No | Yes | Yes | Yes | Yes |
| RHC SPAH | No | Yes | No | – | – | – | Yes | No | Yes | – |
| RHC Hemodynamics | ||||||||||
| Right atrial pressure | 9 | 7 | 9 | – | 21 | – | 3 | 10 | 5 | 4 |
| Pulmonary artery systolic pressure | 45 | 59 | 36 | 46 | 74 | 30 | 48 | 32 | 38 | 75 |
| Pulmonary artery diastolic pressure | 20 | 22 | 17 | 35 | 35 | 12 | 20 | 22 | 25 | 35 |
| mPAP | 28 | 36 | 23 | 39 | 48 | 18 | 33 | 26 | 29 | 48 |
| Pulmonary artery wedge pressure | 20 | 5 | 12 | – | – | – | 10 | 14 | 8 | – |
| Cardiac index | 2.9 | 1.86 | 1.9 | – | – | – | 1.2 | 3.1 | 4.3 | – |
| PVR | 0.9 | 10 | 1.5 | – | – | – | 6.8 | 0.4 | 3.1 | – |
| Systemic hemodynamics | ||||||||||
| LVEDP | – | – | 18 | – | – | – | 9 | 24 | – | – |
| Systolic blood pressure | – | – | 130 | – | – | – | 98 | 125 | 156 | 128 |
| Diastolic blood pressure | – | – | 76 | – | – | – | 70 | 76 | 98 | 87 |
| Mean arterial blood pressure | – | – | 99 | – | – | – | 86 | 96 | 117 | 103 |
Note: (–) data were not obtained.
A vasculopathy of the lung allograft pulmonary arteries and veins is associated with pathologic OB/BOS
We evaluated all of our lung biopsy specimens for pulmonary arteriopathy and venopathy by both H&E and MT/EVG staining. Most LT recipients with pathologic OB/BOS (n = 25) had extensive arteriopathy involving up to one-third of the pulmonary arteries throughout the lung allograft specimens (Figure 2A–C). This arteriopathy predominately involved the intimal and to a lesser extent, the medial layer. More specifically, we found intimal nonlaminar eccentric and concentric proliferation and fibrosis that occurred in nearly all pathologic OB/BOS cases. In most cases, the media demonstrated mild extracellular matrix deposition with smooth muscle atrophy. Plexiform and dilatation lesions were not present in any of the examined specimens. The spectrum of arteriopathy ranged from moderate involvement (85% of the affected vessels) to severe arterio-obliteration (15% of the affected vessels) (Figures 2A–C). Conversely, in LT recipients without pathologic OB/BOS (n = 23), only a single specimen had arteriopathy with the clinical-pathologic diagnosis of spontaneous bacterial endocarditis with septic pulmonary emboli. Overall, an arteriopathy was significantly more prevalent in LT recipients with, as compared to those without, pathologic OB/BOS (84% vs. 4%; respectively, p < 0.0001) (Figure 2D).
Figure 2. Pulmonary allograft arteriopathy occurs during pathologic OB/BOS.
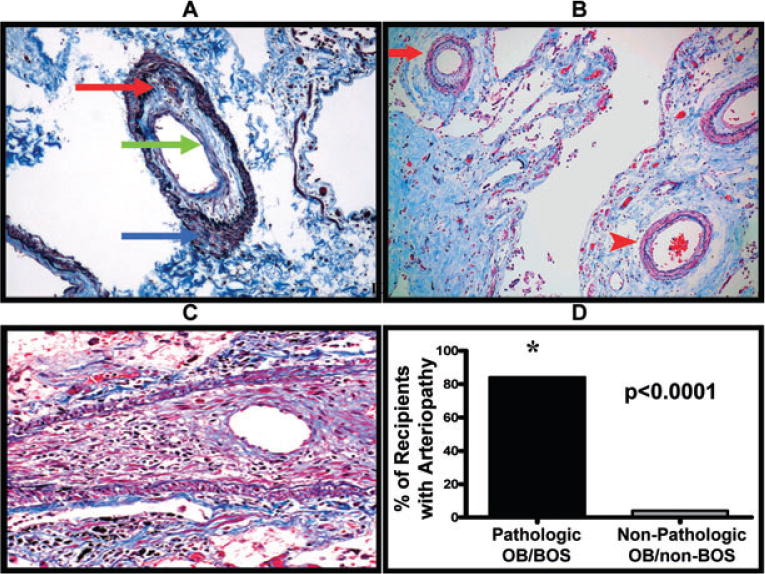
(A) Representative example of a pulmonary artery demonstrating arteriopathy with moderate intimal fibrosis and mononuclear cell infiltration (red and green arrows). Also note the media with mild matrix deposition and no smooth muscle hypertrophy (blue arrow) (Masson’s trichrome/elastic Verhoeff van Gieson stain, original magnification 100×). (B) Representative example of the heterogeneous distribution of the arteriopathy as highlighted by concentric arteriopathy (red arrow) in the same field as a relatively normal artery (red arrowhead); (Masson’s trichrome/elastic Verhoeff van Gieson stain, original magnification 100×). (C) Representative example of a pulmonary artery with severe stenosis due to massive intimal fibrosis and mononuclear cell infiltration. Note the striking medial smooth muscle atrophy; (Masson’s trichrome/elastic Verhoeff van Gieson, original magnification 200×). (D) Lung transplant recipients with pathologic OB/BOS have significantly more arteriopathy as compared to lung transplant recipients without pathologic OB/BOS.
Most LT recipients with pathologic OB/BOS (n = 25) had extensive venopathy involving up to one third of the pulmonary veins/venules (Figure 3A). The venopathy consisted of diffuse intimal fibrosis with partial or subtotal occlusion of pulmonary veins/venules, chronic perivenous mononuclear cell infiltrates and secondary capillary congestion. The spectrum of venopathy ranged from moderate involvement (50% of the affected vessels) to subtotal obliteration (50% of the affected vessels) (Figure 3). From LT recipients without pathologic OB/BOS (n = 23), eight had specimens with venopathy with the following clinical-pathologic diagnoses: acute pulmonary embolism (n = 1), ARDS with multiorgan failure and PE (n = 1), spontaneous bacterial endocarditis with septic PE (n = 1), PGD with CMV pneumonitis (n = 1), antibody-mediated rejection (n = 1), hemothorax with atypical ACR (n = 1), PGD (n = 1), hemothorax with normal lung allograft (n = 1). In all of these cases, the venopathy was mild and paucicellular. Overall, a venopathy was significantly more prevalent in LT recipients with, as compared to those without, pathologic OB/BOS (77% vs. 35%; respectively, p = 0.004) (Figure 3B).
Figure 3. Pulmonary allograft venopathy occurs during pathologic OB/BOS.
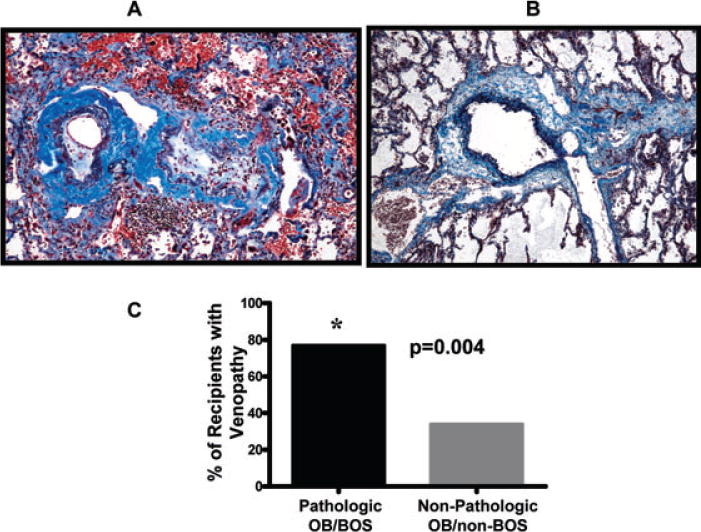
(A) Representative example of two veins in a LT recipient with pathologic OB/BOS demonstrating venopathy with intimal fibrosis and mononuclear cell infiltration; (Masson’s trichrome/elastic Verhoeff van Gieson stain, original magnification 100×). (B) Representative example of a normal pulmonary vein (Masson’s trichrome/elastic Verhoeff van Gieson stain, original magnification 100×). (C) Lung transplant recipients with pathologic OB/BOS have significantly more venopathy when compared to lung transplant recipients without pathologic OB/BOS.
Based on the possibility that lung allografts with time may develop some degree of a vasculopathy without the development of pathologic OB/BOS, we assessed the impact of time posttransplant (in years) on the development of allograft arteriopathy and venopathy. In a univariate logistic regression model, we found a significant association between time posttransplant and allograft arteriopathy (OR 1.32; 95%CI 1.05–1.97) (Table 4). As expected, there was also a significant association between pathologic OB/BOS and allograft arteriopathy, however, the impact of pathologic OB/BOS was much greater (OR 5.70; 95%CI 2.77–13.5 (Table 4). We then constructed a multivariate model to directly compare the impact of time posttransplant and pathologic OB/BOS on the development of allograft arteriopathy. In this model, time posttransplant had no significant impact (OR 0.98; 95%CI 0.79–1.13), however, pathologic OB/BOS had a marked impact (OR 5.93; 95%CI 2.61–16.4) (Table 4).
Table 4.
Univariate and multivariate logistic regression
| Univariate
|
Multivariate
|
|||
|---|---|---|---|---|
| OR | 95%CI | OR | 95%CI | |
| Arteriopathy | ||||
| Time posttransplant (yrs) | 1.32 | 1.05–1.97* | 0.98 | 0.79–1.31 |
| Pathologic OB/BOS | 5.70 | 2.77–13.5* | 5.93 | 2.61–16.4* |
| Venopathy | ||||
| Time posttransplant (yrs) | 1.13 | 0.96–1.49 | 1.01 | 0.83–1.30 |
| Pathologic OB/BOS | 2.24 | 1.23–4.35* | 2.20 | 1.12–4.81* |
Statistically significant.
Using similar logistic regression models for venopathy, we found no association between time posttransplant and allograft venopathy in a univariate analysis (OR 1.13; 95%CI 0.96–1.49) (Table 4). However, we found a strong association between pathologic OB/BOS and allograft venopathy (OR 2.24; 95%CI 1.23–4.35) (Table 4). We then constructed a multivariate model to directly compare the impact of time posttransplant and pathologic OB/BOS on the development of allograft venopathy. In this model, time posttransplant had no significant impact (OR 1.01; 95%CI 0.83–1.30), however, the association between pathologic OB/BOS and allograft venopathy remained significant (OR 2.20; 95%CI 1.12–4.81) (Table 4).
Discussion
Our prior work has demonstrated an association between PH and ILD and that a vascular remodeling of the allograft airway circulation is associated with BOS (4,9). Thus, we hypothesized that PH is associated with post-LT pathologic OB/BOS and a vasculopathy of the allograft pulmonary circulation. To our knowledge, no studies have evaluated the association of PH and pathologic OB/BOS.
We found that PH by RVSP was more prevalent in LT recipients with, as compared to those without, pathologic OB/BOS. We substantiated these findings by evaluating RV dysfunction, since it has been shown to correlate with PH in patients with advanced lung disease (17). We found a higher prevalence of RV dysfunction in LT recipients with, as compared to those without, pathologic OB/BOS and suspect that this RV strain results from continuous exacerbation of underlying PH with exertional activity. Additionally, we corroborated these findings by using RHC data, where available, in place of RVSP and found a significantly higher prevalence of PH in LT recipients with pathologic OB/BOS. Furthermore, all LT recipients with both pathologic OB/BOS and an available RHC (n = 10) had PH with the exception of only two patients with mPAP measurements of 18 mmHg and 23 mmHg. Notably, a RHC was only conducted in a minority of LT recipients with pathologic OB/BOS, specifically only those awaiting a repeat LT for BOS, which inherently excluded patients that died with more severe or rapidly accelerating BOS. Moreover, while there was a higher prevalence of PH in those recipients with, as compared to those without, pathologic OB/BOS, this was despite only five patients in the nonpathologic OB/non-BOS group having a normal lung allograft, underscoring the high prevalence of PH in patients with pathologic OB/BOS. Overall these data suggest a high prevalence of PH in LT recipients with pathologic OB/BOS.
We also determined the incidence of PH resulting from secondary PAH (SPAH) as compared to left heart disease, based on the LT recipients that underwent RHC. In general, post-LT recipients have an increased risk of systemic hypertension, diabetes mellitus and renal failure (21), all of which can contribute to left heart dysfunction and PH. None of our 10 LT recipients undergoing a repeat LT evaluation had significant hypertension, diabetes mellitus or renal failure. Five of eight LT recipients with PH had full hemodynamics available to assess for SPAH. Importantly, 3/5 LT recipients (60%) met criteria for SPAH without evidence for contributing left heart disease and only 2/5 LT recipients had hemodynamic evidence for left heart dysfunction with PH. Importantly, none of the LT recipients with pathologic OB/BOS had clinical or pathologic evidence of acute or chronic thromboembolic disease. Collectively, these data demonstrate that lung transplant recipients with pathologic OB/BOS can develop SPAH as well as PH secondary to left heart dysfunction.
Patients with inflammatory airway/parenchymal disease can have PH out of proportion to the degree of the underlying lung disease and treatment targeting the PH may improve symptoms and survival (22–25,6). Clinically, the question remains whether the deterioration in LT patients with pathologic OB/BOS is explained exclusively by the airway disease or if there is an independent pulmonary vascular process that might be amenable to PH directed therapies. In an effort to begin answering this question, we evaluated all available pathologic lung specimens with a focus on the pulmonary vasculature. We found that all but one of the LT recipients with pathologic OB/BOS had an arteriopathy that involved the intimal and medial layers without plexiform lesions. The intimal component consisted of nonlaminar eccentric and concentric proliferation and fibrosis. The degree of these intimal changes is consistent with the vascular changes described in PH complicating ILD (26). Surprisingly, the medial layer involvement in our cohort demonstrated mild matrix deposition with smooth muscle atrophy and not the hallmark findings of smooth muscle hypertrophy and hyperplasia that are felt to drive most other forms of PH (27). In the context of pathologic OB/BOS after LT, this novel finding might direct some of our attention away from smooth muscle biology to consider the role of fibrocyte and (myo)fibroblast biology as a contributing cause of the PH. Future studies of PH found in the setting of pathologic OB/BOS, as compared to other causes of PH, will be necessary to fully evaluate the contributions and interplay of both matrix deposition and smooth muscle biology during the pathogenesis of PH after LT.
Pulmonary venopathy in the form of PVOD is a rare cause of PH (16). Interestingly, our LT recipients with pathologic OB/BOS have venous changes consistent with PVOD including diffuse venular fibrotic stenoses and perivenular mononuclear cell infiltrates. Remarkably, four of our LT recipients with PH and pathologic OB/BOS also had a marked capillary duplication seen on H&E that was confirmed with a reticulin stain and is similar to that found in patients with PVOD and PCH (15) (Figure 4). Additionally, these vascular changes are consistent with those previously described in the setting of PH associated with mitral stenosis, ILD and connective tissue disease (28–30) and suggest that persistent elevated pressure and chronic inflammatory lung diseases can directly affect the venous circulation and contribute to PH.
Figure 4. Alveolar wall capillary duplication.
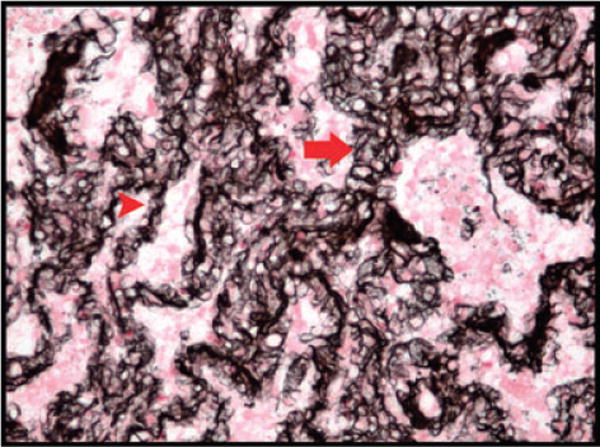
Representative example of significant capillary duplication (arrow) as compared to a normal capillary in an alveolar wall (arrowhead); (reticulin stain, original magnification 200×).
There are recent studies supporting a role for chronic inflammation contributing to PH (31). For instance, autoimmune destruction of pulmonary vasculature cells has been described in the setting of PH associated with connective tissue disease (32) and might be amenable to anti-inflammatory therapy (33). Additionally, studies have demonstrated an interplay between the alloimmune system and allograft vessels that produce the vasculopathy found in chronic renal and cardiac rejection, which parallels that found in our LT recipients with pathologic OB/BOS (34). Moreover, vessel wall cells during alloinjury have the ability to augment their expression of HLA, cytokines, chemokines and growth factors that can produce a vasculopathy similar to what we are describing in LT recipients with pathologic OB/BOS (34). Collectively, these studies suggest that allospecific injury plays an important role in inducing the PH found during the pathogenesis of pathologic OB/BOS.
Based on the possibility that lung allografts with time may develop some degree of vasculopathy without the development of pathologic OB/BOS, we assessed the impact of time posttransplant as compared to pathologic OB/BOS on the development of allograft vasculopathy. Duration posttransplant appeared to be associated with allograft vasculopathy in univariate models. However, multivariate models showed that the duration posttransplant had no significant effect on either allograft arteriopathy or venopathy and that pathologic OB/BOS strongly correlated with arteriopathy and venopathy independent of duration posttransplant.
LT recipients with pathologic OB/BOS and a full hemodynamic evaluation demonstrate that there is true secondary PAH that occurs in patients with pathologic OB/BOS. This is corroborated by a spectrum of allograft vasculopathy found in up to a third of the vessels by histopathology. The spectrum of vasculopathy ranged from moderate involvement to severe arterio/veno-obliteration. Importantly, the one-third vascular involvement (both arteries and veins) in our study is similar to if not greater than that previously reported in patients with severe PH associated with PVOD or mitral stenosis (30). These findings are also supported by a recent study involving patients with severe PH associated with connective tissue disease, which found that approximately 50% of lobular arteries and septal/preseptal veins were affected and included a spectrum of vasculopathy ranging from mild intimal thickening to subtotal occlusion (28). Furthermore, a study involving 58 patients with severe PAH demonstrated that the pulmonary arteries/arterioles had a spectrum of disease including isolated medial hypertrophy (32%), concentric laminar intimal fibrosis (18%), eccentric intimal fibrosis (25%), and plexiform lesions (7%) (35). Collectively, these studies suggest that the degree of PH found in the LT recipients with pathologic OB/BOS is compatible with the degree of allograft vasculopathy found on histopathology.
Although alveolar hypoxia may play some role in the PH of LT recipients with pathologic OB/BOS, our data suggest that the PH is not simply the result of chronic alveolar hypoxia. While none of our LT recipients with RHC were challenged with acute vasodilators, the persistence of PH despite oxygen supplementation indicates that acute hypoxic vasoconstriction is not the only determinant. Furthermore, neomuscularization of the pulmonary arterioles is a prominent pathologic finding under chronic hypoxic conditions, a feature not seen in any of our LT recipients. The absence of medial smooth muscle hypertrophy and the presence of medial smooth muscle atrophy suggest that the allospecific immune response may be a primary driving force of PH found during BOS. Future studies will be required to determine the role of chronic hypoxia and alloimmune injury during the pathogenesis of PH during pathologic OB/BOS.
We acknowledge the limitations of our study that are inherent to a retrospective analysis. Nonetheless, this study demonstrates that PH is associated with pathologic OB/BOS. Furthermore, our data demonstrate that these patients can develop a SPAH and PH secondary to left heart dysfunction. The allograft histopathology reveals the possible cause of the SPAH, an underlying arteriopathy and venopathy. With regard to this vasculopathy, the LT recipient immune system first interacts with the donor lung via the vascular endothelium making the vasculature a prime target for the alloreactive response. Thus, it is no surprise that human and animal studies of acute lung allograft rejection have demonstrated that the rejection process begins with injury to the pulmonary venous and arterial circulations and only then extends out to involve the lung parenchyma and airways (6,13). We suspect that intermittent and/or persistent perivascular inflammation involves allograft pulmonary arteries, veins, capillaries and the airway microcirculation, all resulting in pulmonary vascular remodeling with eventual PH, parenchymal fibrosis and allograft airway injury, respectively. Ultimately, these processes lead to the physiologic changes described in pathologic OB/BOS. The patchy distribution of the vasculopathy perhaps explains why the PH during pathologic OB/BOS appears mild to moderate in severity; however, targeted therapy that protects the vascular endothelium from alloinjury may be a novel way to prevent PH as well as the continuum of acute to chronic lung allograft rejection.
Acknowledgments
This work was supported, in part, by grants from the National Institutes of Health (HL080206 and HL086491 to J.A.B.
References
- 1.Nadrous HF, Pellikka PA, Krowka MJ, et al. The impact of pulmonary hypertension on survival in patients with idiopathic pulmonary fibrosis. Chest. 2005;128(6 Suppl):616S–617S. doi: 10.1378/chest.128.6_suppl.616S-a. [DOI] [PubMed] [Google Scholar]
- 2.Shorr AF, Davies DB, Nathan SD. Outcomes for patients with sarcoidosis awaiting lung transplantation. Chest. 2002;122:233–238. doi: 10.1378/chest.122.1.233. [DOI] [PubMed] [Google Scholar]
- 3.Weitzenblum E, Hirth C, Ducolone A, Mirhom R, Rasaholinjanahary J, Ehrhart M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax. 1981;36:752–758. doi: 10.1136/thx.36.10.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zisman DA, Ross DJ, Belperio JA, et al. Prediction of pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Med. 2007;101:2153–2159. doi: 10.1016/j.rmed.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zisman DA, Karlamangla AS, Ross DJ, et al. High-resolution chest computed tomography findings do not predict the presence of pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2007;132:773–779. doi: 10.1378/chest.07-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collard HR, Anstrom KJ, Schwarz MI, Zisman DA. Sildenafil improves walk distance in idiopathic pulmonary fibrosis. Chest. 2007;131:897–899. doi: 10.1378/chest.06-2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Estenne M, Maurer JR, Boehler A, et al. Bronchiolitis obliterans syndrome 2001: An update of the diagnostic criteria. J Heart Lung Transplant. 2002;21:297–310. doi: 10.1016/s1053-2498(02)00398-4. [DOI] [PubMed] [Google Scholar]
- 8.Trulock EP, Edwards LB, Taylor DO, Boucek MM, Keck BM, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: Twenty-third official adult lung and heart-lung transplantation report–2006. J Heart Lung Transplant. 2006;25:880–892. doi: 10.1016/j.healun.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Belperio JA, Keane MP, Burdick MD, et al. Role of CXCR2/CXCR2 ligands in vascular remodeling during bronchiolitis obliterans syndrome. J Clin Invest. 2005;115:1150–1162. doi: 10.1172/JCI24233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belperio JA, Keane MP, Burdick MD, et al. Critical role for CXCR3 chemokine biology in the pathogenesis of bronchiolitis obliterans syndrome. J Immunol. 2002;169:1037–1049. doi: 10.4049/jimmunol.169.2.1037. [DOI] [PubMed] [Google Scholar]
- 11.Belperio JA, Burdick MD, Keane MP, et al. The role of the CC chemokine, RANTES, in acute lung allograft rejection. J Immunol. 2000;165:461–472. doi: 10.4049/jimmunol.165.1.461. [DOI] [PubMed] [Google Scholar]
- 12.Belperio JA, Keane MP, Burdick MD, et al. Role of CXCL9/CXCR3 chemokine biology during pathogenesis of acute lung allograft rejection. J Immunol. 2003;171:4844–4852. doi: 10.4049/jimmunol.171.9.4844. [DOI] [PubMed] [Google Scholar]
- 13.Yousem SA, Berry GJ, Cagle PT, et al. Revision of the 1990 working formulation for the classification of pulmonary allograft rejection: Lung Rejection Study Group. J Heart Lung Transplant. 1996;15(1 Pt 1):1–15. [PubMed] [Google Scholar]
- 14.Stewart S, Fishbein MC, Snell GI, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant. 2007;26:1229–1242. doi: 10.1016/j.healun.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 15.Lantuejoul S, Sheppard MN, Corrin B, Burke MM, Nicholson AG. Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis: A clinicopathologic study of 35 cases. Am J Surg Pathol. 2006;30:850–857. doi: 10.1097/01.pas.0000209834.69972.e5. [DOI] [PubMed] [Google Scholar]
- 16.Mandel J, Mark EJ, Hales CA. Pulmonary veno-occlusive disease. Am J Respir Crit Care Med. 2000;162:1964–1973. doi: 10.1164/ajrccm.162.5.9912045. [DOI] [PubMed] [Google Scholar]
- 17.Arcasoy SM, Christie JD, Ferrari VA, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003;167:735–740. doi: 10.1164/rccm.200210-1130OC. [DOI] [PubMed] [Google Scholar]
- 18.Torbicki A, Skwarski K, Hawrylkiewicz I, Pasierski T, Miskiewicz Z, Zielinski J. Attempts at measuring pulmonary arterial pressure by means of Doppler echocardiography in patients with chronic lung disease. Eur Respir J. 1989;2:856–860. [PubMed] [Google Scholar]
- 19.NETT. Rationale and design of the National Emphysema Treatment Trial (NETT). A prospective randomized trial of lung volume reduction surgery. J Thorac Cardiovasc Surg. 1999;118:518–528. doi: 10.1016/s0022-5223(99)70191-1. [DOI] [PubMed] [Google Scholar]
- 20.McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):14S–34S. doi: 10.1378/chest.126.1_suppl.14S. [DOI] [PubMed] [Google Scholar]
- 21.Trulock EP, Christie JD, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-fourth official adult lung and heart-lung transplantation report-2007. J Heart Lung Transplant. 2007;26:782–795. doi: 10.1016/j.healun.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Nunes H, Humbert M, Capron F, et al. Pulmonary hypertension associated with sarcoidosis: Mechanisms, haemodynamics and prognosis. Thorax. 2006;61:68–74. doi: 10.1136/thx.2005.042838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fartoukh M, Humbert M, Capron F, et al. Severe pulmonary hypertension in histiocytosis X. Am J Respir Crit Care Med. 2000;161:216–223. doi: 10.1164/ajrccm.161.1.9807024. [DOI] [PubMed] [Google Scholar]
- 24.Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129:746–752. doi: 10.1378/chest.129.3.746. [DOI] [PubMed] [Google Scholar]
- 25.Leuchte HH, Neurohr C, Baumgartner R, et al. Brain natriuretic peptide and exercise capacity in lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2004;170:360–365. doi: 10.1164/rccm.200308-1142OC. [DOI] [PubMed] [Google Scholar]
- 26.Girgis RE, Mathai SC. Pulmonary hypertension associated with chronic respiratory disease. Clin Chest Med. 2007;28:219–232. doi: 10.1016/j.ccm.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 27.Pietra GG, Capron F, Stewart S, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 28.Dorfmuller P, Humbert M, Perros F, et al. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol. 2007;38:893–902. doi: 10.1016/j.humpath.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 29.Colombat M, Mal H, Groussard O, et al. Pulmonary vascular lesions in end-stage idiopathic pulmonary fibrosis: Histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum Pathol. 2007;38:60–65. doi: 10.1016/j.humpath.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Chazova I, Robbins I, Loyd J, et al. Venous and arterial changes in pulmonary veno-occlusive disease, mitral stenosis and fibrosing mediastinitis. Eur Respir J. 2000;15:116–122. doi: 10.1183/09031936.00.15111600. [DOI] [PubMed] [Google Scholar]
- 31.Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–363. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Visscher D, Rihal C, Aubry MC. Pulmonary veno-occlusive disease as a primary cause of pulmonary hypertension in a patient with mixed connective tissue disease. Rheumatol Int. 2007;27:1163–1165. doi: 10.1007/s00296-007-0362-1. [DOI] [PubMed] [Google Scholar]
- 33.Jais X, Launay D, Yaici A, et al. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: A retrospective analysis of twenty-three cases. Arthritis Rheum. 2008;58:521–531. doi: 10.1002/art.23303. [DOI] [PubMed] [Google Scholar]
- 34.Libby P, Pober JS. Chronic rejection. Immunity. 2001;14:387–397. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 35.Pietra GG, Edwards WD, Kay JM, et al. Histopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, Primary Pulmonary Hypertension Registry. Circulation. 1989;80:1198–1206. doi: 10.1161/01.cir.80.5.1198. [DOI] [PubMed] [Google Scholar]
- 36.Flax M. The role of vascular injury in pulmonary allograft rejection. Transplantation. 1966;4:66–78. [Google Scholar]