

Abstract
An efficient one pot condensation of naphthols (1), 2,5-disubstituted indole-3-carboxaldehydes (2), and secondary amines (3) has been achieved using dichloromethane as a solvent, stirring at room temperature. Some of the new [(disubstituted amino)(5-substituted 2-phenyl-1H-indol-3-yl)methyl]naphthalene-ols (4) derivatives were prepared in good yields. The significant features of this method are simple work-up procedure, inexpensive nontoxic solvent, shorter reaction times, and excellent product yields. The structures of newly synthesized compounds (4a–r) are confirmed by their elemental analysis, FTIR, 1H and 13C NMR, and mass spectral data. These compounds were screened for their in vitro antioxidant, antimicrobial, antitubercular, and anticancer activities. Among the synthesized compounds (4a–r), the compound 4e exhibited highest activity for radical scavenging and ferric ions reducing antioxidant power activities; compounds 4b, 4h, and 4k showed good metal chelating activity. Compounds 4n and 4q showed excellent antimicrobial activities with MIC value 08 µg/mL against tested strains. Compounds 4h, 4k, 4n, and 4q exhibited promising antitubercular activity with MIC value 12.5 µg/mL. Compounds 4k and 4q exhibited 100% cell lysis at concentration 10 µg/mL against MDA-MB-231 (human adenocarcinoma mammary gland) cell lines.
1. Introduction
The search and evaluation of chemical compounds and their derivatives with a specific pharmacological activity are a demanding task in the drug discovery process. During last few years, the synthesis of aminoalkylnaphthols has received special attention from the scientific community because of their significant biological [1] and catalytic [2] properties. Multicomponent reactions (MCRs) have emerged as an efficient and powerful tool in modern organic chemistry for the synthesis of biologically potent molecules from readily available substrates without isolation of intermediates in minimal time with maximum selectivity. MCRs have proved to be very powerful and efficient bond-forming tool in synthetic chemistry; the reactions are flexible, of high atom economy, and of high purity with excellent yields [3].
Oxidative stress is a well-known mechanism that is responsible for the development of vascular damage. Oxidative stress results from an imbalance between (i) an overproduction of reactive oxygen species (ROS) by the different cellular sources such as the mitochondrial respiratory chain, nicotinamide adenine dinucleotide phosphate hydride oxidases (NADPHOXs or NOX), xanthine oxidase, lipoxygenases, cytochrome-P450, and other oxidases and (ii) decreased cellular and plasma antioxidant defenses [4]. Antioxidants inhibit the generation of ROS and the subsequent formation of lipid peroxidation products, thereby preventing both oxidative and carbonyl stress. Most antioxidants prevent down low-density lipoproteins (LDL) oxidation in cell-free systems and delay the formation of atherosclerotic lesions in animal models for atherosclerosis such as apo E-/-mice [5]. Discrepancies exist about their efficiency against atherosclerosis in humans, this resulting at least in part from the bioavailability within the plaque, their chemical nature, and inability to scavenge reactive carbonyl compounds (RCCs) once adducts are formed on proteins [6]. However, antioxidants remain very efficient in preventing the early atherosclerotic lesions and inflammatory events implicated in the evolution of lesions towards more advanced states [5, 6].
There are some life-threatening diseases; among them tuberculosis (TB) is one of the major life-threatening chronic infections. TB is a specific communicable disease caused by Mycobacterium tuberculosis (Mtb). It affects both the pulmonary and the nonpulmonary tissues. These bacilli are microscopic and were first discovered in 1882. This disease is general or local, acute or chronic. According to WHO report, around one-third of the world's population is infected with TB, resulting in 2-3 million people are going to die annually [7]. The individuals with HIV-positive patients are highly susceptible to Mtb with 50-fold risk increases over HIV-negative patients [8]. Every year, about 0.35 million people living with HIV die from TB. Another major problem in TB therapy is the fact that Mtb became more resistant to the antituberculosis (anti-TB) drugs [9]. Furthermore, in recent times, the appearance of multidrug-resistant TB (MDR-TB) occurs, which is a form of TB that does not respond to the first- and second-line drugs [10]. TB drugs have become a serious task to TB control and its treatment. Hence, there is an increased demand to develop new tuberculosis agents effective against pathogens resistant to current treatment.
Among women worldwide, breast cancer is the most common cause of cancer death. The latest statistic indicated that about 600,000 women die from the breast cancer disease annually worldwide [11]. MDA-MB 231 cells have high invasive ability that can contribute to metastasis [12, 13]. Most chemotherapeutic drugs induce apoptosis in cancer cell [14]. Apoptosis is considered as a significant form of cancer cell death after treatment with cytotoxic drugs and has been recognized as a standard strategy for the selection of anticancer drug [15, 16]. Thus, there is a great need for new alternative agents for the prevention and treatment of breast cancer.
The indole framework is a medicinally relevant scaffold and has been widely identified as a privileged structure of pharmacophore [17]. Indole scaffold is present in thousands of isolated natural products and also synthetic compounds constitute an important class of therapeutic agents in medicinal chemistry such as antimicrobial [18], antioxidant [19], antiviral [20], anti-HIV, antimalarial [21], and antituberculosis products [22]. Naturally occurring indole derivatives melatonin (I), serotonin (II), tryptophan (III), and indole-3-propionic acid (IV) influence many important biochemical processes, for example, acting as an antioxidant and playing an important role in the immune system [23–28].
In view of Figure 10 and in continuation of our research on the synthesis of biologically active molecules [29–32], in present investigation, we report the synthesis, antioxidant, antimicrobial, antitubercular, and anticancer activities of novel indole derivatives.
Figure 10.
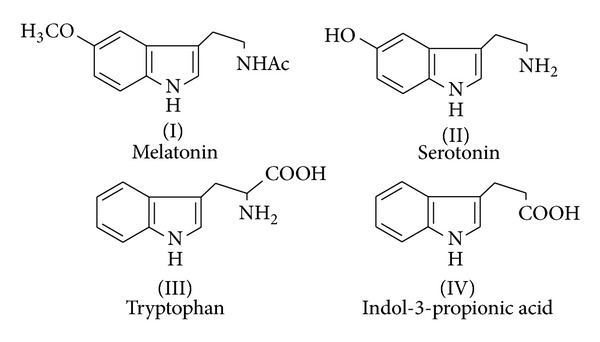
Indole derivatives with antioxidant activity.
2. Result and Discussion
2.1. Chemistry
The one pot synthesis of titled compounds 1-[(5-substituted-2-phenyl-1H-indol-3-yl)(piperidin-1-yl)methyl]naphthalene-2-ol and 1-[(dimethylamino)(5-substituted-2-phenyl-1H-indol-3-yl)methyl]naphthalene-2-ol and their -1-ol isomers was achieved by condensation of 1- or 2-naphthol and secondary amines with 2,5-disubstituted indole-3-carboxaldehydes in dichloromethane stirring at room temperature (Scheme 1 and Table 1). The progress of the reaction was monitored by TLC. After the completion of the reaction as checked by TLC, dichloromethane was pumped out by rotary evaporation. The crude product was purified directly by crystallization from ethanol.
Scheme 1.
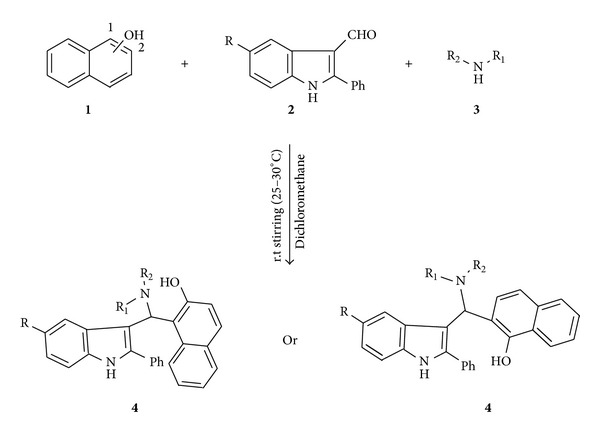
One pot synthesis of indole derivatives.
Table 1.
Synthesis of [(disubstituted amino)(5-substituted 2-phenyl-1H-indol-3-yl)methyl]naphthalene-ols (4a–4r).
| Starting materials | Products (4a–4r) | Time (h) | Yield (%) isolated | |||
|---|---|---|---|---|---|---|
| Entry | Amines | R | Naphthol | |||
| 1 | Piperidine | H | 2-Naphthol | 4a | 3 | 82 |
| 2 | Piperidine | Cl | 2-Naphthol | 4b | 3 | 85 |
| 3 | Piperidine | CH3 | 2-Naphthol | 4c | 2 | 79 |
| 4 | Piperidine | H | 1-Naphthol | 4d | 2 | 89 |
| 5 | Piperidine | Cl | 1-Naphthol | 4e | 3 | 86 |
| 6 | Piperidine | CH3 | 1-Naphthol | 4f | 3 | 87 |
| 7 | Diethyl amine | H | 2-Naphthol | 4g | 3 | 90 |
| 8 | Diethyl amine | Cl | 2-Naphthol | 4h | 2 | 89 |
| 9 | Diethyl amine | CH3 | 2-Naphthol | 4i | 3 | 89 |
| 10 | Diethyl amine | H | 1-Naphthol | 4j | 3 | 78 |
| 11 | Diethyl amine | Cl | 1-Naphthol | 4k | 3 | 82 |
| 12 | Diethyl amine | CH3 | 1-Naphthol | 4l | 2 | 84 |
| 13 | Dimethyl amine | H | 2-Naphthol | 4m | 3 | 89 |
| 14 | Dimethyl amine | Cl | 2-Naphthol | 4n | 3 | 89 |
| 15 | Dimethyl amine | CH3 | 2-Naphthol | 4o | 3 | 85 |
| 16 | Dimethyl amine | H | 1-Naphthol | 4p | 3 | 88 |
| 17 | Dimethyl amine | Cl | 1-Naphthol | 4q | 3 | 84 |
| 18 | Dimethyl amine | CH3 | 1-Naphthol | 4r | 2 | 82 |
The structures of all newly synthesized compounds (4a–r) were confirmed from their elemental analysis, IR, 1H NMR, 13C NMR, and mass spectral data. Compound 4b in its IR spectrum exhibited characteristic absorption bands at 3126 and 3049 cm−1 due to –OH and indole-NH functions, respectively. In its 1H NMR spectrum, the downfield signal appeared at 12.60 ppm as singlet was assigned to –OH (confirmed by deuterium exchange) signal at 9.90 ppm, as singlet attributed to the indole-NH, whereas the fourteen aromatic protons resonated as multiplet between 7.30 and 7.82 ppm. The singlet at 5.01 ppm was assigned to single proton of N–C–H, signal at 3.37 ppm appeared due to four protons of two chemically equivalent –CH2 groups of piperidine ring, signal at 2.55 ppm was assigned to triplet due to two protons of –CH2 group of piperidine ring, and one more signal at 1.11 ppm exhibited due to multiplet four protons of chemically equivalent two CH2 groups of piperidine ring. In its 13C NMR spectrum, the signal appeared at 150.44 ppm due to C–OH, whereas carbon C–Cl resonated at 128.51 ppm. Further the structure of 4b is confirmed by its isotopic molecular ion peak which is in agreement with molecular weight and nitrogen rule.
2.2. Antioxidant Activity
2.2.1. 1-Diphenyl-2-picrylhydrazyl (DPPH) Radical Scavenging Activity (RSA)
The synthesized compounds (4a–r) were screened for their free radical scavenging activity using DPPH method [33]. This model of radical scavenging activity by DPPH radical is extensively applied to evaluate the antioxidant activity in shorter time. The odd electron in the DPPH free radical gives a strong absorption band at 517 nm, which is purple in color. This property makes it suitable for spectrometric studies. The DPPH assay has often been used to estimate the antiradical activity of antioxidant. The free radical scavenging capacities of the synthesized compounds were measured at different concentrations (25, 50, 75, and 100 μg/mL in methanol) in presence of freshly prepared solution of stable free radical DPPH in methanol. The synthesized compounds react with DPPH radical and convert it into 1,1-diphenyl-2-picrylhydrazine. The extent of decolorization is an indicative potentiality of antioxidant behavior of a particular compound. Butylated hydroxyanisole (BHA), tertiary butylated hydroquinone (TBHQ), and ascorbic acid (AA) are used as reference standards.
The analysis of results indicated that (Figures 1, 2, and 3) compound 4e exhibited highest radical scavenging activity 86.17% at a concentration of 75 μg/mL. Compounds 4n, 4q, and 4r showed excellent radical scavenging ability of 85.20, 83.27, and 83.25% at concentrations 25, 75, and 75 μg/mL, respectively, and the rest of the compounds showed moderate activity when compared to standards. It is found that IC50 values (Table 2) of all the compounds are <25 which is equivalent to all the three tested standards.
Figure 1.
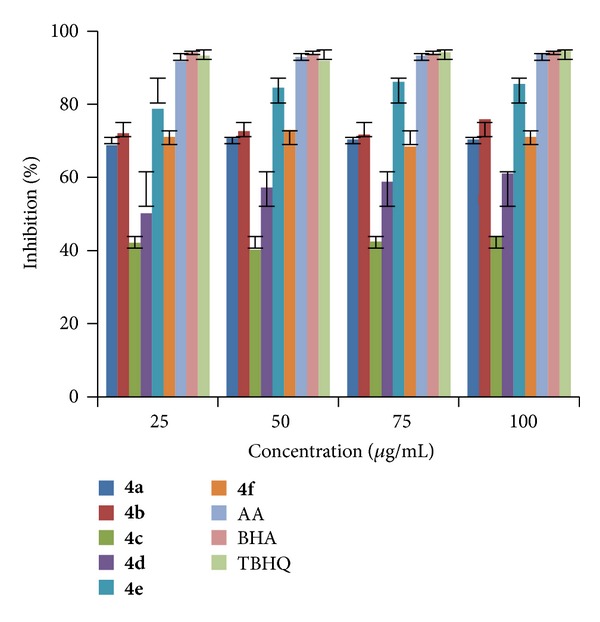
RSA of compounds (4a–f).
Figure 2.
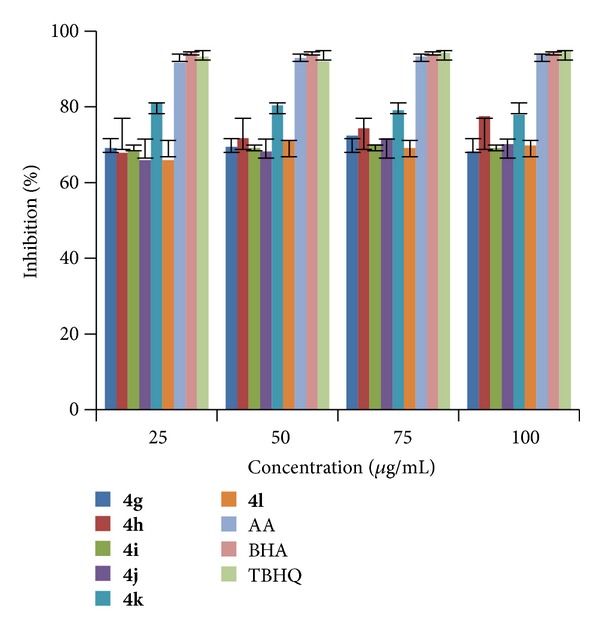
RSA of compounds (4g–l).
Figure 3.
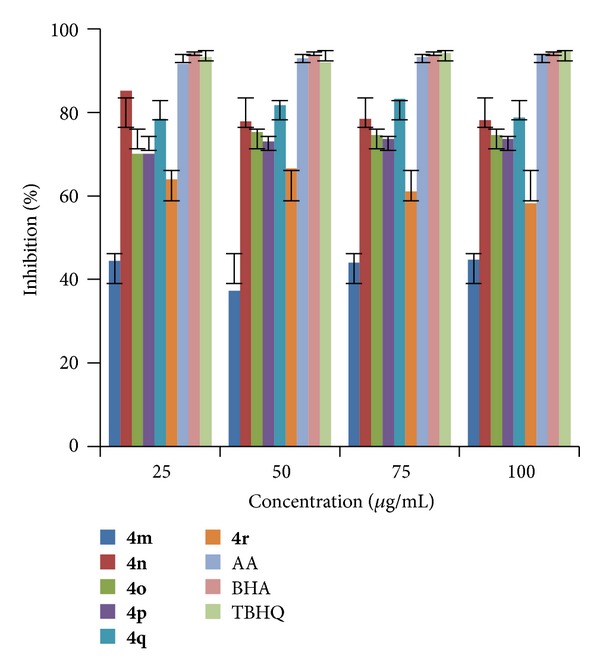
RSA of compounds (4m–r).
Table 2.
RSA of the test compounds (4a–4r) and standards using DPPH scavenging method.
| Compounds | Concentration | ||||
|---|---|---|---|---|---|
| 25 μg/mL (%) | 50 μg/mL (%) | 75 μg/mL (%) | 100 μg/mL (%) | IC50 (μg/mL) | |
| 4a | 68.81 | 70.73 | 70.41 | 70.41 | <25 |
| 4b | 72.05 | 72.66 | 71.70 | 75.88 | <25 |
| 4c | 42.12 | 40.19 | 42.44 | 44.05 | <25 |
| 4d | 50.16 | 57.23 | 58.84 | 61.09 | <25 |
| 4e | 78.77 | 84.56 | 86.17 | 85.53 | <25 |
| 4f | 71.06 | 72.99 | 68.48 | 71.06 | <25 |
| 4g | 69.13 | 69.45 | 72.34 | 68.16 | <25 |
| 4h | 67.84 | 71.70 | 74.27 | 77.49 | <25 |
| 4i | 68.16 | 69.13 | 70.09 | 69.13 | <25 |
| 4j | 65.91 | 68.16 | 71.70 | 70.09 | <25 |
| 4k | 81.02 | 80.38 | 79.09 | 77.81 | <25 |
| 4l | 65.91 | 71.06 | 69.13 | 69.77 | <25 |
| 4m | 44.37 | 37.29 | 44.05 | 44.69 | <25 |
| 4n | 85.20 | 77.81 | 78.45 | 78.13 | <25 |
| 4o | 70.09 | 75.24 | 74.59 | 74.59 | <25 |
| 4p | 70.09 | 72.99 | 73.63 | 73.64 | <25 |
| 4q | 78.45 | 81.67 | 83.27 | 78.77 | <25 |
| 4r | 78.45 | 81.67 | 83.27 | 78.77 | <25 |
| BHA | 94.21 | 93.56 | 93.89 | 94.53 | <25 |
| TBHQ | 93.24 | 91.96 | 94.21 | 94.85 | <25 |
| AA | 91.63 | 92.92 | 93.24 | 93.89 | <25 |
2.2.2. Ferric Ions (Fe3+) Reducing Antioxidant Power (FRAP)
The ferric ion (Fe3+) is a relatively biologically inactive form of iron. However, it can be reduced to the active ferrous ion (Fe2+) depending on condition particularly pH [34] and oxidizing back through Fenton type reaction with the production of hydroxyl radical or Haber-Weiss reaction with superoxide anions. Reducing power is to measure the reductive ability of an antioxidant and it is evaluated by the transformation of Fe3+ to Fe2+ by donation of an electron in the presence of test compounds. Therefore, the Fe2+ can be monitored by measuring the formation of Perl's Prussian blue at 700 nm.
The FRAP of synthesized compounds (4a–r) was determined at different concentrations (25, 50, 75, and 100 μg/mL in methanol) at pH 6.6 using literature method [35]. The increase in absorbance at 700 nm indicates the increase in reducing ability of a compound. The results shown in Figures 4, 5, and 6 indicated that the compounds 4e, 4n, and 4q exhibited good reducing power activity at concentration 100 μg/mL.
Figure 4.
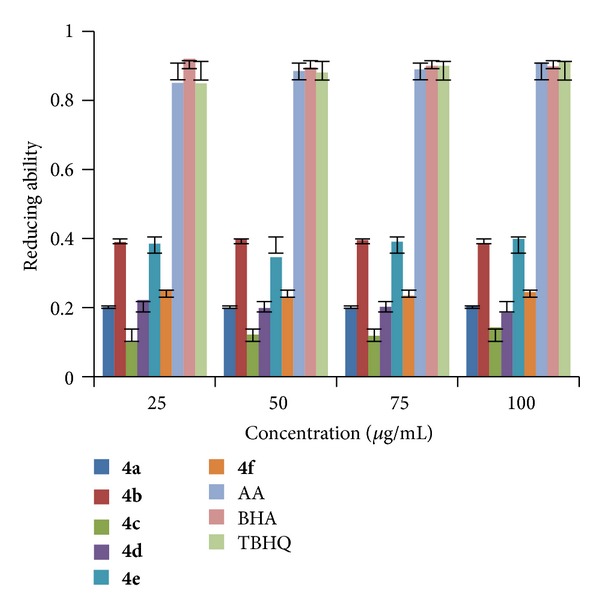
FRAP of compounds (4a–f).
Figure 5.
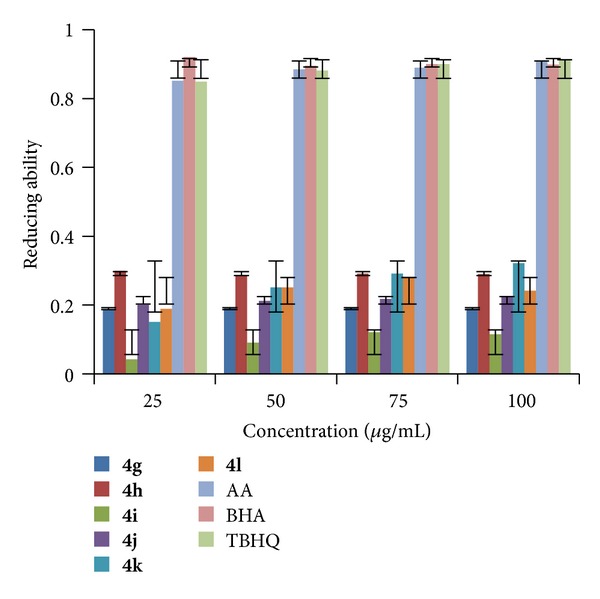
FRAP of compounds (4g–l).
Figure 6.
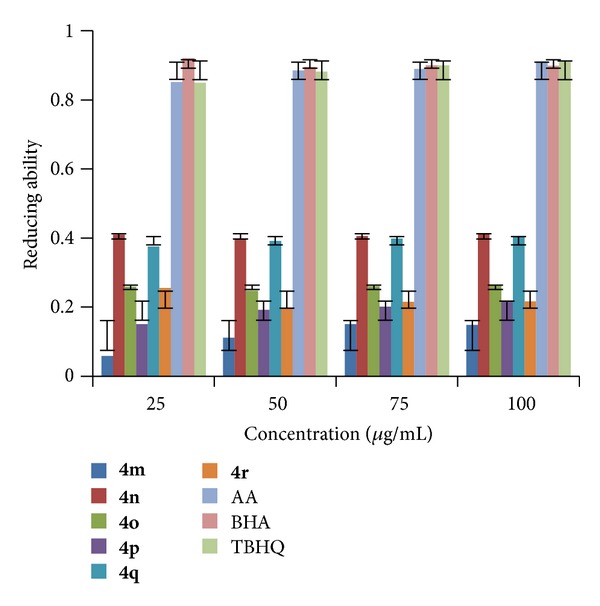
FRAP of compounds (4m–r).
2.2.3. Ferrous (Fe2+) Metal Ion Chelating Activity
Among the transition metals, iron is known as the most important lipid oxidation prooxidant due to its high reactivity. The effective ferrous ions chelators may also afford protection against oxidative damage by removing iron (Fe2+) that may otherwise participate in hydroxyl radical generating Fenton type reactions [36]:
| (1) |
Ferric (Fe3+) ions also produce radicals from peroxides although the rate is tenfold less than that of ferrous (Fe2+) ions [37]. Ferrous ion is the prooxidant among the various species of metal ions [38]. Minimizing ferrous (Fe2+) ion may afford protection against oxidative damage by inhibiting production of reactive oxygen species (ROS) and lipid production. Ferrozine can quantitatively form complex with ferrous ions in this method. In the presence of chelating agents, the complex formation is disrupted resulting in a decrease in red color of the complex. Measurement of color reduction therefore allows estimating the metal chelating activity of the coexisting chelators. Lower absorbance indicates higher metal chelating activity.
All the newly synthesized indole derivatives (4a–r) were screened for their metal chelating activity at concentrations 25, 50, 75, and 100 μg/mL in methanol using reported method [39]. The results were compared with the results obtained for standards BHA, TBHQ, and AA. The results shown in Figures 7, 8, and 9 indicated that the compounds 4b, 4h, and 4k exhibited good metal chelating activity of 72.33, 72.05, and 72.87% at 50, 100, and 75 μg/mL, respectively, whereas other compounds exhibited either moderate or poor chelating activity. These results suggested that the compounds which exhibited good chelating activity interfered with the formation of ferrous and ferrozine complex.
Figure 7.
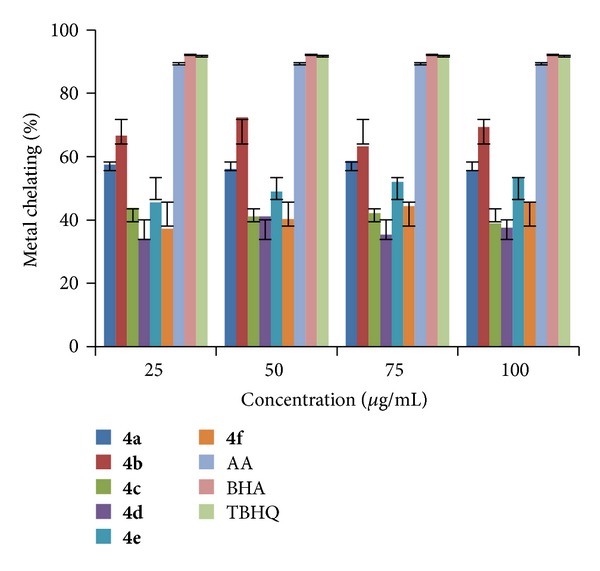
Metal chelating activity of compounds (4a–f).
Figure 8.
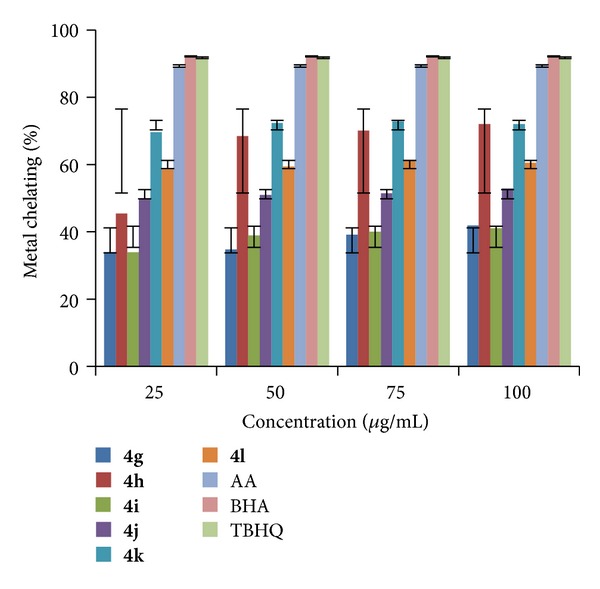
Metal chelating activity of compounds (4g–l).
Figure 9.
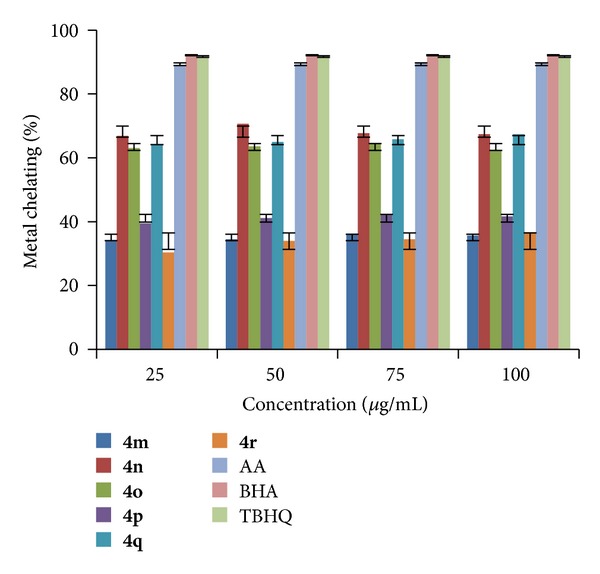
Metal chelating activity of compounds (4m–r).
2.3. Antimicrobial Activity
All the newly synthesized compounds (4a–r) were assessed for their in vitro antibacterial activity against four representative bacterial species, namely, Escherichia coli (MTCC-723), Staphylococcus aureus (ATCC-29513), Klebsiella pneumonia (NCTC-13368), and Pseudomonas aeruginosa (MTCC-1688) using gentamycin as reference. Determination of MIC was done using the serial dilution method [40, 41]. The materials used were 96-well plates, suspension of microorganism (0.5 McFarland), Muller-Hinton broth (Himedia), and stock solutions of each substance to be tested (2048 μg/mL in DMSO). The following concentrations of the substances to be tested were obtained in the 96-well plates: 1024, 512, 128, 64, 32, 16, 8, 4, and 2 μg/mL. After incubation at 37°C for 18–24 h, the MIC for each tested substance was determined by Bio-Rad Elisa reader (microplate reader S/N 12883). The results are tabulated in Table 3.
Table 3.
In vitro antimicrobial activities of compounds (4a–4r).
| Compound code | Antibacterial activity (MIC μg/mL) | Antifungal activity (MIC μg/mL) | ||||||
|---|---|---|---|---|---|---|---|---|
| ECa | SAb | KPc | PAd | AOe | ANf | AFg | ATh | |
| 4a | 512 | 1024 | 512 | 1024 | 512 | 256 | 256 | 256 |
| 4b | 64 | 32 | 64 | 128 | 128 | 128 | 32 | 64 |
| 4c | 128 | 128 | 256 | 128 | 256 | 128 | 128 | 256 |
| 4d | 256 | 512 | 1024 | 512 | 1024 | 512 | 256 | 256 |
| 4e | 64 | 64 | 128 | 64 | 64 | 128 | 64 | 128 |
| 4f | 128 | 512 | 256 | 256 | 128 | 256 | 128 | 256 |
| 4g | 1024 | 256 | 512 | 512 | 256 | 256 | 256 | 256 |
| 4h | 16 | 16 | 64 | 64 | 16 | 16 | 64 | 64 |
| 4i | 256 | 128 | 256 | 256 | 128 | 256 | 128 | 256 |
| 4j | 512 | 1024 | 512 | 512 | 256 | 512 | 512 | 512 |
| 4k | 16 | 16 | 32 | 32 | 16 | 16 | 64 | 32 |
| 4l | 256 | 256 | 128 | 128 | 256 | 256 | 256 | 256 |
| 4m | 512 | 512 | 512 | 1024 | 512 | 256 | 256 | 512 |
| 4n | 16 | 16 | 08 | 16 | 08 | 08 | 16 | 08 |
| 4o | 128 | 128 | 64 | 128 | 128 | 64 | 128 | 128 |
| 4p | 256 | 256 | 256 | 512 | 128 | 128 | 256 | 256 |
| 4q | 16 | 08 | 16 | 08 | 32 | 08 | 16 | 16 |
| 4r | 64 | 32 | 64 | 32 | 32 | 32 | 32 | 32 |
| Gentamycin | 02 | 02 | 02 | 02 | — | — | — | — |
| Fluconazole | — | — | — | — | 02 | 02 | 02 | 02 |
aEC: Escherichia coli (MTCC-723), bSA: Staphylococcus aureus (ATCC-29513), cKP: Klebsiella pneumonia (NCTC-13368), dPA: Pseudomonas aeruginosa (MTCC-1688), eAO: Aspergillus oryzae (MTCC-3567T), fAN: Aspergillus niger (MTCC-281), gAF: Aspergillus flavus (MTCC-1973), and hAT: Aspergillus terreus (MTCC-1782).
In vitro antifungal activity of the synthesized compounds (4a–r) was assessed against four representative fungal species, namely, Aspergillus oryzae (MTCC-3567T), Aspergillus niger (MTCC-281), Aspergillus flavus (MTCC-1973), and Aspergillus terreus (MTCC-1782) using fluconazole as a reference, by serial dilution method [42, 43].
The minimal inhibitory concentration (MIC) values are obtained by the broth microdilution method and were tabulated in Table 4. Synthesized compounds have comparable and similar inhibitory effects (low to moderate MIC values 08 and 512 μg/mL). The antibacterial activity results revealed that compound 4n showed excellent activity with MIC 08 μg/mL against K. pneumonia and MIC 16 μg/mL against E. coli, S. aureus, and P. aeruginosa. Compound 4q showed good activity with MIC 08 μg/mL against S. aureus and P. aeruginosa andMIC 16 μg/mL against E. coli and K. pneumonia. Compounds 4h and 4k exhibited good activity with MIC 16 μg/mL against E. coli and S. aureus.
Table 4.
Antitubercular activity of compounds (4a–4r) against Mycobacterium tuberculosis H37Rv.
| Compound number | MICa values (μg/mL) |
|---|---|
| 4a | 50 |
| 4b | 25 |
| 4c | 50 |
| 4d | 50 |
| 4e | 25 |
| 4f | 25 |
| 4g | 50 |
| 4h | 12.5 |
| 4i | 25 |
| 4j | 25 |
| 4k | 12.5 |
| 4l | 25 |
| 4m | 25 |
| 4n | 12.5 |
| 4o | 25 |
| 4p | 50 |
| 4q | 6.25 |
| 4r | 12.5 |
| Pyrazinamide | 3.125 |
| Streptomycin | 6.25 |
On the other hand, the antifungal activity results revealed that compound 4n exhibited excellent activity with MIC 08 μg/mL against A. oryzae, A. niger, and A. terreus and MIC 16 μg/mL against A. flavus and compound 4q exhibited MIC 08, 16, and 16 μg/mL against A. niger, A. flavus, and A. terreus, respectively. The rest of the compounds showed moderate activity against all the tested fungi.
2.4. Antitubercular Activity
The antitubercular activity of compounds (4a–r) was assessed against M. tuberculosis (ATTC-27294) using the microplate almar blue dye assay (MABA) [44]. The final drug concentrations tested were 100 to 0.2 μg/mL compared with standards pyrazinamide 3.125 μg/mL and streptomycin 6.25 μg/mL. The MIC was defined as the lowest drug concentration which prevented a color change from blue to pink. The results are shown in Table 4.
Compounds 4h, 4k, 4n, and 4q exhibited promising activity with MIC 12.5 μg/mL. And the rest of the compounds exhibited moderate activity with MIC 25 and 50 μg/mL.
2.5. Anticancer Activity
MTT Solution Preparation. 10 mg MTT in 10 mL of Hanks balanced solution was prepared.
Cell Culture. The cells were maintained in 96-well microtiter plate containing MEM media supplemented with 10% heat inactivated fetal calf serum (FCS), containing 5% of mixture of gentamycin, penicillin (100 units/mL), and streptomycin (100 μg/mL) in the presence of 5% CO2 at 37°C for 3-4 days. After 3-4 days, the supernatant was removed, and MEM media were replaced with Hanks balanced solution supplemented with gentamycin, penicillin, and streptomycin and incubated overnight.
Cytotoxicity Assay. In vitro growth effect of test compound was assessed by calorimetric method [45]. Determination of conversion of MTT into “Formazon blue” by living cells was done. The supernatant was removed from the plate, and then fresh Hanks balanced salt solution was added and treated with different concentrations of compounds diluted with DMSO. Control group contains only DMSO. After 24 h incubation at 37°C in a humidified atmosphere of 5% CO2, the medium was replaced with MTT solution (100 μg/mL, 1 mg/mL in sterile Hanks balanced solution) and kept for 4 h for incubation. The supernatant was carefully aspirated, the precipitated crystals of “Formazon blue” were solubilized by adding DMSO (200 μg/mL), and absorbance was measured at λ 570 nm.
The results represent the mean of three readings. The concentration at which the absorbance of treated cells was reduced by 50% with respect to the untreated control was calculated using the following formula:
| (2) |
Based on the antioxidant result, some of the potent compounds were screened for their cytotoxic activity against MDA-MB-231 (human adenocarcinoma mammary gland) cell lines using standard drugs (Table 5). In vitro growth effect of test compounds revealed that compounds 4k and 4q exhibited 100% cell lysis at concentration 10 μg/mL. The compound 4n exhibited 100% cell lysis at concentration 20 μg/mL, and the rest of screened compounds showed moderate activity against MDA-MB-231 (human adenocarcinoma mammary gland) cell lines at concentration of 30 μg/mL.
Table 5.
Anticancer activity of compounds (4a–4r).
| Compound | Concentration (μg/mL) | OD at 492 nm | % of cell lysis | IC50 (μg/mL) |
|---|---|---|---|---|
| 4b | 10 | 0.799 | 75% | <10 μG |
| 4b | 20 | 0.885 | >75% | |
| 4b | 30 | 1.559 | 100% | |
| 4e | 10 | 0.701 | >50% | <10 μG |
| 4e | 20 | 0.915 | 75% | |
| 4e | 30 | 1.010 | >75% | |
| 4h | 10 | 0.453 | <50% | 20 μG |
| 4h | 20 | 0.550 | 50% | |
| 4h | 30 | 0.799 | 75% | |
| 4k | 10 | 1.333 | 100% | Very <10 μG |
| 4k | 20 | 1.548 | 100% | |
| 4k | 30 | 1.873 | 100% | |
| 4n | 10 | 1.143 | >75% | <10 μG |
| 4n | 20 | 1.734 | 100% | |
| 4n | 30 | 1.822 | 100% | |
| 4q | 10 | 0.956 | 100% | Very <10 μG |
| 4q | 20 | 1.392 | 100% | |
| 4q | 30 | 1.752 | 100% | |
| 4r | 10 | 0.377 | No lysis | 20 μG |
| 4r | 20 | 0.606 | 50% | |
| 4r | 30 | 0.700 | >50% | |
| Control | — | 0.349 | No lysis |
Cell line-MDA-MB-human adenocarcinoma, mammary gland.
3. Experimental
3.1. Analysis and Physical Measurements
Elemental analysis was obtained from Perkin Elmer 2400 CHN elemental analyzer which is microprocessor based instrument. All the compounds gave C, H, and N analysis within ±0.4%. IR spectra of the synthesized compounds were recorded as KBr pellets on a Perkin-Elmer Spectrum RX-IFTIR instrument covering the range 4000–400 cm−1. The 1H NMR and 13C NMR spectra were recorded using DMSO-d 6 as a solvent with a BRUKER NMR 500 MHz and 125 MHz spectrometer, respectively. The chemical shift values are expressed in ppm (δ scale) using tetramethylsilane as an internal standard. The mass spectral measurements were carried out by electron impact method on JEOL GC mate spectrometer at 70 eV.
3.2. Methods
General Procedures. Laboratory chemicals were supplied by Merck and Himedia Ltd. and were of high purity grade; solvents were distilled and dried before use. Melting points of the synthesized compounds were determined by electrothermal apparatus using open capillary tubes. The purity of the compounds was checked by TLC using silica gel-G coated aluminium plates (Merck) and spots were visualized by exposing the dry plates to iodine vapors. The precursors 2,5-disubstituted indole-3-carboxaldehydes (2a–c) were prepared by the literature method [46].
3.3. Preparation Method and Physical Data of Synthesized Compounds (4a–r)
2,5-Disubstituted indole-3-carboxaldehydes (0.001 mol), secondary amine (0.0011 mol), 1- or 2-naphthol (0.001 mol), and dichloromethane (5 mL) were introduced in a 50 mL round bottom flask. The resulting mixture was stirred vigorously with a magnetic bar on a magnetic stirrer for 2-3 hours at room temperature (25–30°C). The progress of the reaction was monitored by TLC. After the completion of the reaction as checked by TLC, dichloromethane was pumped out by rotary evaporation. The crude product was purified directly by crystallization from ethanol (Scheme 1) (Table 1).
3.3.1. Preparation of 1-[(2-Phenyl-1H-indol-3-yl)(piperidin-1-yl)methyl]naphthalene-2-ol (4a)
Yield = 82%. m.p. 186–188°C; Anal. Calcd. (%) for C30H28N2O: (Mol. Wt. = 432) C, 83.30; H, 6.48; N, 6.48. Found (%): C, 83.34; H, 6.52; N, 6.50. FT-IR (KBr, cm−1): 3133 br, ν(OH); 3099 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.61 (s, 1H, phenolic OH, exchangeable with D2O); 9.94 (s, 1H, indole-NH); 7.31–7.80 (m, 15H, ArH); 4.98 (s, 1H, N–CH); 3.39 (t, 4H, 2x CH2); 2.50 (m, 2H, CH2); 1.21 (m, 4H, 2x CH2).
3.3.2. Preparation of 1-[(5-Chloro-2-phenyl-1H-indol-3-yl)(piperidin-1-yl)methyl]naphthalene-2-ol (4b)
Yield = 85%; m.p. 258–260°C; Anal. Calcd. (%) for C30H27ClN2O: (Mol. Wt. = 466) C, 77.17; H, 5.78; N, 6.00. Found (%): C, 77.16; H, 5.83; N, 6.02. FT-IR (KBr, cm−1): 3126 br, ν(OH); 3049 ν(indole-NH); 750, ν(C–Cl). 1H NMR (d 6-DMSO, ppm): 12.60 (s, 1H, phenolic OH, exchangeable with D2O); 9.96 (s, 1H, indole-NH); 7.30–7.82 (m, 14H, ArH); 5.01 (s, 1H, N–CH); 3.37 (t, 4H, 2x CH2); 2.55 (m, 2H, CH2); 1.11 (m, 4H, 2x CH2). 13C NMR (d 6-DMSO, ppm): 185.99 (C–Cl) 150.45 (C–OH), 134.91, 130.58, 130.33, 129.81, 129.50, 127.47, 127.41, 124.18, 120.58, 114.14, 113.43, 40.49, 40.33, 40.25, 40.16, 39.99, 39.83, 39.66, 39.49. MS (EI) m/z: (M+, M+2): 466, 488 (4.1%, 1.1%).
3.3.3. Preparation of 1-[(5-Methyl-2-phenyl-1H-indol-3-yl)(piperidin-1-yl)methyl]naphthalene-2-ol (4c)
Yield = 79%; m.p. 252–254°C; Anal. Calcd. (%) for C31H30N2O: (Mol. Wt. = 446) C, 83.37; H, 6.77; N, 6.27. Found (%): C, 83.40; H, 6.72; N, 6.28. FT-IR (KBr, cm−1): 3118 br, ν(OH); 3051 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.59 (s, 1H, phenolic OH, exchangeable with D2O); 9.90 (s, 1H, indole-NH); 7.32–7.89 (m, 14H, ArH); 4.98 (s, 1H, N–CH); 3.35 (t, 4H, 2x CH2); 2.45 (m, 2H, CH2); 2.33 (s, 3H, C–CH3); 1.11 (m, 4H, 2x CH2).
3.3.4. Preparation of 2-[(2-Phenyl-1H-indol-3-yl)(piperidin-1-yl)methyl]naphthalene-1-ol (4d)
Yield = 89%; m.p. 166–168°C; Anal. Calcd. (%) for C30H28N2O: (Mol. Wt. = 432) C, 83.30; H, 6.52; N, 6.48. Found (%): C, 83.35; H, 6.48; N, 6.42. FT-IR (KBr, cm−1): 3120 br, ν(OH); 3035 ν(indole-NH); 2900, ν(Ar CH–Str); 2811, ν(CH–Str); 1559, ν(ter Amine N–C). 1H NMR (d 6-DMSO, ppm): 12.59 (s, 1H, phenolic OH, exchangeable with D2O); 9.99 (s, 1H, indole-NH); 7.32–7.79 (m, 15H, ArH); 4.99 (s, 1H, N–CH); 3.35 (t, 4H, 2x CH2); 2.50 (m, 2H, CH2); 1.18 (m, 4H, 2x CH2).
3.3.5. Preparation of 2-[(5-Chloro-2-phenyl-1H-indol-3-yl)(piperidin-1-yl)methyl]naphthalene-1-ol (4e)
Yield = 86%; m.p. 224–226°C; Anal. Calcd. (%) for C30H27ClN2O: (Mol. Wt. = 466) C, 77.17; H, 5.78; N, 6.00. Found (%): C, 77.15; H, 5.80; N, 5.98. FT-IR (KBr, cm−1): 3127 br, ν(OH); 3044 ν(indole-NH); 751, ν(C–Cl). 1H NMR (d 6-DMSO, ppm): 12.62 (s, 1H, phenolic OH, exchangeable with D2O); 9.95 (s, 1H, indole-NH); 7.31–7.81 (m, 14H, ArH); 4.99 (s, 1H, N–CH); 3.40 (t, 4H, 2x CH2); 2.50 (m, 2H, CH2); 1.13 (m, 4H, 2x CH2). 13C NMR (d 6-DMSO, ppm): 186.00 (C–Cl), 150.51 (C–OH), 134.91, 130.60, 130.33, 129.80, 129.52, 127.47, 127.41, 124.20, 120.57, 114.16, 113.42, 40.50, 40.42, 40.33, 40.25, 40.16, 40.09, 40.00, 39.83, 39.66, 39.49. MS (EI) m/z: (M+, M+2): 466, 488 (5%, 1.5%).
3.3.6. Preparation of 2-[(5-Methyl-2-phenyl-1H-indol-3-yl)(piperidin-1-yl)methyl]naphthalene-1-ol (4f)
Yield = 87%; m.p. 216–218°C; Anal. Calcd. (%) for C31H30N2O: (Mol. Wt. = 446) C, 83.37; H, 6.77; N, 6.27. Found (%): C, 83.41; H, 6.74; N, 6.24. FT-IR (KBr, cm−1): 3125 br, ν(OH); 3075 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.49 (s, 1H, phenolic OH, exchangeable with D2O); 9.92 (s, 1H, indole-NH); 7.30–7.85 (m, 14H, ArH); 5.02 (s, 1H, N–CH); 3.39 (t, 4H, 2x CH2); 2.44 (m, 2H, CH2); 2.35 (s, 3H, C–CH3); 1.19 (m, 4H, 2x CH2).
3.3.7. Preparation of 1-[(Diethylamino)(2-phenyl-1H-indol-3-yl)methyl]naphthalene-2-ol (4g)
Yield = 85%; m.p. 281–283°C; Anal. Calcd. (%) for C29H28N2O: (Mol. Wt. = 420) C, 82.85; H, 6.66; N, 6.66. Found (%): C, 82.82; H, 6.40; N, 6.56. FT-IR (KBr, cm−1): 3137 br, ν(OH); 3051 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.51 (s, 1H, phenolic OH, exchangeable with D2O); 9.92 (s, 1H, indole-NH); 7.32–7.78 (m, 15H, ArH); 5.01 (s, 1H, N–CH); 2.59 (q, 4H, 2x CH2); 1.20 (t, 6H, 2x CH3).
3.3.8. Preparation of 1-[(5-Chloro-2-phenyl-1H-indol-3-yl)(diethylamino)methyl]naphthalene-1-ol (4h)
Yield = 89%; m.p. > 300°C; Anal. Calcd. (%) for C29H27ClN2O: (Mol. Wt. = 454) C, 76.55; H, 5.98; N, 6.16. Found (%): C, 76.56; H, 5.94; N, 6.15. FT-IR (KBr, cm−1): 3126 br, ν(OH); 3045 ν(indole-NH); 750, ν(C–Cl). 1H NMR (d 6-DMSO, ppm): 12.60 (s, 1H, phenolic OH, exchangeable with D2O); 9.96 (s, 1H, indole-NH); 7.30–7.81 (m, 14H, ArH); 4.97 (s, 1H, N–CH); 2.45 (q, 4H, 2x CH2); 1.25 (t, 6H, 2x CH3). 13C NMR (d 6-DMSO, ppm): 185.99 (C–Cl), 150.50 (C–OH), 134.91, 130.58, 130.33, 129.81, 129.51, 127.47, 127.41, 124.19, 120.58, 114.14, 113.44, 40.50, 40.42, 40.33, 40.25, 40.16, 40.09, 39.99, 39.83, 39.66, 39.49. MS (EI) m/z: (M+, M+2): 454, 456 (4.5%, 1.1%).
3.3.9. Preparation of 1-[(Diethylamino)(5-methyl-2-phenyl-1H-indol-3-yl)methyl]naphthalene-2-ol (4i)
Yield = 89%; m.p. 272–274°C; Anal. Calcd. (%) for C30H30N2O: (Mol. Wt. = 434) C, 82.91; H, 6.96; N, 6.45. Found (%): C, 82.94; H, 6.91; N, 6.44. FT-IR (KBr, cm−1): 3119 br, ν(OH); 3051 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.51 (s, 1H, phenolic OH, exchangeable with D2O); 9.92 (s, 1H, indole-NH); 7.32–7.78 (m, 15H, ArH); 5.01 (s, 1H, N–CH); 2.59 (q, 4H, 2x CH2); 1.20 (t, 6H, 2x CH3).
3.3.10. Preparation of 2-[(Diethylamino)(2-phenyl-1H-indol-3-yl)methyl]naphthalene-1-ol (4j)
Yield = 78%; m.p. 248–250°C; Anal. Calcd. (%) for C29H28N2O: (Mol. Wt. = 420) C, 82.82; H, 6.66; N, 6.66. Found (%): C, 82.84; H, 6.67; N, 6.65. FT-IR (KBr, cm−1): 3121 br, ν(OH); 3049 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.49 (s, 1H, phenolic OH, exchangeable with D2O); 9.97 (s, 1H, indole-NH); 7.30–7.79 (m, 15H, ArH); 5.05 (s, 1H, N–CH); 2.48 (q, 4H, 2x CH2); 1.19 (t, 6H, 2x CH3).
3.3.11. Preparation of 2-[(5-Chloro-2-phenyl-1H-indol-3-yl)(diethylamino)methyl]naphthalene-2-ol (4k)
Yield = 82%; m.p. 288–290°C; Anal. Calcd. (%) for C29H27ClN2O: (Mol. Wt. = 454) C, 76.55; H, 5.98; N, 6.16. Found (%): C, 76.57; H, 5.95; N, 6.16. FT-IR (KBr, cm−1): 3125 br, ν(OH); 3044 ν(indole-NH); 752, ν(C–Cl). 1H NMR (d 6-DMSO, ppm): 12.59 (s, 1H, phenolic OH, exchangeable with D2O); 9.95 (s, 1H, indole-NH); 7.31–7.81 (m, 14H, ArH); 5.00 (s, 1H, N–CH); 2.41 (q, 4H, 2x CH2); 1.15 (t, 6H, 2x CH3). 13C NMR (d 6-DMSO, ppm): 186.00 (C–Cl), 150.51 (C–OH), 134.91, 130.59, 130.33, 129.81, 129.51, 127.47, 127.42, 124.20, 120.57, 114.15, 113.43, 40.50, 40.43, 40.33, 40.26, 40.17, 40.09, 39.99, 39.83, 39.67, 39.50. MS (EI) m/z: (M+, M+2): 454, 456 (5%, 1.4%).
3.3.12. Preparation of 1-[(Diethylamino)(5-methyl-2-phenyl-1H-indol-3-yl)methyl]naphthalene-2-ol (4l)
Yield = 84%; m.p. 232–234°C; Anal. Calcd. (%) for C30H30N2O: (Mol. Wt. = 434) C, 82.91; H, 6.96; N, 6.45. Found (%): C, 82.95; H, 6.92; N, 6.46. FT-IR (KBr, cm−1): 3120 br, ν(OH); 3040 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.50 (s, 1H, phenolic OH, exchangeable with D2O); 9.93 (s, 1H, indole-NH); 7.30–7.77 (m, 14H, ArH); 5.03 (s, 1H, N–CH); 2.49 (q, 4H, 2x CH2); 1.19 (t, 6H, 2x CH3).
3.3.13. Preparation of 1-[(Dimethylamino)(2-phenyl-1H-indol-3-yl)methyl]naphthalene-2-ol (4m)
Yield = 89%; m.p. 256–258°C; Anal. Calcd. (%) for C27H24N2O: (Mol. Wt. = 392) C, 82.62; H, 6.16; N, 7.14. Found (%): C, 82.65; H, 6.12; N, 7.15. FT-IR (KBr, cm−1): 3133 br, ν(OH); 3051 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.45 (s, 1H, phenolic OH, exchangeable with D2O); 9.90 (s, 1H, indole-NH); 7.34–8.00 (m, 15H, ArH); 5.05 (s, 1H, N–CH); 2.25 (s, 6H, 2x CH3).
3.3.14. Preparation of 1-[(5-Chloro-2-phenyl-1H-indol-3-yl)(dimethylamino)methyl]naphthalene-2-ol (4n)
Yield = 89%; m.p. 265–267°C; Anal. Calcd. (%) for C27H28ClN2O: (Mol. Wt. = 426) C, 75.96; H, 5.46; N, 6.56. Found (%): C, 75.95; H, 5.39; N, 6.55. FT-IR (KBr, cm−1): 3160 br, ν(OH); 3045 ν(indole-NH); 751, ν(C–Cl). 1H NMR (d 6-DMSO, ppm): 12.60 (s, 1H, phenolic OH, exchangeable with D2O); 9.96 (s, 1H, indole-NH); 7.29–7.76 (m, 14H, ArH); 5.12 (s, 1H, N–CH); 2.38 (s, 6H, 2x CH3). 13C NMR (d 6-DMSO, ppm): 185.99 (C–Cl), 150.50 (C–OH), 134.92, 130.58, 130.29, 129.82, 129.51, 127.48, 127.41, 124.18, 120.58, 114.14, 113.44, 40.49, 40.33, 40.25, 40.16, 39.9, 39.83, 39.66, 39.49. MS (EI) m/z: (M+, M+2): 454, 456 (4%, 1%).
3.3.15. Preparation of 1-[(Dimetylamino)(5-methyl-2-phenyl-1H-indol-3-yl)methyl]naphthalene-2-ol (4o)
Yield = 84%; m.p. 281–283°C; Anal. Calcd. (%) for C28H26N2O: (Mol. Wt. = 406) C, 82.73; H, 6.45; N, 6.89. Found (%): C, 82.75; H, 6.40; N, 6.86. FT-IR (KBr, cm−1): 3137 br, ν(OH); 3051 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.55 (s, 1H, phenolic OH, exchangeable with D2O); 9.98 (s, 1H, indole-NH); 7.32–7.79 (m, 14H, ArH); 5.09 (s, 1H, N–CH); 2.49 (s, 3H, CH3); 1.91 (s, 6H, 2x CH3).
3.3.16. Preparation of 2-[(Dimethylamino)(2-phenyl-1H-indol-3-yl)methyl]naphthalene-1-ol (4p)
Yield = 88%; m.p. 228–230°C; Anal. Calcd. (%) for C27H24N2O: (Mol. Wt. = 392) C, 82.62; H, 6.16; N, 7.14. Found (%): C, 82.64; H, 6.13; N, 7.14. FT-IR (KBr, cm−1): 3140 br, ν(OH); 3041 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.40 (s, 1H, phenolic OH, exchangeable with D2O); 9.91 (s, 1H, indole-NH); 7.33–7.99 (m, 15H, ArH); 5.02 (s, 1H, N–CH); 2.20 (s, 6H, 2x CH3).
3.3.17. Preparation of 2-[(5-Chloro-2-phenyl-1H-indol-3-yl)(dimethylamino)methyl]naphthalene-1-ol (4q)
Yield = 84%; m.p. 240–242°C; Anal. Calcd. (%) for C27H28ClN2O: (Mol. Wt. = 426) C, 75.96; H, 5.46; N, 6.56. Found (%): C, 75.93; H, 5.39; N, 6.55. FT-IR (KBr, cm−1): 3125 br, ν(OH); 3043 ν(indole-NH); 751, ν(C–Cl). 1H NMR (d 6-DMSO, ppm): 12.59 (s, 1H, phenolic OH, exchangeable with D2O); 9.95 (s, 1H, indole-NH); 7.30–7.81 (m, 14H, ArH); 5.10 (s, 1H, N–CH); 2.19 (s, 6H, 2x CH3). 13C NMR (d 6-DMSO, ppm) 185.99 (C–Cl), 150.50 (C–OH), 134.92, 130.58, 130.33, 129.81, 129.51, 127.48, 127.41, 124.18, 120.58, 114.14, 113.44, 40.49, 40.33, 40.25, 40.16, 40.09, 39.98, 39.83, 39.66, 39.49. MS (EI) m/z: (M+, M+2):: 454, 456 (5%, 1.6%).
3.3.18. Preparation of 2-[(Dimetylamino)(5-methyl-2-phenyl-1H-indol-3-yl)methyl]naphthalene-1-ol (4r)
Yield = 82%; m.p. 254–256°C; Anal. Calcd. (%) for C28H26N2O: (Mol. Wt. = 406) C, 82.73; H, 6.46; N, 6.89. Found (%): C, 82.75; H, 6.40; N, 6.88. FT-IR (KBr, cm−1): 3120 br, ν(OH); 3039 ν(indole-NH). 1H NMR (d 6-DMSO, ppm): 12.49 (s, 1H, phenolic OH, exchangeable with D2O); 9.97 (s, 1H, indole-NH); 7.30–7.78 (m, 14H, ArH); 5.04 (s, 1H, N–CH); 2.39 (s, 3H, CH3); 1.93 (s, 6H, 2x CH3).
4. Conclusions
In the conclusion, the present study revealed that the compounds having methyl and chlorosubstituent exhibited good antioxidant, antimicrobial, and cytotoxic activity.
Acknowledgments
Authors are thankful to the Chairman, Department of Chemistry, Gulbarga University, Gulbarga, for providing laboratory facilities, to the Chairman, Department of Microbiology, Gulbarga University, Gulbarga, for providing laboratory facilities to carry out antimicrobial activity, and to the Principal, Maratha Mandals NGH Institute of Dental Science and Research Center, Belgum-10, Karnataka (India), for carrying out antitubercular and cytotoxic activity, and they are also thankful to the Director of Indian Institute of technology, Chennai, for providing 1H NMR, 13C NMR, and mass, as well as the Chairman, Department of Material science, for providing IR spectral data.
Conflict of Interests
Since the authors have procured the IR, NMR, and mass spectra of the synthesized compounds from the National Research Centre, namely, The Indian Institute of Technology, Madras, Chennai, India, as per the condition of institution, they should acknowledge their services in the research paper while publishing the work, which includes the data provided by them in the research paper. The same has been acknowledged in the Acknowledgements section. The authors do not have any agreement, financial assistance, or sponsorship from Perkin-Elmer spectrum, Bruker NMR, and so forth. These names are mentioned in the experimental protocol as these are the instrument models, and it is mandatory for authors to mention the instrument models used to scan the spectra of unknown compounds. Otherwise, the corresponding author or coauthors have no direct financial relationship with the commercial identity mentioned in their paper in any form.
References
- 1.Gerlach M, Maul C. Substituted 1- and 2-naphthol Mannich bases. U.S. Patent 7.202.242 B2, 2007.
- 2.Lu J, Xu X, Wang C, He J, Hu Y, Hu H. Synthesis of chiral ligands derived from the Betti base and their use in the enantioselective addition of diethylzinc to aromatic aldehydes. Tetrahedron Letters. 2002;43(46):8367–8369. [Google Scholar]
- 3.Zhu J, Bienayme H. Multicomponent Reactions. Weinheim, Germany: Wiley-VCH; 2005. [Google Scholar]
- 4.Sies H. Oxidative stress: from basic research to clinical application. The American Journal of Medicine C. 1991;91(3) doi: 10.1016/0002-9343(91)90281-2. [DOI] [PubMed] [Google Scholar]
- 5.Stocker R. Dietary and pharmacological antioxidants in atherosclerosis. Current Opinion in Lipidology. 1999;10(6):589–597. doi: 10.1097/00041433-199912000-00014. [DOI] [PubMed] [Google Scholar]
- 6.Brigelius-Flohé R, Kluth D, Banning A. Is there a future for antioxidants in atherogenesis? Molecular Nutrition and Food Research. 2005;49(11):1083–1089. doi: 10.1002/mnfr.200500094. [DOI] [PubMed] [Google Scholar]
- 7.World Health Organization. WHO Report. Geneva, Switzerland: WHO Press; 2008. Global tuberculosis control: surveillance, planning, financing. [Google Scholar]
- 8.Smith PG, Moss AR. Epidemiology of tuberculosis. In: Bloom BR, editor. Tuberculosis: Pathogenesis, Protection and Control. Washington, DC, USA: ASM Press; 1994. p. p. 47. [Google Scholar]
- 9.World Health Organization. Global Tuberculosis Control. Lyon, France: WHO; 2011. [Google Scholar]
- 10.Suresh Kumar GV, Rajendraprasad Y, Mallikarjuna BP, Chandrashekar SM, Kistayya C. Synthesis of some novel 2-substituted-5-[isopropylthiazole] clubbed 1,2,4-triazole and 1,3,4-oxadiazoles as potential antimicrobial and antitubercular agents. European Journal of Medicinal Chemistry. 2010;45(5):2063–2074. doi: 10.1016/j.ejmech.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 11.LoBue P. Extensively drug-resistant tuberculosis. Current Opinion in Infectious Diseases. 2009;22:167–173. doi: 10.1097/qco.0b013e3283229fab. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Zhang J, Wu AH, Pike MC, Deapen D. Invasive breast cancer incidence trends by detailed race/ethnicity and age. International Journal of Cancer. 2012;130(2):395–404. doi: 10.1002/ijc.26004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheikh MS, Garcia M, Pujol P, Fontana JA, Rochefort H. Why are estrogen-receptor-negative breast cancers more aggressive than the estrogen-receptor-positive breast cancers? Invasion and Metastasis. 1995;14(1–6):329–336. [PubMed] [Google Scholar]
- 14.Balduyck M, Zerimech F, Gouyer V, et al. Specific expression of matrix metalloproteinases 1, 3, 9 and 13 associated with invasiveness of breast cancer cells in vitro . Clinical and Experimental Metastasis. 2000;18(2):171–178. doi: 10.1023/a:1006762425323. [DOI] [PubMed] [Google Scholar]
- 15.Choi B, Kim H, Lee K, Cho Y, Kong G. Clofilium, a potassium channel blocker, induces apoptosis of human promyelocytic leukemia (HL-60) cells via Bcl-2-insensitive activation of caspase-3. Cancer Letters. 1999;147(1-2):85–93. doi: 10.1016/s0304-3835(99)00280-3. [DOI] [PubMed] [Google Scholar]
- 16.Fleischer A, Ghadiri A, Dessauge F, et al. Modulating apoptosis as a target for effective therapy. Molecular Immunology. 2006;43(8):1065–1079. doi: 10.1016/j.molimm.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Bhalla K, Hill C, Prlest DG. Evidence for involvement of tyrosine phosphorylation in taxol-induced apoptosis in a human ovarian tumor cell line. Biochemical Pharmacology. 1994;48(6):1265–1272. doi: 10.1016/0006-2952(94)90164-3. [DOI] [PubMed] [Google Scholar]
- 18.Horton DA, Bourne GT, Smythe ML. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chemical Reviews. 2003;103(3):893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 19.Bouchikhi F, Rossignol E, Sancelme M, et al. Synthesis and biological evaluation of diversely substituted indolin-2-ones. European Journal of Medicinal Chemistry. 2008;43(11):2316–2322. doi: 10.1016/j.ejmech.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Talaz O, Gülçin I, Göksu S, Saracoglu N. Antioxidant activity of 5,10-dihydroindeno[1,2-b]indoles containing substituents on dihydroindeno part. Bioorganic & Medicinal Chemistry. 2009;17(18):6583–6589. doi: 10.1016/j.bmc.2009.07.077. [DOI] [PubMed] [Google Scholar]
- 21.Pajouhesh H, Parson R, Popp FD. Potential anticonvulsants. VI: condensation of isatins with cyclohexanone and other cyclic ketones. Journal of Pharmaceutical Sciences. 1983;72(3):318–321. doi: 10.1002/jps.2600720330. [DOI] [PubMed] [Google Scholar]
- 22.Frederich M, Tits M, Angenot L. Potential antimalarial activity of indole alkaloids. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2008;102(1):11–19. doi: 10.1016/j.trstmh.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Karali N, Gürsoy A, Kandemirli F, et al. Synthesis and structure-antituberculosis activity relationship of 1H-indole-2,3-dione derivatives. Bioorganic and Medicinal Chemistry. 2007;15(17):5888–5904. doi: 10.1016/j.bmc.2007.05.063. [DOI] [PubMed] [Google Scholar]
- 24.Liebmann PM, Wölfler A, Felsner P, Hofer D. Melatonin and the immune system. International Archives of Allergy and Immunology. 1997;112(3):203–211. doi: 10.1159/000237455. [DOI] [PubMed] [Google Scholar]
- 25.Lezoualc’h F, Skutella T, Widmann M, Behl C. Melatonin prevents oxidative stress-induced cell death in hippocampal cells. NeuroReport. 1996;7(13):2071–2077. doi: 10.1097/00001756-199609020-00003. [DOI] [PubMed] [Google Scholar]
- 26.Poon AMS, Liu ZM, Pang CS, Brown GM, Pang SF. Evidence for a direct action of melatonin in the immune system. Biological Signals. 1994;3(2):107–117. doi: 10.1159/000109532. [DOI] [PubMed] [Google Scholar]
- 27.Volko M, Morris H, Cronin MT. Review metals, toxicity and oxidativestress. Current Medicinal Chemistry. 2005;12(10):1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 28.Ragavendran JV, Sriram D, Patel SK, et al. Design and synthesis of anticonvulsants from a combined phthalimide-GABA-anilide and hydrazone pharmacophore. European Journal of Medicinal Chemistry. 2007;42(2):146–151. doi: 10.1016/j.ejmech.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Saundane AR, Kirankumar NM, Annapurna H, Prabhaker W. Synthesis and biological evaluation of some new pyrazole, chromen incorporated indole derivatives. Journal of Applied Chemistry. 2014;3(1):117–128. [Google Scholar]
- 30.Saundane AR, Walmik P. Synthesis, antioxidant, antimicrobial, antimycobacterial, and cytotoxic activities of azetidinone and thiazolidinone moieties linked to indole nucleus. Journal of Chemistry. 2013;2013:9 pages.543815 [Google Scholar]
- 31.Saundane AR, Katkar V, Vaijinath AV. Synthesis antioxidant and antimicrobial activities of N-[(5′- Substituted 2′-phenyl- 1H -indol-3′-yl)methylene]-5-(pyridin-4-yl)- 1,3,4-oxadiazol-2-amines. Journal of Chemistry. 2013;2013:9 pages.718937 [Google Scholar]
- 32.Saundane AR, Katkar V, Vaijinath AV, Prabhaker W. Synthesis, antioxidant and antimicrobial activities of N-[(5′-substituted 2′-phenyl-1H-indol-3′-yl)methylene]-5-(pyridin-4-yl)-1, 3, 4-oxadiazol-2-amines. Medicinal Chemistry Research. 2013;22(2):806–817. [Google Scholar]
- 33.Hatano T, Kagawa H, Yasuhara T, Okuda T. Two new flavonoids and other constituents in licorice root: their relative astringency and radical scavenging effects. Chemical and Pharmaceutical Bulletin. 1988;36(6):2090–2097. doi: 10.1248/cpb.36.2090. [DOI] [PubMed] [Google Scholar]
- 34.Strlič M, Radovič T, Kolar J, Pihlar B. Anti- and prooxidative properties of gallic acid in fenton-type systems. Journal of Agricultural and Food Chemistry. 2002;50(22):6313–6317. doi: 10.1021/jf025636j. [DOI] [PubMed] [Google Scholar]
- 35.Oyaizu M. Studies on products of the browning reaction. Antioxidative activities of browning reaction products prepared from glucosamine. The Japanese Journal of Nutrition. 1986;44:307–315. [Google Scholar]
- 36.Ca̧liş I, Hosny M, Khalifa T, Nishibe S. Secoiridoids from Fraxinus angustifolia. Phytochemistry. 1993;33(6):1453–1456. [Google Scholar]
- 37.Miller DD. Fennema’s Food Chemistry. New York, NY, USA: Marcel Dekker; 1996. Mineral; pp. 618–649. [Google Scholar]
- 38.Halliwell B, Gutteridge JMC. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochemical Journal. 1984;219(1):1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dinis TCP, Madeira VMC, Almeida LM. Action of phenolic derivatives (acetaminophen, salicylate, and 5-aminosalicylate) as inhibitors of membrane lipid peroxidation and as peroxyl radical scavengers. Archives of Biochemistry and Biophysics. 1994;315(1):161–169. doi: 10.1006/abbi.1994.1485. [DOI] [PubMed] [Google Scholar]
- 40.Barry AL. Procedure for testing antimicrobial agents in agar media. In: Corian VL, editor. Antibiotics in Laboratory Medicine. Baltimore, Md, USA: Williams & Wilkins; 1980. pp. 1–23. [Google Scholar]
- 41.D. James. Detailed methodology and implementation of a semiautomated serial dilution microtechnique for antimicrobial susceptibility testing. Applied Microbiology. 1970;20(1):46–53. doi: 10.1128/am.20.1.46-53.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arthington-Skaggs BA, Motley M, Warnock DW, Morrison CJ. Comparative evaluation of PASCO and national committee for clinical laboratory standards M27-A broth microdilution methods for antifungal drug susceptibility testing of yeasts. Journal of Clinical Microbiology. 2000;38(6):2254–2260. doi: 10.1128/jcm.38.6.2254-2260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verma RS, Khan ZK, Sing AP. Antimicrobial Agents: Past, Present and Future Prospects. Lucknow, India: National Academy of Chemistry and Biology; 1998. [Google Scholar]
- 44.Lourenço MCS, de Souza MVN, Pinheiro AC, et al. Evaluation of anti-tubercular activity of nicotinic and isoniazid analogues. Arkivoc. 2007;2007(15):181–191. [Google Scholar]
- 45.Dolly A, Griffiths JB. Cell and Tissue Culture for Medical Research. New York, NY, USA: John Wiley & Sons; 2000. [Google Scholar]
- 46.Hiremath SP, Biradar JS, Purohit MG. A new route to indolo [3, 2-b]isoquinolines. Indian Journal of Chemistry B. 1982;21:249–253. [Google Scholar]