

Abstract
On the basis of recently reported abyssinone II and olympicin A, a series of chemically modified flavonoid phytochemicals were synthesized and evaluated against Mycobacterium tuberculosis and a panel of Gram-positive and -negative bacterial pathogens. Some of the synthesized compounds exhibited good antibacterial activities against Gram-positive pathogens including methicillin resistant Staphylococcus aureus with minimum inhibitory concentration as low as 0.39 μg/mL. SAR analysis revealed that the 2-hydrophobic substituent and the 4-hydrogen bond donor/acceptor of the 4-chromanone scaffold together with the hydroxy groups at 5- and 7-positions enhanced antibacterial activities; the 2′,4′-dihydroxylated A ring and the lipophilic substituted B ring of chalcone derivatives were pharmacophoric elements for antibacterial activities. Mode of action studies performed on selected compounds revealed that they dissipated the bacterial membrane potential, resulting in the inhibition of macromolecular biosynthesis; further studies showed that selected compounds inhibited DNA topoisomerase IV, suggesting complex mechanisms of actions for compounds in this series.
Introduction
Because of the emergence and spread of multidrug resistant microorganisms and pathogenic bacterial infections, novel chemotype antibacterial agents demonstrating distinct modes of action from existing antibiotics are urgently needed. Natural products are known as rich sources of bioactive molecules and chemical diversity and have thus provided invaluable chemical scaffolds as well as served as an inspiration toward antibacterial drug discovery and development.1−4 In this context, synthesis and evaluation of natural-product-inspired compound libraries represent an attractive approach for discovering novel antibacterial agents.5
Flavonoids are a large family of polyphenolic phytochemicals, which widely exist in the plant kingdom.6 As such, flavonoids have been the focus of numerous basic biomedical research as well as clinical investigation.7,8 As examples, high dietary intake of flavonoids may offer potential to reduce the risk of various cancers according to a number of epidemiological studies.9−13 In addition, flavonoids have been reported to display a broad spectrum of pharmacological activities, such as antimicrobial,14−16 anti-inflammatory,17,18 cancer preventive19,20 and anticancer,21,22 and antioxidant activities.23,24 It is also noteworthy that some widely investigated flavonoids, such as flavone acetic acid (FAA),25 flavopiridol,26−28 silibinin (silybin),29,30 and quercetin31 and its derivatives32 (Figure 1), have progressed to various stages of clinical trials.33 In this regard, plant-derived phytochemicals including chemically modified flavonoids and derivatives continue to attract great interest in the development of novel antibiotics.34
Figure 1.

Skeleton structures of chalcones, 4-chromanones, and representative structures of naturally occurring flavonoids including abyssinone II and olympicin A.
Furthermore, chalcones (1,3-diaryl-2-propen-1-ones), one subclass of structural analogues of flavonoids, have been reported to exhibit diverse biological activities,35−38 in which the enone functional group and the 2′-hydroxy group constitute important structural motifs for antibiotic activity. From a chemistry point of view, chalcones and 4-chromanones are structurally related, and 2′-hydroxychalcones serve as important synthetic precursors for the synthesis of 4-chromanones following an intramolecular conjugate addition of the phenol on the α,β-unsaturated system.39 Notably, the 4-chromanone derivatives containing an aromatic substituent at the 2-position, so-called flavanones, have been identified as an important class of bioactive heterocycles.40−42
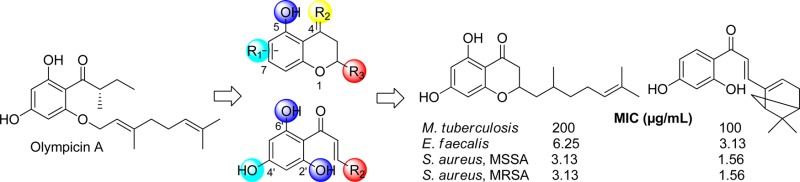
As a result of our longstanding interest in developing natural-product-inspired new antibacterial agents, we recently reported the identification of abyssinone II as a promising antibacterial lead by screening a focused flavonoid and resveratrol library.43 In addition, olympicin A, a member of the natural acylphloroglucinol chemical class, was recently isolated from the plant Hypericum olympicum and reported to exhibit potent antibacterial activity against a panel of multidrug-resistant (MDR) strains of clinically relevant Staphylococcus aureus, with minimum inhibitory concentration (MIC) values ranging from 0.5 to 1 μg/mL.44,45 Very recently, we have shown that synthetic olympicin A also exhibited good activity against Clostridium difficile (MIC = 1–2 μg/mL).46 Inspired by the antibacterial activity of the natural products abyssinone II and olympicin A, in this work we employed the 4-chromanone and chalcone structural scaffolds as chemical starting points to design and synthesize chemically modified flavonoid analogues. Subsequently, several series of structurally related flavonoids were synthesized and evaluated in vitro against a broad set of bacterial pathogens and a detailed structure–activity relationship (SAR) has been obtained. Furthermore, the antibacterial basis of promising lead compounds and their ability to inhibit bacterial topoisomerases such as DNA gyrase or topo IV have also been examined.
Results and Discussion
Synthesis of Olympicin A and Derivatives
The isolation and chemical synthesis of olympicin A (2a) was originally reported by Shiu et al., and its synthesis involved a four-step reaction sequence. However, the overall yield was only 3.3% from 1a.44 To improve the reaction efficiency and develop a modular synthesis toward olympicin A and derivatives, we evaluated diverse protecting schemes including the tert-butyldimethylsilyl (TBDMS), the base-stable methoxymethyl (MOM), and the p-toluenesulfonyl (Ts) groups and developed an improved synthesis of 2a by using the Ts protecting strategy, improving the overall yield to 40% from 1a (Scheme 1). In particular, we found that the reported low yield may be largely due to the instability of the TBDMS protecting group under basic reaction conditions (K2CO3, 80 °C) when introducing the geranyl group. To address this, the base-stable MOM group was next applied instead of TBDMS. Following the O-geranylation reaction, it was found that 2a decomposed during the deprotection of the MOM groups because of the instability of O-geranyl group under the acidic condition, and the deprotected 1a was recovered from the reaction. Subsequently the Ts group was used to protect the hydroxy group,47 and only the tris(tosylate) 1b was obtained as the major product because of the lack of selectivity of the Ts group under the reaction condition. Nevertheless, we found that the tosylate group at the 2-position of 1b was very labile, and the geranyl group could be selectively introduced with sodium hydride as the base. Final removal of the Ts group was performed with excess sodium methoxide in methanol under reflux48 to afford the chiral olympicin A (2a). The spectroscopic data of our synthetic olympicin A were in good agreement with those of the natural product.44 For comparison, the racemic olympicin A (2b) was also synthesized under the same conditions to study the potential effect of stereochemistry on antibacterial property.
Scheme 1. Improved Synthesis of Olympicin A (2a).

Reagents and conditions: (i) SOCl2, 80 °C, 2 h; (ii) AlCl3, CS2, PhNO2, 0.5 h; (iii) TsCl, K2CO3, acetone, 1 h, 73%; (iv) geranyl bromide, NaH, DMF, 1 h; (v) CH3ONa, MeOH, reflux, 8 h, 55% (two-step overall yield); 52% for 2b (two-step overall yield).
Next, to further expand the chemical diversity and investigate the influence of 2-substitution on antibacterial activity in the scaffold, an array of racemic olympicin derivatives (2c–e) and enantiomeric form 2f were designed and synthesized (Scheme 2). Compounds 1a′ and 1a were first protected by reacting with MOMCl in the presence of diisopropylethylamine (DIPEA) to provide 1c and 1c′ in moderate yields. The O-alkylation reaction was subsequently carried out with appropriate alkyl bromide using sodium hydride as a base, after which the MOM groups were removed with hydrochloric acid to give 2c–f in moderate to high yields (66–93%).
Scheme 2. Synthesis of Olympicin A Analogues.

Reagents and conditions: (i) MOMCl, DIPEA, DCM, 1 h; (ii) R-Br, NaH, DMF; (iii) HCl, MeOH, overnight.
Synthesis of 2-Substituted 4-Chromanone and Derivatives
4-Chromanone derivatives bearing an aryl substituent in the 2-position are normally synthesized by reacting an acetophenone with an arylaldehyde under strong basic or acidic conditions.49,50 However, because of the side reaction of self-condensation of the aliphatic aldehyde,51 these conditions are not ideal for the synthesis of 2-alkyl substituted 4-chromanones. Several different synthetic methods to prepare such 4-chromanone derivatives have been reported,51−53 either involving microwave irradiation or requiring very long reaction times. Here, a sealed pressure tube was introduced as a reaction vessel for the preparation of 2-alkyl substituted 4-chromanones. On the basis of the scaffold, several modification strategies (Figure 2) were applied to develop chemical diversity and further evaluate the SAR.
Figure 2.

Modifications based on the 4-chromanone scaffold.
As shown in Scheme 3, a series of 2-alkyl-4-chromanone derivatives 3a–f were synthesized by reacting 2,4-dihydroxyacetophenone (1d) with an appropriate aliphatic aldehyde in the presence of pyrrolidine in ethanol. This reaction was performed in a sealed pressure tube at 150 °C for 1 h, obtaining yields over 30%. Furthermore, the reaction time was shortened significantly. In contrast, a lower yield (12%) was afforded despite a longer reaction time (72 h) for the synthesis of 3b under the conditions (ethanol, 60 °C, pyrrolidine). To optimize the reaction conditions, a panel of different amine bases was screened. We noted that under the same conditions, no product was detected using DIPEA and only traces of product were formed when using morpholine as a base monitored by HPLC, respectively. To investigate the potential effects of the 4-carbonyl group as a hydrogen bond acceptor, the 4-chromanones 3a–d were further reduced by NaBH4 in methanol to provide 4-chromanol derivatives 4a–d in moderate to good yields (64–81%).54
Scheme 3. Synthesis of 2-Alkylated 4-Chromanones and Derivatives.

Reagents and conditions: (i) pyrrolidine, EtOH, 150 °C, pressure tube, 1 h; (ii) NaBH4, MeOH, rt, 24 h.
Accordingly, when the bicyclic (−)-myrtenal (1f) was used as the substrate under these reaction conditions (Scheme 3, step i), no product was obtained. Subsequently, a lower temperature (75 °C) was applied (Scheme 4) to optimize the reaction conditions. The corresponding chalcone intermediate 3g was obtained in low yield (9.5%). To improve the reaction yield, 1d was first regioselectively protected by reacting with MOMCl in the presence of DIPEA to provide the MOM-protected acetophenone 1e in 86% yield. Again, when the reaction was performed at 150 °C, only traces of 3h were detected by HPLC. In contrast, a much higher yield (48%) was obtained when the reaction temperature was reduced to 75 °C. The MOM-protected 3h can be further cyclized in ethanol in the presence of sodium acetate to yield 4-chromanone 3i as a mixture of two diastereomers.55 Final deprotection of the O-MOM group was performed in methanol using concentrated HCl at room temperature to give 3j in 90% yield.
Scheme 4. Reactions of Acetophenone and (−)-Myrtenal.

Reagents and conditions: (i) DIPEA, MOMCl, 0 °C, 1 h; (ii) pyrrolidine, EtOH, 75 °C, pressure tube, 1 h; (iii) NaOAc, EtOH, reflux, 24 h; (iv) concentrated HCl, MeOH, rt, overnight.
Next, on the basis of the promising antibacterial activity of 3f, a focused set of 4-chromanone analogues (Scheme 5) were subsequently designed and synthesized to investigate the antibacterial effect of the phenol free hydroxy group at different positions. Compound 3f was resynthesized from the MOM-protected 1e by using diethylamine (DEA) as a base in an overall 58% yield following O-MOM deprotection. Accordingly, 3k,l with the 5- or 6-hydroxy group were also synthesized using appropriate MOM-protected 1g,h in moderate yields (58–75%).
Scheme 5. Synthesis of 4-Chromanone Analogues 3f, 3k, and 3l.

Reagents and conditions: (i) DEA, EtOH, 150 °C, pressure tube, 1 h; (ii) concentrated HCl, MeOH, rt, overnight.
To further expand the SAR and evaluate the influence of the 5-hydroxy group, an array of 5,7-dihydroxy-4-chromanones were synthesized. Accordingly, 2,4,6-trihydroxyacetophenone (1j) was used to prepare 4-chromanone derivatives 5 (Scheme 6). Unfortunately, no product was obtained under the reaction conditions of pyrrolidine in ethanol at 150 °C in a pressure tube. Therefore, the bis-MOM-protected acetophenone 1k was next prepared in 79% yield.56 The corresponding MOM-protected 4-chromanones 5 were obtained by reacting 1k with an appropriate aldehyde in the presence of DEA in ethanol in a pressure tube at 150 °C in moderate yields (55–70%). Subsequent removal of the MOM groups using concentrated HCl in methanol at room temperature afforded the corresponding 4-chromanones in 75–94% yields. Next, to evaluate the role of the carbonyl group at the 4-position and further explore the chemical diversity of the 4-chromanone scaffold, a panel of 4-oximinochromanes 6a–f were produced by reacting the corresponding 5 with an appropriate hydroxylamine in ethanol in the presence of pyridine in high yields (80–90%). Notably, the reaction time in this series differed from 26 to 72 h, and the electron donating group such as MeO in 6b greatly facilitated the reaction and reduced the reaction time (26 h).
Scheme 6. Synthesis of 2-Substituted 4-Chromanones and Oxime Derivatives.

Reagents and conditions: (i) DIPEA, MOMCl, 0 °C, 1 h, 79%; (ii) DEA, EtOH, 150 °C, pressure tube, 1 h; (iii) concentrated HCl, MeOH, rt, 24 h; (iv) pyridine, EtOH, rt, 26–72 h.
Without isolating the MOM-protected intermediate.
As illustrated in Scheme 7, to synthesize the bicyclic compound 5n bearing the myrtenal motif, the reaction time needed to be extended to 4 h under the reaction conditions, and the chalcone 5m was obtained in overall two-step 34% yield after O-MOM deprotection. Subsequent intramolecular conjugate addition of the phenol on the α,β-unsaturated system was performed under microwave irradiation in the presence of catalytic amounts of concentrated HCl to give 5n in 72% yield as a mixture of two diastereomers.49 We also noted that the reaction was completed in 0.5 h, which was a great improvement in comparison to the condition of refluxing with sodium acetate in ethanol in Scheme 4.
Scheme 7. Synthesis of Compounds 5m and 5n.

Reagents and conditions: (i) DEA, EtOH, 150 °C, pressure tube, 4 h; (ii) HCl, MeOH, rt, 16 h; (iii) HCl, EtOH, microwave irradiation, 150 °C, 0.5 h.
Next, 2-spiro-4-chromanones (7a–c) were synthesized by reacting 1e with an appropriate cycloketone in a pressure tube at 150 °C for 2–16 h in the presence of pyrrolidine (Scheme 8). The following O-MOM deprotection was performed in one pot with excess concentrated HCl. The spiro compounds 7a–c were obtained in moderate to high yields, with 7c giving the lowest yield (53%) probably due to the steric hindrance of the seven-membered ring.
Scheme 8. Synthesis of 2-Spiro-4-chromanones 7a–c.

Reagents and conditions: (i) pyrrolidine, EtOH, 150 °C, pressure tube, 2–16 h; (ii) HCl, rt, overnight.
Synthesis of 2-Substituted Aromatic Chalcone and Flavanone Derivatives
To systematically investigate the SAR of the chalcone and 4-chromanone scaffolds on antibacterial activities, diverse aromatic aldehydes were introduced to synthesize a series of 2-aryl chalcone and flavanone derivatives (Scheme 9). Accordingly, the Claisen–Schmidt aldol condensations were performed at room temperature with the addition of 60% KOH aqueous solution to the mixture of appropriate MOM-protected acetophenone and aldehyde in methanol.55 Subsequent removal of the MOM-protecting group with concentrated HCl afforded the chalcone derivatives 8a–h. Chalcone 8f was synthesized as a comparison under the same conditions to verify the potential effect of 2′-hydroxy group on antibacterial activity. Additionally, in comparison to 8a with the ortho-substituted allyloxy group, regioisomers 8g and 8h (with the allyloxy group at the meta- and para-position, respectively) were next synthesized to investigate the influence of the substitution at the R2 position in the scaffold. Subsequent intramolecular conjugate additions of the phenol on the α,β-unsaturated system of 8a–e were carried out under microwave irradiation in the presence of catalytic amounts of concentrated HCl to yield corresponding flavanones 8i–m in 50–75% yields.
Scheme 9. Synthesis of 2-Substituted Aromatic Chalcones and Flavanones.

Reagents and conditions: (i) 60% KOH aq, MeOH, rt, 60 h; (ii) concentrated HCl, rt, overnight; (iii) microwave, HCl, EtOH, 150 °C, 1.5 h.
Without 2′-OH function for 8f.
All the synthesized compounds were characterized by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry (HRMS), and purity was analyzed by reverse phase HPLC. The structures of 3g and 8a were confirmed by X-ray crystallography, and their ORTEP drawings are shown in Figure S1 (Supporting Information).
All the synthesized compounds were evaluated against Mycobacterium tuberculosis (H37Rv) and a wide set of clinically relevant Gram-positive and -negative bacterial pathogens including Enterococcus faecalis (ATCC 33186), Staphylococcus aureus (ATCC 29213 and NRS 70), Escherichia coli (K12 and ΔtolC), Klebsiella pneumoniae (ATCC 33495), and Pseudomonas aeruginosa (PAO1). Their antitubercular and antibacterial activities are summarized in Tables 1–3.
Table 1. Antibacterial Activities (MIC, μg/mL) of Olympicin A and Derivativesa.
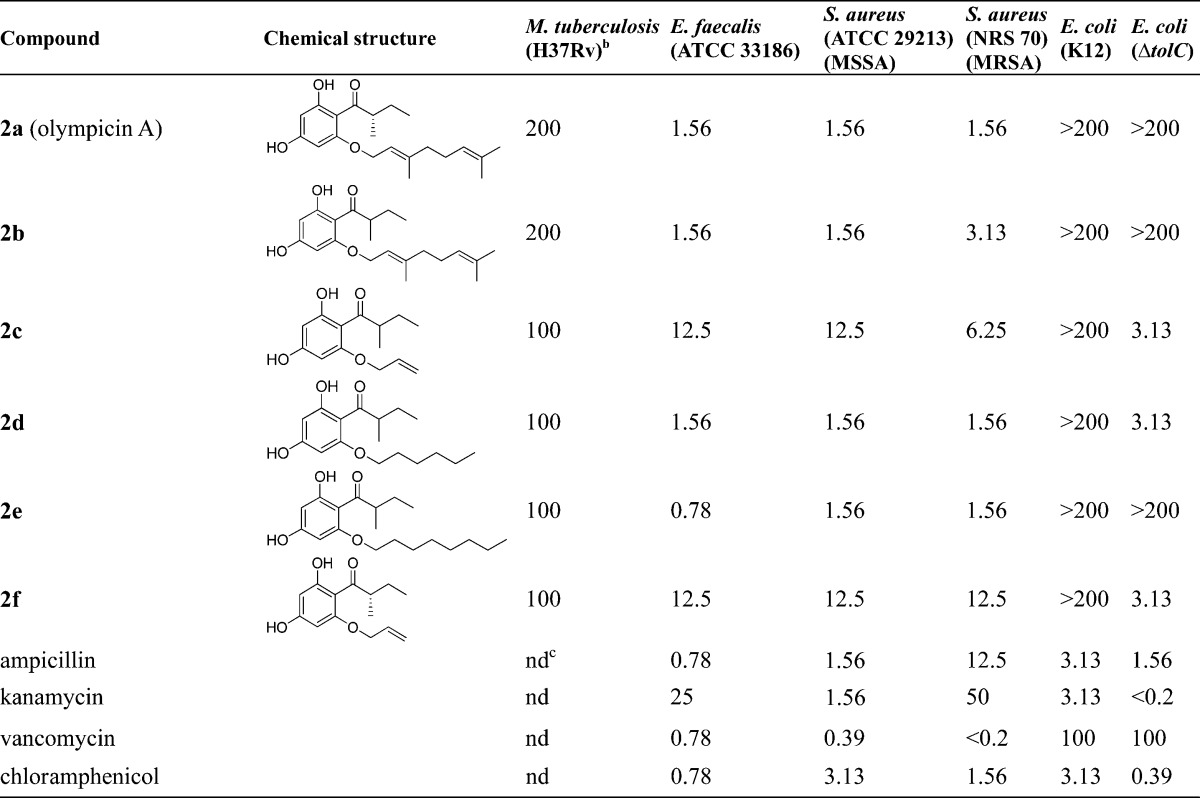
No test compounds were active (>200 μg/mL) against other Gram-negative bacteria including K. pneumonia (ATCC 33495) and P. aeruginosa (PAO1).
Positive controls isoniazid and rifampin inhibited M. tuberculosis at 0.03 and 0.05 μg/mL, respectively.
nd = not determined.
Table 3. Antibacterial Activities (MIC, μg/mL) of Chalcones and Derivativesa.
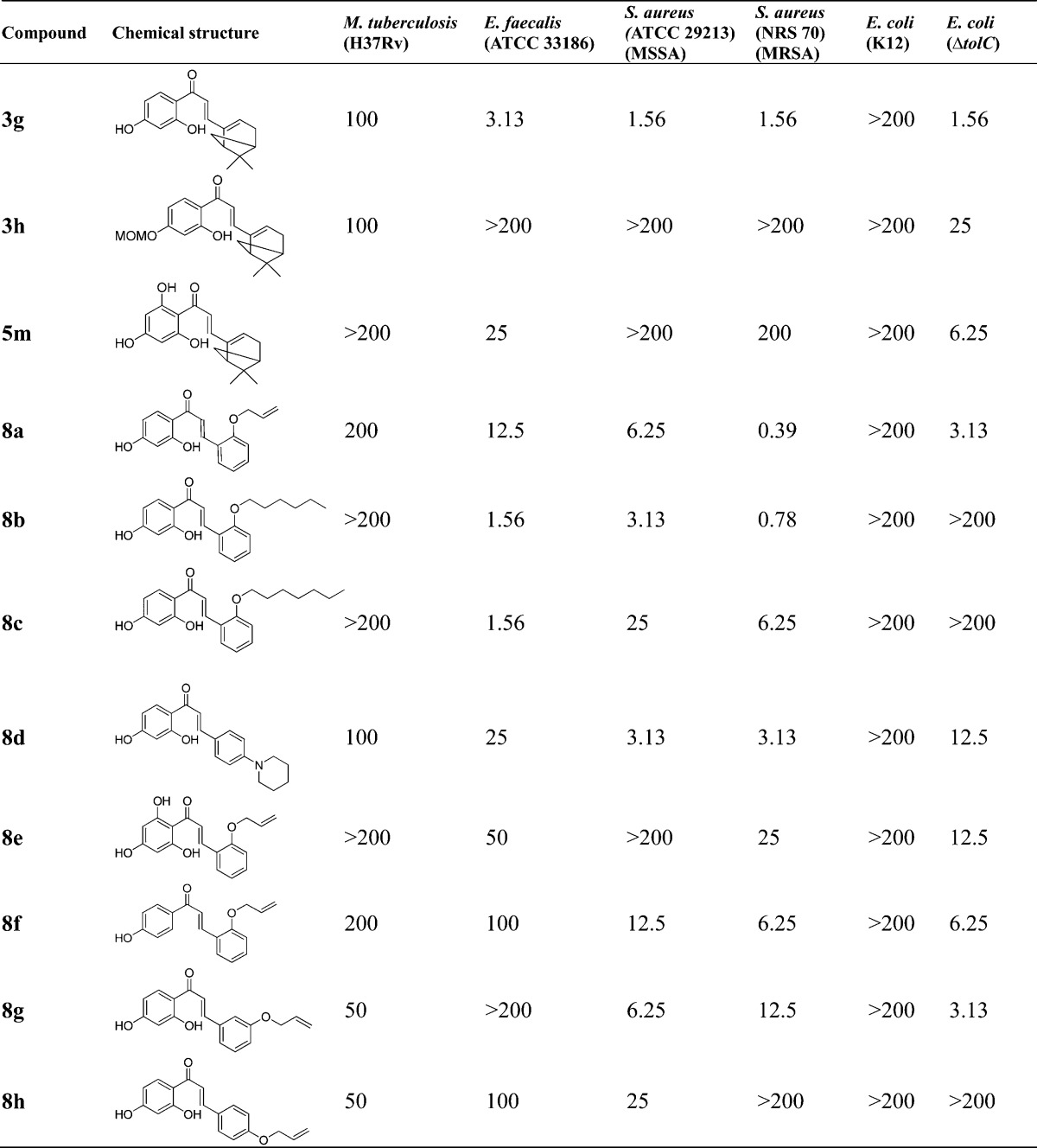
No test compounds were active (>200 μg/mL) against other Gram-negative bacteria including K. pneumonia (ATCC 33495) and P. aeruginosa (PAO1). The MIC values of control antibiotics used in this study are shown in Table 1.
Olympicin A Series
In the olympicin A series, olympicin A (2a) and analogues (2b–f) showed weak antitubercular activity with MICs of 100–200 μg/mL (Table 1). The observed weak antituberculosis activity may be attributed to the general polar nature of this chemical series and decreased membrane penetration. In contrast, the olympicin derivatives with geranyloxy (2a and 2b), n-hexyloxy (2d), and n-octyloxy (2e) groups showed good to potent anti-Gram-positive activity against E. faecalis and S. aureus strains (MIC = 0.78–3.13 μg/mL). However, the less lipophilic olympicin derivatives 2c and 2f with a shorter allyloxy chain exhibited about 8- to 16-fold decrease of antibacterial activity (MIC = 6.25–12.5 μg/mL). In terms of stereochemistry effect, the racemic olympicin A (2b) and allyloxy derivative (2c) showed largely the same antituberculosis and anti-Gram-positive activity compared to their corresponding chiral S-isomers 2a and 2f, respectively. Notably, the anti S. aureus activity (1.56 μg/mL) of our synthetic sample (2a) of olympicin A is consistent with the previously reported anti S. aureus activity (0.5–1 μg/mL) of natural olympicin A.44 In addition, none of these olympicin analogues were active against Gram-negative microorganisms except for the reengineered E. coli strain (ΔtolC) with deficient efflux activity.
4-Chromanone and Chalcone Series. Antituberculosis Activity
In the 4-chromanone and chalcone chemical series (Tables 2 and 3), the majority of compounds exhibited weak antituberculosis activity. The 2-propyl-4-chromanol 4a showed the most potent activity in the entire series with a MIC of 12.5 μg/mL. The reduced 4-chromanol variants 4a and 4c (4-OH, 12.5 and 25 μg/mL, respectively) showed more potent antituberculosis activity than their corresponding 4-chromanones 3a (200 μg/mL) and 3c (50 μg/mL). In addition, the 4-oximinochromane 6a (=NOH, 100 μg/mL) displayed greater potency than 6b (=NOMe, 200 μg/mL) and 6c (=NOBn, >200 μg/mL). Taken together, these results indicate that the small, polar, and hydrophilic groups (e.g., −OH and =NOH) are more favorable at the 4-position of the flavonoid scaffold for antituberculosis activity, suggesting these polar groups may function as hydrogen bond donors when interacting with potential biological cellular target.
Table 2. Antibacterial Activities (MIC, μg/mL) of 4-Chromanone Derivativesa.
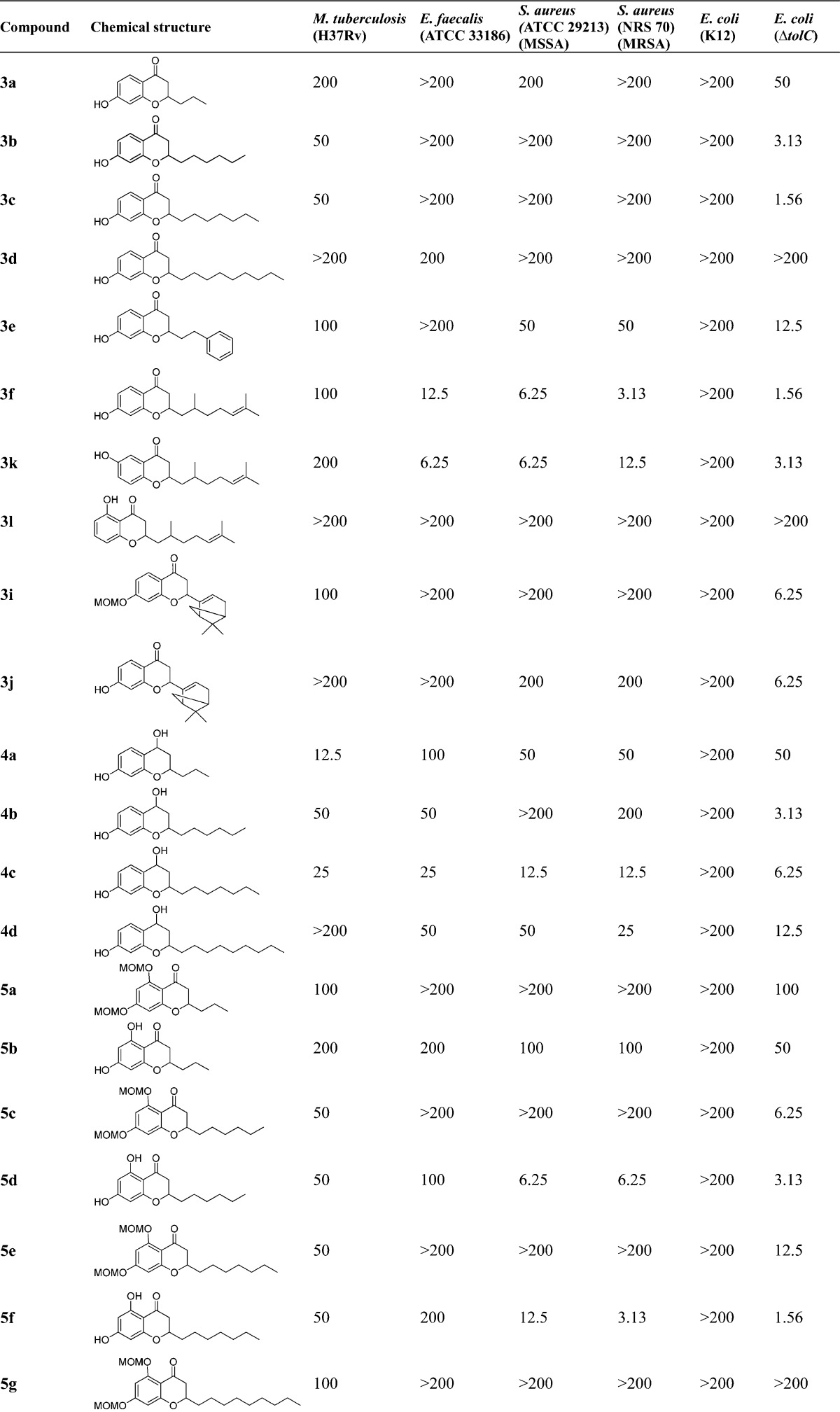
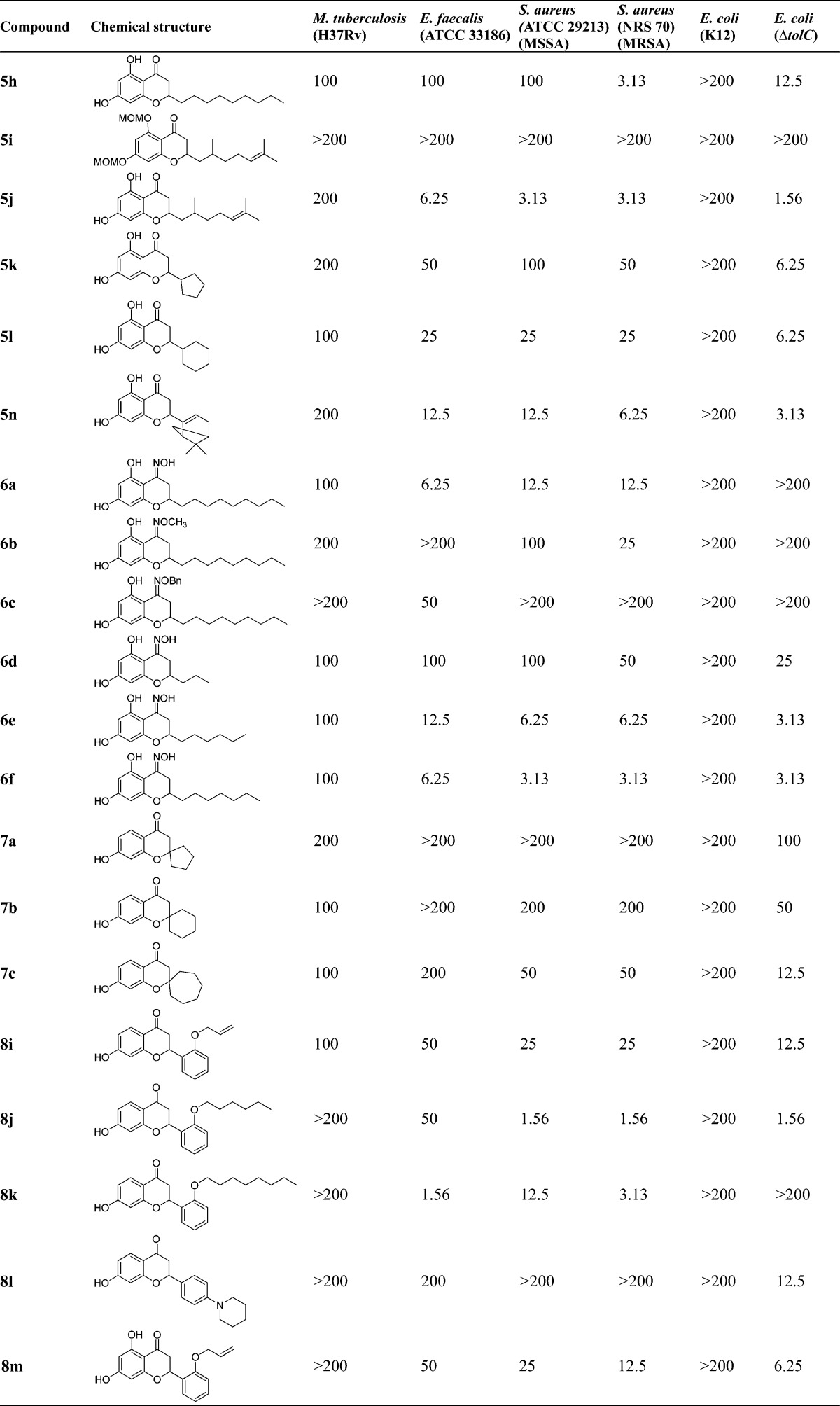
No test compounds were active (>200 μg/mL) against other Gram-negative bacteria including K. pneumonia (ATCC 33495) and P. aeruginosa (PAO1). The MIC values of control antibiotics used in this study are shown in Table 1.
Different lengths of the 2-alkyl side chains in the scaffold displayed an important relationship with antituberculosis activity as well. Compared to 3a (three-carbon, 200 μg/mL), 3b (six-carbon, 50 μg/mL), and 3c (seven-carbon, 50 μg/mL), compound 3d with a nine-carbon linear chain was inactive, suggesting the nine-carbon alkyl group is too long and bulky. To investigate the impact of the substituent at the 5-position in the scaffold, a hydrogen bond donor and/or acceptor (hydroxy group) was introduced. These data showed that the compounds bearing 5,7-dihydroxy functionalities (5b, 5d, and 5f) exhibited similar MIC values to the sole 7-hydroxychromanones 3a–c. However, 5h (5,7-di-OH, nine-carbon chain, 100 μg/mL) was more potent than the corresponding 3d (7-OH, nine-carbon chain, >200 μg/mL). In addition, the position of the sole hydroxy group in the scaffold also played a notable role in their antituberculosis activity by comparing 3f (7-OH, 100 μg/mL) to 3k (6-OH, 200 μg/mL) and 3l (5-OH, >200 μg/mL), with 3f bearing the 7-hydroxy function being the most potent.
In general, 2-aryl substituted compounds together with 2-spiro derivatives possessing a cyclic ring system were less active against M. tuberculosis than 2-alkylated derivatives in the entire series. Moreover, the MOM protected derivatives exhibited comparable or more potent antitubercular activity than their corresponding free phenol parent molecules. The SAR analysis above demonstrates that the 2-alkyl hydrophobic substituents as well as the small and polar functionalities at the 4-position (e.g., hydrogen bond donor groups OH and =NOH) play important roles in antituberculosis activities in the 4-chromanone scaffold.
General Antibacterial Spectra
In addition to antituberculosis evaluation, antimicrobial assessment against representative clinical pathogens revealed that the majority of compounds from these series exhibited notable anti-Gram-positive bacteria activities including against E. faecalis and S. aureus (MSSA and MRSA) and poor activity against Gram-negative bacteria including E. coli, K. pneumoniae, and P. aeruginosa.
Compared with the 4-chromanone flavonoid series, the chalcone series generally exhibited more potent anti-Gram-positive activity than their corresponding cyclized derivatives. The 2′,4′-di-OH chalcone compound 8a having an appended 2-allyloxy group exhibited the best activity (MIC of 0.39–6.25 μg/mL) against MSSA and MRSA, and the o-hydroxy group appeared to have a beneficial effect on anti-Gram-positive bacterial activity by comparing 8a (2′,4′-di-OH, 2-allyloxy, 0.39–12.5 μg/mL) to 8f (4′-OH, 2-allyloxy, 6.25–100 μg/mL) (Table 3). Notably, this observation regarding the importance of the o-hydroxy group in the chalcone scaffold is also in agreement with that found in the 4-chromanone flavonoid series, whereas the corresponding hydroxy group at the 5-position of the 4-chromanone scaffold enhanced the antibacterial activity. However, the 2′,4′,6′-tri-OH chalcone compounds possessing an additional 6′-hydroxy group 5m (25 to >200 μg/mL) and 8e (25 to >200 μg/mL) showed significantly decreased activities against Gram-positive microorganisms compared to the 2′,4′-di-OH chalcones 3g (1.56–3.13 μg/mL) and 8a (0.39–12.5 μg/mL), respectively. The notable decreased activity for trihydroxy compounds is likely due to increased hydrophilicity and polarity properties and thus decreased bacterial membrane penetration. Taken together, these data suggest that two free phenol hydroxy groups on the 4-chromanone and chalcone scaffolds are optimal for Gram-positive antibacterial activity.
Furthermore, in the chalcone series (Table 3), the lipophilic O-alkyl substituent at the 2-position of aromatic chalcones also had a great impact on antibacterial activities, since the chalcones bearing diverse side chains such as the allyloxy (8a), n-hexyloxy (8b), and n-octyloxy (8c) exhibited notable differences in their activities; compound 8b bearing the n-hexyloxy group showed optimal antibacterial activity against E. faecalis, MSSA, and MRSA (1.56, 3.13, and 0.78 μg/mL, respectively). Compound 8a bearing the 2-allyloxy function demonstrated 2-fold more potent activity against MRSA and an 8-fold reduction in activity against E. faecalis than 8b.
In the flavonoid series, the length of 2-alkyl substitutions in the 4-chromanone scaffold plays an important role in determining antibacterial activities. The 5,7-dihydroxy-4-chromanones with long aliphatic alkyl chains 5d, 5f, 5h, and 5j (six to nine-carbon chain, 3.13–6.25 μg/mL) showed better activity against MRSA than the shorter chain derivative 5b (three-carbon chain, 100 μg/mL), and notably, the 2-(2,6-dimethyl-5-heptenyl) substituted 5j with a branched and unsaturated alkyl chain displayed the best potency among these five 2-alkylated derivatives against three Gram-positive bacteria tested (E. faecalis, MSSA, and MRSA; 6.25, 3.13, and 3.13 μg/mL, respectively). These results suggest that the branched unsaturated substitution in 5j may play an important role in enhancing interactions and binding affinity with cellular biological target because of its favorable lipophilicity and high conformational flexibility, as previously noted for the prenylated derivatives.57,58 We also observed that the introduction of an additional 5-OH group to the 7-hydroxy-4-chromanones 5b (100 μg/mL), 5d (6.25 μg/mL), 5f (3.13 μg/mL), 5h (3.13 μg/mL), and 5n (6.25 μg/mL) significantly improved antibacterial activity against MRSA, compared to their corresponding 7-OH-4-chromanones 3a–d (>200 μg/mL) and 3j (200 μg/mL) except that 5j maintained anti-MRSA activity (3.13 μg/mL) compared to 3f. These data demonstrate the importance of the 5-OH group in the 7-OH-4-chromanone scaffold for antibacterial activity.
In addition, the reduced variants 4-chromanols 4a–d (in particular 2-n-heptyl-7-OH-4-chromanol 4c, 12.5–25 μg/mL; and 2-n-nonyl-7-OH-4-chromanol 4d, 25–50 μg/mL) also displayed improved antibacterial activities compared to their corresponding 4-chromanones 3a–d, which were not active against Gram-positive bacteria tested. In contrast, the 4-oximinochromanes (6a and 6d-f: =NOH at the 4-position) showed more potent activity against E. faecalis and MSSA and comparable activity against MRSA than their corresponding 4-chromanones (5h, 5b, 5d, and 5f), suggesting the free oxime =NOH functionality is preferred compared to the carbonyl group. Furthermore, by comparison of 6a with the free 4-oxime functionality (=NOH, 12.5 μg/mL), compounds 6b (=NOMe, 25–100 μg/mL) and 6c (=NOBn, >200 μg/mL) showed decreased antibacterial activity against S. aureus. This observation is also consistent with the trend toward the antituberculosis activity found in this series, indicating that the small, polar, and hydrogen bond donor functionalities (e.g., −OH and =NOH groups) at the 4-position may be more favorable for antibacterial properties, together with the results based upon 4-chromanols (4a–d).
To further evaluate the importance of the 5-, 6-, and 7-OH functionality, a set of 4-chromanone derivatives 3f (7-OH), 3k (6-OH), and 3l (5-OH) were subsequently synthesized. Biological evaluation revealed that the 6- or 7-hydroxy group also proved to be a determining factor for antibacterial activities, since 3f (7-OH, 3.13–12.5 μg/mL) and 3k (6-OH, 6.25–12.5 μg/mL) demonstrated almost equally potent antibacterial activities against Gram-positive bacteria tested. However, their regioisomer 3l with the 5-OH substitution was not active. The lack of antibacterial activity of 3l may be due to the presence of intramolecular hydrogen bonding between the carbonyl group at the 4-position and the hydroxy group at the 5-position. It should be noted that all the MOM protected derivatives (3h, 3i, 5a, 5c, 5e, 5g, and 5i) completely lost anti-Gram-positive bacterial activities against E. faecalis and S. aureus, demonstrating the importance of the free phenol hydroxy functionality and its weakly acidic nature in the scaffold. This observation is consistent with our previous report.43 Additionally, among the chemical series bearing a cyclic/bicyclic ring system at the 2-position (5k, 2-cyclopentyl; 5l, 2-cyclohexyl; 5n, 2-myrtenyl; and the 2-spiro compounds 7a–c), compound 5n bearing the myrtenyl motif showed relatively good antibacterial activity (6.25–12.5 μg/mL) against Gram-positive bacteria tested. In contrast, the 7-OH and 2-myrtenyl substituted flavonoid 3j was largely inactive. Interestingly, its corresponding ring-opened chalcone derivative 3g demonstrated very good anti-Gram-positive activity (1.56–3.13 μg/mL).
A detailed SAR of the 4-chromanone and chalcone series is summarized in Figure 3.
Figure 3.

General SAR of 4-chromanone and chalcone derivatives.
Solubility and Cytotoxicity Determination
Cytotoxicity against mammalian (Vero epithelial) cells and solubility in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) were evaluated for a selected panel of 4-chromanone and chalcone lead compounds, and the results are shown in Table 4. Overall, all the tested compounds had good solubility (≥100 μg/mL) in the DMEM/FBS medium used in the cytotoxicity assay except for the 4-oxime derivative 6a (37.5 ± 17.7 μg/mL). The selectivity indices (SI) for the compounds were calculated as the ratio of the IC50 value of cytotoxicity against Vero monkey kidney cell line and the MIC value against tested MRSA. Notably, 4-chromanone derivatives 5f (SI = 10.5) and 8j (SI = 15.7) and the chalcone derivative 8a (SI = 54.4) possessed a more favorable selectivity index (SI > 10). On the basis of these promising antibacterial lead structures, advanced medicinal chemistry will be applied to produce compounds with improved potency and decreased cytotoxicity.
Table 4. Cytotoxicity and Solubility Profiles of Selected Lead Compoundsa.
| compd | molecular weight (g/mol) | lipophilicity (cLogP)b | solubility (μg/mL) | cytotoxicity IC50 (μg/mL)c | MIC against MRSA (μg/mL) | selectivity index (SI)d |
|---|---|---|---|---|---|---|
| 3f | 288.2 | 3.77 | >200 | 28.8 ± 1.8 | 3.13 | 9.2 |
| 3g | 284.1 | 3.19 | >200 | 6.9 ± 0.3 | 1.56 | 4.4 |
| 5f | 278.2 | 3.20 | 150.0 | 33.0 ± 2.1 | 3.13 | 10.5 |
| 5h | 306.2 | 4.03 | >200 | 8.6 ± 2.7 | 3.13 | 2.8 |
| 5j | 304.2 | 3.38 | >200 | 27.0 ± 2.8 | 3.13 | 8.6 |
| 6a | 321.2 | 4.42 | 37.5 ± 17.7 | 10.1 ± 1.9 | 12.5 | 0.8 |
| 6f | 293.2 | 3.59 | >200 | 9.6 ± 0.1 | 3.13 | 3.1 |
| 8a | 296.1 | 3.37 | >200 | 21.2 ± 5.4 | 0.39 | 54.4 |
| 8b | 340.2 | 4.76 | >200 | 7.2 ± 0.4 | 0.78 | 9.2 |
| 8d | 323.2 | 3.82 | 100.0 | 16.7 ± 0.1 | 3.13 | 5.3 |
| 8j | 340.2 | 4.36 | >200 | 24.5 ± 2.1 | 1.56 | 15.7 |
| 8k | 368.2 | 5.19 | >200 | 17.0 ± 0.0 | 3.13 | 5.4 |
| thioridazine | nde | nd | nd | 5.3 ± 1.3 | nd | nd |
| verapamil | nd | nd | nd | 57.3 ± 3.2 | nd | nd |
| DMSOf | nd | nd | nd | 177.5 ± 10.6 | nd | nd |
DMEM/FBS: Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
The cLogP values of compounds were calculated using ChemBioOffice Ultra, version 12.0, from CambridgeSoft Corporation.
Cytotoxicity IC50, concentration that reduces viability of Vero kidney cells by 50%.
Selectivity index = (cytotoxic IC50)/(MIC against MRSA).
nd = not determined.
Carrier effect.
Time Kill Experiments and Mutation Selection
The bactericidal activities of compounds 3g, 5j, 8a, and 8d were examined against S. aureus Newman (Figure 4). The most effective compound was the chalcone derivative 3g, which killed more than 6 log of cells in just 2 h at 4× its MIC, but at its MIC (1.56 μg/mL against S. aureus Newman) up to 24 h was required to achieve a 3 log reduction in cells. Compound 3g thus exhibited concentration-dependent killing. Compound 8a (MIC = 6.25 μg/mL) was also rapidly bactericidal at 4× its MIC and achieved a 6 log reduction in 6 h. Interestingly, compound 8d (6.25 μg/mL) was entirely bacteriostatic and failed to kill more than 3 log of culture at 4 × its MIC, even up to 24 h of exposure. At concentrations between 1× and 4× their MICs, spontaneous mutants of S. aureus Newman could not be selected with 8a, 3g, and 5j, but mutants arose to 8d at a frequency of 10–10. The controls mupirocin and vancomycin were either bacteriostatic or slowly bactericidal over a 24 h period, and mutants could only be selected with mupirocin at frequencies of 10–7–10–9.
Figure 4.

Time kill studies against S. aureus Newman exposed to 4× MIC of compounds. Each point represents the average of two biological replicates.
Effects on Macromolecular Synthesis and Membrane Potential
Several key biosynthetic processes were simultaneously inhibited in S. aureus Newman exposed to the compounds 2a, 5j, 8a, 3g, and 8d (Figure 5). These effects are consistent with the bacterial membrane being the primary target site of action, resulting in multiple nonspecific cellular effects. Surprisingly, the bacteriostatic compound 8d displayed the same time-dependent effects on macromolecular synthesis as the bactericidal compounds. To determine if the compounds dissipated the membrane potential of S. aureus, the fluorescent probe DiOC2(3) was used. Compounds 2a, 5j, 3g, and 8d all dissipated the membrane potential of S. aureus Newman in a concentration-dependent manner. Maximum dissipation occurred at 4× their MICs and was similar to the control CCCP (carbonyl cyanide m-chlorophenylhydrazone) (Figure 6); as expected, vancomycin (at 4× MIC) failed to affect the membrane potential in S. aureus.
Figure 5.

Effects of 2a, 5j, 3g, 8a, and 8d and indicated positive controls at 4× their MICs on macromolecular synthesis in S. aureus Newman. The standard error of the mean (SEM) is shown for three biological replicates. ERY = erythromycin (MIC = 0.78 μg/mL); RMP = rifampicin (0.12 μg/mL); CIP = ciprofloxacin (0.25 μg/mL).
Figure 6.

Dissipation of the staphylococcal membrane potential by 2a, 5j, 3g, and 8d. A representative of three biological replicates is shown. Vancomycin (MIC = 0.8 μg/mL) and CCCP (MIC = 6.25 μg/mL) were used as negative and positive controls.
Inhibition of Bacterial Topoisomerase IV and DNA Gyrase
Clinically, DNA gyrase and topo IV are validated and attractive antibacterial targets for fluoroquinolone and novobiocin antibiotics. However, because of the emergence of target-based bacterial resistance with fluoroquinolone class and safety concerns of novobiocin, novel DNA gyrase and topoisomerase inhibitors are urgently needed for the treatment of pathogenic and resistant bacterial infections.59 Thus, development of new chemotype bacterial topo inhibitors has attracted great interest in the scientific community, and recent examples include bisbenzimidazoles,60 anziaic acid, and its analogues61,62 as bacterial topo IA inhibitors, and pyridylureas63 and pyrrolamides59 as topo II inhibitors. Previously, flavonoids have been identified as bacterial topoisomerase inhibitors,64−66 and we next tested to see if these promising antibacterial compounds from olympicin A, 4-chromanone, and chalcone series inhibited E. coli topo I, topo IV, and DNA gyrase. The results are shown in Table 5 and Figure S2 (Supporting Information). The E. coli topo I assay (relaxation of negatively supercoiled plasmid DNA) was performed at both 0.5 and 5 mM magnesium chloride concentrations, and no significant inhibition was observed against topo I at 0.5 mM compound concentration. The assay against E. coli gyrase (supercoiling of relaxed plasmid DNA) and E. coli topo IV (decatenation of catenated kinetoplast DNA) was subsequently performed. Again for DNA gyrase, inhibition was not observed or was weak with 8a, 3g, and 2a (MIC values of 0.39–6.25 μg/mL against S. aureus) exhibiting IC50 values of >0.25 mM. However, the E. coli topo IV showed more significant sensitivity toward 2a and 2e (MIC values of 0.78–1.56 μg/mL against E. faecalis and S. aureus) with IC50 values of 30–60 μM (Table 5 and Figure S2, Supporting Information). These results showed that the olympicin A (2a) and its analogue 2e may serve as a promising and novel scaffold for topo IV inhibitors. Interestingly, olympicin A was also recently reported as a moderate ATP-dependent Mycobacterium tuberculosis MurE ligase inhibitor with an IC50 value of 75 μM.45 Studies to determine if the olympicin A analogues 2c–f inhibit the MurE pathway in M. tuberculosis remain to be performed. Finally, the antibacterial mechanism of the 4-chromanone flavanone compounds 8k and 8j (MIC values of 1.56–12.5 μg/mL against S. aureus) may not be likely to involve topoisomerase inhibition, as they were inactive in these topo enzyme inhibition assays (Table 5). Collectively, no correlations between whole-cell-based activity and topoisomerase inhibition were observed for selected compounds, suggesting that the membrane is likely the primary biological target responsible for antibacterial activity and topo IV is a secondary or alternative target. Further mechanistic studies are warranted to define the exact mechanism of antibacterial action of these chemically modified flavonoid and polyphenol compounds.
Table 5. E. coli Topoisomerases and DNA Gyrase Inhibition of Selected Analogues.
| compd | topo I IC50 (μM) | DNA gyrase IC50 (μM) | topo IV IC50 (μM) |
|---|---|---|---|
| 2a | >500 | 500–1000 | 30–60 |
| 2e | >500 | >1000 | 30–60 |
| 3g | >500 | 250–500 | 60–120 |
| 5f | >500 | >1000 | 120–250 |
| 8a | >500 | 500–1000 | 250–500 |
| 8b | >500 | >1000 | 60–120 |
| 8d | >500 | >1000 | 500–1000 |
| 8j | >500 | >1000 | >1000 |
| 8k | >500 | >1000 | >1000 |
| known inhibitors | 14–19a | 0.2,b 100c | 2.1,b 806c |
Conclusions
In summary, 58 olympicin A, 4-chromanone, and chalcone derivatives containing various functionalities were synthesized and evaluated against Mycobacterium tuberculosis and a panel of clinically relevant Gram-positive and -negative bacterial pathogens. Bacterial evaluation showed that this class of compounds generally exhibited good activities against Gram-positive bacteria tested. Systematic SAR study revealed that the phenol hydroxy groups at the 5- and 7-position of the 4-chromanone scaffold were essential for antibacterial activities. Additionally, the hydrogen bond donor/acceptor functionality at the 4-position together with the lipophilic 2-alkyl moiety in the scaffold also played important roles in antibacterial activities. The flavanone derivatives bearing the lipophilic substituent on the 2-phenyl ring showed good antibacterial properties as well. In the chalcone chemical series, both hydroxy groups at 2′- and 4′-position, as well as the bicyclic myrtenyl motif and the 2-alkyloxy substitution on the aromatic ring, favored anti-Gram-positive bacterial activities. The selected compounds generally possessed favorable solubility, and 5f, 8a, and 8j had more desirable selectivity indices ranging from 10.5 to 54.4. In addition, compounds 2a, 5j, 3g, and 8d were found to disrupt bacterial membrane potential and have secondary inhibitory effect on macromolecular biosynthesis of DNA, RNA, and protein. Further evaluation of selected compounds against bacterial topoisomerases and DNA gyrase revealed that 2a and 2e inhibited topoisomerase IV (IC50 = 30–60 μM). Taken together, the antibacterial agents identified from this study provide chemically modified flavonoid phytochemicals as promising antibacterial leads for further medicinal chemistry optimization in an effort to identify advanced experimental candidates with antimicrobial therapeutic potential.
Experimental Section
Chemical Synthesis. General Procedures
Solvents and reagents were supplied from Aldrich, Acros, or Fisher and used without further purification. NMR spectra were recorded on a Bruker AM-400 (400 MHz) spectrometer. High-resolution mass spectra were obtained on an Agilent 6530 Accurate Mass Q-TOF LC/MS instrument. Reactions in pressure tube were carried out using a Q-Tube reactor from Q Labtech. Microwave reactions were conducted using a Biotage Initiator reactor. All reactions were monitored by either TLC or HPLC. Compounds were purified by flash chromatography on silica gel on a Biotage Isolera One system. The purity of compounds was determined by analytical HPLC (Shimadzu LC-20A series) using a Gemini, 3 μm, C18, 110 Å column (50 mm × 4.6 mm, Phenomenex) and flow rate of 1 mL/min. Gradient conditions were the following: solvent A (0.1% trifluoroacetic acid in water) and solvent B (acetonitrile), 0–2.0 min 100% A, 2.0–7.0 min 0–100% B (linear gradient), 7.0–8.0 min 100% B, UV detection at 254 and 220 nm. All the tested compounds were obtained with ≥95% purity by HPLC. NMR standards used are as follows. 1H NMR: CDCl3, 7.26 ppm; CD3OD, 3.31 ppm; DMSO-d6, 2.50 ppm. 13C NMR: CDCl3, 77.16 ppm; CD3OD, 49.00 ppm; DMSO-d6, 39.52 ppm. MOM-protected acetophenones were synthesized as described in the literature.56,69,70
(S)-2-(2-Methylbutanoyl)benzene-1,3,5-triyl Tris(4-methylbenzenesulfonate) (1b)
To a solution of 1a (150 mg, 0.715 mmol) in acetone (11 mL) were added p-toluenesulfonic chloride (408 mg, 2.14 mmol) and K2CO3 (845 mg, 6.15 mmol) successively. The resulting mixture was stirred under reflux for 1 h, and then acetone was removed under reduced pressure. The residue was diluted with water and DCM, and the organic layer was washed with 1 M HCl aq, water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc (first 85/15, then 75/25) to give the product (350 mg, 0.52 mmol, 73%) as clear oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.69 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 8.4 Hz, 4H), 7.36 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.3 Hz, 4H), 7.0 (s, 2H), 2.64–2.59 (m, 1H), 2.44 (s, 9H), 1.56–1.50 (m, 1H), 1.24–1.14 (m, 1H), 0.90 (d, J = 7.0 Hz, 3H), 0.76 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 201.2, 149.8, 146.8, 146.5, 146.4, 131.7, 131.3, 130.2, 130.0, 128.5, 127.1, 114.3, 48.6, 24.5, 21.80, 21.79, 14.2, 11.2. HRMS calculated for C32H32O10S3 (M + H)+ 673.1230, found (M + H)+ 673.1229. HPLC purity: 98.6% (254 nm), tR = 8.17 min; 99.5% (220 nm), tR = 8.17 min.
1-(2-Hydroxy-4,6-bis(methoxymethoxy)phenyl)-2-methylbutan-1-one (1c)
To a suspension of 1a′ (1.12 g, 5.33 mmol) in DCM (11 mL) at 0 °C was added DIPEA (2.78 mL, 15.9 mmol) carefully. After stirring for 10 min, MOMCl (1.21 mL, 15.9 mmol) was added to the solution dropwise. The resulting mixture was stirred for 1 h. Afterward, the solution was poured into sat. NH4Cl aq, and then water and DCM were added into the mixture. The organic layer was washed with water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc (90/10) to give the product (1.02 g, 3.42 mmol, 64%) as pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 13.74 (s, 1H), 6.27 (s, 2H), 5.24 (s, 2H), 5.16 (br s, 2H), 3.65–3.60 (m, 1H), 3.51 (s, 3H), 3.46 (s, 3H), 1.87–1.80 (m, 1H), 1.43–1.36 (m, 1H), 1.16 (d, J = 6.8 Hz, 3H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 210.3, 167.2, 163.2, 160.1, 106.7, 97.6, 94.9, 94.4, 94.1, 56.9, 56.6, 46.6, 27.1, 16.7, 12.2. HRMS calculated for C15H22O6 (M + H)+ 299.1489, found (M + H)+ 299.1483. HPLC purity: 99.8% (254 nm), tR = 7.42 min; 99.8% (220 nm), tR = 7.42 min.
(S)-1-(2-Hydroxy-4,6-bis(methoxymethoxy)phenyl)-2-methylbutan-1-one (1c′)
Synthesis and purification were performed as described in compound 1c. Product: 1.03 g, 3.46 mmol, 65%. 1H NMR (400 MHz, CDCl3): δ (ppm) 13.74 (s, 1H), 6.27 (s, 2H), 5.24 (s, 2H), 5.16 (br s, 2H), 3.65–3.60 (m, 1H), 3.52 (s, 3H), 3.46 (s, 3H), 1.87–1.80 (m, 1H), 1.43–1.36 (m, 1H), 1.16 (d, J = 6.8 Hz, 3H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 210.3, 167.2, 163.2, 160.1, 106.7, 97.6, 94.9, 94.4, 94.1, 56.8, 56.6, 46.6, 27.1, 16.7, 12.1. HRMS calculated for C15H22O6 (M + H)+ 299.1489, found (M + H)+ 299.1475. HPLC purity: 99.9% (254 nm), tR = 7.38 min; 100% (220 nm), tR = 7.38 min.
General Procedure for the Synthesis of 2a,b
To a solution of 1b (410 mg, 0.61 mmol) in DMF (4 mL) at 0 °C was added NaH (49 mg, 1.21 mmol) carefully. After stirring for 30 min, geranyl bromide (198 mg, 0.91 mmol) in DMF (1 mL) was added dropwise. The resulting mixture was stirred at room temperature for 1 h. Afterward, the reaction was quenched with water and extracted with EtOAc twice. The organic layer was washed with water, brine and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was dissolved in MeOH (10 mL), followed by the careful addition of sodium methoxide (659 mg, 12.2 mmol). The resulting mixture was stirred under reflux for 8 h, and then MeOH was removed. The residue was diluted with 1 M HCl aq and extracted with EtOAc twice. The organic layer was washed with water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to give the product.
(S,E)-1-(2-((3,7-Dimethylocta-2,6-dien-1-yl)oxy)-4,6-dihydroxyphenyl)-2-methylbutan-1-one (Olympicin A, 2a)
Purified with hexane/EtOAc (90/10) to give the product (116 mg, 0.34 mmol, 55%, two-step overall yield) as pale yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 5.98 (d, J = 2.2 Hz, 1H), 5.91 (d, J = 2.2 Hz, 1H), 5.54 (br, 1H), 5.52–5.49 (m, 1H), 5.11–5.09 (m, 1H), 4.56 (d, J = 6.6 Hz, 2H), 3.67–3.63 (m, 1H), 2.14–2.08 (m, 4H), 1.81–1.78 (m, 1H), 1.74 (s, 3H), 1.68 (s, 3H), 1.61 (s, 3H), 1.40–1.33 (m, 1H), 1.12 (d, J = 6.7 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 210.8, 167.3, 163.1, 162.8, 142.6, 132.1, 123.7, 118.3, 105.8, 96.6, 92.1, 65.8, 46.1, 39.6, 27.0, 26.4, 25.8, 17.8, 16.73, 16.67, 12.0. HRMS calculated for C21H30O4 (M + H)+ 347.2217, found (M + H)+ 347.2210. HPLC purity: 99.7% (254 nm), tR = 8.18 min; 99.6% (220 nm), tR = 8.18 min.
(E)-1-(2-((3,7-Dimethylocta-2,6-dien-1-yl)oxy)-4,6-dihydroxyphenyl)-2-methylbutan-1-one (2b)
Purified with hexane/EtOAc (90/10) to give the product (110 mg, 0.32 mmol, 52% over two steps) as pale yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 5.98 (d, J = 2.2 Hz, 1H), 5.91 (d, J = 2.2 Hz, 1H), 5.54 (br, 1H), 5.52–5.49 (m, 1H), 5.11–5.09 (m, 1H), 4.56 (d, J = 6.6 Hz, 2H), 3.67–3.63 (m, 1H), 2.14–2.08 (m, 4H), 1.81–1.78 (m, 1H), 1.74 (s, 3H), 1.68 (s, 3H), 1.61 (s, 3H), 1.40–1.33 (m, 1H), 1.12 (d, J = 6.7 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 210.8, 167.3, 163.1, 162.8, 142.6, 132.1, 123.7, 118.3, 105.8, 96.6, 92.1, 65.8, 46.1, 39.6, 27.0, 26.4, 25.8, 17.8, 16.74, 16.68, 12.0. HRMS calculated for C21H30O4 (M + H)+ 347.2217, found (M + H)+ 347.2214. HPLC purity: 99.7% (254 nm), tR = 8.17 min; 99.6% (220 nm), tR = 8.17 min.
General Procedure for the Synthesis of 2c–f
To a solution of 1c (200 mg, 0.67 mmol) in DMF (4 mL) at 0 °C was added NaH (54 mg, 1.34 mmol) carefully. After stirring for 30 min, appropriate alkyl bromide (1 mmol) in DMF (1 mL) was added dropwise. The resulting mixture was stirred at room temperature until the complete consumption of 1c (1–7 h). Afterward, the reaction was quenched with water and extracted with EtOAc twice. The organic layer was washed with water, brine and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was dissolved in MeOH (10 mL), followed by the addition of concentrated HCl (510 μL, 6 mmol) carefully. The resulting mixture was stirred at room temperature overnight, and the solvent was removed under reduced pressure. The crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to give the product.
1-(2-(Allyloxy)-4,6-dihydroxyphenyl)-2-methylbutan-1-one (2c)
Purified with hexane/EtOAc (85/15) to give the product (157 mg, 0.63 mmol, 93% over two steps) as pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.11–6.02 (m, 1H), 6.00 (d, J = 2.1 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.86 (br, 1H), 5.42 (d, J = 17.2 Hz, 1H), 5.35 (d, J = 10.4 Hz, 1H), 4.57 (d, J = 5.6 Hz, 2H), 3.69–3.64 (m, 1H), 1.84–1.79 (m, 1H), 1.42–1.35 (m, 1H), 1.13 (d, J = 6.7 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 210.9, 167.2, 163.1, 162.3, 132.1, 119.2, 105.7, 96.9, 92.3, 70.0, 46.1, 27.0, 16.7, 11.9. HRMS calculated for C14H18O4 (M – H)− 249.1132, found (M – H)− 249.1102. HPLC purity: 99.7% (254 nm), tR = 7.16 min; 99.9% (220 nm), tR = 7.16 min.
1-(2-(Hexyloxy)-4,6-dihydroxyphenyl)-2-methylbutan-1-one (2d)
Purified with reverse phase C18 silica gel chromatography with H2O/MeCN (45/55) to give the product (130 mg, 0.44 mmol, 66% over two steps) as pale solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.01 (s, 1H), 5.92 (s, 1H), 3.96 (t, J = 6.5 Hz, 2H), 3.74–3.69 (m, 1H), 1.85–1.75 (m, 3H), 1.46–1.33 (m, 7H), 1.14 (d, J = 6.8 Hz, 3H), 0.92–0.87 (m, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 210.8, 167.3, 163.2, 163.0, 105.6, 96.6, 91.9, 69.2, 46.0, 31.6, 29.1, 26.8, 26.1, 22.7, 17.0, 14.1, 11.9. HRMS calculated for C17H26O4 (M – H)− 293.1758, found (M – H)− 293.1713. HPLC purity: 99.2% (254 nm), tR = 7.93 min; 99.9% (220 nm), tR = 7.92 min.
1-(2,4-Dihydroxy-6-(octyloxy)phenyl)-2-methylbutan-1-one (2e)
Purified with reverse phase C18 silica gel chromatography with H2O/MeCN (40/60) to give the product (151 mg, 0.47 mmol, 70% over two steps) as pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.33 (br, 1H), 6.01 (d, J = 1.5 Hz, 1H), 5.91 (d, J = 1.5 Hz, 1H), 3.97 (t, J = 6.5 Hz, 2H), 3.74–3.69 (m, 1H), 1.85–1.77 (m, 3H), 1.46–1.28 (m, 11H), 1.14 (d, J = 6.8 Hz, 3H), 0.90–0.87 (m, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 210.7, 167.3, 163.2, 163.0, 105.6, 96.6, 91.9, 69.2, 46.1, 31.9, 29.4, 29.3, 29.1, 26.8, 26.4, 22.8, 17.0, 14.2, 11.9. HRMS calculated for C19H30O4 (M + H)+ 323.2217, found (M + H)+ 323.2213. HPLC purity: 98.8% (254 nm), tR = 8.31 min; 99.9% (220 nm), tR = 8.31 min.
(S)-1-(2-(Allyloxy)-4,6-dihydroxyphenyl)-2-methylbutan-1-one (2f)
Purified with hexane/EtOAc (85/15) to give the product (152 mg, 0.61 mmol, 90% over two steps) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.11–6.02 (m, 1H), 6.00 (d, J = 2.1 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.86 (br, 1H), 5.42 (d, J = 17.2 Hz, 1H), 5.35 (d, J = 10.4 Hz, 1H), 4.57 (d, J = 5.6 Hz, 2H), 3.69–3.64 (m, 1H), 1.84–1.79 (m, 1H), 1.42–1.35 (m, 1H), 1.13 (d, J = 6.7 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 210.8, 167.3, 163.0, 162.3, 132.1, 119.3, 105.7, 96.9, 92.3, 70.1, 46.2, 27.0, 16.7, 11.9. HRMS calculated for C14H18O4 (M – H)− 249.1132, found (M – H)− 249.1104. HPLC purity: 99.1% (254 nm), tR = 7.17 min; 99.6% (220 nm), tR = 7.17 min.
General Procedure for the Synthesis of 3a–e
To a solution of 1d (152 mg, 1 mmol) in ethanol (2.5 mL) were added pyrrolidine (220 mg, 3.1 mmol) and corresponding aldehyde (6 mmol) successively. The resulting mixture was stirred at 150 °C in a pressure tube for 1 h. Afterward, the solution was diluted with ethyl acetate and the organic layer was washed with 10% HCl, water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to give the product.
7-Hydroxy-2-propylchroman-4-one (3a)
Purified with hexane/EtOAc (87/13) to give the product (85 mg, 0.41 mmol, 41%) as light brown solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.13 (br, 1H), 7.79 (d, J = 8.6 Hz, 1H), 6.55 (d, J = 8.4 Hz, 1H), 6.43 (s, 1H), 4.42 (br, 1H), 2.67–2.63 (m, 2H), 1.85–1.80 (m, 1H), 1.68–1.44 (m, 3H), 0.99–0.95 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 193.0, 164.32, 164.29, 129.5, 114.6, 110.8, 103.4, 78.0, 42.6, 37.1, 18.3, 14.0. HRMS calculated for C12H14O3 (M – H)− 205.0870, found (M – H)− 205.0863. HPLC purity: 97.3% (254 nm), tR = 6.31 min; 98.6% (220 nm), tR = 6.31 min.
2-Hexyl-7-hydroxychroman-4-one (3b)
Purified with hexane/EtOAc (90/10) to give the product (92 mg, 0.37 mmol, 37%) as light brown solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.78 (d, J = 8.7 Hz, 1H), 6.55 (dd, J = 8.6, 1.7 Hz, 1H), 6.44 (d, J = 1.7 Hz, 1H), 4.43–4.39 (m, 1H), 2.66–2.63 (m, 2H), 1.87–1.82 (m, 1H), 1.67–1.64 (m, 1H), 1.53–1.30 (m, 8H), 0.90–0.87 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 193.1, 164.34, 164.30, 129.4, 114.6, 110.8, 103.4, 78.3, 42.6, 35.0, 31.8, 29.2, 25.0, 22.7, 14.2. HRMS calculated for C15H20O3 (M – H)− 247.1340, found (M – H)− 247.1331. HPLC purity: 98.4% (254 nm), tR = 7.06 min; 99.1% (220 nm), tR = 7.06 min.
2-Heptyl-7-hydroxychroman-4-one (3c)
Purified with hexane/EtOAc (90/10) to give the product (91 mg, 0.35 mmol, 35%) as light brown solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.78 (d, J = 8.7 Hz, 1H), 6.55 (dd, J = 8.7, 2.3 Hz, 1H), 6.43 (d, J = 2.2 Hz, 1H), 4.43–4.39 (m, 1H), 2.69–2.60 (m, 2H), 1.88–1.80 (m, 1H), 1.70–1.63 (m, 1H), 1.53–1.25 (m, 10H), 0.89–0.86 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 193.0, 164.4, 164.3, 129.4, 114.6, 110.9, 103.4, 78.3, 42.6, 35.0, 31.9, 29.5, 29.3, 25.0, 22.8, 14.2. HRMS calculated for C16H22O3 (M – H)− 261.1496, found (M – H)− 261.1479. HPLC purity: 95.0% (254 nm), tR = 7.28 min; 96.1% (220 nm), tR = 7.28 min.
7-Hydroxy-2-nonylchroman-4-one (3d)
Purified with hexane/EtOAc (90/10) to give the product (98 mg, 0.33 mmol, 33%) as light brown solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.23 (br, 1H), 7.79 (d, J = 8.7 Hz, 1H), 6.56 (dd, J = 8.7, 2.1 Hz, 1H), 6.43 (d, J = 2.1 Hz, 1H), 4.45–4.38 (m, 1H), 2.70–2.61 (m, 2H), 1.88–1.80 (m, 1H), 1.70–1.63 (m, 1H), 1.55–1.25 (m, 14H), 0.89–0.86 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 193.1, 164.4, 129.5, 114.6, 110.9, 103.4, 78.3, 42.5, 35.0, 32.0, 29.7, 29.6, 29.5, 29.4, 25.0, 22.8, 14.2. HRMS calculated for C18H26O3 (M – H)− 289.1809, found (M – H)− 289.1787. HPLC purity: 99.1% (254 nm), tR = 7.71 min; 99.3% (220 nm), tR = 7.71 min.
7-Hydroxy-2-phenethylchroman-4-one (3e)
Purified with reverse phase C18 silica gel chromatography with H2O/MeCN (45/55) to give the product (54 mg, 0.20 mmol, 20%) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.79 (d, J = 8.6 Hz, 1H), 7.27–7.25 (m, 2H), 7.20–7.16 (m, 3H), 6.53 (dd, J = 8.7, 1.8 Hz, 1H), 6.45 (d, J = 1.7 Hz, 1H), 4.43–4.37 (m, 1H), 2.91–2.76 (m, 2H), 2.71–2.59 (m, 2H), 2.22–2.14 (m, 1H), 2.01–1.94 (m, 1H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.7, 164.3, 164.2, 141.0, 129.5, 128.7, 128.6, 126.3, 114.6, 111.0, 103.4, 77.1, 42.6, 36.6, 31.2. HRMS calculated for C17H16O3 (M – H)− 267.1027, found (M – H)− 267.1012. HPLC purity: 100% (254 nm), tR = 6.69 min; 99.5% (220 nm), tR = 6.69 min.
General Procedure for the Synthesis of 3f, 3k, and 3l
To a solution of corresponding MOM-protected acetophenone (196 mg, 1 mmol) in ethanol (2.5 mL) was added DEA (154 mg, 2.1 mmol) and (±)-citronellal (309 mg, 2 mmol) successively. The resulting mixture was stirred at 150 °C in a pressure tube for 1 h. The resulting solution was cooled to room temperature and diluted with EtOAc. The organic layer was washed with 10% HCl aq, water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the residue was dissolved in methanol (14 mL), followed by the careful addition of concentrated HCl (340 μL, 4 mmol), and the resulting mixture was stirred at room temperature overnight. Afterward, the solution was concentrated and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to give the product.
2-(2,6-Dimethylhept-5-en-1-yl)-7-hydroxychroman-4-one (3f)
Purified with hexane/EtOAc (90/10) to obtain a mixture of two diastereomers (167 mg, 0.58 mmol, 58% over two steps) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.79 (d, J = 8.6 Hz, 2H), 6.55 (d, J = 8.7 Hz, 2H), 6.43 (s, 2H), 5.98 (br, 2H), 5.11–5.08 (m, 2H), 4.55–4.50 (m, 2H), 2.69–2.56 (m, 4H), 2.04–1.90 (m, 6H), 1.82–1.69 (m, 2H), 1.68 (s, 6H), 1.60 (s, 6H), 1.44–1.32 (m, 4H), 1.26–1.19 (m, 2H), 0.97–0.94 (m, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.95, 192.86, 164.4, 164.32, 164.28, 131.7, 131.6, 129.4, 124.6, 124.5, 114.6, 110.9, 103.4, 76.2, 43.2, 42.9, 42.3, 42.2, 37.5, 36.9, 28.9, 28.5, 25.9, 25.5, 25.4, 20.0, 19.4, 17.8. HRMS calculated for C18H24O3 (M – H)− 287.1653, found (M – H)− 287.1632. HPLC purity: 97.8% (254 nm), tR = 7.32 min; 98.0% (220 nm), tR = 7.32 min.
2-(2,6-Dimethylhept-5-en-1-yl)-6-hydroxychroman-4-one (3k)
Purified with hexane/EtOAc (92/8) to obtain a mixture of two diastereomers (216 mg, 0.75 mmol, 75% over two steps) as yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.40 (d, J = 3.1 Hz, 2H), 7.07 (dd, J = 8.9, 3.1 Hz, 2H), 6.91 (br, 2H), 6.87 (dd, J = 8.9, 2.6 Hz, 2H), 5.11–5.08 (m, 2H), 4.51–4.45 (m, 2H), 2.71–2.58 (m, 4H), 2.04–1.72 (m, 8H), 1.68 (s, 6H), 1.60 (s, 6H), 1.45–1.17 (m, 6H), 0.97–0.94 (m, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 194.2, 194.1, 156.44, 156.38, 150.5, 131.61, 131.56, 125.3, 124.61, 124.56, 121.0, 119.4, 119.3, 111.1, 76.6, 76.1, 43.7, 43.4, 42.30, 42.26, 37.5, 36.9, 28.9, 28.6, 25.8, 25.5, 25.4, 20.0, 19.4, 17.8. HRMS calculated for C18H24O3 (M – H)− 287.1653, found (M – H)− 287.1613. HPLC purity: 99.8% (254 nm), tR = 7.40 min; 99.7% (220 nm), tR = 7.38 min.
2-(2,6-Dimethylhept-5-en-1-yl)-5-hydroxychroman-4-one (3l)
Purified by reverse phase C18 silica gel chromatography with H2O/MeCN (30/70) to obtain a mixture of two diastereomers (170 mg, 0.59 mmol, 59% over two steps) as pale yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 11.69 (s, 2H), 7.33 (t, J = 8.3 Hz, 2H), 6.48 (dd, J = 8.3, 0.8 Hz, 2H), 6.41 (ddd, J = 8.3, 2.2, 0.8 Hz, 2H), 5.11–5.07 (m, 2H), 4.52–4.48 (m, 2H), 2.77–2.62 (m, 4H), 2.07–1.63 (m, 8H), 1.68 (s, 6H), 1.61 (s, 6H), 1.47–1.18 (m, 6H), 0.98–0.95 (m, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 198.8, 198.7, 162.2, 161.84, 161.79, 138.24, 138.23, 131.7, 131.6, 124.54, 124.48, 109.2, 108.4, 107.48, 107.47, 76.2, 75.6, 43.0, 42.7, 42.21, 42.15, 37.5, 36.8, 28.9, 28.6, 25.9, 25.5, 25.4, 20.0, 19.4, 17.8. HRMS calculated for C18H24O3 (M – H)− 287.1653, found (M – H)− 287.1604. HPLC purity: 99.9% (254 nm), tR = 8.00 min; 100% (220 nm), tR = 7.99 min.
(E)-3-(6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)-1-(2-hydroxy-4-(methoxymethoxy)phenyl)prop-2-en-1-one (3h)
To a solution of 1e (196 mg, 1 mmol) in ethanol (2.5 mL) were added pyrrolidine (149 mg, 2.1 mmol) and (−)-myrtenal (300 mg, 2 mmol) successively. The resulting mixture was stirred at 75 °C in a pressure tube for 1 h. Afterward, the solution was diluted with ethyl acetate, and the organic layer was washed with 10% HCl, water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc (97/3) to give compound 3h (160 mg, 0.48 mmol, 48%) as yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 13.37 (s, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.52 (d, J = 15.1 Hz, 1H), 6.88 (d, J = 15.1 Hz, 1H), 6.61 (d, J = 2.4 Hz, 1H), 6.54 (dd, J = 8.9, 2.4 Hz, 1H), 6.21 (m, 1H), 5.20 (s, 2H), 3.47 (s, 3H), 2.68–2.67 (m, 1H), 2.55–2.41 (m, 3H), 2.19–2.18 (m, 1H), 1.39 (s, 3H), 1.18 (d, J = 9.0 Hz, 1H), 0.80 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.9, 166.2, 163.5, 146.5, 145.6, 137.3, 131.3, 116.7, 115.2, 108.1, 104.0, 94.2, 56.5, 41.6, 40.8, 38.1, 33.1, 31.3, 26.3, 21.0. HRMS calculated for C20H24O4 (M + H)+ 329.1747, found (M + H)+ 329.1735. HPLC purity: 98.8% (254 nm), tR = 7.98 min; 98.5% (220 nm), tR = 7.98 min.
(E)-1-(2,4-Dihydroxyphenyl)-3-(6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl)prop-2-en-1-one (3g)
To a solution of 3h (130 mg, 0.396 mmol) in methanol (6 mL) was added concentrated HCl (154 μL, 1.78 mmol) dropwise. The resulting mixture was stirred at room temperature for 24 h, after which the solvent was removed. The crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc (90/10) to obtain compound 3g (100 mg, 0.35 mmol, 89%) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 13.56 (s, 1H), 7.72 (d, J = 9.5 Hz, 1H), 7.51 (d, J = 15.1 Hz, 1H), 6.87 (d, J = 15.2 Hz, 1H), 6.43–6.41 (m, 2H), 6.20 (m, 1H), 2.67 (t, J = 5.4 Hz, 1H), 2.54–2.41 (m, 3H), 2.18 (br, 1H), 1.38 (s, 3H), 1.18 (d, J = 9.0 Hz, 1H), 0.79 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 193.0, 166.2, 163.1, 146.5, 145.8, 137.5, 132.1, 116.6, 114.5, 108.0, 103.8, 41.5, 40.8, 38.1, 33.1, 31.3, 26.3, 21.0. HRMS calculated for C18H20O3 (M – H)− 283.1340, found (M – H)− 283.1313. HPLC purity: 99.3% (254 nm), tR = 7.48 min; 99.2% (220 nm), tR = 7.48 min.
2-(6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)-7-(methoxymethoxy)chroman-4-one (3i)
To a solution of 3h (60 mg, 0.183 mmol) in ethanol (3 mL) was added sodium acetate (300 mg, 3.6 mmol). The resulting mixture was refluxed for 24 h, after which the solvent was removed. The residue was dissolved in ethyl acetate, and the organic layer was washed with water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc (95/5) to obtain a mixture of two diastereomers 3i (28 mg, 0.085 mmol, 47%) as clear oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.82–7.79 (m, 2H), 6.66–6.62 (m, 2H), 6.61 (s, 2H), 5.67–5.64 (m, 2H), 5.19 (s, 2H), 5.18 (s, 2H), 4.88–4.77 (m, 2H), 3.47 (s, 3H), 3.46 (s, 3H), 2.82–2.72 (m, 2H), 2.66–2.53 (m, 2H), 2.51–2.38 (m, 3H), 2.34–2.23 (m, 5H), 2.12 (br, 2H), 1.32–1.30 (m, 6H), 1.28–1.14 (m, 2H), 0.87 (s, 3H), 0.74 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 191.6, 191.5, 163.59, 163.58, 163.50, 163.48, 145.4, 145.3, 128.72, 128.67, 122.1, 120.7, 115.9, 115.8, 111.0, 103.63, 103.58, 94.15, 94.14, 80.3, 79.8, 77.4, 56.5, 56.4, 42.6, 42.2, 41.0, 40.9, 40.8, 38.2, 38.0, 31.8, 31.7, 31.38, 31.36, 26.24. 26.20, 21.40, 21.35. HRMS calculated for C20H24O4 (M + H)+ 329.1747, found (M + H)+ 329.1736. HPLC purity: 99.6% (49.0% + 50.6%) (254 nm), tR = 7.66 and 7.68 min; 98.8% (220 nm), tR = 7.66 min. The split HPLC product peaks further supported the product is a mixture of two diastereomers (see Supporting Information for the details of HPLC trace).
2-(6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)-7-hydroxychroman-4-one (3j)
To a solution of 3i (38 mg, 0.116 mmol) in methanol (2 mL) was added concentrated HCl (45 μL, 0.52 mmol) carefully. The resulting mixture was stirred at room temperature for 24 h, after which the solvent was removed. The crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc (85/15) to obtain a mixture of two diastereomers 3j (30 mg, 0.106 mmol, 90%) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.79–7.76 (m, 2H), 7.67 (br, 2H), 6.54–6.51 (m, 2H), 6.43–6.42 (m, 2H), 5.65–5.64 (m, 2H), 4.87–4.77 (m, 2H), 2.84–2.75 (m, 2H), 2.67–2.54 (m, 2H), 2.49–2.22 (m, 8H), 2.11 (br, 2H), 2.00 (br, 2H), 1.31–1.29 (m, 6H), 1.26–1.12 (m, 2H), 0.85 (s, 3H), 0.73 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.39, 192.36, 164.0, 163.7, 145.3, 145.2, 129.4, 129.3, 122.2, 120.9, 114.89, 114.86, 110.62, 110.61, 103.5, 80.1, 79.6, 77.4, 77.1, 42.6, 42.2, 40.9, 40.83, 40.79, 40.7, 38.2, 38.0, 31.8, 31.7, 31.39, 31.36, 26.24, 26.20, 21.38, 21.36. HRMS calculated for C18H20O3 (M – H)− 283.1340, found (M – H)− 283.1310. HPLC purity: 99.4% (49.7% + 49.7%) (254 nm), tR = 7.09 and 7.11 min; 99.6% (50.6% + 49.0%) (220 nm), tR = 7.09 and 7.11 min. The split HPLC product peaks further supported the product is a mixture of two diastereomers (see Supporting Information for the details of HPLC trace).
General Procedure for the Synthesis of 4a–d
To a solution of appropriate 4-chromanone 3a–d (0.22 mmol) in methanol (3 mL) was added sodium borohydride (16.6 mg, 0.44 mmol) every 1.5 h for a total of 9 h. The resulting mixture was allowed to stir at room temperature overnight. The mixture was cooled to 0 °C, quenched with saturated NH4Cl aq carefully, and extracted with EtOAc twice. The combined organic layer was washed with water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to give the product.
2-Propylchroman-4,7-diol (4a)
Purified with hexane/EtOAc (85/15) to give the product (29 mg, 0.14 mmol, 64%) as light brown oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.19 (d, J = 8.4 Hz, 1H), 6.64 (br, 1H), 6.34 (dd, J = 8.4, 2.4 Hz, 1H), 6.24 (d, J = 2.4 Hz, 1H), 4.83–4.79 (m, 1H), 4.04–4.98 (m, 1H), 2.68 (br, 1H), 2.33 (br, 1H), 2.23–2.17 (m, 1H), 1.74–1.62 (m, 2H), 1.59–1.39 (m, 3H), 0.94 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 156.6, 155.8, 128.3, 118.3, 108.6, 103.1, 75.2, 65.5, 38.0, 37.6, 18.4, 14.1. HRMS calculated for C12H16O3 (M – H2O + H)+ 191.1067, found (M – H2O + H)+ 191.1058. HPLC purity: 95.3% (220 nm), tR = 5.91 min.
2-Hexylchroman-4,7-diol (4b)
Purified with hexane/EtOAc (85/15) to give the product (42 mg, 0.17 mmol, 77%) as light brown oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.18 (d, J = 8.4 Hz, 1H), 6.72 (br, 1H), 6.34 (dd, J = 8.4, 2.4 Hz, 1H), 6.25 (d, J = 2.4 Hz, 1H), 4.82–4.80 (m, 1H), 4.03–3.97 (m, 1H), 2.75–2.73 (m, 1H), 2.43 (br, 1H), 2.24–2.19 (m, 1H), 1.76–1.52 (m, 3H), 1.50–1.30 (m, 7H), 0.90–0.87 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 156.5, 155.8, 128.3, 118.3, 108.6, 103.1, 75.5, 65.6, 38.0, 35.5, 31.9, 29.3, 25.2, 22.7, 14.2. HRMS calculated for C15H22O3 (M – H2O + H)+ 233.1536, found (M – H2O + H)+ 233.1534. HPLC purity: 97.8% (220 nm), tR = 6.73 min.
2-Heptylchroman-4,7-diol (4c)
Purified with hexane/EtOAc (85/15) to give the product (39 mg, 0.15 mmol, 68%) as light brown oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.25 (d, J = 8.4 Hz, 1H), 6.39 (dd, J = 8.4, 2.5 Hz, 1H), 6.26 (d, J = 2.4 Hz, 1H), 5.82 (br, 1H), 4.88–4.82 (m, 1H), 4.07–4.01 (m, 1H), 2.29–2.23 (m, 1H), 2.11–2.09 (m, 1H), 1.92 (br, 1H), 1.78–1.38 (m, 5H), 1.33–1.28 (m, 9H), 0.90–0.87 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 156.6, 155.9, 128.3, 118.5, 108.5, 103.1, 75.5, 65.6, 38.2, 35.6, 32.0, 29.7, 29.4, 25.2, 22.8, 14.2. HRMS calculated for C16H24O3 (M – H)− 263.1653, found (M – H)− 263.1632. HPLC purity: 95.7% (220 nm), tR = 6.97 min.
2-Nonylchroman-4,7-diol (4d)
Purified with hexane/EtOAc (85/15) to give the product (52 mg, 0.18 mmol, 81%) as light brown oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.19 (d, J = 8.4 Hz, 1H), 6.54 (br, 1H), 6.34 (dd, J = 8.4, 2.4 Hz, 1H), 6.25 (d, J = 2.4 Hz, 1H), 4.83–4.81 (m, 1H), 4.02–3.99 (m, 1H), 2.61–2.59 (m, 1H), 2.32 (br, 1H), 2.25–2.20 (m, 1H), 1.75–1.53 (m, 2H), 1.49–1.27 (m, 14H), 0.90–0.87 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 156.5, 155.8, 128.3, 118.3, 108.6, 103.1, 75.5, 65.6, 38.1, 35.5, 32.0, 29.72, 29.70, 29.5, 25.2, 22.8, 14.2. HRMS calculated for C18H28O3 (M – H)− 291.1966, found (M – H)− 291.1926. HPLC purity: 97.7% (220 nm), tR = 7.41 min.
General Procedure for the Synthesis of 5a–l
To a solution of 1h (256 mg, 1 mmol) in ethanol (2.5 mL) were added DEA (154 mg, 2.1 mmol) and corresponding aldehyde (2 mmol) successively. The resulting mixture was stirred at 150 °C in a pressure tube for 1 h. Afterward, the solution was diluted with ethyl acetate and the organic layer was washed with 10% HCl, water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to give the MOM-protected product.
To a solution of appropriate MOM-protected compound (0.22 mmol) in methanol (4 mL) was added concentrated HCl (172 μL, 1.98 mmol) at room temperature, and the resulting mixture was stirred overnight. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to give the corresponding deprotected product.
5,7-Bis(methoxymethoxy)-2-propylchroman-4-one (5a)
Purified with hexane/EtOAc (87/13) to give the product (195 mg, 0.63 mmol, 63%) as clear oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.37 (s, 1H), 6.29 (s, 1H), 5.24 (s, 2H), 5.16 (s, 2H), 4.39–4.35 (m, 1H), 3.51 (s, 3H), 3.46 (s, 3H), 2.64–2.53 (m, 2H), 1.83–1.79 (m, 1H), 1.66–1.43 (m, 3H), 0.96 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 190.1, 164.8, 163.2, 159.5, 107.5, 97.8, 97.2, 95.1, 94.1, 77.3, 56.64, 56.57, 44.3, 37.0, 18.3, 14.0. HRMS calculated for C16H22O6 (M + H)+ 311.1489, found (M + H)+ 311.1481. HPLC purity: 98.0% (254 nm), tR = 6.78 min; 96.3% (220 nm), tR = 6.78 min.
5,7-Dihydroxy-2-propylchroman-4-one (5b)
Purified with hexane/EtOAc (85/15) to give the product (46 mg, 0.207 mmol, 94%) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 12.03 (s, 1H), 7.24 (br, 1H), 5.97 (s, 1H), 5.93 (s, 1H), 4.41–4.36 (m, 1H), 2.73–2.57 (m, 2H), 1.86–1.79 (m, 1H), 1.69–1.42 (m, 3H), 0.97 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 197.0, 165.3, 164.2, 163.7, 103.2, 96.5, 95.5, 41.6, 36.9, 18.2, 14.0. HRMS calculated for C12H14O4 (M – H)− 221.0819, found (M – H)− 221.0802. HPLC purity: 96.7% (254 nm), tR = 6.54 min; 100% (220 nm), tR = 6.54 min.
2-Hexyl-5,7-bis(methoxymethoxy)chroman-4-one (5c)
Purified with hexane/EtOAc (90/10) to give the product (220 mg, 0.63 mmol, 63%) as clear oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.37 (s, 1H), 6.30 (s, 1H), 5.24 (s, 2H), 5.16 (s, 2H), 4.39–4.32 (m, 1H), 3.51 (s, 3H), 3.47 (s, 3H), 2.65–2.53 (m, 2H), 1.84–1.77 (m, 1H), 1.67–1.60 (m, 1H), 1.55–1.30 (m, 8H), 0.90–0.87 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 190.0, 164.8, 163.2, 159.5, 107.5, 97.8, 97.3, 95.1, 94.1, 77.6, 56.6, 56.5, 44.3, 34.9, 31.8, 29.2, 24.9, 22.7, 14.2. HRMS calculated for C19H28O6 (M + H)+ 353.1959, found (M + H)+ 353.1954. HPLC purity: 99.5% (254 nm), tR = 7.49 min; 99.3% (220 nm), tR = 7.49 min.
2-Hexyl-5,7-dihydroxychroman-4-one (5d)
Purified with hexane/EtOAc (85/15) to give the product (44 mg, 0.165 mmol, 75%) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 12.02 (s, 1H), 6.99 (br, 1H), 5.96 (d, J = 1.9 Hz, 1H), 5.93 (d, J = 1.9 Hz, 1H), 4.40–4.33 (m, 1H), 2.72–2.57 (m, 2H), 1.86–1.78 (m, 1H), 1.69–1.62 (m, 1H), 1.54–1.30 (m, 8H), 0.90–0.87 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 197.0, 165.3, 164.2, 163.7, 103.2, 96.5, 95.5, 77.7, 41.6, 34.9, 31.8, 29.1, 24.9, 22.7, 14.2. HRMS calculated for C15H20O4 (M – H)− 263.1289, found (M – H)− 263.1264. HPLC purity: 97.9% (254 nm), tR = 7.26 min; 99.1% (220 nm), tR = 7.26 min.
2-Heptyl-5,7-bis(methoxymethoxy)chroman-4-one (5e)
Purified with hexane/EtOAc (90/10) to give the product (220 mg, 0.60 mmol, 60%) as clear oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.37 (s, 1H), 6.29 (s, 1H), 5.24 (s, 2H), 5.16 (s, 2H), 4.38–4.31 (m, 1H), 3.51 (s, 3H), 3.46 (s, 3H), 2.64–2.52 (m, 2H), 1.85–1.76 (m, 1H), 1.68–1.59 (m, 1H), 1.54–1.27 (m, 10H), 0.89–0.86 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 190.0, 164.8, 163.2, 159.6, 107.5, 97.8, 97.3, 95.1, 94.2, 77.6, 56.64, 56.57, 44.3, 34.9, 31.9, 29.5, 29.3, 25.0, 22.8, 14.2. HRMS calculated for C20H30O6 (M + H)+ 367.2115, found (M + H)+ 367.2114. HPLC purity: 99.6% (254 nm), tR = 7.69 min; 98.8% (220 nm), tR = 7.69 min.
2-Heptyl-5,7-dihydroxychroman-4-one (5f)
Purified with hexane/EtOAc (90/10) to give the product (49 mg, 0.176 mmol, 80%) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 12.03 (s, 1H), 6.57 (br, 1H), 5.96 (s, 1H), 5.93 (s, 1H), 4.39–4.34 (m, 1H), 2.72–2.57 (m, 2H), 1.86–1.79 (m, 1H), 1.69–1.62 (m, 1H), 1.54–1.26 (m, 10H), 0.90–0.87 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 196.9, 165.0, 164.3, 163.6, 103.3, 96.5, 95.4, 77.8, 41.7, 34.9, 31.9, 29.4, 29.3, 25.0, 22.8, 14.2. HRMS calculated for C16H22O4 (M – H)− 277.1445, found (M – H)− 277.1420. HPLC purity: 98.6% (254 nm), tR = 7.47 min; 98.8% (220 nm), tR = 7.47 min.
5,7-Bis(methoxymethoxy)-2-nonylchroman-4-one (5g)
Purified with hexane/EtOAc (90/10) to give the product (218 mg, 0.55 mmol, 55%) as clear oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.37 (s, 1H), 6.29 (s, 1H), 5.24 (s, 2H), 5.16 (s, 2H), 4.37–4.31 (m, 1H), 3.51 (s, 3H), 3.47 (s, 3H), 2.64–2.53 (m, 2H), 1.81–1.76 (m, 1H), 1.67–1.59 (m, 1H), 1.54–1.26 (m, 14H), 0.89–0.85 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 190.1, 164.8, 163.2, 159.6, 107.5, 97.8, 97.3, 95.1, 94.2, 77.6, 56.64, 56.57, 44.3, 34.9, 32.0, 29.63, 29.63, 29.5, 29.4, 25.0, 22.8, 14.2. HRMS calculated for C22H34O6 (M + H)+ 395.2428, found (M + H)+ 395.2407. HPLC purity: 99.9% (254 nm), tR = 8.09 min; 99.5% (220 nm), tR = 8.09 min.
5,7-Dihydroxy-2-nonylchroman-4-one (5h)
Purified with hexane/EtOAc (90/10) to give the product (55 mg, 0.18 mmol, 82%) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 12.05 (s, 1H), 5.95 (s, 1H), 5.92 (s, 1H), 5.44 (br, 1H), 4.40–4.35 (m, 1H), 2.72–2.58 (m, 2H), 1.86–1.80 (m, 1H), 1.69–1.61 (m, 1H), 1.54–1.26 (m, 14H), 0.90–0.88 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 197.1, 165.4, 164.2, 163.7, 103.2, 96.5, 95.5, 77.7, 41.6, 34.8, 32.0, 29.62, 29.58, 29.5, 29.4, 24.9, 22.8, 14.2. HRMS calculated for C18H26O4 (M – H)− 305.1758, found (M – H)− 305.1719. HPLC purity: 99.7% (254 nm), tR = 7.87 min; 99.2% (220 nm), tR = 7.87 min.
2-(2,6-Dimethylhept-5-en-1-yl)-5,7-bis(methoxymethoxy)chroman-4-one (5i)
Purified with hexane/EtOAc (90/10) to obtain a mixture of two diastereomers (275 mg, 0.70 mmol, 70%) as clear oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.35 (s, 2H), 6.27 (s, 2H), 5.22 (s, 4H), 5.14 (s, 4H), 5.07–5.05 (m, 2H), 4.46–4.41 (m, 2H), 3.49 (s, 6H), 3.44 (s, 6H), 2.61–2.45 (m, 4H), 1.99–1.68 (m, 8H), 1.65 (s, 6H), 1.57 (s, 6H), 1.41–1.14 (m, 6H), 0.93–0.91 (m, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 189.9, 189.8, 164.72, 164.67, 163.1, 159.49, 159.48, 131.5, 131.4, 124.54, 124.48, 107.5, 97.8, 97.22, 97.21, 95.0, 94.1, 76.1, 75.5, 56.53, 56.46, 44.9, 44.5, 42.11, 42.06, 37.4, 36.8, 28.7, 28.4, 25.8, 25.4, 25.3, 19.9, 19.4, 17.7. HRMS calculated for C22H32O6 (M + H)+ 393.2272, found (M + H)+ 393.2263. HPLC purity: 98.8% (254 nm), tR = 7.71 min; 98.4% (220 nm), tR = 7.71 min.
2-(2,6-Dimethylhept-5-en-1-yl)-5,7-dihydroxychroman-4-one (5j)
Purified with hexane/EtOAc (90/10) to obtain a mixture of two diastereomers (54 mg, 0.176 mmol, 80%) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 12.02 (s, 2H), 7.25 (br, 2H), 5.97 (s, 2H), 5.94 (s, 2H), 5.09–5.08 (m, 2H), 4.51–4.46 (m, 2H), 2.73–2.56 (m, 4H), 2.04–1.70 (m, 8H), 1.68 (s, 6H), 1.60 (s, 6H), 1.53–1.32 (m, 4H), 1.25–1.18 (m, 2H), 0.98–0.94 (m, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 197.1, 197.0, 165.4, 164.2, 163.67, 163.65, 131.72, 131.67, 124.54, 124.48, 103.2, 96.6, 95.62, 95.60, 76.3, 75.7, 42.3, 42.14, 42.08, 41.9, 37.5, 36.8, 28.9, 28.5, 25.8, 25.5, 25.4, 20.0, 19.4, 17.8. HRMS calculated for C18H24O4 (M – H)− 303.1602, found (M – H)− 303.1564. HPLC purity: 98.4% (254 nm), tR = 7.50 min; 98.4% (220 nm), tR = 7.50 min.
2-Cyclopentyl-5,7-dihydroxychroman-4-one (5k)
Purified with hexane/EtOAc (85/15) to give the product (174 mg, 0.70 mmol, 70% over two steps) as white solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 12.12 (s, 1H), 10.73 (s, 1H), 5.83 (s, 2H), 4.26–4.20 (m, 1H), 2.80–2.73 (m, 1H), 2.61–2.56 (m, 1H), 2.18–2.12 (m, 1H), 1.84–1.78 (m, 1H), 1.72–1.42 (m, 6H), 1.32–1.27 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 196.4, 166.5, 163.4, 163.0, 101.8, 95.6, 94.8, 80.7, 43.3, 40.1, 28.3, 27.7, 25.0, 24.9. HRMS calculated for C14H16O4 (M – H)− 247.0976, found (M – H)− 247.0954. HPLC purity: 99.6% (254 nm), tR = 6.84 min; 99.6% (220 nm), tR = 6.84 min.
2-Cyclohexyl-5,7-dihydroxychroman-4-one (5l)
Purified with hexane/EtOAc (85/15) to give the product (168 mg, 0.64 mmol, 64% over two steps) as white solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 12.12 (s, 1H), 10.74 (s, 1H), 5.84 (s, 2H), 4.24–4.18 (m, 1H), 2.85–2.77 (m, 1H), 2.55–2.51 (m, 1H), 1.91–1.88 (m, 1H), 1.75–1.63 (m, 5H), 1.28–1.01 (m, 5H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 196.7, 166.5, 163.4, 163.0, 101.8, 95.6, 94.7, 80.9, 40.9, 38.3, 27.58, 27.56, 25.9, 25.43, 25.36. HRMS calculated for C15H18O4 (M – H)− 261.1132, found (M – H)− 261.1106. HPLC purity: 99.1% (254 nm), tR = 7.05 min; 99.5% (220 nm), tR = 7.05 min.
(E)-3-(6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)-1-(2,4,6-trihydroxyphenyl)prop-2-en-1-one (5m)
To a solution of 1h (256 mg, 1 mmol) in ethanol (2.5 mL) were added DEA (154 mg, 2.1 mmol) and (−)-myrtenal (300 mg, 2 mmol) successively. The resulting mixture was stirred at 150 °C in a pressure tube for 4 h. Afterward, the solution was diluted with ethyl acetate and the organic layer was washed with 10% HCl, water, brine and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was dissolved in methanol (15 mL), followed by the addition of concentrated HCl (765 μL, 9 mmol) dropwise. The resulting mixture was stirred at room temperature for 16 h, after which the solvent was removed. The crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc (75/25) to obtain compound 5m (102 mg, 0.34 mmol, 34%) as yellow solid. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.55 (d, J = 15.4 Hz, 1H), 7.38 (d, J = 15.4 Hz, 1H), 6.11 (s, 1H), 5.81 (s, 2H), 2.65–2.64 (m, 1H), 2.57–2.47 (m, 3H), 2.18–2.17 (m, 1H), 1.38 (s, 3H), 1.18 (d, J = 9.0 Hz, 1H), 0.82 (s, 3H). 13C NMR (100 MHz, CD3OD): δ (ppm) 194.6, 166.2, 165.9, 148.3, 144.2, 135.5, 125.5, 105.9, 96.0, 42.9, 42.0, 38.7, 33.7, 32.0, 26.6, 21.2. HRMS calculated for C18H20O4 (M – H)− 299.1289, found (M – H)− 299.1240. HPLC purity: 99.6% (254 nm), tR = 7.29 min; 99.9% (220 nm), tR = 7.29 min.
2-(6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)-5,7-dihydroxychroman-4-one (5n)
To a suspension of 5m (57 mg, 0.19 mmol) in ethanol (2.5 mL) was added catalytic amount of concentrated HCl. The resulting mixture was stirred at 150 °C under microwave irradiation for 30 min. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc (75/25) to obtain a mixture of two diastereomers 5n (41 mg, 0.136 mmol, 72%) as pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 12.05 (br, 2H), 5.98–5.94 (m, 4H), 5.68–5.67 (m, 2H), 4.86–4.75 (m, 2H), 2.89–2.80 (m, 2H), 2.69–2.55 (m, 2H), 2.53–2.45 (m, 2H), 2.43–2.26 (m, 6H), 2.16–2.15 (m, 2H), 1.34–1.33 (m, 6H), 1.29–1.16 (m, 2H), 0.88 (s, 3H), 0.78 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 196.82, 196.80, 165.2, 164.2, 163.5, 145.0, 144.8, 122.4, 121.2, 103.21, 103.18, 96.55, 96.54, 95.7, 79.5, 79.0, 42.5, 42.2, 40.9, 40.8, 39.8, 39.7, 38.2, 38.0, 31.7, 31.6, 31.4, 31.3, 26.20, 26.16, 21.4, 21.3. HRMS calculated for C18H20O4 (M – H)− 299.1289, found (M – H)− 299.1247. HPLC purity: 99.7% (254 nm), tR = 7.53 min; 99.9% (220 nm), tR = 7.52 min.
General Procedure for the Synthesis of 6a–f
To a suspension of corresponding hydroxyamine (0.8 mmol) in ethanol (1 mL) was added pyridine (64 mg, 0.8 mmol). After stirring at room temperature for 15 min, the solution of appropriate 4-chromanone (0.1 mmol) in ethanol (2 mL) was added. The resulting mixture was stirred for 26–72 h at room temperature, after which the solvent was removed. EtOAc was added to the residue, and the organic layer was washed with water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to give oxime compound.
5,7-Dihydroxy-2-nonylchroman-4-one oxime (6a)
Purified with hexane/EtOAc (85/15) to give the product (29 mg, 0.09 mmol, 90%) as white solid. 1H NMR (400 MHz, CD3OD): δ (ppm) 5.89 (d, J = 2.3 Hz, 1H), 5.85 (d, J = 2.3 Hz, 1H), 3.99–3.93 (m, 1H), 3.24 (dd, J = 17.1, 3.5 Hz, 1H), 2.32 (dd, J = 17.1, 11.4 Hz, 1H), 1.79–1.70 (m, 1H), 1.67–1.59 (m, 1H), 1.57–1.30 (m, 14H), 0.92–0.88 (m, 3H). 13C NMR (100 MHz, CD3OD): δ (ppm) 161.5, 160.7, 159.5, 154.7, 98.8, 97.3, 96.2, 76.2, 36.1, 33.1, 30.69, 30.68, 30.62, 30.5, 29.0, 26.2, 23.7, 14.5. HRMS calculated for C18H27NO4 (M – H)− 320.1867, found (M – H)− 320.1818. HPLC purity: 99.9% (254 nm), tR = 7.62 min; 99.5% (220 nm), tR = 7.62 min.
5,7-Dihydroxy-2-nonylchroman-4-one O-Methyloxime (6b)
Purified with hexane/EtOAc (90/10) to give the product (27 mg, 0.081 mmol, 81%) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 10.96 (s, 1H), 6.04 (d, J = 2.3 Hz, 1H), 5.96 (d, J = 2.3 Hz, 1H), 5.60 (br, 1H), 4.03–3.98 (m, 1H), 3.94 (s, 3H), 3.18 (dd, J = 17.2, 3.0 Hz, 1H), 2.36 (dd, J = 17.2, 11.6 Hz, 1H), 1.82–1.73 (m, 1H), 1.66–1.57 (m, 1H), 1.54–1.27 (m, 14H), 0.90–0.86 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 159.7, 159.1, 158.7, 154.1, 98.3, 97.0, 95.8, 75.1, 62.5, 35.0, 32.0, 29.67, 29.66, 29.59, 29.5, 28.6, 25.2, 22.8, 14.3. HRMS calculated for C19H29NO4 (M – H)− 334.2024, found (M – H)− 334.1977. HPLC purity: 99.8% (254 nm), tR = 8.13 min; 99.1% (220 nm), tR = 8.13 min.
5,7-Dihydroxy-2-nonylchroman-4-one O-Benzyloxime (6c)
Purified with hexane/EtOAc (92/8) to give the product (34 mg, 0.083 mmol, 83%) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 10.89 (s, 1H), 7.39–7.31 (m, 5H), 6.01 (d, J = 2.4 Hz, 1H), 5.94 (d, J = 2.3 Hz, 1H), 5.45 (br, 1H), 5.12 (s, 2H), 4.02–3.96 (m, 1H), 3.22 (dd, J = 17.2, 3.0 Hz, 1H), 2.40 (dd, J = 17.2, 11.6 Hz, 1H), 1.80–1.72 (m, 1H), 1.65–1.57 (m, 1H), 1.54–1.27 (m, 14H), 0.91–0.87 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 159.7, 159.1, 158.7, 154.5, 137.0, 128.72, 128.68, 128.5, 98.4, 96.9, 95.8, 76.7, 75.0, 35.0, 32.0, 29.67, 29.65, 29.58, 29.4, 28.9, 25.1, 22.8, 14.3. HRMS calculated for C25H33NO4 (M + H)+ 412.2482, found (M + H)+ 412.2472. HPLC purity: 99.1% (254 nm), tR = 8.36 min; 98.7% (220 nm), tR = 8.37 min.
5,7-Dihydroxy-2-propylchroman-4-one Oxime (6d)
Purified with hexane/EtOAc (83/17) to give the product (20 mg, 0.084 mmol, 84%) as white solid. 1H NMR (400 MHz, CD3OD): δ (ppm) 5.90 (s, 1H), 5.85 (s, 1H), 3.98 (br, 1H), 3.24 (d, J = 17.0, 1H), 2.33 (dd, J = 17.0, 11.4 Hz, 1H), 1.75–1.72 (m, 1H), 1.63–1.47 (m, 3H), 1.0–0.96 (m, 3H). 13C NMR (100 MHz, CD3OD): δ (ppm) 161.5, 160.7, 159.5, 154.8, 98.8, 97.3, 96.2, 76.0, 38.2, 28.0, 19.4, 14.3. HRMS calculated for C12H15NO4 (M – H)− 236.0928, found (M – H)− 236.0908. HPLC purity: 99.9% (254 nm), tR = 6.35 min; 100% (220 nm), tR = 6.34 min.
2-Hexyl-5,7-dihydroxychroman-4-one Oxime (6e)
Purified with hexane/EtOAc (86/14) to give the product (22 mg, 0.08 mmol, 80%) as white solid. 1H NMR (400 MHz, CD3OD): δ (ppm) 5.90 (d, J = 2.3 Hz, 1H), 5.85 (d, J = 2.3 Hz, 1H), 3.97–3.91 (m, 1H), 3.24 (dd, J = 17.1, 3.1 Hz, 1H), 2.32 (dd, J = 17.1, 11.4 Hz, 1H), 1.78–1.69 (m, 1H), 1.65–1.49 (m, 2H), 1.45–1.32 (m, 7H), 0.92–0.89 (m, 3H). 13C NMR (100 MHz, CD3OD): δ (ppm) 161.4, 160.6, 159.5, 154.7, 98.8, 97.3, 96.2, 76.2, 36.0, 32.9, 30.3, 29.0, 26.2, 23.6, 14.4. HRMS calculated for C15H21NO4 (M – H)− 278.1398, found (M – H)− 278.1354. HPLC purity: 99.8% (254 nm), tR = 7.02 min; 99.9% (220 nm), tR = 7.01 min.
2-Heptyl-5,7-dihydroxychroman-4-one Oxime (6f)
Purified with hexane/EtOAc (90/10) to give the product (25 mg, 0.085 mmol, 85%) as white solid. 1H NMR (400 MHz, CD3OD): δ (ppm) 5.90 (s, 1H), 5.85 (s, 1H), 3.95 (br, 1H), 3.24 (d, J = 17.0 Hz, 1H), 2.32 (dd, J = 17.0, 11.4 Hz, 1H), 1.75–1.73 (m, 1H), 1.63–1.33 (m, 11H), 0.90 (br, 3H). 13C NMR (100 MHz, CD3OD): δ (ppm) 161.5, 160.7, 159.5, 154.7, 98.8, 97.3, 96.2, 76.2, 36.0, 33.0, 30.6, 30.3, 29.0, 26.2, 23.7, 14.4. HRMS calculated for C16H23NO4 (M – H)− 292.1554, found (M – H)− 292.1512. HPLC purity: 99.9% (254 nm), tR = 7.22 min; 99.8% (220 nm), tR = 7.22 min.
General Procedure for the Synthesis of 7a–c
To a solution of 1e (196 mg, 1 mmol) in ethanol (2.5 mL) were added pyrrolidine (149 mg, 2.1 mmol) and corresponding cycloketone (2 mmol) successively. The resulting mixture was stirred at 150 °C in a pressure tube for 2–16 h. The resulting solution was cooled to room temperature, and concentrated HCl (1.02 mL, 12 mmol) was added. The resulting mixture was then stirred at room temperature overnight. Afterward, the solution was diluted with EtOAc, and the organic layer was washed with water, brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to give the product.
7-Hydroxyspiro[chroman-2,1′-cyclopentan]-4-one (7a)
Purified with hexane/EtOAc (90/10) to give the product (170 mg, 0.78 mmol, 78% over two steps) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.78 (d, J = 8.6 Hz, 1H), 7.12 (br, 1H), 6.49 (dd, J = 8.6, 2.3 Hz, 1H), 6.36 (d, J = 2.3 Hz, 1H), 2.78 (s, 2H), 2.09–2.04 (m, 2H), 1.87–1.83 (m, 2H), 1.72–1.59 (m, 4H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.5, 163.7, 162.8, 129.2, 114.9, 110.1, 104.0, 90.5, 46.8, 37.7, 24.0. HRMS calculated for C13H14O3 (M – H)− 217.0870, found (M – H)− 217.0854. HPLC purity: 99.1% (254 nm), tR = 6.26 min; 98.7% (220 nm), tR = 6.26 min.
7-Hydroxyspiro[chroman-2,1′-cyclohexan]-4-one (7b)
Purified with hexane/EtOAc (90/10) to give the product (207 mg, 0.89 mmol, 89% over two steps) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.77 (d, J = 8.6 Hz, 1H), 7.45 (br, 1H), 6.50 (dd, J = 8.6, 2.2 Hz, 1H), 6.42 (d, J = 2.2 Hz, 1H), 2.66 (s, 2H), 1.99–1.96 (m, 2H), 1.72–1.59 (m, 3H), 1.50–1.44 (m, 4H), 1.34–1.25 (m, 1H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.6, 164.0, 162.2, 129.0, 114.6, 110.1, 103.9, 80.5, 47.9, 35.0, 25.3, 21.6. HRMS calculated for C14H16O3 (M – H)− 231.1027, found (M – H)− 231.1007. HPLC purity: 98.2% (254 nm), tR = 6.47 min; 98.8% (220 nm), tR = 6.47 min.
7-Hydroxyspiro[chroman-2,1′-cycloheptan]-4-one (7c)
Purified by reverse phase C18 silica gel chromatography with H2O/MeCN (65/35) to give the product (130 mg, 0.53 mmol, 53% over two steps) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.26 (br, 1H), 7.75 (d, J = 8.6 Hz, 1H), 6.51 (dd, J = 8.6, 2.2 Hz, 1H), 6.40 (d, J = 2.2 Hz, 1H), 2.70 (s, 2H), 2.09–2.03 (m, 2H), 1.77–1.61 (m, 6H), 1.57–1.50 (m, 2H), 1.43–1.36 (m, 2H). 13C NMR (100 MHz, CDCl3): δ (ppm) 193.1, 164.6, 162.5, 128.9, 114.3, 110.2, 103.9, 84.7, 48.5, 38.4, 29.4, 22.1. HRMS calculated for C15H18O3 (M – H)− 245.1183, found (M – H)− 245.1154. HPLC purity: 97.4% (254 nm), tR = 6.67 min; 97.0% (220 nm), tR = 6.67 min.
General Procedure for the Synthesis of 8a–h
To a solution of corresponding acetophenone (1 mmol) and appropriate benzaldehyde (1.5 mmol) in methanol (10 mL) was added 60% KOH aqueous solution (1.5 mL) dropwise at room temperature. The resulting solution was stirred for 60 h at room temperature, after which the reaction mixture was neutralized with 10% HCl aq and extracted with ethyl acetate twice. The organic layer was washed with water, brine and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was dissolved in methanol (14 mL), followed by the careful addition of concentrated HCl (340 μL, 4 mmol). The obtained mixture was stirred at room temperature until the MOM-protected chalcone was gone, and then the mixture was concentrated. The crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to afford the chalcones.
(E)-3-(2-(Allyloxy)phenyl)-1-(2,4-dihydroxyphenyl)prop-2-en-1-one (8a)
Purified with hexane/EtOAc (90/10) to give the product (175 mg, 0.59 mmol, 59% over two steps) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 13.49 (s, 1H), 8.18 (d, J = 15.6 Hz, 1H), 7.81 (d, J = 9.4 Hz, 1H), 7.74 (d, J = 15.6 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.36 (t, J = 8.4 Hz, 1H), 7.0 (t, J = 7.5 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.43–6.41 (m, 2H), 6.22 (s, 1H), 6.16–6.09 (m, 1H), 5.47 (d, J = 17.2 Hz, 1H), 5.35 (d, J = 10.6 Hz, 1H), 4.65 (d, J = 5.3 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.8, 166.5, 162.9, 158.1, 140.5, 133.0, 132.2, 132.0, 130.1, 124.2, 121.4, 121.2, 118.4, 114.8, 112.8, 107.9, 103.9, 69.5. HRMS calculated for C18H16O4 (M – H)− 295.0976, found (M – H)− 295.0955. HPLC purity: 100% (254 nm), tR = 7.20 min; 99.9% (220 nm), tR = 7.20 min.
(E)-1-(2,4-Dihydroxyphenyl)-3-(2-(hexyloxy)phenyl)prop-2-en-1-one (8b)
Purified by reverse phase C18 silica gel chromatography with H2O/MeCN (43/57) to give the product (170 mg, 0.50 mmol, 50% over two steps) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 13.61 (s, 1H), 8.11 (d, J = 15.6 Hz, 1H), 7.81–7.77 (m, 2H), 7.57 (dd, J = 7.6, 1.2 Hz, 1H), 7.35 (t, J = 7.9 Hz, 1H), 6.99–6.92 (m, 3H), 6.45–6.43 (m, 2H), 4.08–4.04 (m, 2H), 1.93–1.86 (m, 2H), 1.56–1.49 (m, 2H), 1.39–1.32 (m, 4H), 0.92–0.88 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 193.0, 166.4, 163.2, 158.8, 141.1, 132.14, 132.06, 130.7, 123.8, 121.3, 120.7, 114.6, 112.3, 108.1, 103.8, 68.7, 31.7, 29.4, 26.1, 22.7, 14.1. HRMS calculated for C21H24O4 (M – H)− 339.1602, found (M – H)− 339.1550. HPLC purity: 99.6% (254 nm), tR = 8.00 min; 98.9% (220 nm), tR = 8.01 min.
(E)-1-(2,4-Dihydroxyphenyl)-3-(2-(octyloxy)phenyl)prop-2-en-1-one (8c)
Purified by reverse phase C18 silica gel chromatography with H2O/MeCN (45/55) to give the product (195 mg, 0.53 mmol, 53% over two steps) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 13.64 (s, 1H), 8.12 (d, J = 15.6 Hz, 1H), 7.80 (s, 1H), 7.77 (d, J = 6.2 Hz, 1H), 7.57 (d, J = 7.5 Hz, 1H), 7.35 (t, J = 8.2 Hz, 1H), 7.14 (br, 1H), 6.97 (t, J = 7.5 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.46–6.44 (m, 2H), 4.06–4.03 (m, 2H), 1.92–1.85 (m, 2H), 1.55–1.48 (m, 2H), 1.39–1.27 (m, 8H), 0.88–0.85 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 193.0, 166.3, 163.3, 158.8, 141.1, 132.2, 132.1, 130.6, 123.8, 121.2, 120.7, 114.6, 112.3, 108.2, 103.8, 68.7, 31.9, 29.5, 29.4, 29.3, 26.4, 22.8, 14.2. HRMS calculated for C23H28O4 (M + H)+ 369.2060, found (M + H)+ 369.2057. HPLC purity: 99.5% (254 nm), tR = 8.36 min; 99.4% (220 nm), tR = 8.36 min.
(E)-1-(2,4-Dihydroxyphenyl)-3-(4-(piperidin-1-yl)phenyl)prop-2-en-1-one (8d)
Purification was performed without chromatography because of the poor solubility of product. The crude solid was washed with small amount of methanol three times to give the product (210 mg, 0.65 mmol, 65% over two steps) as yellow solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.19 (d, J = 9.0 Hz, 1H), 7.98–7.92 (m, 3H), 7.77 (d, J = 15.4 Hz, 1H), 7.67 (br, 2H), 6.45 (dd, J = 8.9, 2.2 Hz, 1H), 6.33 (d, J = 2.2 Hz, 1H), 3.49–3.46 (m, 4H), 1.88 (br, 4H), 1.64 (br, 2H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 191.7, 166.3, 165.8, 133.5, 131.0, 113.5, 108.8, 103.1, 24.0. HRMS calculated for C20H21NO3 (M – H)− 322.1449, found (M – H)− 322.1407. HPLC purity: 99.0% (254 nm), tR = 5.99 min; 98.8% (220 nm), tR = 5.98 min.
(E)-3-(2-(Allyloxy)phenyl)-1-(2,4,6-trihydroxyphenyl)prop-2-en-1-one (8e)
Purified with hexane/EtOAc (70/30) to give the product (106 mg, 0.34 mmol, 34% over two steps) as yellow solid. 1H NMR (400 MHz, CD3OD): δ (ppm) 8.25 (d, J = 15.8 Hz, 1H), 8.10 (d, J = 15.8 Hz, 1H), 7.61 (dd, J = 7.7, 1.3 Hz, 1H), 7.34–7.30 (m, 1H), 7.0–6.94 (m, 2H), 6.16–6.09 (m, 1H), 5.86 (s, 2H), 5.45 (dd, J = 17.3, 1.6 Hz, 1H), 5.29 (dd, J = 10.6, 1.4 Hz, 1H), 4.63 (d, J = 5.2 Hz, 2H). 13C NMR (100 MHz, CD3OD): δ (ppm) 194.5, 166.4, 166.1, 158.9, 138.2, 134.6, 132.4, 129.5, 129.1, 126.0, 122.0, 117.8, 113.9, 106.0, 96.0, 70.3. HRMS calculated for C18H16O5 (M – H)− 311.0925, found (M – H)− 311.0877. HPLC purity: 97.5% (254 nm), tR = 6.93 min; 97.2% (220 nm), tR = 6.93 min.
(E)-3-(2-(Allyloxy)phenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one (8f)
Purified with hexane/EtOAc (80/20) to give the product (205 mg, 0.73 mmol, 73% over two steps) as pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.42 (br, 1H), 8.16 (d, J = 15.8 Hz, 1H), 8.01–7.99 (m, 2H), 7.70 (d, J = 15.8 Hz, 1H), 7.61 (dd, J = 7.7, 1.4 Hz, 1H), 7.36–7.31 (m, 1H), 7.03–6.96 (m, 3H), 6.91 (d, J = 8.3 Hz, 1H), 6.11–6.02 (m, 1H), 5.43 (dd, J = 17.2, 1.4 Hz, 1H), 5.29 (dd, J = 10.6, 1.3 Hz, 1H), 4.61–4.59 (m, 2H). 13C NMR (100 MHz, CDCl3): δ (ppm) 190.8, 161.6, 158.0, 140.6, 132.9, 131.8, 131.5, 130.5, 129.6, 124.3, 122.8, 121.1, 118.1, 115.9, 112.7, 69.3. HRMS calculated for C18H16O3 (M – H)− 279.1027, found (M – H)− 279.0988. HPLC purity: 99.6% (254 nm), tR = 6.99 min; 99.8% (220 nm), tR = 6.99 min.
(E)-3-(3-(Allyloxy)phenyl)-1-(2,4-dihydroxyphenyl)prop-2-en-1-one (8g)
Purified with hexane/EtOAc (90/10) to give the product (113 mg, 0.38 mmol, 38% over two steps) as yellow solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 13.40 (s, 1H), 10.77 (br, 1H), 8.23 (d, J = 9.0 Hz, 1H), 7.98 (d, J = 15.4 Hz, 1H), 7.76 (d, J = 15.4 Hz, 1H), 7.53 (s, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.37 (t, J = 7.7 Hz, 1H), 7.05 (dd, J = 8.0, 2.0 Hz, 1H), 6.43 (dd, J = 8.9, 2.3 Hz, 1H), 6.30 (d, J = 2.2 Hz, 1H), 6.11–6.03 (m, 1H), 5.44 (dd, J = 17.3, 1.4 Hz, 1H), 5.29 (d, J = 10.5 Hz, 1H), 4.65 (d, J = 5.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 191.5, 165.9, 165.3, 158.6, 143.7, 136.0, 133.6, 133.3, 130.0, 122.2, 121.5, 117.7, 117.4, 114.1, 113.0, 108.3, 102.6, 68.4. HRMS calculated for C18H16O4 (M + H)+ 297.1121, found (M + H)+ 297.1107. HPLC purity: 99.7% (254 nm), tR = 7.22 min; 99.6% (220 nm), tR = 7.22 min.
(E)-3-(4-(Allyloxy)phenyl)-1-(2,4-dihydroxyphenyl)prop-2-en-1-one (8h)
Purified with hexane/EtOAc (90/10) to give the product (130 mg, 0.44 mmol, 44% over two steps) as yellow solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 13.54 (s, 1H), 10.70 (br, 1H), 8.19 (d, J = 9.0 Hz, 1H), 7.87–7.75 (m, 4H), 7.04 (d, J = 8.7 Hz, 2H), 6.41 (dd, J = 8.9, 2.3 Hz, 1H), 6.29 (d, J = 2.3 Hz, 1H), 6.10–6.01 (m, 1H), 5.42 (dd, J = 17.3, 1.4 Hz, 1H), 5.28 (d, J = 10.5 Hz, 1H), 4.65 (d, J = 5.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 191.5, 165.8, 165.1, 160.4, 143.7, 133.4, 133.0, 131.0, 127.4, 118.6, 117.8, 115.1, 113.0, 108.2, 102.6, 68.4. HRMS calculated for C18H16O4 (M + H)+ 297.1121, found (M + H)+ 297.1108. HPLC purity: 99.8% (254 nm), tR = 7.18 min; 99.6% (220 nm), tR = 7.18 min.
General Procedure for the Synthesis of 8i–m
The obtained appropriate chalcone (0.2 mmol) was dissolved in ethanol (2.5 mL) with a catalytic amount of concentrated HCl and then heated at 150 °C under microwave irradiation for 1.5 h. The solvent was removed under reduced pressure and the crude material was adsorbed onto silica gel and subjected to silica gel chromatography with hexane/EtOAc to afford the corresponding flavanones.
2-(2-(Allyloxy)phenyl)-7-hydroxychroman-4-one (8i)
Purified with hexane/EtOAc (90/10) to give the product (30 mg, 0.1 mmol, 50%) as pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.85 (d, J = 8.7 Hz, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.54 (br, 1H), 7.30 (t, J = 8.5 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 6.58 (dd, J = 8.7, 2.2 Hz, 1H), 6.51 (d, J = 2.1 Hz, 1H), 6.03–5.95 (m, 1H), 5.87 (dd, J = 11.3, 5.0 Hz, 1H), 5.34 (d, J = 17.2 Hz, 1H), 5.24 (d, J = 10.6 Hz, 1H), 4.61–4.51 (m, 2H), 2.97–2.86 (m, 2H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.6, 164.5, 163.9, 154.9, 133.0, 129.6, 129.5, 127.8, 126.7, 121.2, 117.8, 114.8, 112.0, 111.0, 103.6, 75.0, 69.0, 43.4. HRMS calculated for C18H16O4 (M – H)− 295.0976, found (M – H)− 295.0949. HPLC purity: 98.0% (254 nm), tR = 6.76 min; 99.0% (220 nm), tR = 6.76 min.
2-(2-(Hexyloxy)phenyl)-7-hydroxychroman-4-one (8j)
Purified by reverse phase C18 silica gel chromatography with H2O/MeCN (50/50) to give the product (35 mg, 0.1 mmol, 52%) as pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.86 (d, J = 8.7 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.31 (t, J = 8.5 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.59 (dd, J = 8.7, 2.2 Hz, 1H), 6.52 (d, J = 2.1 Hz, 1H), 5.86–5.82 (m, 1H), 4.03–3.93 (m, 2H), 2.96–2.87 (m, 2H), 1.79–1.72 (m, 2H), 1.45–1.37 (m, 2H), 1.31–1.27 (m, 4H), 0.87–0.84 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.8, 164.5, 164.1, 155.5, 129.52, 129.50, 127.5, 126.5, 120.7, 114.7, 111.5, 111.0, 103.6, 75.1, 68.3, 43.3, 31.6, 29.2, 25.9, 22.6, 14.1. HRMS calculated for C21H24O4 (M – H)− 339.1602, found (M – H)− 339.1563. HPLC purity: 99.9% (254 nm), tR = 7.65 min; 99.6% (220 nm), tR = 7.65 min.
7-Hydroxy-2-(2-(octyloxy)phenyl)chroman-4-one (8k)
Purified by reverse phase C18 silica gel chromatography with H2O/MeCN (42/58) to give the product (37 mg, 0.1 mmol, 50%) as pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.86 (d, J = 8.7 Hz, 1H), 7.58 (dd, J = 7.5, 1.0 Hz, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.60 (dd, J = 8.7, 2.2 Hz, 1H), 6.53 (d, J = 2.2 Hz, 1H), 5.86–5.82 (m, 1H), 4.03–3.93 (m, 2H), 2.97–2.87 (m, 2H), 1.79–1.72 (m, 2H), 1.44–1.37 (m, 2H), 1.32–1.24 (m, 8H), 0.87–0.83 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 192.9, 164.6, 164.2, 155.5, 129.53, 129.49, 127.5, 126.5, 120.7, 114.6, 111.5, 111.0, 103.6, 75.1, 68.3, 43.3, 31.9, 29.4, 29.3, 29.2, 26.2, 22.7, 14.2. HRMS calculated for C23H28O4 (M + H)+ 369.2060, found (M + H)+ 369.2056. HPLC purity: 99.9% (254 nm), tR = 8.05 min; 99.9% (220 nm), tR = 8.05 min.
7-Hydroxy-2-(4-(piperidin-1-yl)phenyl)chroman-4-one (8l)
Purified with hexane/EtOAc (84/16) to give the product (37 mg, 0.11 mmol, 57%) as pale yellow solid. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.73 (d, J = 8.7 Hz, 1H), 7.37–7.35 (m, 2H), 7.02–6.99 (m, 2H), 6.50 (dd, J = 8.7, 2.2 Hz, 1H), 6.36 (d, J = 2.2 Hz, 1H), 5.39 (dd, J = 12.9, 2.9 Hz, 1H), 3.19–3.16 (m, 4H), 3.06 (dd, J = 16.9, 12.9 Hz, 1H), 2.70 (dd, J = 16.9, 3.0 Hz, 1H), 1.75–1.69 (m, 4H), 1.63–1.59 (m, 2H). 13C NMR (100 MHz, CD3OD): δ (ppm) 193.6, 166.8, 165.6, 153.8, 131.2, 129.9, 128.5, 117.7, 115.1, 115.0, 111.7, 103.8, 81.0, 51.9, 44.8, 26.8, 25.4. HRMS calculated for C20H21NO3 (M – H)− 322.1449, found (M – H)− 322.1401. HPLC purity: 97.4% (254 nm), tR = 5.22 min; 98.7% (220 nm), tR = 5.22 min.
2-(2-(Allyloxy)phenyl)-5,7-dihydroxychroman-4-one (8m)
Purified with hexane/EtOAc (85/15) to give the product (47 mg, 0.15 mmol, 75%) as white solid. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.54 (d, J = 7.6 Hz, 1H), 7.30 (td, J = 7.9, 1.6 Hz, 1H), 7.03–6.99 (m, 2H), 6.11–6.01 (m, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.90 (d, J = 2.1 Hz, 1H), 5.73 (dd, J = 12.7, 3.2 Hz, 1H), 5.38 (dd, J = 17.3, 1.6 Hz, 1H), 5.25 (dd, J = 10.6, 1.4 Hz, 1H), 4.65–4.56 (m, 2H), 2.99–2.91 (m, 1H), 2.83–2.78 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 197.7, 168.3, 165.5, 165.0, 156.4, 134.6, 130.5, 128.8, 127.5, 122.0, 117.6, 113.2, 103.3, 97.2, 96.2, 75.7, 70.0, 43.1. HRMS calculated for C18H16O5 (M – H)− 311.0925, found (M – H)− 311.0877. HPLC purity: 99.8% (254 nm), tR = 7.14 min; 99.6% (220 nm), tR = 7.14 min.
MIC Determination
MIC values were determined against M. tuberculosis (H37Rv) and other bacteria using the standard microbroth dilution method exactly as previously described,71 which is based on the methods by the Clinical and Laboratory Standards Institute.72,73 The maximum test concentration used was 200 μg/mL.
Cytotoxicity Assays
Cytotoxicity was assessed in vitro using Vero cells (kidney epithelial cells, ATCC CCL-81) as described previously.74 In brief, monolayers of cells cultured in Dulbecco’s modified Eagle medium (DMEM)/10% fetal bovine serum (FBS) were trypsinized, seeded at approximately 10% confluence in white-walled 96-well plates, and incubated overnight to allow adherence. Medium was replaced with DMEM/FBS containing 2-fold serial dilutions of test compounds. Detection was performed using MTT (CellTiter96, Promega) with overnight solubilization according to the manufacturer’s instructions.
Approximation of Solubility in Cell Culture Media
The 2-fold serial dilutions in clear, round-bottom 96-well plates were prepared in DMEM with 5% FBS. Plates were incubated at 37 °C overnight. The highest compound concentration that did not result in visible precipitation of compound was used to approximate the solubility limit in cell culture medium.
Time Kill Experiments
The ability of compounds to kill S. aureus Newman was evaluated using standard approaches.75 Overnight cultures were diluted 1:25 in fresh Mueller–Hinton broth and grown to OD600nm ≈ 0.3 before being exposed to compounds at various increments of their MICs. The number of viable bacteria over time was then determined.
Macromolecular Synthesis
The effects of compounds on key macromolecular processes were determined as described,76 using radiolabeled precursors from American Radiolabeled Chemicals, Inc.: [methyl-3H]thymidine, [5,6-3H]uridine, and [4,5-3H]leucine for DNA, RNA, and protein, respectively. Experiments were performed on three independent mid-logarithmic cultures (OD600nm ≈ 0.3). The antibiotics ciprofloxacin (DNA, from Sigma-Aldrich), rifampicin (RNA, from TCI America), and erythromycin (protein, from Calbiochem) were used as positive controls.
Analysis of the Membrane Potential
The effects of compounds on the membrane potential of S. aureus were evaluated by flow cytometry using the fluorescent probe diethyloxacarbocyanine dye DiOC2(3). The emission of red fluorescence of DiOC2(3) from cells is dependent on the membrane potential, while green fluorescence emission is independent of the membrane potential. Therefore, the ratio of red to green fluorescence provides a measure of the effects of compounds on the membrane potential. These experiments were performed as previously described,76 using S. aureus Newman grown to OD600nm ≈ 0.3 in Mueller–Hinton II. Samples were analyzed using BD LSR II flow cytometer and the effects of compounds on the membrane potential evaluated using the software FlowJo X 10.0.7 as described.76 Carbonyl cyanide m-chlorophenylhydrazone (CCCP; Sigma-Aldrich) and vancomycin were used as positive and negative controls, respectively.
Assay of Relaxation Activity of E. coli Topoisomerase I
Recombinant E. coli topoisomerase I (EcTopI) was purified with published procedures.77 CsCl gradient purified plasmid DNA (160 ng) was added to relaxation reaction buffer containing 10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 0.1 mg/mL gelatin, and 0.5 mM MgCl2. The resulting solution was divided into 9.5 μL aliquots, to which 0.5 μL of the 20 mM compounds dissolved in DMSO was added for final compound concentrations of 500 μM. The compound control reaction received only DMSO. EcTopI (20 ng) in 10 μL of the relaxation reaction buffer was then added to each DNA/compound reaction mixture. The reaction mixtures were then incubated at 37 °C for 30 min before the reaction was terminated, and the mixtures were loaded onto a 1% agarose gel for electrophoretic analysis in TAE buffer (40 mM Tris–acetate, pH 8.1, 2 mM EDTA) as described previously.78
E. coli DNA Gyrase Supercoiling Assay
The E. coli DNA gyrase supercoiling assay was performed according to the manufacturer’s protocol. Briefly, 1.25 U of E. coli DNA gyrase (New England Biolabs) was added to 20 μL of DNA gyrase reaction mixture (35 mM Tris-HCl, pH 7.5, 24 mM KCl, 4 mM MgCl2, 2 mM DTT, 1.75 mM ATP, 5 mM spermidine, 0.1 mg/mL BSA, 6.5% glycerol) containing 300 ng of relaxed plasmid DNA substrate (New England Biolabs) in the presence of indicated concentration of compound. After incubation at 37 °C for 30 min, the reactions were stopped and analyzed by agarose gel electrophoresis similarly as the topoisomerase I relaxation assay.78 IC50 values (compound concentrations at which only 50% of the input DNA was converted to supercoiled form) were calculated by spot densitometry analysis with AlphaView, AlphaImager Mini (Protein Simple). The IC50 values represent the average from at least two experiments performed separately.
E. coli Topoisomerase IV Decatenation Assay
For the assay of decatenation activity of topoisomerase IV,79 0.25 U of E. coli topoisomerase IV (Topogen) was added to 20 μL of decatenation reaction mixture (40 mM HEPES-KOH, pH 8.0, 100 mM potassium glutamate, 10 mM magnesium acetate, 10 mM dithiothreitol, 50 μg of BSA/mL) containing 290 ng of kinetoplast DNA substrate (Topogen) and 1 mM ATP in the presence of indicated concentration of compound. After incubation at 37 °C for 30 min, the reactions were stopped by the addition of stop buffer (50 mM EDTA, 50% glycerol, and 0.5% (v/v) bromophenol blue). The DNA samples were electrophoresed in a 1% agarose gel containing 0.5 μg/mL ethidium bromide in TAE buffer also containing 0.5 μg/mL ethidium bromide. Visualization of the bands was carried out by exposure to UV light. IC50 values (compound concentrations at which only 50% of kinetoplast DNA was converted into monomeric products) were calculated by spot densitometry analysis with AlphaView, AlphaImager Mini (Protein Simple). The IC50 values represent the average from at least two experiments performed separately.
Acknowledgments
We thank financial support from the National Institutes of Health Grants P20GM103466 and R15AI092315 and Hawaii Community Foundation’s Leahi Fund to Treat & Prevent Pulmonary Disease Grant 12ADVC-57499 (D.S.), National Institutes of Health Grants R01AI069313 (Y.-C.T.-D.) and R01AT006732 (J.G.H.), and the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital (R.E.L.). We thank Dr. Benjamin Clark for his assistance with the measurement of high-resolution mass spectra.
Glossary
Abbreviations Used
- ATCC
American Type Culture Collection
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- Cip
ciprofloxacin
- DEA
diethylamine
- DIPEA
diisopropylethylamine
- DMEM
Dulbecco’s modified Eagle medium
- DMSO
dimethylsulfoxide
- FBS
fetal bovine serum
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- MIC
minimum inhibitory concentration
- MOM
methoxymethyl
- MSSA
methicillin-sensitive Staphylococcus aureus
- MRSA
methicillin-resistant Staphylococcus aureus
- NMR
nuclear magnetic resonance
- SAR
structure–activity relationship
- SEM
standard error of mean
- SI
selectivity index
- Topo
topoisomerase
- TBDMS
tert-butyldimethylsilyl
- Ts
p-toluenesulfonyl
Supporting Information Available
NMR and HRMS spectra and HPLC profiles of all synthetic compounds; ORTEP views of 3g and 8a; inhibition data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# M.M.M and Md.Z.A contributed equally to the work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cushnie T. P. T.; Lamb A. J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Lugo M.-T.; Bewley C. A. Natural products, small molecules, and genetics in tuberculosis drug development. J. Med. Chem. 2008, 51, 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negi A. S.; Kumar J. K.; Luqman S.; Saikia D.; Khanuja S. P. S. Antitubercular potential of plants: a brief account of some important molecules. Med. Res. Rev. 2010, 30, 603–645. [DOI] [PubMed] [Google Scholar]
- Walsh C. Where will new antibiotics come from?. Nat. Rev. Microbiol. 2003, 1, 65–70. [DOI] [PubMed] [Google Scholar]
- Shang S.; Tan D. S. Advancing chemistry and biology through diversity-oriented synthesis of natural product-like libraries. Curr. Opin. Chem. Biol. 2005, 9, 248–258. [DOI] [PubMed] [Google Scholar]
- Havsteen B. Flavonoids, a class of natural products of high pharmacological potency. Biochem. Pharmacol. 1983, 32, 1141–1148. [DOI] [PubMed] [Google Scholar]
- Harborne J. B.; Williams C. A. Advances in flavonoid research since 1992. Phytochemistry 2000, 55, 481–504. [DOI] [PubMed] [Google Scholar]
- Middleton E. Jr.; Kandaswami C.; Theoharides T. C. The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [PubMed] [Google Scholar]
- Knekt P.; Jarvinen R.; Seppanen R.; Heliovaara M.; Teppo L.; Pukkala E.; Aromaa A. Dietary flavonoids and the risk of lung cancer and other malignant neoplasms. Am. J. Epidemiol. 1997, 146, 223–230. [DOI] [PubMed] [Google Scholar]
- Knekt P.; Kumpulainen J.; Jarvinen R.; Rissanen H.; Heliovaara M.; Reunanen A.; Hakulinen T.; Aromaa A. Flavonoid intake and risk of chronic diseases. Am. J. Clin. Nutr. 2002, 76, 560–568. [DOI] [PubMed] [Google Scholar]
- Le Marchand L.; Murphy S. P.; Hankin J. H.; Wilkens L. R.; Kolonel L. N. Intake of flavonoids and lung cancer. J. Natl. Cancer Inst. 2000, 92, 154–160. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Stevens V. L.; Shah R.; Peterson J. J.; Dwyer J. T.; Gapstur S. M.; McCullough M. L. Dietary flavonoid and proanthocyanidin intakes and prostate cancer risk in a prospective cohort of US men. Am. J. Epidemiol. 2014, 179, 974–986. [DOI] [PubMed] [Google Scholar]
- Andrews L. Dietary flavonoids for the prevention of colorectal cancer. Clin. J. Oncol. Nurs. 2013, 17, 671–672. [DOI] [PubMed] [Google Scholar]
- Gradisar H.; Pristovsek P.; Plaper A.; Jerala R. Green tea catechins inhibit bacterial DNA gyrase by interaction with its ATP binding site. J. Med. Chem. 2006, 50, 264–271. [DOI] [PubMed] [Google Scholar]
- Favela-Hernández J. M. J.; García A.; Garza-González E.; Rivas-Galindo V. M.; Camacho-Corona M. R. Antibacterial and antimycobacterial lignans and flavonoids from Larrea tridentata. Phytother. Res. 2012, 26, 1957–1960. [DOI] [PubMed] [Google Scholar]
- Cushnie T. P. T.; Lamb A. J. Recent advances in understanding the antibacterial properties of flavonoids. Int. J. Antimicrob. Agents 2011, 38, 99–107. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. F.; Sanz M. J.; Ubeda A.; Miranda M. A.; Iborra S.; Paya M.; Alcaraz M. J. Synthesis and pharmacological evaluation of 2′-hydroxychalcones and flavones as inhibitors of inflammatory mediators generation. J. Med. Chem. 1995, 38, 2794–2797. [DOI] [PubMed] [Google Scholar]
- Wu J.; Li J.; Cai Y.; Pan Y.; Ye F.; Zhang Y.; Zhao Y.; Yang S.; Li X.; Liang G. Evaluation and discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J. Med. Chem. 2011, 54, 8110–8123. [DOI] [PubMed] [Google Scholar]
- Takemura H.; Sakakibara H.; Yamazaki S.; Shimoi K. Breast cancer and flavonoids—a role in prevention. Curr. Pharm. Des. 2013, 19, 6125–6132. [DOI] [PubMed] [Google Scholar]
- Park E. J.; Pezzuto J. M. Flavonoids in cancer prevention. Anti-Cancer Agents Med. Chem. 2012, 12, 836–851. [DOI] [PubMed] [Google Scholar]
- Echeverria C.; Santibanez J. F.; Donoso-Tauda O.; Escobar C.; Ramirez-Tagle R. Structural antitumoral activity relationships of synthetic chalcones. Int. J. Mol. Sci. 2009, 10, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loa J.; Chow P.; Zhang K. Studies of structure–activity relationship on plant polyphenol-induced suppression of human liver cancer cells. Cancer Chemother. Pharmacol. 2009, 63, 1007–1016. [DOI] [PubMed] [Google Scholar]
- Stevenson D.; Hurst R. Polyphenolic phytochemicals—just antioxidants or much more?. Cell. Mol. Life Sci. 2007, 64, 2900–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erasto P.; Bojase-Moleta G.; Majinda R. R. T. Antimicrobial and antioxidant flavonoids from the root wood of Bolusanthus speciosus. Phytochemistry 2004, 65, 875–880. [DOI] [PubMed] [Google Scholar]
- Weiss R. B.; Greene R. F.; Knight R. D.; Collins J. M.; Pelosi J. J.; Sulkes A.; Curt G. A. Phase I and clinical pharmacology study of intravenous flavone acetic acid (NSC 347512). Cancer Res. 1988, 48, 5878–5882. [PubMed] [Google Scholar]
- Senderowicz A. M. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest. New Drugs 1999, 17, 313–320. [DOI] [PubMed] [Google Scholar]
- Burdette-Radoux S.; Tozer R. G.; Lohmann R. C.; Quirt I.; Ernst D. S.; Walsh W.; Wainman N.; Colevas A. D.; Eisenhauer E. A. Phase II trial of flavopiridol, a cyclin dependent kinase inhibitor, in untreated metastatic malignant melanoma. Invest. New Drugs 2004, 22, 315–322. [DOI] [PubMed] [Google Scholar]
- Jones J. A.; Rupert A. S.; Poi M.; Phelps M. A.; Andritsos L.; Baiocchi R.; Benson D. M.; Blum K. A.; Christian B.; Flynn J.; Penza S.; Porcu P.; Grever M. R.; Byrd J. C. Flavopiridol can be safely administered using a pharmacologically derived schedule and demonstrates activity in relapsed and refractory non-Hodgkin’s lymphoma. Am. J. Hematol. 2014, 89, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loguercio C.; Festi D. Silybin and the liver: from basic research to clinical practice. World J. Gastroenterol. 2011, 17, 2288–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C. W. Y.; Gibbons N.; Johnson D. W.; Nicol D. L. Silibinin—a promising new treatment for cancer. Anti-Cancer Agents Med. Chem. 2010, 10, 186–195. [DOI] [PubMed] [Google Scholar]
- Ferry D. R.; Smith A.; Malkhandi J.; Fyfe D. W.; deTakats P. G.; Anderson D.; Baker J.; Kerr D. J. Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin. Cancer Res. 1996, 2, 659–668. [PubMed] [Google Scholar]
- Mulholland P. J.; Ferry D. R.; Anderson D.; Hussain S. A.; Young A. M.; Cook J. E.; Hodgkin E.; Seymour L. W.; Kerr D. J. Pre-clinical and clinical study of QC12, a water-soluble, pro-drug of quercetin. Ann. Oncol. 2001, 12, 245–248. [DOI] [PubMed] [Google Scholar]
- Bast A.; Haenen G.; Bruynzeel A.; Van der Vijgh W. Protection by flavonoids against anthracycline cardiotoxicity: from chemistry to clinical trials. Cardiovasc. Toxicol. 2007, 7, 154–159. [DOI] [PubMed] [Google Scholar]
- Savoia D. Plant-derived antimicrobial compounds: alternatives to antibiotics. Future Microbiol. 2012, 7, 979–990. [DOI] [PubMed] [Google Scholar]
- Jandial D. D.; Blair C. A.; Zhang S.; Krill L. S.; Zhang Y. B.; Zi X. Molecular targeted approaches to cancer therapy and prevention using chalcones. Curr. Cancer Drug Targets 2014, 14, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Kumar M.; Kumar A.; Singh S. K. Chalcone and curcumin derivatives: a way ahead for malarial treatment. Mini-Rev. Med. Chem. 2013, 13, 2116–2133. [DOI] [PubMed] [Google Scholar]
- Bukhari S. N. A.; Franzblau S. G.; Jantan I.; Jasamai M. Current prospects of synthetic curcumin analogs and chalcone derivatives against Mycobacterium tuberculosis. Med. Chem. 2013, 9, 897–903. [DOI] [PubMed] [Google Scholar]
- Lin Y.-M.; Zhou Y.; Flavin M. T.; Zhou L.-M.; Nie W.; Chen F.-C. Chalcones and flavonoids as anti-tuberculosis agents. Bioorg. Med. Chem. 2002, 10, 2795–2802. [DOI] [PubMed] [Google Scholar]
- Cabrera M.; Simoens M.; Falchi G.; Lavaggi M. L.; Piro O. E.; Castellano E. E.; Vidal A.; Azqueta A.; Monge A.; de Ceráin A. L.; Sagrera G.; Seoane G.; Cerecetto H.; González M. Synthetic chalcones, flavanones, and flavones as antitumoral agents: Biological evaluation and structure–activity relationships. Bioorg. Med. Chem. 2007, 15, 3356–3367. [DOI] [PubMed] [Google Scholar]
- Chiari M. E.; Vera D. M. A.; Palacios S. M.; Carpinella M. C. Tyrosinase inhibitory activity of a 6-isoprenoid-substituted flavanone isolated from Dalea elegans. Bioorg. Med. Chem. 2011, 19, 3474–3482. [DOI] [PubMed] [Google Scholar]
- Tsuchiya H.; Sato M.; Miyazaki T.; Fujiwara S.; Tanigaki S.; Ohyama M.; Tanaka T.; Iinuma M. Comparative study on the antibacterial activity of phytochemical flavanones against methicillin-resistant Staphylococcus aureus. J. Ethnopharmacol. 1996, 50, 27–34. [DOI] [PubMed] [Google Scholar]
- Pouget C.; Fagnere C.; Basly J.-P.; Besson A.-E.; Champavier Y.; Habrioux G.; Chulia A.-J. Synthesis and aromatase inhibitory activity of flavanones. Pharm. Res. 2002, 19, 286–291. [DOI] [PubMed] [Google Scholar]
- Sun D.; Hurdle J. G.; Lee R.; Lee R.; Cushman M.; Pezzuto J. M. Evaluation of flavonoid and resveratrol chemical libraries reveals abyssinone II as a promising antibacterial lead. ChemMedChem 2012, 7, 1541–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiu W. K. P.; Rahman M. M.; Curry J.; Stapleton P.; Zloh M.; Malkinson J. P.; Gibbons S. Antibacterial acylphloroglucinols from Hypericum olympicum. J. Nat. Prod. 2012, 75, 336–343. [DOI] [PubMed] [Google Scholar]
- Shiu W. K. P.; Malkinson J. P.; Rahman M. M.; Curry J.; Stapleton P.; Gunaratnam M.; Neidle S.; Mushtaq S.; Warner M.; Livermore D. M.; Evangelopoulos D.; Basavannacharya C.; Bhakta S.; Schindler B. D.; Seo S. M.; Coleman D.; Kaatz G. W.; Gibbons S. A new plant-derived antibacterial is an inhibitor of efflux pumps in Staphylococcus aureus. Int. J. Antimicrob. Agents 2013, 42, 513–518. [DOI] [PubMed] [Google Scholar]
- Wu X.; Alam M. Z.; Feng L.; Tsutsumi L. S.; Sun D.; Hurdle J. G. Prospects for flavonoid and related phytochemicals as nature-inspired treatments for Clostridium difficile infection. J. Appl. Microbiol. 2014, 116, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon O.; Reux B.; La Clair J. J.; Lear M. J. Total synthesis confirms laetirobin as a formal Diels–Alder adduct. Chem.—Asian J. 2010, 5, 342–351. [DOI] [PubMed] [Google Scholar]
- Sarmah M. P.; Shashidhar M. S.; Sureshan K. M.; Gonnade R. G.; Bhadbhade M. M. Sulfonate protecting groups. Synthesis of O- and C-methylated inositols: d- and l-ononitol, d- and l-laminitol, mytilitol and scyllo-inositol methyl ether. Tetrahedron 2005, 61, 4437–4446. [Google Scholar]
- Wallen E. A. A.; Dahlen K.; Grotli M.; Luthman K. Synthesis of 3-aminomethyl-2-aryl-8-bromo-6-chlorochromones. Org. Lett. 2007, 9, 389–391. [DOI] [PubMed] [Google Scholar]
- Poonia N. S.; Chhabra K.; Kumar C.; Sharma T. C.; Bhagwat V. W. Coordinative role of alkali cations in organic synthesis. 2. The chalcone–flavanone system. J. Org. Chem. 1977, 42, 3311–3313. [Google Scholar]
- Fridén-Saxin M.; Pemberton N.; da Silva Andersson K.; Dyrager C.; Friberg A.; Grotli M.; Luthman K. Synthesis of 2-alkyl-substituted chromone derivatives using microwave irradiation. J. Org. Chem. 2009, 74, 2755–2759. [DOI] [PubMed] [Google Scholar]
- Ji H.; Zhang W.; Zhang M.; Kudo M.; Aoyama Y.; Yoshida Y.; Sheng C.; Song Y.; Yang S.; Zhou Y.; Lu J.; Zhu J. Structure-based de novo design, synthesis, and biological evaluation of non-azole inhibitors specific for lanosterol 14α-demethylase of fungi. J. Med. Chem. 2003, 46, 474–485. [DOI] [PubMed] [Google Scholar]
- Kabbe H.-J.; Widdig A. Synthesis and reactions of 4-chromanones. Angew. Chem., Int. Ed. 1982, 21, 247–256. [Google Scholar]
- Pouget C.; Fagnere C.; Basly J.-P.; Leveque H.; Chulia A.-J. Synthesis and structure of flavan-4-ols and 4-methoxyflavans as new potential anticancer drugs. Tetrahedron 2000, 56, 6047–6052. [Google Scholar]
- Maiti A.; Cuendet M.; Croy V. L.; Endringer D. C.; Pezzuto J. M.; Cushman M. Synthesis and biological evaluation of abyssinone II and its analogues as aromatase inhibitors for chemoprevention of breast cancer. J. Med. Chem. 2007, 50, 2799–2806. [DOI] [PubMed] [Google Scholar]
- Vogel S.; Heilmann J. Synthesis, cytotoxicity, and antioxidative activity of minor prenylated chalcones from Humulus lupulus. J. Nat. Prod. 2008, 71, 1237–1241. [DOI] [PubMed] [Google Scholar]
- Jung D. H.; Lee Y. R.; Kim S. H. New synthetic routes to biologically interesting geranylated flavanones and geranylated chalcones: first total synthesis of (±)-prostratol F, xanthoangelol, and (±)-lespeol. Helv. Chim. Acta 2010, 93, 635–647. [Google Scholar]
- Tello M.; Kuzuyama T.; Heide L.; Noel J.; Richard S. The ABBA family of aromatic prenyltransferases: broadening natural product diversity. Cell. Mol. Life Sci. 2008, 65, 1459–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basarab G. S.; Hill P. J.; Garner C. E.; Hull K.; Green O.; Sherer B. A.; Dangel P. B.; Manchester J. I.; Bist S.; Hauck S.; Zhou F.; Uria-Nickelsen M.; Illingworth R.; Alm R.; Rooney M.; Eakin A. E. Optimization of pyrrolamide topoisomerase II inhibitors toward identification of an antibacterial clinical candidate (AZD5099). J. Med. Chem. 2014, 57, 6060–6082. [DOI] [PubMed] [Google Scholar]
- Nimesh H.; Sur S.; Sinha D.; Yadav P.; Anand P.; Bajaj P.; Virdi J. S.; Tandon V. Synthesis and biological evaluation of novel bisbenzimidazoles as Escherichia coli topoisomerase IA inhibitors and potential antibacterial agents. J. Med. Chem. 2014, 57, 5238–5257. [DOI] [PubMed] [Google Scholar]
- Cheng B.; Cao S.; Vasquez V.; Annamalai T.; Tamayo-Castillo G.; Clardy J.; Tse-Dinh Y.-C. Identification of anziaic acid, a lichen depside from Hypotrachyna sp., as a new topoisomerase poison inhibitor. PLoS One 2013, 8, e60770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.; Annamalai T.; Bansod P.; Tse-Dinh Y.-C.; Sun D. Synthesis and antibacterial evaluation of anziaic acid and its analogues as topoisomerase I inhibitors. MedChemComm 2013, 4, 1613–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basarab G. S.; Manchester J. I.; Bist S.; Boriack-Sjodin P. A.; Dangel B.; Illingworth R.; Sherer B. A.; Sriram S.; Uria-Nickelsen M.; Eakin A. E. Fragment-to-hit-to-lead discovery of a novel pyridylurea scaffold of ATP competitive dual targeting type II topoisomerase inhibiting antibacterial agents. J. Med. Chem. 2013, 56, 8712–8735. [DOI] [PubMed] [Google Scholar]
- Plaper A.; Golob M.; Hafner I.; Oblak M.; Šolmajer T.; Jerala R. Characterization of quercetin binding site on DNA gyrase. Biochem. Biophys. Res. Commun. 2003, 306, 530–536. [DOI] [PubMed] [Google Scholar]
- Russo P.; Del Bufalo A.; Cesario A. Flavonoids acting on DNA topoisomerases: recent advances and future perspectives in cancer therapy. Curr. Med. Chem. 2012, 19, 5287–5293. [DOI] [PubMed] [Google Scholar]
- Constantinou A.; Mehta R.; Runyan C.; Rao K.; Vaughan A.; Moon R. Flavonoids as DNA topoisomerase antagonists and poisons: structure–activity relationships. J. Nat. Prod. 1995, 58, 217–225. [DOI] [PubMed] [Google Scholar]
- Black M. T.; Stachyra T.; Platel D.; Girard A.-M.; Claudon M.; Bruneau J.-M.; Miossec C. Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 2008, 52, 3339–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K.; Kitamura A.; Morrissey I.; Sato K.; Kato J.; Ikeda H. Comparison of inhibition of Escherichia coli topoisomerase IV by quinolones with DNA gyrase inhibition. Antimicrob. Agents Chemother. 1994, 38, 2623–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa H.; Shigematsu A.; Ohta S.; Harigaya Y. Preparative monohydroxyflavanone syntheses and a protocol for gas chromatography-mass spectrometry analysis of monohydroxyflavanones. Chem. Pharm. Bull. 2005, 53, 547–554. [DOI] [PubMed] [Google Scholar]
- Kuo S.-C.; Lee H.-Z.; Juang J.-P.; Lin Y.-T.; Wu T.-S.; Chang J.-J.; Lednicer D.; Paull K. D.; Lin C. M.; Hamel E.; Lee K.-H. Synthesis and cytotoxicity of 1,6,7,8-substituted 2-(4′-substituted phenyl)-4-quinolones and related compounds: identification as antimitotic agents interacting with tubulin. J. Med. Chem. 1993, 36, 1146–1156. [DOI] [PubMed] [Google Scholar]
- Lee R. E.; Hurdle J. G.; Liu J.; Bruhn D. F.; Matt T.; Scherman M. S.; Vaddady P. K.; Zheng Z.; Qi J.; Akbergenov R.; Das S.; Madhura D. B.; Rathi C.; Trivedi A.; Villellas C.; Lee R. B.; Rakesh; Waidyarachchi S. L.; Sun D.; McNeil M. R.; Ainsa J. A.; Boshoff H. I.; Gonzalez-Juarrero M.; Meibohm B.; Bottger E. C.; Lenaerts A. J. Spectinamides: a new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat. Med. 2014, 20, 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes: Approved Standard. Clinical and Laboratory Standards Institute (CLSI), Wayne, PA, U.S., 2003; Vol. 23, M24-A. [PubMed] [Google Scholar]
- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard, 7th ed.; Clinical and Laboratory Standards Institute (CLSI), Wayne, PA, U.S., 2006; Vol. 26, M7-A7. [Google Scholar]
- Rakesh; Bruhn D.; Madhura D. B.; Maddox M.; Lee R. B.; Trivedi A.; Yang L.; Scherman M. S.; Gilliland J. C.; Gruppo V.; McNeil M. R.; Lenaerts A. J.; Meibohm B.; Lee R. E. Antitubercular nitrofuran isoxazolines with improved pharmacokinetic properties. Bioorg. Med. Chem. 2012, 20, 6063–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurdle J. G.; O’Neill A. J.; Chopra I. Anti-staphylococcal activity of indolmycin, a potential topical agent for control of staphylococcal infections. J. Antimicrob. Chemother. 2004, 54, 549–552. [DOI] [PubMed] [Google Scholar]
- Cherian P. T.; Wu X.; Maddox M. M.; Singh A. P.; Lee R. E.; Hurdle J. G. Chemical modulation of the biological activity of reutericyclin: a membrane-active antibiotic from Lactobacillus reuteri. Sci. Rep. 2014, 4, 4721. 10.1038/srep04721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin E. P.; Cheng B.; Rathi S.; Aedo S. J.; Abrenica M. V.; Tse-Dinh Y.-C. Inhibition of Mg2+ binding and DNA religation by bacterial topoisomerase I via introduction of an additional positive charge into the active site region. Nucleic Acids Res. 2008, 36, 4788–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.-X.; Tse-Dinh Y.-C. The acidic triad conserved in type IA DNA topoisomerases is required for binding of Mg(II) and subsequent conformational change. J. Biol. Chem. 2000, 275, 5318–5322. [DOI] [PubMed] [Google Scholar]
- Peng H.; Marians K. J. Escherichia coli topoisomerase IV. Purification, characterization, subunit structure, and subunit interactions. J. Biol. Chem. 1993, 268, 24481–24490. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.