

Abstract
Staphylococcus aureus is a rapidly growing health threat in the U.S., with resistance to several commonly prescribed treatments. A high-throughput screen identified the antihistamine terfenadine to possess, previously unreported, antimicrobial activity against S. aureus and other Gram-positive bacteria. In an effort to repurpose this drug, structure–activity relationship studies yielded 84 terfenadine-based analogues with several modifications providing increased activity versus S. aureus and other bacterial pathogens, including Mycobacterium tuberculosis. Mechanism of action studies revealed these compounds to exert their antibacterial effects, at least in part, through inhibition of the bacterial type II topoisomerases. This scaffold suffers from hERG liabilities which were not remedied through this round of optimization; however, given the overall improvement in activity of the set, terfenadine-based analogues provide a novel structural class of antimicrobial compounds with potential for further characterization as part of the continuing process to meet the current need for new antibiotics.
Introduction
The ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) are responsible for 51% of all U.S. hospital-acquired infections (HAIs).1 Among these, S. aureus accounts for 16% of HAIs and is responsible for more deaths in the U.S. annually than HIV/AIDS.1−3 The organism’s high morbidity and mortality rates are, in part, attributable to the fact that S. aureus has developed resistance to currently available antibiotics.4 The quinolone class of antibiotics was once a predominant treatment option for S. aureus infections;5 however, due to increasing quinolone resistance, these drugs continue to have diminishing efficacy.6,7
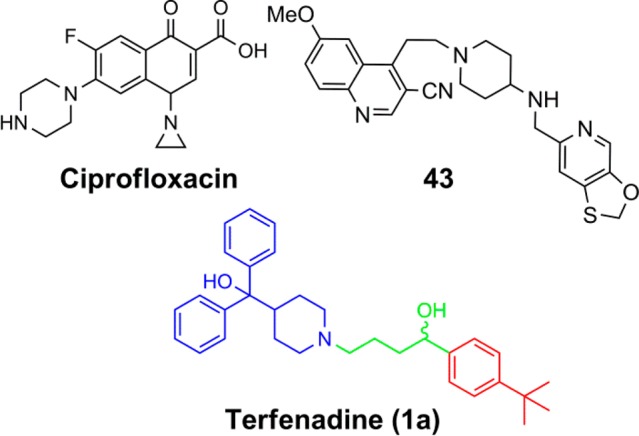
The antimicrobial activity of the quinolones and fluoroquinolones, such as ciprofloxacin (Figure 1) and levofloxacin, is thought to be mediated by their ability to inhibit the DNA religation activity of the bacterial type II topoisomerases, DNA gyrase and topoisomerase IV. Resistance can arise from decreased access to these cellular targets or by mutations within the type II topoisomerases.8,9 Despite the rise in resistance to quinolones, their previous success validates the type II topoisomerases as valuable targets in searching for novel antimicrobial scaffolds. Indeed, academic and industrial laboratories have devoted much effort toward developing novel bacterial type II topoisomerase inhibitors (NBTIs) featuring compound scaffolds chemically distinct from those of the quinolone class of antibiotics,10−15 including the N-linked piperidine 6-(methyloxy)-4-(2-{4-[([1,3]oxathiolo[5,4-c]pyridin-6-ylmethyl)amino]-1-piperidinyl}ethyl)-3-quinolinecarbonitrile dihydrochloride 43 (GSK299423).16 However, despite NBTI development efforts, pharmaceutical pipelines are still severely lacking quality antibiotic candidates,17 and the loss of several antibiotic groups in the pharmaceutical industry only exacerbates the problem.18,19 This, coupled with increasing prevalence of quinolone-resistant S. aureus, makes it paramount that novel small molecule inhibitors are discovered for type II topoisomerases for the therapeutic intervention of staphylococcal disease and possibly other bacterial pathogens.
Figure 1.
Structures of ciprofloxacin, NBTI 43, and terfenadine (1a).
The practice of drug repurposing, in which a drug previously developed to treat one disease is then identified for possibly treating a second disease, has emerged as an attractive alternative for drug discovery research.20,21 The repurposed drug most likely has already been optimized for physicochemical and pharmacokinetic properties,22 providing a more attractive starting point as far as these factors are concerned. Recently, the National Center for Advancing Translational Science (NCATS) has invested in this area with the formation of the NIH Chemical Genomic Center Pharmaceutical Collection as both an informatics and screening resource for drug repurposing research,23 lending credence to the potential and popularity of drug repurposing.
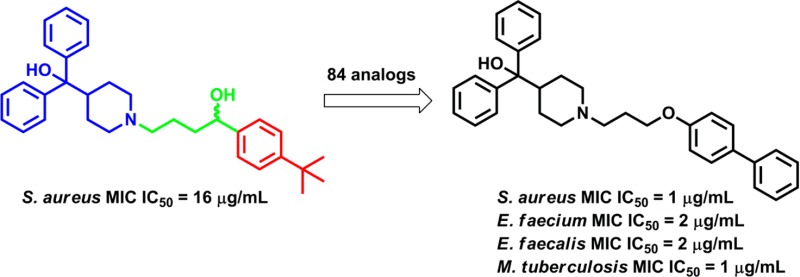
Given the need for novel antimicrobials and the attractive features of drug repurposing, we previously performed a high-throughput screen (HTS) to identify FDA approved drugs with bactericidal activity toward the ESKAPE pathogens.24 Results identified a set of compounds that were developed for other indications but displayed previously unreported antimicrobial activity. One particular hit, the antihistamine terfenadine (Figure 1), was found to possess antimicrobial activity versus the planktonic, biofilm, and small-colony variant forms of S. aureus. Given the activity and relatively convenient synthetic route to analogues, terfenadine provided an attractive starting point for studying the structure–activity relationship (SAR) of S. aureus antimicrobial activity. However, terfenadine is not without its flaws. The clinical use of the drug was discontinued in favor of its active metabolite fexofenadine (Allegra) because a segment of the patient population exhibited cardiac arrhythmia, attributed to prolonged QT interval,25,26 due to inhibition of the human ether-á-go-go related gene (hERG) potassium channel.27 Nonetheless, it has been shown previously that it is possible to reduce hERG liabilities via an SAR strategy13 and given the encouraging results from the HTS, we decided it would be beneficial to embark on an SAR-optimization study of terfenadine (1a) and its analogues for inhibition of S. aureus and those results are reported herein.
Results and Discussion
Chemistry
A total of 84 terfenadine-based analogues were synthesized for optimization of antimicrobial activity against S. aureus strain UAMS-1,14 a well-studied osteomyelitis clinical isolate, by standard CLSI methods.28 The majority of analogues were synthesized by one of two routes, while several required alternate routes or further modification. The first route employs a substitution reaction with diphenyl(piperidin-4-yl)methanol (7) and corresponding substituted chloro-phenylbutanones (8) followed by subsequent reduction of the ketone intermediate (9) yielding analogues 1a–1h and 1j–1l (Scheme 1). An alternate pathway was used to synthesize analogue 1i in which the methyl 4-(4-chlorobutanoyl)benzoate 8i was prepared according to a previously reported procedure,29 reduced, and subjected to a Finkelstein reaction with 7 to yield the desired analogue (Scheme 2). This ester was then hydrolyzed to the corresponding carboxylic acid 1m. Compound 1n was synthesized by Suzuki–Miyaura coupling using a procedure adapted from Moseley et al.30 (Scheme 3A). The final analogue in this set, the known metabolite of terfenadine (1p also known as fexofenadine),31 was generated according to a previously published procedure32 (Scheme S2 in Supporting Information).
Scheme 1. General Synthetic Route for Terfenadine (1a) and Analogues Series 1.
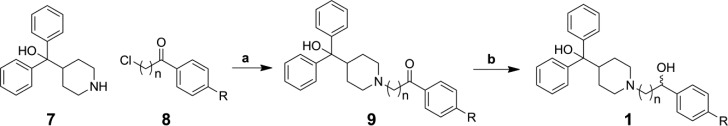
Reagents and conditions: (a) NaHCO3, 2-butanone/water, 85 °C, 16 h, 23–95%; (b) NaBH4, MeOH, rt, 3 h, 52–95%.
Scheme 2. Synthetic Route for Analogues 1i and 1m.

Reagents and conditions: (a) 1,3-propanedithiol, CH2Cl2, rt, 1.5 h then BF3·OEt2, 0 °C to rt, 18 h, 86%; (b) NaHMDS, THF, −78 °C then 1-chloro-3-iodopropane, rt, 18 h, 31%; (c) bis(trifluoroacetoxy)iodobenzene, CH3CN/water, rt, 1 h, 69%; (d) NaBH4, MeOH, rt, 3 h, 87%; (e) NaHCO3, NaI, CH3CN, reflux, 18 h, 37%; (g) LiOH, THF/water, rt, 3 h, 47%.
Scheme 3. Synthetic Routes for Analogues 1n (A), 3l–3n (B).

Reagents and conditions: (a) R-B(OH)2, K2CO3, CH3CN/water, 60 °C, 18 h, 30–88%; (b) NaBH4, MeOH, rt, 3 h, 94%;
The second route to a majority of analogues was via nucleophilic substitution in which 7 was coupled with various substituted phenyl alkyl halides or tosylates (10) yielding analogues 2a–2d, 3a–3i, 4a–4r, 4t, 4w, and 4y–4bb (Scheme 4). Benzyl bromides were not available for four desired analogues, thus reductive amination was employed for analogues 4s, 4u, 4v, and 4x with the corresponding aldehydes 11a–11d (Scheme 4). A number of analogues required further modification such as reduction of the 4-nitro group of 3i, providing the 4-amino derivative 3j followed by subsequent dimethylation, yielding the 4-dimethylamino analogue 3k (Scheme 5). Analogues 3l–3n were synthesized from 3e using the aforementioned Suzuki–Miyaura cross-coupling procedure (Scheme 3B). Saponification of methyl esters 4y–4aa resulted in carboxylic acids 4cc–4ee. A set of five-membered heterocycles at the 4-position were synthesized in which the 4-iodo derivative 4bb was subjected to Suzuki–Miyaura cross coupling with the corresponding boronic acids or potassium trifluoroborate salts to provide 4ff–4ii, while a copper mediated Ullman type coupling of pyrrole33 in acetonitrile afforded the pyrrole derivative 4jj (Scheme 6).
Scheme 4. General Synthetic Route for Analogue Series 2, 3, and 4.
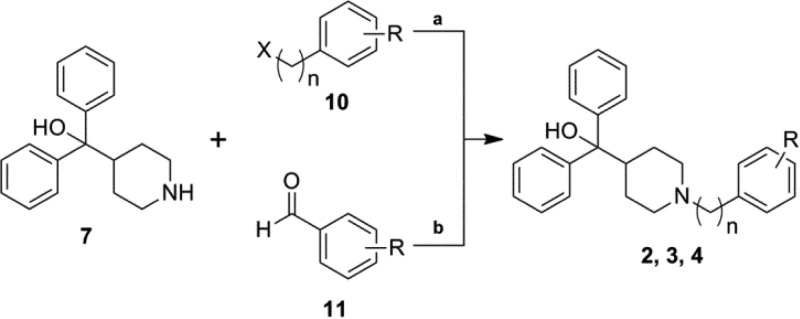
Reagents and conditions: (a) 7, K2CO3 or Et3N, acetonitrile, reflux,18 h, 20–94%; (b) For analogues 4s, 4u, 4v, and 4x: 7, Na(OAc)3BH, THF, rt to 65 °C, 3 h, 16–29%.
Scheme 5. Synthetic Route for Analogues 3j and 3k.

Reagents and conditions: (a) Raney nickel, NaBH4, CH2Cl2/MeOH, 0 °C – rt, 28 h, 61%; (b) paraformaldehyde, NaBH3CN, AcOH, rt, 20 h, 84%.
Scheme 6. Synthetic Route for Analogues 4ff–4jj.

Reagents and conditions: for 4ff and 4hh (a) R-B(OH)2, K2CO3, CH3CN/water, 60 °C, 18 h, 85–89%; for 4gg and 4ii (b) R-BF3K, Pd(OAc)2, RuPhos, NaHCO3, 100 °C, μW, 60 min, 68–69%; for 4jj (c) Cu, pyrrole, Cs2CO3, CH3CN, 80 °C, 21 h, 47%.
Compounds 2e and 2f were prepared by alkylation of 7 with 3-bromopropan-1-ol, providing the alcohol intermediate 12, which was subsequently tosylated and subjected to substitution conditions with 4-tert-butylaniline or 4-tert-butylphenol, respectively (Scheme 7). Analogue 2g was synthesized via reductive amination of 9a to arrive at the benzylic amine. Each enantiomer for terfenadine, 2h and 2i, were synthesized via a previously reported procedure (Supporting Information).34
Scheme 7. Synthetic Route to Analogues 2e, 2f, and 6.

Reagents and conditions: 3-bromopropanol, Et3N, acetonitrile, reflux, 3 h, 65%; (b) for 2e and 2f, TsCl, Et3N, CH2Cl2, rt, 20 h, 33%; (c) for 2e, 4-tert-butylaniline, for 2f, 4-tert-butylphenol, Et3N, acetonitrile, reflux, 18 h, 15–45%; (d) for 6, 4-phenylphenol, triphenylphosphine, DIAD, THF, rt, 18 h, 77%.
A slightly more polar tert-butyl isostere, methyl oxetane,35 was prepared using an adapted procedure from Wuitschik et al.36 The key step being a rhodium catalyzed coupling of 4-(2-hydroxyethyl)phenylboronic acid with the Michael acceptor intermediate. Further modification resulted in analogue 3o (Scheme 8).
Scheme 8. Synthetic Route for Analogue 3o.

Reagents and conditions: (a) BuLi, THF, 0 °C, 45 min then diethyl chlorophosphate, 0 °C, 30 min then cool to −78 °C, oxetan-3-one, 2 h, 75%; (b) [Rh(cod)Cl]2, KOH, 4-(2-hydroxyethyl) phenylboronic acid, 1,4-dioxane, 100 °C, μW, 30 min, 76%; (c) Et3N, CH2Cl2, TsCl, rt, 20 h, 27%; (d) 7, Et3N, CH3CN, reflux, 18 h, 49%; (e) Mg, MeOH, 50 °C, 4.5 h, 25%.
Analogues 5a and 5b (Figure 2) were created by utilizing the same substitution and subsequent reduction conditions described for previous series. The diphenyl piperazine intermediate was synthesized using a previously reported procedure,37 followed by substitution with 8a and reduction to arrive at 5c. Finally, 5d and 5e were created by reducing the alcohol of 7 in either TFA alone or in the presence of sodium borohydride.38 The reduced intermediates were then subjected to the same substitution/reduction procedure as described for previous series (Supporting Information).
Figure 2.

Analogues 5a–5e displaying modifications to diphenyl piperidine region.
The final analogue combined favorable changes to the linker and pendant phenyl in an attempt to further enhance potency. This compound was synthesized via Mitsunobu chemistry in which the intermediate 12 was coupled with 4-phenylphenol, providing 6 (Scheme 7).39
Design and SAR for Terfenadine Analogues
The SAR for all terfenadine analogues was studied using antimicrobial potency toward planktonic S. aureus UAMS-1 cells, as measured by minimum inhibitory concentration testing (MIC). Data from these assays helped drive iterative analogue design and synthesis. Mechanism of action studies on terfenadine were performed in parallel. The most promising analogues were carried into MIC assays testing across a spectrum of Gram-positive and Gram-negative pathogens as well as M. tuberculosis. Furthermore, a common strategy for reducing hERG activity is to lower the log P of the compound,40,41 thus attempts at incorporating polar functional groups while optimizing for potency were undertaken with this goal in mind.
Series 1 compounds were designed to gain initial SAR information at the 4-position of the pendant phenyl (red region in Figure 1) of 1a while preserving the linker (Table 1). A scan of lipophilic steric bulk showed that anti-S. aureus activity correlated with the size of the lipophilic groups (4-tert-butyl (1a) > 4-iso-propyl (1b) > 4-methyl (1c) > 4-H (1d)). A halogen scan at the para-position also displayed a similar trend (4-bromo (1g) > 4-chloro (1f) > 4-fluoro (1e)). The 4-phenyl analogue 1n further supports this observation, as it displayed a modest increase in potency (MIC to 8 μg/mL). Unfortunately, the introduction of polarity at this position resulted in loss of antimicrobial activity as observed for the 4-methoxy (1h), 4-methyl carboxylate, and its corresponding carboxylic acid (1i and 1m) as well as for the primary metabolite of 1a, carboxylic acid 1p and the methyl ester precursor 1o (Table 1). These results suggest lipophilicity and steric bulk at the 4-position of the pendant phenyl are optimal for antistaphylococcal activity.
Table 1. MIC Values for Series 1 and 2.
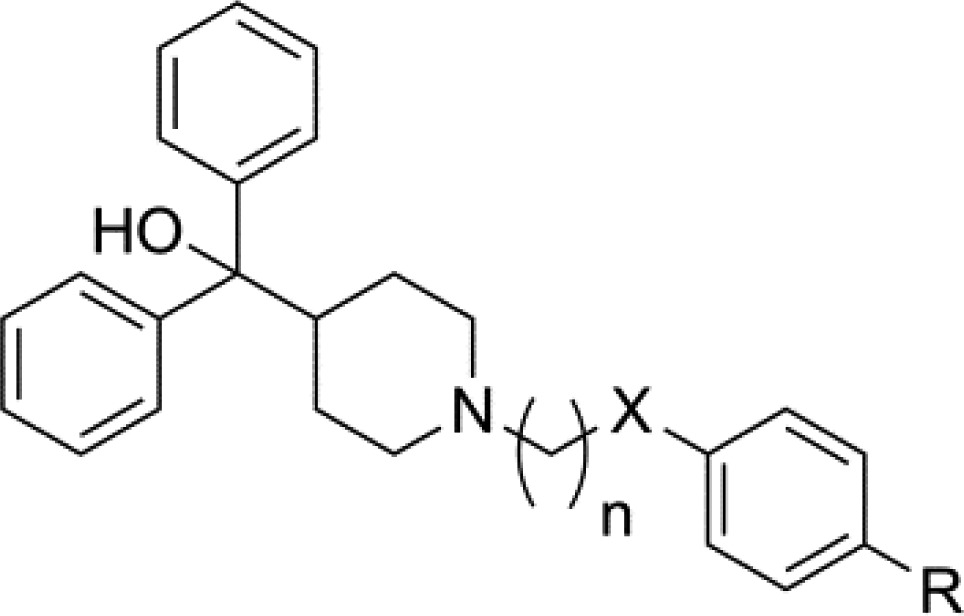
| compd | n | X | R | S. aureus MIC (μg/mL) |
|---|---|---|---|---|
| 1a | 3 | CH(OH) | tert-butyl | 16 |
| 1b | 3 | CH(OH) | iso-propyl | 32 |
| 1c | 3 | CH(OH) | CH3 | 128 |
| 1d | 3 | CH(OH) | H | >256 |
| 1e | 3 | CH(OH) | F | 128 |
| 1f | 3 | CH(OH) | Cl | 64 |
| 1g | 3 | CH(OH) | Br | 32 |
| 1h | 3 | CH(OH) | OMe | >256 |
| 1i | 3 | CH(OH) | CO2Me | >256 |
| 1j | 4 | CH(OH) | tert-butyl | 16 |
| 1k | 2 | CH(OH) | tert-butyl | 8 |
| 1l | 1 | CH(OH) | tert-butyl | 16 |
| 1m | 3 | CH(OH) | CO2H | >256 |
| 1n | 3 | CH(OH) | phenyl | 8 |
| 1o | 3 | CH(OH) | C(CH3)2CO2Me | >256 |
| 1p | 3 | CH(OH) | C(CH3)2CO2H | >256 |
| 2a | 3 | CH2 | tert-butyl | 8 |
| 2b | 2 | CH2 | tert-butyl | 8 |
| 2c | 1 | CH2 | tert-butyl | 8 |
| 2d | 0 | CH2 | tert-butyl | 8 |
| 2e | 3 | NH | tert-butyl | 8 |
| 2f | 3 | O | tert-butyl | 8 |
| 2g | 3 | CH(NH2) | tert-butyl | 16 |
| 2h | 3 | CH(OH) (S) | tert-butyl | 16 |
| 2i | 3 | CH(OH) (R) | tert-butyl | 16 |
| 9a | 3 | CO | tert-butyl | 8 |
| 9k | 2 | CO | tert-butyl | 32 |
| 9l | 1 | CO | tert-butyl | >256 |
| 9m | 0 | CO | tert-butyl | >256 |
The second set of analogues was designed to study modifications to the linker (green region in Figure 1) while maintaining the 4-tert-butyl group on the pendant phenyl. Compounds 1j–1l scanned linker length while maintaining the benzylic alcohol and showed that changes to the length were tolerated and did not reduce activity (MICs = 8 and 16 μg/mL). A study of linker length while altering the oxidation state on the benzylic carbon provided similar results. Analogues 2a–2d all contained fully reduced benzylic carbons, scanning from four to one carbon linkers, respectively, and provided MICs = 8 μg/mL. The ketone precursor to the hit (9a) maintained activity (MIC = 8 μg/mL), while the three-carbon derivative (9l) showed a slight reduction in activity compared to 1a (MIC = 32 μg/mL). The ketone containing two-carbon and amide-containing linkers resulted in a loss of activity, possibly due to restriction of movement for the phenyl group, at least in terms of amide linker. Further exploration of the necessity of the alcohol led to a benzylic amino derivative (2g) and the S-OH (2h) and R-OH (2i) enantiomers, all of which exhibited no change in activity compared to the hit. The final two analogues in this set replaced the benzylic carbon with nitrogen (2e) and oxygen (2f), resulting in slight increases in activity to MICs = 8 μg/mL for each. The SAR of the linker region suggests the secondary alcohol is not necessary for S. aureus antimicrobial activity while shortening the linker in some cases led to a small, yet consistent, improvement in activity.
Given the modest success of the one- and two-carbon linkers from the previous set, the next two series further studied SAR on the pendant phenyl while utilizing shorter, more accessible linkers. The two carbon linker set provided mixed results as far as potency but maintained the trend of favoring lipophilic bulk at the 4-position (Table 2). Moving the tert-butyl group around the pendent phenyl (3a and 3b, MIC = 16 μg/mL for each) was tolerated and did not alter activity significantly compared to 2c. The same was observed for bulkier derivatives as the 4-bromo (3e), 4-trifluoromethyl (3f) displayed MICs = 16 μg/mL for each, and 4-phenyl analogue 3l had an MIC = 8 μg/mL. Smaller and more polar substitutions in this set led to varying ranges of reduction in activity or a complete loss.
Table 2. MIC Values for Series 3 and 4.
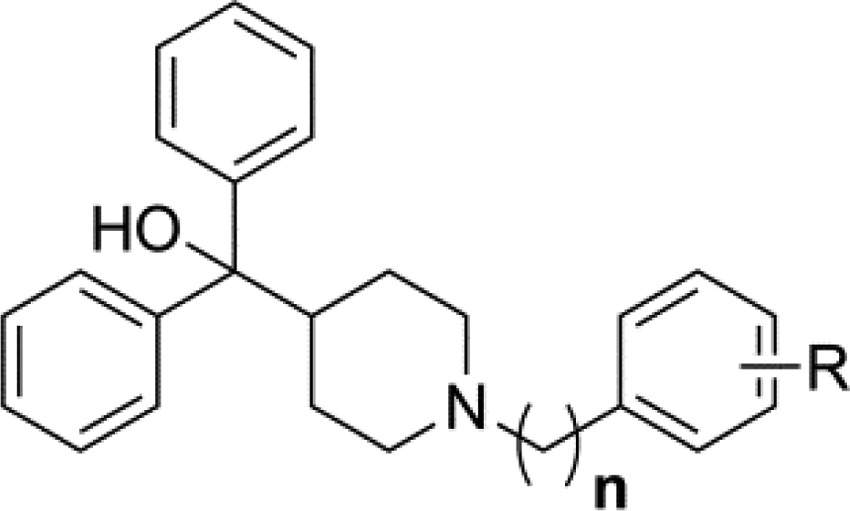
| compd | n | R | S. aureus MIC (μg/mL) | compd | n | R | S. aureus MIC (μg/mL) |
|---|---|---|---|---|---|---|---|
| 3a | 2 | 3-tert-butyl | 16 | 4k | 1 | 3-CN | >256 |
| 3b | 2 | 2-tert-butyl | 16 | 4l | 1 | 2-CN | >256 |
| 3c | 2 | 4-F | 128 | 4m | 1 | 4-CF3 | >256 |
| 3d | 2 | 4-Cl | 32 | 4n | 1 | 3-CF3 | >256 |
| 3e | 2 | 4-Br | 16 | 4o | 1 | 2-CF3 | >256 |
| 3f | 2 | 4-CF3 | 16 | 4p | 1 | 4-F | >256 |
| 3g | 2 | 4-OMe | >256 | 4q | 1 | 3-F | 128 |
| 3h | 2 | 3-pyridyl-4-tert-butyla | 64 | 4r | 1 | 2-F | >256 |
| 3i | 2 | 4-NO2 | 64 | 4s | 1 | 4-OMe | 128 |
| 3j | 2 | 4-NH2 | >256 | 4t | 1 | 3-OMe | 128 |
| 3k | 2 | 4-N(CH3)2 | >256 | 4u | 1 | 2-OMe | 128 |
| 3l | 2 | 4-phenyl | 8 | 4v | 1 | 4-OH | 128 |
| 3m | 2 | 4-(4-pyridyl) | 32 | 4w | 1 | 3-OH | >256 |
| 3n | 2 | 4-(3-pyridyl) | 32 | 4x | 1 | 2-OH | 64 |
| 3o | 2 | 4-C(CH2OCH2)CH3 | >256 | 4y | 1 | 4-CO2Me | >256 |
| 4a | 1 | 3-tert-butyl | 32 | 4z | 1 | 3-CO2Me | >256 |
| 4b | 1 | 2-tert-butyl | >256 | 4aa | 1 | 2-CO2Me | >256 |
| 4c | 1 | 4-iso-propyl | 32 | 4cc | 1 | 4-CO2H | >256 |
| 4d | 1 | 4-Me | 128 | 4dd | 1 | 3-CO2H | >256 |
| 4e | 1 | 3-Me | 128 | 4ee | 1 | 2-CO2H | >256 |
| 4f | 1 | 2-Me | 128 | 4ff | 1 | 4-(3-thiophene) | 16 |
| 4g | 1 | 4-phenyl | 4 | 4gg | 1 | 4-(2-thiophene) | 32 |
| 4h | 1 | 3-phenyl | 64 | 4hh | 1 | 4-(3-furan) | 32 |
| 4i | 1 | 2-phenyl | >256 | 4ii | 1 | 4-(2-furan) | 32 |
| 4j | 1 | 4-CN | >256 | 4jj | 1 | 4-(1-pyrrole) | 32 |
Analogue 3h employs a pendant 3-pyridine in place of the pendant phenyl.
These trends continued in the one-carbon linker series as the 4-phenyl analogue 4g was the most active, with an MIC = 4 μg/mL, a 4-fold increase in potency compared to the hit (Table 2). Unfortunately, all polar modifications at the 2-, 3-, or 4-positions led to a significant reduction or complete loss of S. aureus antimicrobial activity. A 4– 8-fold reduction in activity was observed for heterocycles at the para-position, as indicated by the 3- and 2-thiophene analogues 4ff and 4gg, 3- and 2-furan analogues 4hh and 4ii, and the N-linked pyrrole 4jj, compared to 4g, suggesting the slight increase in polarity for these common phenyl isosteres affect their activity.
A small set of analogues were designed to explore more global changes to the piperidinyl diphenylmethanol (blue region in Figure 1) side of 1a (Figure 2). Analogues 5a–5d showed a complete loss of activity toward S. aureus; however, the removal of the alcohol while maintaining sp3 character of the carbon (5e) was tolerated (Table 3). This provides some evidence, however small, that future work could establish SAR trends on this side of the molecule.
Table 3. MIC Values for Series 5.
| compd | S. aureus MIC (μg/mL) |
|---|---|
| 5a | >256 |
| 5b | >256 |
| 5c | >256 |
| 5d | >256 |
| 5e | 8 |
In summary, there appears to be a trend in which lipophilicity may be driving the potency of this scaffold, at least in general. A scatter plot of log P vs MIC (Chart S1 in Supporting Information) displays this trend, with the average log P generally decreasing as potency decreases. However, there are a few analogues that lie outside of the trend, such as 4g, which has a lower calculated log P (estimated by ALOGPS)42,43 than 1a but displays a 4-fold increase in potency. Therefore, while the trend is general, there are a few instances in which functional group identity or placement still might play a role in the potency. A strong relationship was noticed at the 4-position of the pendant phenyl, with lipophilic bulk being beneficial for potency against S. aureus. Downsizing the tert-butyl group of the hit 1a resulted in a reduction of potency, however slight, on all occasions while the addition of a phenyl group in this position was the lone substitution, leading to a modest increase of potency.
The addition of hetercyclic isosteres (pyridinyl, thiophenyl, and furyl moieties) are comparable in size to the phenyl, however, contain slightly larger polar surface areas. The benefit of steric bulk in these cases may have been mostly negated due to increased polarity, however slight, ultimately resulting in an overall reduction of potency but not a complete loss. Modifications, either polar or nonpolar, to other positions of the pendant phenyl were unfavorable, leading to reduction or loss of potency. Therefore, SAR on the pendant phenyl, while relatively steep for the ortho- and meta-positions, did offer some room for improvement at the para-position as far as potency against S. aureus is concerned. However, further optimization to the piperidinyl diphenylmethanol side of 1a may be required for reduction of log P while maintaining or improving potency.
Several modifications to the linker region provided a slight increase in potency. The benzylic alcohol was not necessary for activity, as indicated by the removal of the hydroxy group, leading to moderately increased antimicrobial potency across the four different linker lengths (2a–2d). It was also shown that substituting an amine for the alcohol or resolving the enantiomers had no effect on activity. Furthermore, substituting the benzylic carbon for an amine or ether linkage resulted in a 2-fold increase in activity compared to the hit. Given these data, it was evident that modifications to the linker could lead to an analogue with enhanced activity.
A final analogue was designed to combine the favorable ether linker modification from 2f and the 4-phenyl substitution on the pendant group to yield 6. The combination of these modifications resulted in a synergistic boost in potency, with an MIC = 1 μg/mL against S. aureus comparable to an MIC = 0.5 μg/mL for the widely prescribed fluoroquinoline, ciprofloxacin, in the same assay (Figure 3). The success of the combined modifications offers optimism that more potent analogues may be within reach with future SAR studies. However, compound properties such as solubility and log P remain an issue and will be addressed with future work.
Figure 3.

MIC values for ciprofloxacin, 4g, and 6.
Mechanism of Action
Transcription profiling was used to compare the cellular response of S. aureus strain UAMS-1 following challenge with a subinhibitory concentration (0.5× MIC) of terfenadine with that of cells treated with 0.5× MIC of the known topoisomerase II inhibitor, ciprofloxacin, or antibiotics with independent mechanisms of action. Results (Figure S1 in Supporting Information) revealed that terfenadine-treated cells elicit a cellular response most similar to that of ciprofloxacin and cells treated with the DNA damaging agent mitomycin C in comparison to cells treated with subinhibitory concentrations of cell wall active agents, RNA synthesis inhibitors, or general stress responses such as cold-shock conditions and metal depletion. For instance, 221 (43%) of the 509 genes that were differentially expressed within ciprofloxacin treated cells were also differentially expressed within terfenadine-treated cells. A total of 149 (67%) of these 221 genes were also differentially expressed within mitomycin treated cells, but none of the other stress conditions evaluated. Of direct relevance to this work, while the majority of these genes (55%) are annotated as hypothetical proteins, two key components of the organism’s SOS response, lexA and umuC, as well as 12 phage-associated replication proteins, were identified and known to be responsive to the topoisomerase inhibitor, ciprofloxacin.44,45 The similarities between the cellular response of terfenadine-, ciprofloxacin-, and mitomycin-challenged cells provided an initial indication that the scaffold’s antimicrobial activity may be mediated via topoisomerase II inactivation.
As a direct test of whether the antimicrobial properties of terfenadine correlate with type II topoisomerase inhibition, S. aureus DNA gyrase and topoisomerase IV inhibition assays were used to measure each enzyme’s activity in the absence and presence of various concentrations of terfenadine. As shown in Figure 4A (lanes 1 and 2), the addition of 0.5 units (U) of S. aureus DNA gyrase catalyzed supercoiling of relaxed circular plasmid pBR322 DNA, resulting in increased substrate mobility in an agarose gel, confirming that the assay conditions were appropriate to measure gyrase activity. Similar assays in the presence of increasing concentrations of terfenadine (62.5, 125, 250, and 500 μM), demonstrated that the compound was a relatively mild inhibitor of S. aureus gyrase with an apparent IC50 value of 190 μM. Similarly, terfenadine appeared to exhibit moderate topoisomerase IV inhibitory activity (Figure 5A).
Figure 4.

DNA gyrase supercoiling inhibition assays (A) dose–response gel for 1a; (B) gel showing inhibition of DNA gyrase with analogues 1a, 4g, and 6 with the positive control ciprofloxacin at 100 μM.
Figure 5.

Topoisomerase IV inhibition assay (A) dose–response gel for 1a; (B) gel showing inhibition of topoisomerase IV with analogues 1a, 4g, and 6 with the positive control ciprofloxacin at 100 μM.
While these values are admittedly modest, they are comparable to the topoisomerase II inhibitor, ciprofloxacin, in these assays, which displayed IC50 values of 110 and 4 μM for S. aureus DNA gyrase and topoisomerase IV, respectively (data not shown). This, combined with the fact that 1a did not appear to affect the activity of other enzymes assessed (S. aureus RNase J2 and RnpA; data not shown), supports the hypothesis that the compound’s antimicrobial activity may, in part, correspond to its topoisomerase II inhibitory activity.
Accordingly, IC50 values were measured for the first 48 analogues synthesized (Table 4) to determine whether improvements to the compound’s antimicrobial activity tracked with enhanced enzyme inhibition. Results of those studies revealed two overarching features of the analogue series. First, most compounds (11 of 12 in total) displaying modest improvement in antimicrobial activity (MIC ≤ 8 μg/mL), in comparison to terfenadine, also exhibited at least modest improvement in potency toward S. aureus DNA gyrase and/or topoisomerase IV. Second, most compounds exhibiting reduced antimicrobial activity (MIC ≥ 32 μg/mL) displayed decreased potency toward the enzymes. Compounds 4g and 6, which exhibited the most improved antimicrobial properties toward S. aureus (MIC values of 4 and 1 μg/mL, respectively), also displayed modest improvements toward S. aureus DNA gyrase inhibition (IC50s = 130 and 50 μM, respectively) when compared to 1a. Indeed, as shown in Figure 4B, when tested at 100 μM, 4g, 6, and ciprofloxacin, all displayed improved inhibition of S. aureus DNA gyrase supercoiling activity, in comparison to the parent molecule, terfenadine. This suggests that even if the gains in potency for 4g and 6 over terfenadine are modest, they do appear to display a real improvement in DNA gyrase inhibition. Moreover, both derivatives maintained S. aureus topoisomerase IV inhibition activity that approximated the inhibitory activity of that of the parent molecule, 1a (Figure 5B).
Table 4. DNA Gyrase and Topoisomerase IV IC50 Measurements.
| compd | DNA Ggyrase IC50 (μM) | topoisomerase IV IC50 (μM) | compd | DNA gyrase IC50 (μM) | topoisomerase IV IC50 (μM) |
|---|---|---|---|---|---|
| ciprofloxacin | 110 | 4 | 2h | 127 | 100 |
| 1a (terfenadine) | 190 | 206 | 2i | 410 | 110 |
| 1b | 127 | 273 | 9a | 93 | 110 |
| 1c | >500 | >333 | 9k | 90 | 133 |
| 1d | >500 | >333 | 9l | 73 | 247 |
| 1e | >500 | >333 | 9m | 93 | >333 |
| 1f | 133 | >333 | 3a | 93 | >333 |
| 1g | >500 | >333 | 3c | >333 | >333 |
| 1h | >500 | >333 | 3d | 260 | 267 |
| 1i | 440 | >333 | 3e | 247 | 260 |
| 1j | 100 | 133 | 3f | 17 | >333 |
| 1k | 133 | 333 | 3g | >333 | >333 |
| 1l | 73 | 100 | 3h | >333 | >333 |
| 1m | >500 | >333 | 3i | 263 | >333 |
| 1n | 13 | 100 | 3j | 93 | >333 |
| 1o | >500 | >333 | 3k | >333 | >333 |
| 1p | >500 | >333 | 3l | 80 | 133 |
| 2a | 93 | 320 | 3m | 93 | 227 |
| 2b | 93 | 100 | 3n | 250 | 280 |
| 2c | 93 | 147 | 3o | >333 | >333 |
| 2d | 90 | 213 | 4g | 125 | 160 |
| 2e | 193 | 250 | 5a | >333 | >333 |
| 2f | 100 | >333 | 5b | >333 | >333 |
| 2g | >500 | >333 | 6 | 50 | 160 |
Even though the terfenadine scaffold seems to be inhibiting the type II topoisomerases, it appears there may be other mechanisms contributing to the overall bactericidal effect. It is well noted that receptor promiscuity is correlated with lipophilicity.46 Therefore, it is not out of the question that these compounds could be binding to multiple targets, and the combined effect of these targets may be exerting the antibacterial effect. Analogues 2g, 2i, 3d, 3e, and 3n all are shown to have activity versus S. aureus, with MICs ranging from 16–32 μg/mL. However, these analogues possess reduced or no activity in the DNA gyrase and topoisomerase IV assays. Therefore, they must be exerting an antimicrobial effect via other means, supporting the multiple target hypothesis. Moreover, attempts to isolate resistant strains of S. aureus provided no stable resistant organisms, also supporting a multiple target hypothesis. It should be noted that given the above correlations, expense, and laborious nature of the enzyme assays and the fact that no improvements in antimicrobial activity (≥32 μg/mL) were observed for the remaining compounds synthesized, their IC50 values were not measured.
DNA Gyrase Modeling Studies
As was shown in the previous section, the terfenadine-based scaffolds are likely to, at least in part, exert their antibacterial effect via inhibition of the type II topoisomerases. Recently, NBTIs such as 43 have been reported.16 These NBTIs also feature a piperidine, with aromatic regions linked to the N- and 4-position. Therefore, we hypothesized that the terfenadine-based analogues may be binding in the same region as 43. A docking study was carried out using the software Surflex module of SYBYL from Certara. The receptor protein (PDB ID 2XCS) was prepared by removing the ligand, 43, and taking the corresponding ligand binding pocket defined as a 20 Å sphere around the GSK ligand. Out of 30 poses, the best pose was selected on the basis of Combined CScore (ChemScore, G_Score, D_Score, and PMF_Score), and the scores were 8.85 for 43, 7.65 for 6, and 5.99 for 1a, respectively. The scores generally correlated with the experimentally determined DNA gyrase activity (43 = 14 nM, 6 = 50 μM, and 1a = 190 μM, respectively).
To assess the potential of the terfenadine analogues for binding to other known sites on DNA gyrase, the same docking study was performed with a solved structure of ciprofloxacin (PDB ID 2XCT),16 and the docking score of the best poses for 43 and 6 were 2.22 and 0.72, respectively. The same modeling was performed at the ATP site on the GyrB subunit, in which a pyrrolamide ligand was extracted from the ATP-binding site in a structure published by Eakin et al.47 (PDB ID 3U2K). Both 43 and 6 were docked and provided similar docking scores (5.05 and 5.23, respectively). These in silico results suggest a higher likelihood of the terfenadine-based analogues interacting in the same region as 43 than in the other two known inhibitor binding sites.
Information from the above modeling of the binding site suggests that the aromatic character of the pendant biphenyl moiety intercalates between DNA base pairs similar to 43 (Figure 6A). However, due to the length of the linker, the diphenyl methanol portion may be able to interact with a section of the protein surface adjacent to the pocket in which the oxathiolo-pyridine group of 43 binds. The best pose of 6 suggests that hydrogen bonds may form between the alcohol with an acceptor phosphate group in the DNA backbone and a donor Arg1122, respectively, with additional potential hydrophobic π–π stacking interactions (Figure 6B). While the modeling suggests a similar binding site, further work is needed to validate these results. Future plans include designing analogues to interrogate these possible interactions, competition assays with known inhibitors, and solving a terfenadine-based ligand bound DNA gyrase structure aimed at providing more definitive evidence as to the binding site for this scaffold.
Figure 6.
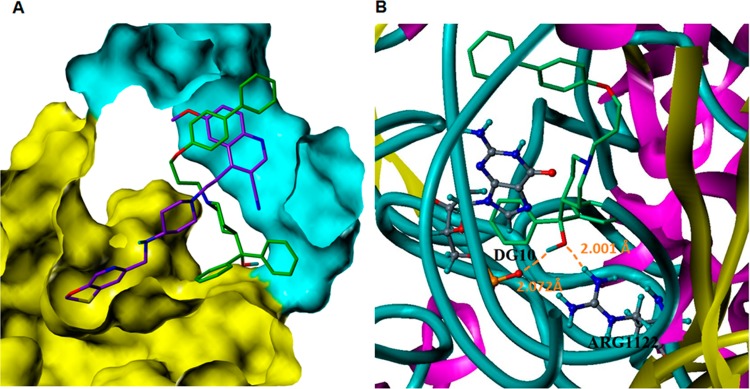
Docking studies utilizing the ligand-bound DNA gyrase structure PDB ID 2XCS (A) Overlay of solved ligand, 43 (purple ligand) and best docked pose for 6 (green ligand) within binding site. Protein surface shaded yellow and DNA surface shaded blue. (B) Potential interactions from the best docked pose of 6 (light-blue ligand). Docking studies in two known alternative binding sites, the ATP and ciprofloxacin binding sites, were also performed using solved ligand-bound structures (PDB IDs 3U2K and 2XCT, respectively), and the binding score were inferior compared to the 43 binding site.
Antimicrobial Spectrum of Activity of Terfenadine, 4g, and 6
As stated above, terfenadine (1a) was found to have antistaphylococcal activity in a HTS campaign using the S. aureus strain UAMS-1, a methicillin-susceptible strain. To determine the spectrum of antimicrobial activity for 1a, the activity of the scaffold toward genetically diverse S. aureus strains, other Gram-positive and Gram-negative bacterial species of immediate healthcare concern, and Mycobacterium tuberculosis was determined. As shown in Table 5, each compound’s antimicrobial activity was conserved across all Gram-positive strains evaluated. More specifically, the MIC values of 1a, 4g, and 6 were 16, 4, and approximately 2 μg/mL, respectively toward methicillin-resistant (MRSA), vancomycin-intermediate (VISA), vancomycin-resistant (VRSA) S. aureus, as well as fluoroquinolone resistant strains. Likewise, they demonstrated corresponding antimicrobial activity toward Enterococcus faecium, Enterococcus faecalis, and M. tuberculosis. Neither 1a nor its analogues were active against wild-type Gram-negative species tested thus far. The MIC is >256 μg/mL for both K. pneumoniae CKP4 and E. coli 8314. The MIC value for A. baumannii 98-37-09 is 256 μg/mL. However, when tested versus a membrane-compromised, efflux pump-deficient strain of E. coli (tolC, imp), all three compounds did show activity. Therefore, the lack of efficacy versus Gram-negative species may be due to an inability of these compounds to traverse the outer membrane to gain entry to the cellular target and/or a result of expulsion via efflux pumps. Nonetheless, these results indicate that a terfenadine scaffold could potentially be optimized for use as an antimicrobial against Gram-positive organisms and potentially Gram-negative organisms if treated with an efflux pump inhibitor.
Table 5. Antimicrobial Spectrum for Terfenadine, 4g, and 6.
| strain (relevant resistance) | terfenadine (1a) MIC (μg/mL) | 4g MIC (μg/mL) | 6 MIC (μg/mL) |
|---|---|---|---|
| S. aureus U1 | 16 | 4 | 1 |
| S. aureus CRC61 (ciprofloxacin) | 16 | 4 | 2 |
| S. aureus CRC118 (ciprofloxacin) | 16 | 4 | 2 |
| S. aureus USA300 NRS-384 (MRSA) | 16 | 4 | 2 |
| S. aureus USA 300–0114 (MRSA) | 16 | 8 | 2 |
| S. aureus Mu50 (VISA) | 16 | 8 | 2 |
| S. aureus VRSA-1 (VRSA) | 16 | 8 | 2 |
| E. faecium 824–05 | 16 | 8 | 2 |
| E. faecalis OG1RF | 16 | 4 | 2 |
| M. tuberculosis mc26020 | 16 | 4 | 1 |
| A. baumannii 983709 | 256 | >256 | >128 |
| K. pneumoniae CKP4 | >256 | >256 | >128 |
| E. coli 8314 | >256 | >256 | >128 |
| E. coli (tolC, imp−) | 8 | 2 | 4 |
hERG Activity of Terfenadine Analogues
Compounds 4g and 6 emerged as the most promising analogues with MICs = 4 and 1 μg/mL, respectively, against S. aureus. Unfortunately, the SAR did not allow for addition of polar functionality, and the predicted log P for each compound remains relatively high when compared to the hit. The predicted log P values for 1a, 4g, and 6 are 5.89, 5.73, and 6.30, respectively (estimated by ALOGPS). Therefore, it appears no improvement was able to be gained in log P while optimizing for potency in this set of analogues. This observation carries over to the measured hERG activity for each compound. Terfenadine (1a) displayed a hERG IC50 = 130 nM, while analogues 4g and 6 display hERG IC50s = 210 and 140 nM, respectively. While hERG activity was not able to be reduced during this campaign, future SAR work on the piperdinyl diphenylmethanol side of the molecule may still provide opportunities for addition of polarity or reduction of the pKa of the molecule, thus reducing log P and potentially reducing hERG activity.
Conclusions
The project commenced with a phenotypic whole-cell HTS against the ESKAPE pathogens using a library of off-patent FDA-approved drugs. The HTS identified the antihistamine, terfenadine (1a), as a hit possessing previously unreported antimicrobial activity versus S. aureus (MIC = 16 μg/mL). In an effort to repurpose 1a, a total of 84 terfenadine-based analogues were designed and synthesized for optimized antimicrobial activity versus S. aureus. Analogues 4g and 6 displayed improved activity (4 and 1 μg/mL respectively) compared to 1a and promising activity toward other bacterial pathogens of immediate healthcare concern. The SAR study revealed bulky lipophilic substituents at the 4-position of the pendant phenyl, shortening the linker and/or replacing the benzylic carbon with oxygen, enhance activity.
Mechanism of action studies suggest 1a, 4g, and 6 are likely acting as bacterial type II topoisomerase inhibitors targeting both DNA gyrase and topoisomerase IV. However, there is evidence to suggest this scaffold may have multiple targets. Docking studies show these analogues may bind within the same site as the NBTI 43. While antimicrobial activity versus S. aureus strains tested, including fluoroquinolone, methicillin and vancomycin resistant isolates, and M. tuberculosis was improved, the hERG liability and physicochemical properties for this class of compounds still remains an issue. We believe 6, and possibly 4g, provide good starting points for further optimization to the piperidinyl diphenylmethanol side of the molecule. Furthemore, a strategy has been devised to improve antimicrobial activity while reducing hERG activity. In future work, we also plan to confirm the possible DNA gyrase binding site for these analogues via competition assays and crystallography studies.
Experimental Section
Chemistry: General Experimental
Purity of all final compounds was confirmed by HPLC/MS analysis, and all compounds have a final purity of ≥95% purity. 1H and 13C NMR spectra were recorded on a Bruker AM 400 spectrometer (operating at 400 and 101 MHz, respectively) or a Bruker AVIII spectrometer (operating at 500 and 126 MHz, respectively) in CDCl3 with 0.03% TMS as an internal standard or DMSO-d6. The chemical shifts (d) reported are given in parts per million (ppm), and the coupling constants (J) are in hertz (Hz). The spin multiplicities are reported as s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, and m = multiplet. The LCMS analysis was performed on an Agilent 1200 RRL chromatograph with photodiode array UV detection and an Agilent 6224 TOF mass spectrometer. The chromatographic method utilized the following parameters: a Waters Acquity BEH C-18 2.1 mm × 50 mm, 1.7 mm column; UV detection wavelength = 214 nm; flow rate = 0.4 mL/min; gradient = 5–100% acetonitrile over 3 min with a hold of 0.8 min at 100% acetonitrile; the aqueous mobile phase contained 0.15% ammonium hydroxide (v/v). The mass spectrometer utilized the following parameters: an Agilent multimode source which simultaneously acquires ESI+/APCI+, a reference mass solution consisting of purine and hexakis(1H,1H,3H-tetrafluoropropoxy) phosphazine, and a makeup solvent of 90:10:0.1 MeOH:water:formic acid which was introduced to the LC flow prior to the source to assist ionization.
General Method A
1-(4-(tert-Butyl)phenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (9a)
To a vial was added diphenyl(piperidin-4-yl)methanol 7 (1.19 g, 4.45 mmol), 1-(4-(tert-butyl)phenyl)-4-chlorobutan-1-one 8a (1.01 g, 4.24 mmol), and sodium bicarbonate (0.42 g, 5.09 mmol) with water and 2-butanone (18 mL, 1:5). The reaction stirred at 85 °C for 16 h and was then allowed to cooled to rt and diluted with water (50 mL). The reaction was diluted with water (20 mL) and extracted with EtOAc (3 × 50 mL). The organic layers were combined then dried with MgSO4, filtered, concentrated, and purified by MPLC (0–10% MeOH:CH2Cl2) to produce pure 9a (1.37 g, 2.92 mmol, 69% yield) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.90 (d, J = 8.4 Hz, 2H), 7.49–7.45 (m, 6H), 7.31–7.25 (m, 4H), 7.20–7.14 (m, 2H), 2.97–2.92 (m, 4H), 2.45–2.36 (m, 3H), 2.09 (br s, 1H), 2.00–1.87 (m, 4H), 1.49–1.32 (m, 4H), 1.34 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 199.7, 156.5, 146.0, 134.5, 128.1, 128.0, 126.4, 125.7, 125.4, 79.4, 57.9, 43.9, 43.4, 44.1, 36.2, 35.0, 31.0, 26.2, 21.9. LCMS retention time: 4.207 min. LCMS purity 99.5%. HRMS (ESI): m/z calcd for C32H39NO2 [M + H]+ 470.3053, found 470.3076.
General Method B
1-(4-(tert-Butyl)phenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (1a)
To a vial was added 9a (0.20 g, 0.422 mmol) and MeOH (2 mL). The sodium borohydride (0.032 g, 0.844 mmol) was then added and the reaction stirred at rt for 3 h. The reaction was concentrated, water (5 mL) was added, and a white precipitate formed. The precipitate was filtered out and then dissolved in CH2Cl2 (10 mL), dried with MgSO4, filtered, and concentrated to produce pure 1a (0.15 g, 0.320 mmol, 76% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.52–7.46 (m, 4H), 7.33–7.25 (m, 8H), 7.21–7.15 (m, 2H), 4.61–4.56 (m, 1H), 3.16–3.11 (br m, 1H), 3.00–2.94 (m, 1H), 2.51–2.34 (m, 4H), 2.10–1.88 (m, 4H), 1.83–1.75 (m, 1H), 1.70–1.45 (m, 6H), 1.30 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 149.4, 146.1, 146.0, 142.7, 128.2, 128.1, 126.4, 126.3, 125.7, 125.6, 125.3, 125.0, 79.2, 73.4, 58.9, 54.7, 53.3, 44.2, 39.7, 34.4, 31.4, 26.0, 25.9, 24.1. LCMS retention time: 4.137 min. LCMS purity 97.5%. HRMS (ESI): m/z calcd for C32H41NO2 [M + H]+ 472.3209, found 472.3219.
General Method C
(1-([1,1′-Biphenyl]-4-ylmethyl)piperidin-4-yl)diphenylmethanol (4g)
To a vial was added the 4-phenylbenzyl bromide 10t (0.10 g, 0.40 mmol), 7 (0.098 g, 0.37 mmol), and acetonitrile (3 mL). The triethylamine (0.077 mL, 0.55 mmol) was added and the reaction stirred at 70 °C for 4 h. The reaction was cooled to rt and diluted with water then extracted with EtOAc. The EtOAc layer was concentrated, and the crude product was purified by reverse-phase MPLC (10–100% MeCN:water) to produce the pure 4g (0.12 g, 0.27 mmol, 72% yield). 1H NMR (400 MHz, CDCl3): δ 7.61–7.55 (m, 2H), 7.55–7.51 (m, 2H), 7.51–7.46 (m, 4H), 7.46–7.39 (m, 2H), 7.39–7.32 (m, 3H), 7.32–7.26 (m, 4H), 7.22–7.12 (m, 2H), 3.55 (s, 2H), 3.04–2.80 (m, 2H), 2.50–2.36 (m, 1H), 2.11–1.88 (m, 3H), 1.56–1.43 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 140.9, 139.9, 137.1, 129.6, 128.7, 128.1, 127.1, 127.0, 126.8, 126.4, 125.7, 79.5, 62.8, 53.9, 44.1, 26.4. LCMS retention time: 4.101 min. LCMS purity 98.5%. HRMS (ESI): m/z calcd for C31H31NO [M + H]+ 434.2478, found 434.2485.
1-(4-(iso-Propyl)phenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (9b)
Method A: 7 (0.62 g, 2.32 mmol), 4-chloro-1-(4-iso-propylphenyl)butan-1-one (8b) (0.50 g, 2.21 mmol), and sodium bicarbonate (0.22 g, 2.65 mmol) with water and 2-butanone (15 mL, 1:5) to produce pure 9b (0.48 g, 1.06 mmol, 48% yield) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.89 (d, J = 8.3 Hz, 2H), 7.51–7.42 (m, 4H), 7.33–7.26 (m, 6H), 7.20–7.13 (m, 2H), 2.99–2.90 (m, 4H), 2.46–2.33 (m, 3H), 2.08 (s, 1H), 2.00–1.85 (m, 4H), 1.61 (s, 1H), 1.50–1.33 (m, 4H), 1.27 (d, J = 6.8 Hz, 6H).
1-(4-(Methyl)phenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (9c)
Method A: 7 (0.50 g, 1.87 mmol), 4-chloro-1-(4-methylphenyl)butan-1-one (8c) (0.35 g, 1.78 mmol), and sodium bicarbonate (0.18 g, 2.14 mmol) with water and 2-butanone (15 mL, 1:5) to produce pure 9c (0.27 g, 0.64 mmol, 36% yield) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.86 (d, J = 8.2 Hz, 2H), 7.51–7.43 (m, 4H), 7.32–7.26 (m, 4H), 7.24 (d, J = 8.0 Hz, 2H), 7.18 (tt, 2H), 3.02–2.83 (m, 4H), 2.46–2.32 (m, 6H), 2.21 (s, 1H), 2.05–1.81 (m, 4H), 1.57–1.34 (m, 4H).
1-Phenyl-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (9d)
Method A: 7 (0.52 g, 1.96 mmol), 4-chloro-1-phenylbutan-1-one 8d (0.30 mL, 1.87 mmol), and sodium bicarbonate (0.19 g, 2.24 mmol) with water:2-butanone (18 mL, 1:5) to produce pure 9d (0.18 g, 0.43 mmol, 23% yield) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 8.00–7.89 (m, 2H), 7.59–7.51 (m, 1H), 7.50–7.39 (m, 6H), 7.34–7.25 (m, 4H), 7.21–7.14 (m, 2H), 3.03–2.81 (m, 4H), 2.46–2.32 (m, 3H), 2.09 (br s, 1H), 2.01–1.86 (m, 4H), 1.53–1.29 (m, 4H).
1-(4-Fluorophenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (9e)
Method A: 7 (0.39 g, 1.44 mmol), 4-chloro-1-(4-fluorophenyl)butan-1-one 8e (0.28 g, 1.37 mmol), and sodium bicarbonate (0.14 g, 1.64 mmol) with water (3 mL) and 2-butanone (15 mL) to produce 9e (0.17 g, 0.40 mmol, 29% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.00–7.96 (m, 2H), 7.48–7.44 (m, 4H), 7.31–7.26 (m, 4H), 7.20–7.15 (m, 2H), 7.14–7.08 (m, 2H), 2.97–2.94 (m, 4H), 2.46–2.36 (m, 3H), 2.15 (br s, 1H), 2.01–1.88 (m, 4H), 1.52–1.35 (m, 4H).
1-(4-Chlorophenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (9f)
Method A: 7 (0.63 g, 2.34 mmol), 4-chloro-1-(4-chlorophenyl)butan-1-one 8f (0.48 g, 2.23 mmol), and sodium bicarbonate (0.23 g, 2.68 mmol) with water:2-butanone (18 mL, 1:5) to produce pure 9f (0.39 g, 0.88 mmol, 39% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.90 (d, J = 8.4 Hz, 2H), 7.48–7.44 (m, 4H), 7.42 (d, J = 8.6 Hz, 2H), 7.31–7.26 (m, 4H), 7.20–7.15 (m, 2H), 3.96–2.89 (m, 4H), 2.45–2.34 (m, 3H), 2.08 (br s, 1H), 1.99–1.87 (m, 4H), 1.50–1.30 (m, 4H).
1-(4-Bromophenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (9g)
Method A: 7 (0.44 g, 1.64 mmol), 1-(4-bromophenyl)-4-chlorobutan-1-one (8g) (0.41 g, 1.56 mmol), and sodium bicarbonate (0.16 g, 1.88 mmol) with water (3 mL) and 2-butanone (15 mL) to produce pure 9g (0.25 g, 0.50 mmol, 32% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.83–7.79 (m, 2H), 7.60–7.56 (m, 2H), 7.48–7.45 (m, 4H), 7.31–7.26 (m, 4H), 7.20–7.15 (m, 2H), 2.95–2.90 (m, 4H), 2.45–2.35 (m, 3H), 2.17 (br s, 1H), 2.00–1.88 (m, 4H), 1.51–1.33 (m, 4H).
1-(4-Methoxyphenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (9h)
Method A: 7 (0.49 g, 1.83 mmol), 4-chloro-1-(4-methoxyphenyl)butan-1-one (8h) (0.37 g, 1.74 mmol), and sodium bicarbonate (0.18 g, 2.09 mmol) with water (3 mL) and 2-butanone (15 mL) to produce pure 9h (0.28 g, 0.64 mmol, 36% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 8.9 Hz, 2H), 7.51–7.44 (m, 4H), 7.32–7.26 (m, 4H), 7.21–7.12 (m, 2H), 6.92 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H), 3.05–2.82 (m, 4H), 2.40 (q, J = 7.5, 6.6 Hz, 3H), 2.02–1.86 (m, 4H), 1.50–1.24 (m, 5H).
1-(4-(tert-Butyl)phenyl)-5-(4-(hydroxydiphenylmethyl)piperidin-1-yl)pentan-1-one (9j)
Method A: 7 (0.54 g, 2.04 mmol), 1-(4-(tert-butyl)phenyl)-5-chloropentan-1-one (8j) (0.49 g, 1.94 mmol), and sodium bicarbonate (0.20 g, 2.33 mmol) with water:2-butanone (18 mL, 1:5) to produce pure 9j (0.60 g, 1.24 mmol, 64% yield) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.93–7.84 (m, 2H), 7.51–7.42 (m, 6H), 7.34–7.27 (m, 4H), 7.21–7.12 (m, 2H), 3.02–2.91 (m, 4H), 2.43 (dd, J = 10.2, 4.6 Hz, 1H), 2.38–2.29 (m, 2H), 2.14 (s, 1H), 1.94 (td, J = 11.1, 4.0 Hz, 2H), 1.80–1.65 (m, 2H), 1.61–1.40 (m, 6H), 1.34 (s, 9H).
1-(4-(tert-Butyl)phenyl)-3-(4-(hydroxydiphenylmethyl)piperidin-1-yl)propan-1-one (9k)
Method A: 7 (0.54 g, 2.03 mmol), 1-(4-(tert-butyl)phenyl)-3-chloropropan-1-one (8k) (0.44 g, 1.94 mmol), and sodium bicarbonate (0.20 g, 2.32 mmol) with water:2-butanone (18 mL, 1:5) to produce pure 9k (0.84 g, 1.84 mmol, 95% yield) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.89 (d, J = 8.4 Hz, 2H), 7.50–7.45 (m, 6H), 7.32–7.25 (m, 4H), 7.21–7.14 (m, 2H), 3.15 (t, J = 7.1 Hz, 2H), 3.01–2.96 (m, 2H), 2.81 (t, J = 7.1 Hz, 2H), 2.49–2.42 (m, 1H), 2.15–2.04 (m, 3H), 1.56–1.46 (m, 4H), 1.33 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 198.9, 171.1, 156.8, 145.8, 134.3, 128.2, 128.0, 126.5, 125.8, 125.5, 79.4, 60.4, 54.2, 53.4, 44.0, 36.3, 35.1, 31.1, 26.4, 21.1, 14.2. LCMS retention time: 3.968 min. LCMS purity 98.1%. HRMS (ESI): m/z calcd for C31H37NO2 [M + H]+ 456.2896, found 456.2897.
1-(4-(tert-Butyl)phenyl)-2-(4-(hydroxydiphenylmethyl)piperidin-1-yl)ethanone (9l)
Method A: 7 (0.40 g, 1.49 mmol), 1-(4-(tert-butyl)phenyl)-2-chloroethanone (8l) (0.30 g, 1.43 mmol), and sodium bicarbonate (0.14 g, 1.71 mmol) with water (3 mL) and 2-butanone (15 mL) to produce pure 9l (0.45 g, 1.02 mmol, 72% yield) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.93 (d, J = 8.6 Hz, 2H), 7.50–7.43 (m, 6H), 7.32–7.26 (m, 4H), 7.20–7.15 (m, 2H), 3.78 (s, 2H), 3.07–3.01 (m, 2H), 2.47 (tt, J = 11.8 Hz, 3.5 Hz, 1H), 2.26–2.18 (m, 2H), 1.66–1.45 (m, 5H), 1.33 (s, 9H).). 13C NMR (125 MHz, CDCl3): δ 196.1, 157.0, 145.8, 133.5, 129.8, 128.2, 128.0, 126.5, 125.8, 125.5, 125.1, 79.5, 64.2, 54.2, 43.8, 35.1, 31.2, 31.0, 26.2. LCMS retention time: 3.987 min. LCMS purity 98.8%. HRMS (ESI): m/z calcd for C30H35NO2 [M + H]+ 442.2740, found 442.2743.
(4-(tert-Butyl)phenyl)(4-(hydroxydiphenylmethyl)piperidin-1-yl)methanone (9m)
To a vial was added the 7 (0.23 g, 0.87 mmol), acetonitrile (3 mL), and triethylamine (0.18 mL, 1.30 mmol). The 4-(tert-butyl)benzoyl chloride (8m) (0.17 mL, 0.95 mmol) was added and the reaction stirred at 70 °C for 6 h and was then allowed to cool to rt and was diluted with EtOAc (15 mL) then washed with saturated NaHCO3 (15 mL). The EtOAc was collected, dried with MgSO4, filtered, and adsorbed on silica and purified by MPLC (0–30% EtOAc:hexanes) to produce pure 9m (0.30 g, 0.71 mmol, 82% yield) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.50–7.44 (m, 4H), 7.38–7.14 (m, 10H), 4.77 (br s, 1H), 3.84 (br s, 1H), 3.05–2.63 (m, 3H), 2.25–2.17 (m, 1H), 1.75–1.35 (m, 4H), 1.29 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 170.4, 152.7, 145.4, 133.2, 128.3, 126.7, 125.7, 125.2, 79.5, 44.5, 34.7, 31.2. LCMS retention time: 3.774 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C29H33NO2 [M + H]+ 428.2583, found 428.2579.
1-(4-iso-Propylphenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (1b)
Method B: 9b (0.15 g, 0.34 mmol) and MeOH (2 mL) sodium borohydride (0.026 g, 0.68 mmol) to produce pure 1b (0.15 g, 0.32 mmol, 94% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.53–7.42 (m, 4H), 7.35–7.22 (m, 6H), 7.21–7.11 (m, 4H), 4.60 (dd, J = 8.1, 2.8 Hz, 1H), 3.14 (d, J = 11.0 Hz, 1H), 2.97 (d, J = 11.5 Hz, 1H), 2.87 (hept, J = 7.0 Hz, 1H), 2.51–2.33 (m, 3H), 2.29 (s, 1H), 2.11–1.87 (m, 3H), 1.85–1.42 (m, 8H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ 147.2, 146.1, 146.0, 128.2, 128.1, 126.5, 126.4, 126.1, 125.7, 125.7, 79.2, 73.5, 58.9, 54.7, 53.3, 44.2, 39.9, 33.7, 26.0, 25.9, 24.2, 24.1, 24.0. LCMS retention time: 4.056 min. LCMS purity 98.6%. HRMS (ESI): m/z calcd for C31H39NO2 [M + H]+ 458.3053, found 458.3062.
1-(4-Methyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (1c)
Method B: 9c (0.073 g, 0.17 mmol) and MeOH (2 mL) sodium borohydride (0.013 g, 0.34 mmol) to produce pure 1c (0.070 g, 0.16 mmol, 94% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.54–7.43 (m, 4H), 7.34–7.13 (m, 8H), 7.13–7.06 (m, 2H), 4.59 (dd, J = 8.1, 2.9 Hz, 1H), 3.16–3.07 (m, 1H), 3.00–2.88 (m, 1H), 2.54–2.35 (m, 4H), 2.32 (s, 3H), 2.05 (td, J = 11.9, 2.7 Hz, 1H), 2.01–1.85 (m, 2H), 1.85–1.72 (m, 1H), 1.71–1.43 (m, 7H). 13C NMR (125 MHz, CDCl3): δ 146.1, 146.0, 142.9, 136.0, 128.7, 128.1, 128.10, 128.07, 126.39, 126.37, 125.7, 125.64, 125.56, 79.2, 73.4, 58.9, 54.6, 53.4, 44.2, 39.9, 30.9, 26.0, 25.9, 24.0, 21.0. LCMS retention time: 3.809 min. LCMS purity 98.6%. HRMS (ESI): m/z calcd for C29H35NO2 [M + H]+ 430.2740, found 430.2753.
1-Phenyl-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (1d)
Method B: 9d (0.065 g, 0.16 mmol) and MeOH (2 mL) and sodium borohydride (0.012 g, 0.31 mmol) to produce pure 1d (0.048 g, 0.12 mmol, 73% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.53–7.44 (m, 4H), 7.38–7.33 (m, 2H), 7.29 (td, J = 7.6, 4.0 Hz, 6H), 7.23–7.09 (m, 3H), 4.63 (dd, J = 8.2, 2.7 Hz, 1H), 3.24–3.07 (m, 1H), 3.02–2.90 (m, 1H), 2.54–2.26 (m, 4H), 2.13–1.88 (m, 3H), 1.87–1.71 (m, 2H), 1.71–1.42 (m, 6H). 13C NMR (125 MHz, CDCl3): δ 146.0, 145.9, 145.8, 128.2, 128.1, 128.0, 126.6, 126.5, 126.4, 125.6, 125.6, 79.2, 73.6, 58.9, 54.7, 53.3, 44.2, 40.1, 26.0, 25.9, 24.1. LCMS retention time: 3.711 min. LCMS purity 99.8%. HRMS (ESI): m/z calcd for C28H33NO2 [M + H]+ 416.2583, found 416.2592.
1-(4-Fluorophenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (1e)
Method B: 9e (0.064 g, 0.15 mmol) and MeOH (2 mL) and sodium borohydride (0.011 g, 0.30 mmol) to produce pure 1e (0.059 g, 0.14 mmol, 92% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.52–7.43 (m, 4H), 7.34–7.26 (m, 6H), 7.21–7.12 (m, 2H), 6.97 (t, J = 8.7 Hz, 2H), 4.65–4.53 (m, 1H), 3.20–3.08 (m, 1H), 3.00–2.84 (m, 1H), 2.51–2.29 (m, 4H), 2.09 (td, J = 11.9, 2.6 Hz, 1H), 2.01–1.82 (m, 2H), 1.80–1.41 (m, 8H). 13C NMR (125 MHz, CDCl3): δ 161.6 (d, J = 243.8 Hz), 146.0 (d, J = 14.9 Hz), 141.7 (d, J = 3.1 Hz), 128.2, 128.1, 127.2, 127.1, 126.5, 126.4, 125.6, 125.5, 114.77 (d, J = 21.2 Hz), 79.2, 73.0, 58.8, 54.8, 53.1, 44.2, 40.3, 30.9, 26.0, 25.9, 24.1. LCMS retention time: 3.691 min. LCMS purity 98.5%. HRMS (ESI): m/z calcd for C28H32FNO2 [M + H]+ 434.2489, found 434.2482.
1-(4-Chlorophenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (1f)
Method B: 9f (0.16 g, 0.35 mmol) and MeOH (2 mL) sodium borohydride (0.026 g, 0.70 mmol) to produce pure 1f (0.12 g, 0.26 mmol, 75% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.53–7.42 (m, 4H), 7.34–7.23 (m, 8H), 7.21–7.11 (m, 2H), 4.59 (dd, J = 8.0, 2.5 Hz, 1H), 3.19–3.10 (m, 1H), 3.03–2.80 (m, 1H), 2.52–2.41 (m, 1H), 2.41–2.31 (m, 3H), 2.08 (td, J = 12.0, 2.8 Hz, 1H), 2.03–1.84 (m, 2H), 1.83–1.35 (m, 8H). 13C NMR (125 MHz, CDCl3): δ 146.0, 145.9, 144.5, 139.1, 128.19, 128.18, 128.1, 127.1, 126.49, 126.45, 125.62, 125.57, 79.2, 72.9, 58.8, 54.7, 53.2, 44.2, 40.2, 26.0, 25.9, 24.1. LCMS retention time: 3.845 min. LCMS purity 98.4%. HRMS (ESI): m/z calcd for C28H32ClNO2 [M + H]+ 450.2194, found 450.2203.
1-(4-Bromophenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (1g)
Method B: 9g (0.083 g, 0.17 mmol) and MeOH (2 mL) and sodium borohydride (0.013 g, 0.34 mmol) to produce pure 1g (0.053 g, 0.11 mmol, 64% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.52–7.43 (m, 4H), 7.43–7.36 (m, 2H), 7.33–7.26 (m, 4H), 7.24–7.20 (m, 2H), 7.20–7.14 (m, 2H), 4.58 (dd, J = 8.2, 2.4 Hz, 1H), 3.19–3.07 (m, 1H), 3.00–2.86 (m, 1H), 2.46 (tt, J = 11.9, 3.4 Hz, 1H), 2.41–2.34 (m, 3H), 2.08 (td, J = 11.9, 2.7 Hz, 1H), 1.98 (td, J = 11.9, 2.7 Hz, 1H), 1.95–1.86 (m, 1H), 1.78–1.45 (m, 8H). 13C NMR (125 MHz, CDCl3): δ 146.0, 145.9, 145.1, 131.1, 128.2, 128.1, 127.5, 126.5, 126.4, 125.6, 125.6, 120.2, 79.2, 72.9, 58.8, 54.7, 53.2, 44.1, 40.1, 26.0, 25.9, 24.1. LCMS retention time: 3.902 min. LCMS purity 99.1%. HRMS (ESI): m/z calcd for C28H32BrNO2 [M + H]+ 494.1685, found 494.1673.
1-(4-Methoxyphenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (1h)
Method B: 9h (0.11 g, 0.26 mmol) and MeOH (2 mL) and sodium borohydride (0.019 g, 0.51 mmol) to produce pure 1h (0.062 g, 0.14 mmol, 55% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.53–7.44 (m, 4H), 7.32–7.23 (m, 6H), 7.20–7.11 (m, 2H), 6.84 (d, J = 8.7 Hz, 2H), 4.58 (dd, J = 8.3, 2.6 Hz, 1H), 3.78 (s, 3H), 3.19–3.06 (m, 1H), 3.01–2.87 (m, 1H), 2.52–2.29 (m, 4H), 2.06 (td, J = 11.9, 2.6 Hz, 1H), 2.00–1.84 (m, 2H), 1.84–1.70 (m, 1H), 1.70–1.39 (m, 7H). 13C NMR (125 MHz, CDCl3): δ 158.3, 146.1, 146.0, 138.1, 128.1, 128.1, 126.7, 126.4, 126.4, 125.7, 125.6, 113.5, 79.2, 73.2, 58.9, 55.2, 54.7, 53.2, 44.2, 40.0, 26.0, 25.9, 24.1. LCMS retention time: 3.620 min. LCMS purity 98.3%. HRMS (ESI): m/z calcd for C29H35NO3 [M + H]+ 446.2689, found 446.2728.
Methyl 4-(1-Hydroxy-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butyl)benzoate (1i)
Step 1 (method B): 8i (0.042 g, 0.18 mmol) and MeOH with sodium borohydride (0.013 g, 0.35 mmol) to produce methyl 4-(4-chloro-1-hydroxybutyl)benzoate (0.037 g, 0.15 mmol, 87% yield) and was carried into the next reaction. 1H NMR (400 MHz, CDCl3): δ 8.00 (d, J = 8.4 Hz, 2H), 7.40 (d, J = 8.2 Hz, 2H), 4.8–4.6 (m, 1H), 3.90 (s, 3H), 3.58–3.52 (m, 2H), 2.19 (br s, 1H), 1.96–1.78 (m, 4H). Step 2: Methyl 4-(4-chloro-1-hydroxybutyl)benzoate (0.037 g, 0.15 mmol), 7 (0.12 g, 0.46 mmol), sodium bicarbonate (0.026 g, 0.31 mmol), sodium iodide (1.14 mg, 7.6 μmol), and the vial was evacuated with argon 3 times. Dry acetonitrile (2 mL) was added, and the reaction stirred overnight at reflux and was then cooled to rt after 18 h and the solvent was concentrated. The residue was dissolved in CH2Cl2 (5 mL) and washed with 0.1 N HCl (5 mL), water (5 mL), and brine (5 mL). The product was purified by MPLC (0–10% MeOH: CH2Cl2) to produce pure 1i (0.027 g, 0.057 mmol, 37% yield). 1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 8.4 Hz, 2H), 7.52–7.47 (m, 4H), 7.42 (d, J = 7.9 Hz, 2H), 7.32–7.29 (m, 4H), 7.20–7.14 (m, 2H), 4.66 (m, 1H), 3.90 (s, 3H), 3.14 (m, 1H), 2.94 (m, 1H), 2.78 (br s, 1H), 2.52–2.43 (m, 1H), 2.39 (t, J = 4.8 Hz, 2H), 2.13–2.06 (m, 1H), 2.04–1.92 (m, 2H), 1.77–1.47 (m, 8H). 13C NMR (125 MHz, CDCl3): δ 167.2, 151.4, 146.0, 145.9, 129.5, 128.4, 128.2, 128.2, 126.5, 126.5, 125.7, 125.6, 79.2, 73.2, 58.8, 54.7, 51.9, 44.2, 40.0, 26.0, 25.9, 24.0. LCMS retention time: 3.652 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C30H35NO4 [M + H]+ 474.2638, found 474.2646.
1-(4-(tert-Butyl)phenyl)-5-(4-(hydroxydiphenylmethyl)piperidin-1-yl)pentan-1-ol (1j)
Method B: 9j (0.20 g, 0.42 mmol) and MeOH (2 mL) and sodium borohydride (0.032 g, 0.84 mmol) to produce pure 1j (0.16 g, 0.32 mmol, 76% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.50–7.45 (m, 4H), 7.36 (d, J = 8.4 Hz, 2H), 7.31–7.24 (m, 6H), 7.19–7.14 (m, 2H), 4.65–4.60 (m, 1H), 2.98–2.90 (br m, 1H), 2.48–2.38 (m, 1H), 2.30 (t, J = 7.2 Hz, 2H), 2.23 (br s, 1H), 1.97–1.87 (m, 2H), 1.84–1.60 (m, 4H), 1.55–1.35 (m, 8H), 1.31 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 150.3, 146.0, 142.0, 128.1, 126.4, 125.8, 125.5, 125.3, 79.4, 74.1, 58.4, 54.1, 54.0, 44.2, 38.5, 34.5, 31.4, 31.3, 26.5, 26.3, 26.2, 23.7. LCMS retention time: 4.254 min. LCMS purity 96.4%. HRMS (ESI): m/z calcd for C33H43NO2 [M + H]+ 486.3366, found 486.3370.
1-(4-(tert-Butyl)phenyl)-3-(4-(hydroxydiphenylmethyl)piperidin-1-yl)propan-1-ol (1k)
Method B: 9k (0.12 g, 0.26 mmol) and MeOH (2 mL) and sodium borohydride (0.019 g, 0.51 mmol) to produce pure 1k (0.11 g, 0.23 mmol, 91% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.49–7.44 (m, 4H), 7.36–7.25 (m, 8H), 7.22–7.16 (m, 2H), 6.72 (br s, 1H), 4.90–4.85 (m, 1H), 3.21–3.15 (br m, 1H), 3.11–3.05 (br m, 1H), 2.70–2.62 (m, 1H), 2.57–2.40 (m, 2H), 2.14–2.06 (m, 2H), 1.91–1.79 (m, 3H), 1.57–1.45 (m, 4H), 1.31 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 149.6, 145.8, 145.7, 141.9, 128.2, 128.1, 126.6, 126.5, 125.8, 125.7, 125.2, 125.0, 79.4, 75.3, 57.3, 55.2, 53.2, 44.1, 34.4, 33.7, 31.4, 26.7, 26.4. LCMS retention time: 4.006 min. LCMS purity 97.7%. HRMS (ESI): m/z calcd for C31H39NO2 [M + H]+ 458.3053, found 458.3066.
1-(4-(tert-Butyl)phenyl)-2-(4-(hydroxydiphenylmethyl)piperidin-1-yl)ethanol (1l)
Method B: 9l (0.11 g, 0.26 mmol) and MeOH (1 mL) and sodium borohydride (0.019 g, 0.51 mmol) to produce pure 1l (0.11 g, 0.24 mmol, 93% yield) as a solid. 1H NMR (400 MHz, CDCl3): δ 7.50–7.46 (m, 4H), 7.37–7.26 (m, 8H), 7.22–7.17 (M, 2H), 4.67 (m, 1H), 4.01 (br s, 1H), 3.20 (m, 1H), 2.86 (m, 1H), 2.50–2.45 (m, 3H), 2.37–2.30 (m, 1H), 2.10–2.02 (m, 1H), 1.60–1.45 (m, 5H), 1.31 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 150.3, 145.82, 145.78, 139.1, 128.20, 128.2, 126.6, 126.6, 125.7, 125.6, 125.2, 79.5, 68.6, 66.3, 55.8, 53.4, 52.2, 44.1, 34.5, 31.3, 26.8, 26.5. LCMS retention time: 4.083 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C30H37NO2 [M + H]+ 444.2896, found 444.2915.
4-(1-Hydroxy-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butyl)benzoic Acid (1m)
To a vial was added the 1i (0.020 g, 0.041 mmol) and THF (1 mL). The LiOH (6.9 mg, 0.29 mmol) was dissolved in water (1 mL) and added to the reaction. The reaction stirred at rt for 18 h and was then acidified with 1.0 M HCl to pH 2–3 and extracted with CH2Cl2 (3 × 5 mL). The organic layer was concentrated and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 1m (0.009 g, 0.020 mmol, 47% yield). 1H NMR (400 MHz, CD3OD): δ 7.85 (d, J = 8.2 Hz, 2H), 7.55–7.45 (m, 4H), 7.38–7.23 (m, 6H), 7.22–7.11 (m, 2H), 4.70 (t, J = 5.8 Hz, 1H), 3.53–3.42 (m, 2H), 2.99 (t, J = 7.1 Hz, 2H), 2.93–2.74 (m, 3H), 1.86–1.56 (m, 8H). 13C NMR (125 MHz, CD3OD): δ 174.7, 148.2, 147.2, 137.6, 130.4, 129.1, 127.6, 127.0, 126.4, 79.9, 74.0, 53.9, 36.9, 25.5, 21.8. LCMS retention time: 2.490 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C29H33NO4 [M + H]+ 460.2482, found 460.2483.
1-([1,1′-Biphenyl]-4-yl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (1n)
Step 1: To a vial was added the 9g (0.098 g, 0.20 mmol), 1,1′-bis(di-tert-butylphosphino)ferrocene palladium dichloride (2.7 mg, 4.0 μmol), and phenylboronic acid (0.029 g, 0.24 mmol), followed by acetonitrile (1.5 mL). The potassium carbonate (0.041 g, 0.30 mmol) was dissolved in water (1.5 mL) and added the reaction. The reaction stirred at 60 °C for 18 h. The reaction was stopped, and the organic layer was diluted with saturated NaHCO3 (10 mL) and extracted with EtOAc (2 × 15 mL). The organic layers were collected, dried with MgSO4, filtered, and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce the desired intermediate 1-([1,1′-biphenyl]-4-yl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (0.035 g, 0.071 mmol, 36% yield) and was carried into step 2. 1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 8.6 Hz, 2H), 7.67 (d, J = 8.4 Hz, 2H), 7.64–7.61 (m, 2H), 7.50–7.45 (m, 6H), 7.42–7.38 (m, 1H), 7.31–7.26 (m, 4H), 7.19–7.14 (m, 2H), 3.02–2.91 (m, 4H), 2.46–2.37 (m, 3H), 2.10 (br s, 1H), 1.97–1.92 (m, 4H), 1.50–1.35 (m, 4H). Step 2. The intermediate from the previous reaction was carried into method B: 1-([1,1′-biphenyl]-4-yl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one (0.035 g, 0.071 mmol) and MeOH (2 mL) and sodium borohydride (5.4 mg, 0.143 mmol) to produce pure 1n (0.033 g, 0.067 mmol, 94% yield) as on oil. 1H NMR (400 MHz, CDCl3): δ 7.59–7.55 (m, 2H), 7.53–7.46 (m, 6H), 7.43–7.38 (m, 4H), 7.34–7.25 (m, 5H), 7.19–7.13 (m, 2H), 4.65 (dd, J = 8.2 Hz, 2.7 Hz, 1H), 3.16–3.10 (m, 1H), 2.99–2.93 (m, 1H), 2.56 (br s, 1H), 2.50–2.34 (m, 3H), 2.10–1.92 (m, 3H), 1.86–1.76 (m, 1H), 1.70–1.45 (m, 7H).). 13C NMR (125 MHz, CDCl3): δ 146.1, 146.0, 145.0, 141.1, 139.4, 128.6, 128.2, 128.1, 127.0, 126.9, 126.8, 126.4, 126.4, 126.1, 125.7, 125.6, 79.2, 73.3, 58.9, 54.7, 53.3, 44.2, 40.0, 26.0, 25.9, 24.1. LCMS retention time: 4.049 min. LCMS purity 97.6%. HRMS (ESI): m/z calcd for C34H37NO2 [M + H]+ 492.2896, found 492.2893.
Methyl 2-(4-(1-Hydroxy-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butyl)phenyl)-2-methylpropanoate (1o)
Method B: 18 (0.148 g, 0.288 mmol) and MeOH (1 mL) and sodium borohydride (0.016 g, 0.432 mmol) to produce pure 1o (0.11 g, 0.21 mmol, 73% yield). 1H NMR (400 MHz, CDCl3): δ 7.56–7.47 (m, 4H), 7.36–7.25 (m, 8H), 7.20 (m, 2H), 4.63 (dd, J = 8.2, 2.7 Hz, 1H), 3.65 (s, 3H), 3.25–3.09 (m, 1H), 3.07–2.90 (m, 1H), 2.55–2.36 (m, 3H), 2.29 (s, 1H), 2.10 (td, J = 11.9, 2.6 Hz, 1H), 2.02–1.86 (m, 2H), 1.87–1.44 (m, 14H). LCMS retention time: 3.786 min. LCMS purity 98.2%. HRMS (ESI): m/z calcd for C33H41NO4 [M + H]+ 516.3108, found 516.3141.
2-(4-(1-Hydroxy-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butyl)phenyl)-2-methyl propanoic Acid (1p)
To a vial was added the 1o (0.094 g, 0.18 mmol) and THF (3 mL) to form a solution. The LiOH (0.022 g, 0.91 mmol) was dissolved in water (3 mL) and then added to the reaction and stirred at rt for 18 h. The reaction was acidified with 1.0 M HCl in water to pH 3. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), and organic layers were combined and concentrated. The residue was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 2-(4-(1-hydroxy-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butyl)phenyl)-2-methylpropanoic acid 1p (0.038 g, 0.075 mmol, 41% yield). 1H NMR (400 MHz, DMSO-d6): δ 8.22 (s, 1H), 7.55–7.45 (m, 4H), 7.31–7.19 (m, 8H), 7.17–7.07 (m, 2H), 5.28 (s, 1H), 4.47 (t, J = 5.9 Hz, 1H), 2.98–2.82 (m, 2H), 2.36–2.24 (m, 2H), 2.07–1.92 (m, 2H), 1.45 (s, 12H), 1.31–1.19 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 177.7, 163.7, 147.2, 144.4, 143.4, 127.8, 125.8, 125.7, 125.6, 125.1, 78.4, 71.8, 57.7, 53.4, 53.2, 45.5, 43.1, 37.3, 26.5, 25.6, 22.6. LCMS retention time: 2.612 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C32H39NO4 [M + H]+ 502.2951, found 502.2952.
(1-(4-(4-(tert-Butyl)phenyl)butyl)piperidin-4-yl)diphenylmethanol (2a)
To a vial was added the 7 (0.15 g, 0.57 mmol), 1-(tert-butyl)-4-(4-chlorobutyl)benzene (10a) (0.15 g, 0.69 mmol), and potassium carbonate (0.47 g, 3.43 mmol) in acetonitrile (5 mL). The reaction stirred overnight at 85 °C and for 18 h and was then cooled to rt and filtered. The filtrate was then diluted with brine and extracted with EtOAc (3 × 15 mL). The organic layers were combined, dried with MgSO4, filtered, and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 2a (0.18 g, 0.40 mmol, 69% yield) as an oil. 1H NMR (400 MHz, CDCl3): δ 7.38–7.34 (m, 4H), 7.19–7.14 (m, 6H), 7.07–7.02 (m, 2H), 6.99–6.95 (m, 2H), 2.86–2.80 (m, 2H), 2.45 (t, J = 7.3 Hz, 2H), 2.35–2.26 (m, 1H), 2.22–2.17 (m, 2H), 1.84–1.76 (m, 3H), 1.52–1.30 (m, 8H), 1.18 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 148.4, 146.0, 139.4, 128.1, 128.0, 126.4, 125.8, 125.1, 79.5, 58.8, 54.1, 44.2, 35.2, 34.3, 31.4, 29.5, 26.8, 26.4. LCMS retention time: 3.928 min. LCMS purity 98.5%. HRMS (ESI): m/z calcd for C32H41NO [M + H]+ 456.3260, found 456.3282.
(1-(3-(4-(tert-Butyl)phenyl)propyl)piperidin-4-yl)diphenylmethanol (2b)
Same procedure as 2a using 7 (0.14 g, 0.53 mmol), 1-(tert-butyl)-4-(3-chloropropyl)benzene (10b) (0.13 g, 0.64 mmol), and potassium carbonate (0.44 g, 3.18 mmol) in acetonitrile (5 mL). Purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 2b (0.15 g, 0.34 mmol, 65% yield) as an oil. 1H NMR (400 MHz, CDCl3): δ 7.49–7.45 (m, 4H), 7.31–7.24 (m, 6H), 7.19–7.14 (m, 2H), 7.11–7.08 (m, 2H), 2.96 (m, 2H), 2.57 (t, J = 7.7 Hz, 2H), 2.48–2.32 (m, 3H), 2.22 (br s, 1H), 1.97–1.90 (m, 2H), 1.83–1.75 (m, 2H), 1.53–1.45 (m, 4H), 1.29 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 148.5, 145.9, 138.8, 128.1, 127.9, 126.4, 125.7, 125.2, 79.4, 58.2, 53.9, 44.0, 40.9, 34.3, 33.2, 31.2, 28.4, 26.1. LCMS retention time: 2.965 min. LCMS purity 97.2%. HRMS (ESI): m/z calcd for C31H39NO [M + H]+ 442.3104, found 442.3113.
(1-(4-(tert-Butyl)phenethyl)piperidin-4-yl)diphenylmethanol (2c)
Same procedure as 2a using 7 (0.15 g, 0.56 mmol), 1-(tert-butyl)-4-(2-chloroethyl)benzene (10c) (0.11 g, 0.56 mmol), and potassium carbonate (0.47 g, 3.37 mmol) in acetonitrile (5 mL). Purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 2c (0.18 g, 0.421 mmol, 75% yield) as an oil. 1H NMR (400 MHz, CDCl3): δ 7.53–7.45 (m, 4H), 7.35–7.27 (m, 6H), 7.22–7.15 (m, 2H), 7.15–7.08 (m, 2H), 3.09–3.00 (m, 2H), 2.81–2.72 (m, 2H), 2.60–2.53 (m, 2H), 2.51–2.40 (m, 1H), 2.28 (s, 1H), 2.11–2.00 (m, 2H), 1.63–1.44 (m, 4H), 1.30 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 148.8, 145.9, 137.3, 128.3, 128.1, 126.5, 125.8, 125.2, 79.5, 60.8, 54.0, 44.2, 40.9, 34.3, 33.1, 31.4, 26.4. LCMS retention time: 4.384 min. LCMS purity 99.7%. HRMS (ESI): m/z calcd for C30H37NO [M + H]+ 428.2947, found 428.2955.
(1-(4-(tert-Butyl)benzyl)piperidin-4-yl)diphenylmethanol (2d)
Method C: 7 (0.085 mL, 0.39 mmol), 4-tert-butylbenzyl bromide (10d) (0.098 g, 0.43 mmol), triethylamine (0.082 mL, 0.59 mmol), and acetonitrile (2 mL) was then added and the reaction stirred at 70 °C and stirred for 5 h then diluted with EtOAc (15 mL) and washed with saturated NaHCO3 (15 mL). The EtOAc was collected, dried with MgSO4, filtered, and adsorbed to silica and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 2d (0.13 g, 0.32 mmol, 81% yield) as a brown oil. 1H NMR (400 MHz, CDCl3): δ 7.40–7.30 (m, 4H), 7.23–7.11 (m, 6H), 7.11–6.95 (m, 4H), 3.34 (s, 2H), 2.86–2.71 (m, 2H), 2.35–2.20 (m, 1H), 1.99 (s, 1H), 1.89–1.76 (m, 2H), 1.42–1.28 (m, 4H), 1.18 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 149.8, 146.0, 135.1, 128.9, 128.1, 126.4, 125.8, 125.0, 79.5, 62.8, 53.9, 44.2, 34.4, 31.4, 26.5. LCMS retention time: 4.186 min. LCMS purity 99.7%. HRMS (ESI): m/z calcd for C29H35NO [M + H]+ 414.2767, found 414.2786.
4-(4-Benzylpiperidin-1-yl)-1-(4-(tert-butyl)phenyl)butan-1-ol (2e)
Step 1: To a vial was added 12 (0.40 g, 1.21 mmol) and dry CH2Cl2 (15 mL). The methanesulfonyl chloride (0.024 g, 0.35 g, 1.82 mmol) and triethylamine (0.51 mL, 3.64 mmol) were each added to the vial and the reaction stirred at rt for 16 h. The reaction was then diluted with CH2Cl2 (15 mL) and washed with 1% w/v sulfuric acid in water (3 × 25 mL), saturated NaHCO3 (25 mL), and brine (25 mL). The organic layer was dried with MgSO4, filtered, and concentrated to produce 3-(4-(hydroxy diphenylmethyl)piperidin-1-yl)propyl-4-methylbenzenesulfonate (0.19 g, 0.40 mmol, 33% yield), of which a portion was carried into step 2. 1H NMR (400 MHz, CDCl3): δ 7.69–7.66 (m, 2H), 7.46–7.41 (m, 4H), 7.24–7.19 (m, 4H), 7.15–7.05 (m, 4H), 4.26–4.12 (m, 4H), 3.73–3.65 (m, 2H), 3.27–3.18 (m, 2H), 2.68–2.60 (m, 1H), 2.47–2.37 (m, 2H), 2.30 (s, 3H), 1.88–1.76 (m, 3H), 1.52–1.43 (m, 2H). Step 2: To a vial was added the 3-(4-(hydroxydiphenyl methyl)piperidin-1-yl)propyl-4-methylbenzenesulfonate (0.088 g, 0.18 mmol), 4-(tert-butyl) aniline (0.035 mL, 0.22 mmol), and triethylamine (0.038 mL, 0.28 mmol) with acetonitrile (3 mL). The reaction began to stir at 85 °C for 18 h and was then cooled to rt and diluted with saturated NaHCO3 (10 mL) then extracted with EtOAc (2 × 15 mL). The organic layers were combined and concentrated then purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 2e (0.038 g, 0.083 mmol, 45% yield). 1H NMR (400 MHz, CDCl3): δ 7.50–7.46 (m, 4H), 7.33–7.28 (m, 4H), 7.21–7.16 (m, 4H), 6.51 (d, J = 8.7 Hz, 2H), 3.13 (t, J = 6.4 Hz, 2H), 3.02–2.96 (m, 2H), 2.49–2.41 (m, 3H), 2.15 (br s, 1H), 2.00–1.93 (m, 2H), 1.80–1.73 (m, 2H), 1.57–1.43 (m, 5H), 1.27 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 146.4, 145.9, 139.7, 128.2, 126.5, 125.9, 125.8, 112.4, 79.6, 57.4, 54.2, 44.2, 43.7, 33.8, 31.5, 26.6, 26.2. LCMS retention time: 4.208 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C31H40N2O [M + H]+ 457.3213, found 457.3206.
(1-(3-(4-(tert-Butyl)phenoxy)propyl)piperidin-4-yl)diphenylmethanol (2f)
To a vial was added the 3-(4-(hydroxydiphenyl methyl)piperidin-1-yl)propyl 4-methylbenzenesulfonate (0.11 g, 0.22 mmol) (from 2e step 1), 4-(tert-butyl)phenol (0.039 g, 0.26 mmol), and triethylamine (0.046 mL, 0.33 mmol) with acetonitrile (3 mL). The reaction began to stir at 85 °C for 18 h and was then cooled to rt and diluted with saturated NaHCO3 (10 mL) then extracted with EtOAc (2 × 15 mL). The organic layers were combined and concentrated then purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 2f (0.015 g, 0.033 mmol, 15% yield). 1H NMR (400 MHz, CDCl3): δ 7.49–7.46 (m, 4H), 7.31–7.26 (m, 6H), 7.19–7.14 (m, 2H), 6.81 (d, J = 8.8 Hz, 2H), 3.96 (t, J = 6.4 Hz, 2H), 3.01–2.94 (m, 2H), 2.51–2.40 (m, 3H), 1.99–1.89 (m, 5H), 1.52–1.45 (m, 4H), 1.28 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 156.7, 145.9, 143.2, 128.1, 126.5, 126.1, 125.7, 116.3, 113.9, 79.5, 66.3, 60.4, 55.4, 54.1, 44.1, 34.0, 31.5, 29.7, 27.0, 26.4. LCMS retention time: 4.293 min. LCMS purity 95.2%. HRMS (ESI): m/z calcd for C31H39NO2 [M + H]+ 458.3053, found 458.3065.
(1-(4-Amino-4-(4-(tert-butyl)phenyl)butyl)piperidin-4-yl)diphenylmethanol (2g)
To a vial was added the 9a (0.20 g, 0.43 mmol), ammonium acetate (0.33 g, 4.26 mmol), and sodium cyanoborohydride (0.040 g, 0.64 mmol) with MeOH (5 mL). The reaction stirred at rt for 18 h then concentrated and diluted with aqueous ammonium hydroxide (10 mL) then extracted with CH2Cl2 (3 × 10 mL). The organic layers were combined, dried with MgSO4, filtered, and concentrated then purified by reverse-phase MPLC (10–100% CH3CN:water). Fractions containing the desired product were further purified by MPLC (0–10% MeOH (5% NH3OH):CH2Cl2) to produce pure 2g (0.010 g, 0.021 mmol, 5% yield). 1H NMR (400 MHz, CDCl3): δ 7.48–7.44 (m, 4H), 7.34–7.26 (m, 6H), 7.22–7.14 (m, 4H), 3.84 (t, J = 6.7 Hz, 1H), 2.95–2.87 (m, 2H), 2.46–2.37 (m, 1H), 2.28 (t, J = 7.6 Hz, 2H), 1.95–1.85 (m, 2H), 1.70–1.60 (m, 5H), 1.55–1.40 (m, 6H), 1.30 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 149.7, 146.0, 143.3, 128.1, 126.5, 125.9, 125.8, 125.3, 79.5, 58.7, 55.8, 54.2, 54.0, 44.2, 37.5, 34.4, 31.4, 26.4, 24.2. LCMS retention time: 2.397 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C32H42N2O [M + H]+ 471.3369, found 471.3366.
(S)-1-(4-(tert-Butyl)phenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (2h)
To a vial was added 23 (0.14 g, 0.26 mmol), and the vial was evacuated with nitrogen three times. The dry THF (9 mL) was then added. The 1.0 M lithium aluminum hydride (0.26 mL, 0.26 mmol) in THF was added dropwise at rt and the reaction stirred for 5 h. The reaction was quenched slowly with water (10 mL) and then extracted with diethyl ether (2 × 10 mL). The ether layer was dried with MgSO4, filtered, concentrated, and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 2h (0.10 g, 0.21 mmol, 81% yield); [α]D25 −38.8 (c 1, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.52–7.46 (m, 4H), 7.33–7.25 (m, 8H), 7.21–7.15 (m, 2H), 4.61–4.56 (m, 1H), 3.16–3.11 (br m, 1H), 3.00–2.94 (m, 1H), 2.51–2.34 (m, 4H), 2.10–1.88 (m, 4H), 1.83–1.75 (m, 1H), 1.70–1.45 (m, 6H), 1.30 (s, 9H). 13C NMR (126 MHz, CDCl3): δ 149.4, 146.1, 146.0, 142.7, 128.2, 128.1, 126.5, 126.4, 125.7, 125.6, 125.4, 125.0, 79.3, 73.4, 58.9, 54.7, 53.3, 44.2, 39.7, 34.4, 31.4, 26.1, 26.0, 24.1. LCMS retention time: 4.159 min. LCMS purity 99.7%. HRMS (ESI): m/z calcd for C32H41NO2 [M + H]+ 472.3209, found 472.3234.
(R)-1-(4-(tert-Butyl)phenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-ol (2i)
To a vial was added 24 (0.24 g, 0.46 mmol), and this vial was evacuated with nitrogen three times. The dry THF (4.5 mL) was added, and then the 1.0 M lithium aluminum hydride (0.46 mL, 0.46 mmol) in THF was added portionwise at rt for 5 h. The reaction was quenched slowly with water (10 mL) and then extracted with diethyl ether (2 × 10 mL). The ether layer was dried with MgSO4, filtered, concentrated, and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 2i (0.12 g, 0.25 mmol, 53% yield); [α]D25 +38.6 (c 1, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.52–7.46 (m, 4H), 7.33–7.25 (m, 8H), 7.21–7.15 (m, 2H), 4.61–4.56 (m, 1H), 3.16–3.11 (br m, 1H), 3.00–2.94 (m, 1H), 2.51–2.34 (m, 4H), 2.10–1.88 (m, 4H), 1.83–1.75 (m, 1H), 1.70–1.45 (m, 6H), 1.30 (s, 9H). 13C NMR (126 MHz, CDCl3): δ 149.4, 146.1, 146.0, 142.7, 128.2, 128.1, 126.5, 126.4, 125.7, 125.6, 125.4, 125.0, 79.3, 73.4, 58.9, 54.7, 53.3, 44.2, 39.7, 34.4, 31.4, 26.1, 26.0, 24.1. LCMS retention time: 4.156 min. LCMS purity 97.8%. HRMS (ESI): m/z calcd for C32H41NO2 [M + H]+ 472.3209, found 472.3236.
(1-(3-(tert-Butyl)phenethyl)piperidin-4-yl)diphenylmethanol (3a)
Method C: 1-(2-Bromoethyl)-3-(tert-butyl)benzene 10e (0.060 g, 0.18 mmol), 7 (0.048 g, 0.18 mmol), triethylamine (0.038 mL, 0.271 mmol), and acetonitrile (1 mL) were purified by reverse-phase MPLC (10–100% CH3CN:water) to produce the pure 3a (0.065 g, 0.15 mmol, 84% yield). 1H NMR (400 MHz, CDCl3): δ 7.51–7.47 (m, 4H), 7.32–7.27 (m, 4H), 7.23–7.16 (m, 5H), 7.01–6.98 (m, 1H), 3.09–3.04 (m, 2H), 2.81–2.76 (m, 2H), 2.61–2.55 (m, 2H), 2.51–2.43 (m, 1H), 2.28 (br s, 1H), 2.10–2.02 (m, 2H), 1.30 (s, 9H), 1.32–1.26 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 151.2, 145.9, 140.0, 128.1, 128.0, 126.5, 125.8, 125.7, 125.7, 123.0, 79.5, 54.1, 44.2, 34.6, 34.0, 31.6, 31.4, 26.4, 22.6. LCMS retention time: 4.214 min. LCMS purity 96.4%. HRMS (ESI): m/z calcd for C30H37NO [M + H]+ 428.2948, found 428.2970.
(1-(2-(tert-Butyl)phenethyl)piperidin-4-yl)diphenylmethanol (3b)
Method C: 2-(tert-Butyl)phenethyl 4-methylbenzenesulfonate 10f (0.022 g, 0.066 mmol), 7 (0.018 g, 0.066 mmol), triethylamine (0.014 mL, 0.099 mmol), and acetonitrile (1 mL) were purified by reverse-phase MPLC (10–100% CH3CN:water) to produce the pure 3b (0.013 g, 0.030 mmol, 46% yield). 1H NMR (400 MHz, CDCl3): δ 7.51–7.48 (m, 4H), 7.37–7.28 (m, 5H), 7.21–7.09 (m, 5H), 3.15–3.04 (m, 4H), 2.65–2.60 (m, 2H), 2.52–2.42 (m, 1H), 2.17–2.08 (m, 3H), 1.60–1.53 (m, 4H), 1.41 (s. 9H). 13C NMR (125 MHz, CDCl3): δ 147.7, 145.9, 138.5, 132.1, 128.2, 126.5, 126.1, 125.9, 125.8, 125.8, 79.5, 61.7, 54.2, 44.2, 35.7, 31.7, 29.7, 26.4. LCMS retention time: 4.200 min. LCMS purity 99.3%. HRMS (ESI): m/z calcd for C30H37NO [M + H]+ 428.2947, found 428.2948.
(1-(4-Fluorophenethyl)piperidin-4-yl)diphenylmethanol (3c)
Method C: 7 (0.099 g, 0.37 mmol), 1-(2-bromoethyl)-4-fluorobenzene 10g (0.047 mL, 0.34 mmol), triethylamine (0.070 mL, 0.51 mmol), and acetonitrile (10 mL) were purified by MPLC (0–10% MeOH:CH2Cl2) to produce pure 3c (0.12 g, 0.32 mmol, 94% yield). 1H NMR (400 MHz, CDCl3): δ 7.51–7.47 (m, 4H), 7.32–7.27 (m, 4H), 7.21–7.11 (m, 4H), 6.98–6.92 (m, 2H), 3.06–3.00 (m, 2H), 2.78–2.72 (m, 2H), 2.56–2.51 (m, 2H), 2.50–2.42 (m, 1H), 2.21 (br s, 1H), 2.08–1.99 (m, 2H), 1.57–1.48 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 161.2 (d, J = 243.6 Hz), 145.9, 136.5 (d, J = 3.2 Hz), 129.9 (d, J = 7.9 Hz), 128.1, 126.5, 125.8, 115.0 (d, J = 21.2 Hz), 79.5, 60.8, 54.1, 44.2, 33.0, 26.4, 21.0. LCMS retention time: 3.733 min. LCMS purity 96.8%. HRMS (ESI): m/z calcd for C26H28FNO [M + H]+ 390.2228, found 390.2227.
(1-(4-Chlorophenethyl)piperidin-4-yl)diphenylmethanol (3d)
Method C: 7 (0.22 g, 0.83 mmol), 1-(2-bromoethyl)-4-chlorobenzene 10h (0.17 g, 0.75 mmol), triethylamine (0.16 mL, 1.13 mmol), and acetonitrile (4 mL) were purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 3d (0.22 g, 0.55 mmol, 73% yield). 1H NMR (400 MHz, CDCl3): δ 7.39–7.36 (m, 4H), 7.20–7.15 (m, 4H), 7.12–7.09 (m, 2H), 7.08–7.03 (m, 2H), 6.99–6.96 (m, 2H), 2.92–2.87 (m, 2H), 2.64–2.58 (m, 2H), 2.43–2.38 (m, 2H), 2.37–2.30 (m, 1H), 2.22 (br s, 1H), 1.95–1.88 (m, 2H), 1.44–1.35 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 145.8, 138.9, 131.6, 129.9, 128.3, 128.0, 126.4, 125.7, 79.4, 79.3, 60.4, 57.0, 44.1, 33.0, 26.4. LCMS retention time: 3.903 min. LCMS purity 99.6%. HRMS (ESI): m/z calcd for C26H28ClNO [M + H]+ 406.1931, found 406.1930.
(1-(4-Bromophenethyl)piperidin-4-yl)diphenylmethanol (3e)
Method C: 7 (0.50 g, 1.86 mmol), 1-(2-bromoethyl)-4-bromobenzene 10i (0.26 mL, 1.69 mmol), triethylamine (0.353 mL, 2.53 mmol), and acetonitrile (10 mL) were purified by MPLC (0–10% MeOH:CH2Cl2) to produce pure 3e (0.49 g, 1.08 mmol, 64% yield). 1H NMR (400 MHz, CDCl3): δ 7.50–7.46 (m, 4H), 7.38 (d, J = 8.3 Hz, 2H), 7.34–7.27 (m, 4H), 7.18 (tt, J = 7.3 Hz, 1.8 Hz, 2H), 7.06 (d, J = 8.4 Hz, 2H), 3.06–3.00 (m, 2H), 2.77–2.72 (m, 2H), 2.58–2.52 (m, 2H), 2.50–2.41 (m, 1H), 2.15–2.00 (m, 3H), 1.56–1.49 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.8, 139.3, 131.4, 130.4, 128.2, 126.6, 125.8, 119.8, 79.5, 60.4, 54.0, 44.1, 33.1, 26.4, 21.1 LCMS retention time: 3.969 min. LCMS purity 99.8%. HRMS (ESI): m/z calcd for C26H28BrNO [M + H]+ 450.1426, found 452.1403.
(1-(4-Trifluoromethylphenethyl)piperidin-4-yl)diphenylmethanol (3f)
Method C: 7 (0.10 g, 0.37 mmol), 1-(2-bromoethyl)-4-trifluorobenzene 10j (0.057 mL, 0.34 mmol), triethylamine (0.071 mL, 0.51 mmol), and acetonitrile (10 mL) were purified by MPLC (0–10% MeOH:CH2Cl2) to produce pure 3f (0.074 g, 0.17 mmol, 49% yield) as an oil. 1H NMR (400 MHz, CDCl3): δ 7.54–7.47 (m, 6H), 7.33–7.27 (m, 6H), 7.19 (tt, J = 7.3 Hz, 1.8 Hz, 2H), 3.06–3.00 (m, 2H), 2.86–2.80 (m, 2H), 2.60–2.55 (m, 2H), 2.51–2.42 (m, 1H), 2.14 (br s, 1H), 2.10–2.03 (m, 2H), 1.57–1.47 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 144.6, 129.0, 128.3 (q, J = 32 Hz), 128.2, 126.5, 125.8, 125.2 (q, 3.8 Hz), 124.2 (q, J = 271.8 Hz), 79.5, 60.2, 54.1, 44.1, 33.6, 26.4, 21.0. LCMS retention time: 3.877 min. LCMS purity 99.1%. HRMS (ESI): m/z calcd for C27H28F3NO [M + H]+ 440.2195, found 440.2204.
(1-(4-Methoxyphenethyl)piperidin-4-yl)diphenylmethanol (3g)
Method C: 7 (0.38 g, 1.43 mmol), 1-(2-bromoethyl)-4-methoxybenzene 10k (0.25 mL, 1.57 mmol), triethylamine (0.30 mL, 2.14 mmol), and acetonitrile (5 mL) were purified by MPLC (0–10% MeOH:CH2Cl2) to produce pure 3g (0.29 g, 0.72 mmol, 50% yield) as a sticky solid. 1H NMR (400 MHz, CDCl3): δ 7.49–7.46 (m, 4H), 7.32–7.28 (m, 4H), 7.19 (tt, J = 7.3 Hz, 1.9 Hz, 2H), 7.12 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.5 Hz, 2H), 3.77 (s, 3H), 3.29–3.24 (m, 2H), 2.95–2.90 (m, 2H), 2.80–2.75 (m, 2H), 2.58–2.50 (m, 1H), 2.42–2.30 (m, 3H), 1.90–1.77 (m, 2H), 1.65–1.57 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 158.3, 145.5, 129.6, 128.3, 126.7, 125.6, 114.0, 79.3, 55.3, 53.6, 53.4, 43.4, 31.5, 25.1. LCMS retention time: 3.695 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C27H31NO2 [M + H]+ 402.2426, found 402.2428.
(1-(2-(6-(tert-Butyl)pyridin-3-yl)ethyl)piperidin-4-yl)diphenylmethanol (3h)
Method C: 7 (0.25 g, 0.94 mmol), 2-(6-(tert-butyl)pyridin-3-yl)ethyl-4-methylbenzenesulfonate 10l (0.26 g, 0.78 mmol), triethylamine (0.16 mL, 1.17 mmol), and acetonitrile (5 mL) were purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 3h (0.31 g, 0.72 mmol, 92% yield). 1H NMR (400 MHz, CDCl3): δ 8.39–8.38 (m, 1H), 7.51–7.48 (m, 4H), 7.42 (dd, J = 8.1 Hz, 2.4 Hz, 1H), 7.32–7.27 (m, 4H), 7.25–7.22 (m, 1H), 7.20–7.15 (m, 2H), 3.07–3.01 (m, 2H), 2.76–2.71 (m, 2H), 2.57–2.52 (m, 2H), 2.50–2.42 (m, 1H), 2.31 (br s, 1H), 2.09–2.01 (m, 2H), 1.57–1.48 (m, 4H), 1.35 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 166.9, 148.7, 145.9, 136.3, 132.3, 128.1, 126.5, 125.8, 118.6, 79.4, 60.2, 54.0, 44.1, 37.0, 30.3, 30.2, 26.4. LCMS retention time: 3.802 min. LCMS purity 99.5%. HRMS (ESI): m/z calcd for C29H36N2O [M + H]+ 429.2900, found 429.2894.
(1-(4-Nitrophenethyl)piperidin-4-yl)diphenylmethanol (3i)
Method C: 7 (0.51 g, 1.92 mmol), 1-(2-bromoethyl)-4-nitrobenzene 10m (0.40 g, 1.74 mmol), triethylamine (0.37 mL, 2.62 mmol), and acetonitrile (10 mL) were purified by MPLC (0–10% MeOH:CH2Cl2) to produce 3i (0.15 g, 0.35 mmol, 20% yield). 1H NMR (400 MHz, CDCl3): δ 8.13 (d, J = 8.7 Hz, 2H), 7.49–7.46 (m, 4H), 7.35 (d, J = 8.7 Hz, 2H), 7.32–7.27 (m, 4H), 7.21–7.16 (m, 2H), 3.11–3.05 (m, 2H), 2.97–2.91 (m, 2H), 2.69–2.63 (m, 2H), 2.52–2.43 (m, 1H), 2.25 (br s, 1H), 2.20–2.10 (m, 2H), 1.63–1.53 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 147.9, 146.5, 145.7, 129.5, 128.2, 126.6, 125.7, 123.7, 79.4, 59.4, 53.9, 43.9, 33.2, 26.1, 21.1. LCMS retention time: 3.654 min. LCMS purity 98.8%. HRMS (ESI): m/z calcd for C26H28N2O3 [M + H]+ 417.2172, found 417.2163.
(1-(4-Aminophenethyl)piperidin-4-yl)diphenylmethanol (3j)
To a vial was added 3i (0.14 g, 0.32 mmol) with MeOH (1 mL) and CH2Cl2 (1 mL). The reaction was cooled to 0 °C, and the Raney nickel (2 mg, 0.032 mmol) was added. The sodium borohydride (0.031 g, 0.81 mmol) was then added portionwise, and the reaction stirred at rt for 28 h, after which the Raney nickel was filtered through Celite. The reaction was diluted with water (10 mL) and extracted with CH2Cl2 (3 × 10 mL), and the organic layers were combined and dried with MgSO4, filtered, and adsorbed to silica then purified by MPLC (0–15% MeOH:CH2Cl2) to produce 3j (0.077 g, 0.20 mmol, 61% yield). 1H NMR (400 MHz, CDCl3): δ 7.51–7.47 (m, 4H), 7.32–7.27 (m, 4H), 7.18 (tt, J = 7.3 Hz, 1.8 Hz, 2H), 6.97 (d, J = 8.3 Hz, 2H), 6.61 (d, J = 8.3 Hz, 2H), 3.52 (br s, 2H), 3.07–3.01 (m, 2H), 2.70–2.65 (m, 2H), 2.54–2.41 (m, 3H), 2.22 (br s, 1H), 2.07–1.98 (m, 2H), 1.56–1.49 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 144.4, 130.4, 129.4, 128.1, 126.5, 125.8, 115.2, 79.5, 61.2, 54.1, 44.2, 32.8, 26.4, 21.0. LCMS retention time: 3.328 min. LCMS purity 97.0%. HRMS (ESI): m/z calcd for C26H30N2O [M + H]+ 387.2430, found 387.2412.
(1-(4-(Dimethylamino)phenethyl)piperidin-4-yl)diphenylmethanol (3k)
To a vial was added 3j (0.030 g, 0.078 mmol) and acetic acid (1 mL). The paraformaldehyde (0.058 mL, 0.78 mmol) solution in water followed by sodium cyanoborohydride (0.015 g, 0.23 mmol) was then added and the reaction stirred at rt for 20 h. The reaction was concentrated and diluted with saturated NaHCO3 (10 mL) and extracted with EtOAc (3 × 10 mL). The organic layers were dried with MgSO4, filtered, and concentrated. The product was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 3k (0.027 g, 0.065 mmol, 84% yield). 1H NMR (400 MHz, CDCl3): δ 7.50–7.46 (m, 4H), 7.31–7.26 (m, 4H), 7.17 (tt, J = 7.3 Hz, 1.8 Hz, 2H), 7.06 (d, J = 8.3 Hz, 2H), 6.68 (d, J = 8.3 Hz, 2H), 3.08–3.02 (m, 2H), 2.90 (s, 6H), 2.72–2.67 (m, 2H), 2.56–2.50 (m, 2H), 2.48–2.42 (m, 1H), 2.30 (br s, 1H), 2.07–2.00 (m, 2H), 1.56–1.51 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 149.1, 146.0, 129.2, 128.4, 128.1, 126.4, 125.8, 112.9, 79.4, 61.1, 54.0, 44.2, 41.0, 40.8, 32.6, 26.4. LCMS retention time: 2.50 min. LCMS purity 96.1%. HRMS (ESI): m/z calcd for C28H34N2O [M + H]+ 415.2744, found 415.2743.
(1-(2-([1,1′-Biphenyl]-4-yl)ethyl)piperidin-4-yl)diphenylmethanol (3l)
To a vial was added the phenylboronic acid (0.019 g, 0.15 mmol), 1,1′-bis(di-tert-butylphosphino)ferrocene palladium dichloride (4.1 mg, 6.3 μmol), 3e (0.057 g, 0.13 mmol), and potassium carbonate (0.035 g, 0.253 mmol). The vial was then evacuated with argon three times, and acetonitrile (1 mL) was added followed by water (1 mL). The reaction then stirred at 60 °C for 18 h then was diluted with saturated NaHCO3 (5 mL) and extracted with EtOAc (3 × 5 mL). The organic layers were combined and dried with MgSO4, filtered, and concentrated then purified by reverse-phase MPLC (10–100% CH3CN:water) to produce pure 3l (0.045 g, 0.10 mmol, 79% yield) as a clear oil. 1H NMR (400 MHz, CDCl3): δ 7.59–7.45 (m, 2H), 7.53–7.48 (m, 6H), 7.45–7.40 (m, 2H), 7.35–7.25 (m, 7H), 7.22–7.16 (m, 2H), 3.11–3.05 (m, 2H), 2.87–2.81 (m, 2H), 2.65–2.59 (m, 2H), 2.53–2.44 (m, 1H), 2.22 (br s, 1H), 2.12–2.04 (m, 2H), 1.59–1.52 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 141.0, 139.5, 139.0, 129.1, 128.7, 128.2, 127.1, 127.0, 127.0, 126.5, 125.8, 79.5, 60.7, 54.0, 50.8, 44.2, 33.3, 26.4. LCMS retention time: 4.086 min. LCMS purity 99%. HRMS (ESI): m/z calcd for C32H33NO [M + H]+ 448.2634, found 448.2648.
(1-(4-(Pyridin-4-yl)phenethyl)piperidin-4-yl)diphenylmethanol (3m)
Same procedure as 3l with 3e (0.050 g, 0.11 mmol), pyridin-4-ylboronic acid (0.018 g, 0.13 mmol), 1,1′-bis(di-tert-butylphosphino)ferrocenepalladium dichloride (3.6 mg, 5.6 μmol)m and potassium carbonate (0.031 g, 0.22 mmol). Purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 3m (0.015 g, 0.033 mmol, 30% yield) as an oil. 1H NMR (400 MHz, CDCl3): δ 8.64–8.62 (m, 2H), 7.60 (d, J = 8.2 Hz, 2H), 7.51–7.46 (m, 6H), 7.32–7.28 (m, 6H), 7.18 (tt, J = 7.3 Hz, 1.8 Hz, 2H), 3.10–3.04 (m, 2H), 2.88–2.82 (m, 2H), 2.65–2.58 (m, 2H), 2.51–2.43 (m, 1H), 2.19 (br s, 1H), 2.12–2.03 (m, 2H), 1.58–1.51 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 150.2, 148.1, 145.9, 141.7, 135.8, 129.5, 128.2, 127.0, 126.5, 125.8, 121.4, 79.5, 60.5, 54.1, 44.1, 41.0, 33.4, 26.4. LCMS retention time: 3.585 min. LCMS purity 95.9%. HRMS (ESI): m/z calcd for C31H32N2O [M + H]+ 449.2587, found 449.2559.
(1-(4-(Pyridin-3-yl)phenethyl)piperidin-4-yl)diphenylmethanol (3n)
Same procedure as 3l using 3e (0.057 g, 0.13 mmol), pyridin-3-ylboronic acid (0.019 g, 0.15 mmol), 1,1′-bis(di-tert-butylphosphino)ferrocenepalladium dichloride (4.1 mg, 6.3 μmol), and potassium carbonate (0.035 g, 0.253 mmol), acetonitrile (1 mL), and water (1 mL). Purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 3n (0.05 g, 0.11 mmol, 88% yield) as an oil. 1H NMR (400 MHz, CDCl3): δ 8.79 (dd, J = 2.4 Hz, 0.9 Hz, 1H), 8.53 (dd, J = 4.8 Hz, 1.6 Hz, 1H), 7.86–7.83 (m, 1H), 7.51–7.47 (m, 6H), 7.36–7.27 (m, 7H), 7.20–7.15 (m, 2H), 3.10–3.03 (m, 2H), 2.87–2.81 (m, 2H), 2.71 (br s, 1H), 2.63–2.58 (m, 2H), 2.51–2.43 (m, 1H), 2.11–2.03 (m, 2H), 1.58–1.52 (m, 4H).). 13C NMR (125 MHz, CDCl3): δ 148.04, 147.97, 146.0, 140.5, 136.5, 135.5, 134.3, 129.4, 128.1, 127.1, 126.4, 125.8, 123.5, 79.4, 60.5, 54.1, 44.1, 33.2, 26.2. LCMS retention time: 3.582 min. LCMS purity 98.8%. HRMS (ESI): m/z calcd for C31H32N2O [M + H]+ 449.2587, found 449.2595.
(1-(4-(3-Methyloxetan-3-yl)phenethyl)piperidin-4-yl)diphenylmethanol (3o)
To a vial was added the 34 (0.024 g, 0.041 mmol) and MeOH (10 mL), and the reaction was heated to 50 °C. The magnesium was added in three additions (0.016 g, 0.066 mmol) 1.5 h apart. The reaction was removed from heat 1.5 h after the final magnesium addition, cooled to rt, and poured into 1.0 M HCl (10 mL) with ice. The aqueous layer was then extracted with CH2Cl2 (3 × 15 mL), and the organic layers were combined and dried with MgSO4, filtered, and concentrated then purifed by reverse-phase MPLC (10–100% CH3CN:water) to produce 3o (0.005 g, 10 μmol, 25% yield) as a light-brown solid. 1H NMR (400 MHz, CDCl3): δ 7.51–7.47 (m, 4H), 7.32–7.27 (m, 4H), 7.21–7.16 (m, 4H), 7.13–7.10 (m, 2H), 4.95 (d, J = 5.5 Hz, 2H), 4.61 (d, J = 5.5 Hz, 2H), 3.09–3.02 (m, 2H), 2.81–2.75 (m, 2H), 2.59–2.54 (m, 2H), 2.50–2.42 (m, 1H), 2.16 (br s, 1H), 2.09–2.00 (m, 2H), 1.71 (s, 3H), 1.56–1.48 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 144.1, 138.4, 128.8, 128.2, 126.5, 125.8, 125.1, 83.8, 79.5, 60.7, 54.1, 44.1, 43.1, 33.3, 27.8, 26.4. LCMS retention time: 3.652 min. LCMS purity 97.9%. HRMS (ESI): m/z calcd for C30H35NO2 [M + H]+ 442.2740, found 442.2749.
(1-(3-(tert-Butyl)benzyl)piperidin-4-yl)diphenylmethanol (4a)
Method C: 7 (0.030 g, 0.11 mmol), 3-tert-butylbenzyl bromide 10n (0.031 g, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4a (0.025 g, 0.060 mmol, 53% yield). 1H NMR (400 MHz, CDCl3): δ 7.51–7.41 (m, 4H), 7.34–7.26 (m, 6H), 7.26–7.20 (m, 1H), 7.20–7.14 (m, 2H), 7.11 (dt, J = 7.1, 1.6 Hz, 1H), 3.52 (s, 2H), 3.01–2.85 (m, 2H), 2.53–2.34 (m, 1H), 2.34 (br s, 1H), 2.05–1.95 (m, 2H), 1.58–1.44 (m, 4H), 1.32 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 150.9, 146.0, 137.5, 128.1, 127.8, 126.4, 126.3, 126.2, 125.8, 123.8, 79.5, 63.4, 53.8, 44.1, 34.6, 31.4, 26.4. LCMS retention time: 2.79 min. LCMS purity 99%. HRMS (ESI): m/z calcd for C29H35NO [M + H]+ 414.2767, found 414.2765.
(1-(2-(tert-Butyl)benzyl)piperidin-4-yl)diphenylmethanol (4b)
Method C: 7 (0.023 g, 0.084 mmol), 2-tert-butylbenzyl bromide 10o (0.023 g, 0.10 mmol), and triethylamine (0.018 mL, 0.13 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4b (0.014 g, 0.035 mmol, 41% yield). 1H NMR (400 MHz, CDCl3): δ 7.71 (s, 1H), 7.53–7.44 (m, 4H), 7.41–7.34 (m, 1H), 7.32–7.23 (m, 4H), 7.22–7.06 (m, 4H), 3.73 (s, 2H), 2.96 (d, J = 11.2 Hz, 2H), 2.56–2.42 (m, 1H), 2.26–2.06 (m, 3H), 1.58–1.44 (m, 4H), 1.41 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 148.1, 146.0, 130.9, 128.1, 126.6, 126.5, 126.0, 125.8, 125.7, 79.5, 61.3, 54.1, 44.2, 35.9, 31.6, 26.5. LCMS retention time: 2.93 min. LCMS purity 95.7%. HRMS (ESI): m/z calcd for C29H35NO [M + H]+ 414.2780, found 414.2829.
(1-(4-iso-Propylbenzyl)piperidin-4-yl)diphenylmethanol (4c)
Method C: 7 (0.030 g, 0.11 mmol), 4-iso-propylbenzyl bromide 10p (0.017 g, 0.023 mL, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce (4c) (0.030 g, 0.075 mmol, 67% yield). 1H NMR (400 MHz, CDCl3): δ 7.51–7.43 (m, 4H), 7.31–7.26 (m, 4H), 7.23–7.07 (m, 6H), 3.48 (s, 2H), 2.98–2.91 (m, 2H), 2.91–2.83 (m, 1H), 2.54–2.18 (m, 2H), 2.05–1.92 (m, 2H), 1.53–1.42 (m, 4H), 1.25 (s, 3H), 1.23 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 147.6, 146.0, 135.2, 129.2, 128.1, 126.4, 126.1, 125.8, 79.5, 62.9, 53.8, 44.1, 33.7, 26.4, 24.0. LCMS retention time: 4.094 min. LCMS purity 97.9%. HRMS (ESI): m/z calcd for C28H33NO [M + H]+ 400.2634, found 400.2670.
(1-(4-Methylbenzyl)piperidin-4-yl)diphenylmethanol (4d)
Method C: 7 (0.030 g, 0.11 mmol), 4-methylbenzyl bromide 10q (0.025 g, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4d (0.025 g, 0.068 mmol, 61% yield). 1H NMR (400 MHz, CDCl3): δ 7.51–7.41 (m, 4H), 7.33–7.24 (m, 4H), 7.21–7.14 (m, 4H), 7.11 (d, J = 7.8 Hz, 2H), 3.48 (s, 2H), 2.97–2.86 (m, 2H), 2.48–2.36 (m, 2H), 2.33 (s, 3H), 2.05–1.92 (m, 2H), 1.55–1.39 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 146.0, 136.5, 134.8, 129.2, 128.8, 128.1, 126.4, 125.8, 79.5, 62.8, 53.7, 44.1, 26.4. LCMS retention time: 2.51 min. LCMS purity 97.7%. HRMS (ESI): m/z calcd for C26H29NO [M + H]+ 372.2321, found 372.2336.
(1-(3-Methylbenzyl)piperidin-4-yl)diphenylmethanol (4e)
Method C: 7 (0.030 g, 0.11 mmol), 3-methylbenzyl bromide 10r (0.025 g, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4e (0.026 g, 0.070 mmol, 62% yield). 1H NMR (400 MHz, CDCl3): δ 7.52–7.43 (m, 4H), 7.32–7.26 (m, 4H), 7.22–7.14 (m, 3H), 7.11 (br s, 1H), 7.07 (t, J = 7.9 Hz, 2H), 3.48 (s, 2H), 3.00–2.91 (m, 2H), 2.50–2.37 (m, 1H), 2.33 (s, 3H), 2.31 (br s, 1H), 2.05–1.94 (m, 2H), 1.57–1.41 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 146.0, 137.9, 137.7, 130.0, 128.1, 128.0, 127.7, 126.4, 126.3, 125.8, 79.5, 63.2, 53.9, 44.1, 26.4, 21.4. LCMS retention time: 2.51 min. LCMS purity 97.7%. HRMS (ESI): m/z calcd for C26H29NO [M + H]+ 372.2321, found 372.2348.
(1-(2-Methylbenzyl)piperidin-4-yl)diphenylmethanol (4f)
Method C: 7 (0.030 g, 0.11 mmol), 2-methylbenzyl bromide 10s (0.024 g, 0.018 mL, 0.13 mmol), and triethylamine (0.023 mL, 0.17 mmol) in CH3CN (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4f (0.036 g, 0.097 mmol, 89% yield). 1H NMR (400 MHz, CDCl3): δ 7.55–7.43 (m, 4H), 7.35–7.23 (m, 5H), 7.22–7.08 (m, 5H), 3.44 (s, 2H), 2.99–2.86 (m, 2H), 2.51–2.41 (m, 1H), 2.35 (s, 3H), 2.25 (s, 1H), 2.08–1.97 (m, 2H), 1.51–1.38 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 146.0, 137.3, 136.7, 130.1, 129.6, 128.1, 126.8, 126.4, 125.8, 125.9, 79.6, 60.8, 54.1, 44.2, 26.6, 19.2. LCMS retention time: 3.987 min. LCMS purity 98.6%. HRMS (ESI): m/z calcd for C26H29NO [M + H]+ 372.2321, found 372.2327.
(1-([1,1′-Biphenyl]-4-ylmethyl)piperidin-4-yl)diphenylmethanol (4g)
Reported as general method C.
(1-([1,1′-Biphenyl]-3-ylmethyl)piperidin-4-yl)diphenylmethanol (4h)
Method C: 7 (0.030 g, 0.11 mmol), 3-phenylbenzyl bromide 10u (0.033 g, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4h (0.041 g, 0.095 mmol, 84% yield). 1H NMR (400 MHz, CDCl3): δ 7.63–7.57 (m, 2H), 7.54–7.50 (m, 1H), 7.50–7.40 (m, 7H), 7.40–7.32 (m, 2H), 7.32–7.25 (m, 5H), 7.21–7.13 (m, 2H), 3.58 (s, 2H), 3.03–2.86 (m, 2H), 2.50–2.22 (m, 2H), 2.10–1.96 (m, 2H), 1.51 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 146.0, 141.2, 141.1, 138.6, 128.7, 128.5, 128.1, 128.1, 128.0, 127.2, 127.2, 126.5, 125.8, 79.5, 63.2, 53.8, 44.1, 26.4. LCMS retention time: 4.072 min. LCMS purity 99%. HRMS (ESI): m/z calcd for C31H31NO [M + H]+ 434.2478, found 434.2500.
(1-([1,1′-Biphenyl]-2-ylmethyl)piperidin-4-yl)diphenylmethanol (4i)
Method C: 7 (0.030 g, 0.11 mmol), 2-phenylbenzyl bromide 10v (0.025 mL, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in CH3CN (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4i (0.036 g, 0.083 mmol, 74% yield). 1H NMR (400 MHz, CDCl3): δ 7.57–7.50 (m, 1H), 7.49–7.43 (m, 4H), 7.42–7.21 (m, 12H), 7.20–7.12 (m, 2H), 3.41 (s, 2H), 2.91–2.79 (m, 2H), 2.47–2.17 (m, 2H), 1.97–1.83 (m, 2H), 1.52–1.36 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 146.0, 142.5, 141.5, 130.0, 129.9, 129.5, 128.1, 127.8, 127.1, 126.7, 126.6, 126.4, 125.7, 79.5, 59.8, 53.7, 44.1, 26.5. LCMS retention time: 4.136 min. LCMS purity 98.6%. HRMS (ESI): m/z calcd for C31H31NO [M + H]+ 434.2478, found 434.2499.
(1-(4-Cyanobenzyl)piperidin-4-yl)diphenylmethanol (4j)
Method C: 7 (0.030 g, 0.11 mmol), 4-cyanobenzyl bromide 10w (0.026 g, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4j (0.031 g, 0.081 mmol, 72% yield). 1H NMR (400 MHz, CDCl3): δ 7.61–7.53 (m, 2H), 7.51–7.44 (m, 4H), 7.44–7.37 (m, 2H), 7.34–7.26 (m, 4H), 7.22–7.11 (m, 2H), 3.53 (s, 2H), 2.92–2.81 (m, 2H), 2.53–2.36 (m, 1H), 2.25 (s, 1H), 2.09–1.95 (m, 2H), 1.57–1.39 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.8, 144.4, 132.0, 129.4, 128.1, 126.5, 125.7, 119.0, 110.7, 79.5, 62.5, 54.0, 44.0, 26.4. LCMS retention time: 2.26 min. LCMS purity 99%. HRMS (ESI): m/z calcd for C26H26N2O [M + H]+ 383.2117, found 383.2116.
(1-(3-Cyanobenzyl)piperidin-4-yl)diphenylmethanol (4k)
Method C: 7 (0.030 g, 0.11 mmol), 3-cyanobenzyl bromide 10x (0.026 g, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4k (0.035 g, 0.092 mmol, 82% yield). 1H NMR (400 MHz, CDCl3): δ 7.61 (s, 1H), 7.56–7.50 (m, 2H), 7.50–7.44 (m, 4H), 7.38 (t, J = 7.7 Hz, 1H), 7.34–7.26 (m, 4H), 7.23–7.13 (m, 2H), 3.50 (s, 2H), 2.91–2.77 (m, 2H), 2.56–2.33 (m, 1H), 2.22 (s, 1H), 2.07–1.95 (m, 2H), 1.57–1.40 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.8, 140.2, 133.3, 132.3, 130.6, 128.9, 128.1, 126.5, 125.8, 118.9, 112.2, 79.5, 62.2, 53.9, 44.0, 26.4. LCMS retention time: 2.26 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C26H26N2O [M + H]+ 383.2117, found 383.2116.
(1-(2-Cyanobenzyl)piperidin-4-yl)diphenylmethanol (4l)
Method C: 7 (0.030 g, 0.11 mmol), 2-cyanobenzyl bromide 10y (0.026 g, 0.135 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4l (0.027 g, 0.071 mmol, 63% yield). 1H NMR (400 MHz, CDCl3): δ 7.64–7.58 (m, 1H), 7.58–7.51 (m, 2H), 7.51–7.43 (m, 4H), 7.35–7.26 (m, 5H), 7.22–7.11 (m, 2H), 3.69 (s, 2H), 2.97–2.84 (m, 2H), 2.52–2.40 (m, 1H), 2.37–2.07 (m, 3H), 1.57–1.37 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 142.7, 132.7, 132.5, 129.9, 128.1, 127.4, 126.5, 125.7, 117.9, 112.8, 79.5, 60.5, 53.9, 44.0, 26.5. LCMS retention time: 2.28 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C26H26N2O [M + H]+ 383.2117, found 383.2141.
(1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)diphenylmethanol (4m)
Method C: 7 (0.030 g, 0.11 mmol), 4-trifluoromethylbenzyl bromide 10z (0.032 g, 0.021 mL, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4m (0.036 g, 0.085 mmol, 76% yield). 1H NMR (400 MHz, CDCl3): δ 7.55 (d, J = 7.7 Hz, 2H), 7.52–7.43 (m, 4H), 7.41 (d, J = 8.0 Hz, 2H), 7.35–7.25 (m, 4H), 7.23–7.12 (m, 2H), 3.54 (s, 2H), 2.97–2.78 (m, 2H), 2.44 (m, 1H), 2.28 (s, 1H), 2.11–1.95 (m, 2H), 1.50 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 142.6, 129.2 (q, J = 32.0 Hz), 129.2, 128.1, 126.5, 125.8, 125.0 (q, J = 3.9 Hz), 124.2 (q, J = 270.3 Hz), 79.5, 62.5, 53.9, 44.0, 26.4. LCMS retention time: 3.904 min. LCMS purity 98.9%. HRMS (ESI): m/z calcd for C26H26F3NO [M + H]+ 426.2039, found 426.2070.
(1-(3-(Trifluoromethyl)benzyl)piperidin-4-yl)diphenylmethanol (4n)
Method C: 7 (0.030 g, 0.11 mmol), 3-trifluoromethylbenzyl bromide 10aa (0.032 g, 0.021 mL, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4n (0.034 g, 0.080 mmol, 71% yield). 1H NMR (400 MHz, CDCl3): δ 7.56 (s, 1H), 7.53–7.45 (m, 6H), 7.44–7.37 (m, 1H), 7.35–7.26 (m, 4H), 7.22–7.12 (m, 2H), 3.54 (s, 2H), 2.95–2.84 (m, 2H), 2.50–2.38 (m, 1H), 2.24 (s, 1H), 2.08–1.96 (m, 2H), 1.55–1.45 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 139.4, 132.3, 130.5 (q, J = 31.7 Hz), 128.6, 128.2, 126.5, 125.6 (q, J = 3.7 Hz), 124.2 (q, J = 270.7 Hz), 123.8 (q, J = 3.8 Hz), 79.5, 62.6, 53.9, 44.1, 26.4. LCMS retention time: 3.897 min. LCMS purity 99.3%. HRMS (ESI): m/z calcd for C26H26F3NO [M + H]+ 426.2039, found 426.2063.
(1-(2-(Trifluoromethyl)benzyl)piperidin-4-yl)diphenylmethanol (4o)
Method C: 7 (0.030 g, 0.11 mmol), 2-trifluoromethylbenzyl bromide 10bb (0.032 g, 0.020 mL, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4o (0.045 g, 0.11 mmol, 95% yield). 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.54–7.44 (m, 5H), 7.35–7.27 (m, 5H), 7.23–7.15 (m, 2H), 3.64 (d, J = 1.7 Hz, 2H), 2.94–2.82 (m, 2H), 2.55–2.42 (m, 1H), 2.27–2.06 (m, 3H), 1.58–1.43 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 146.0, 138.2, 131.7, 130.1, 128.2, 127.2 (q, J = 262.9 Hz), 126.5, 125.7, 125.5 (q, J = 5.8 Hz), 123.1, 79.6, 58.2 (q, J = 2.1 Hz), 54.2, 44.1, 26.6. LCMS retention time: 4.054 min. LCMS purity 98.1%. HRMS (ESI): m/z calcd for C26H26F3NO [M + H]+ 426.2039, found 426.2081.
(1-(4-Fluorobenzyl)piperidin-4-yl)diphenylmethanol (4p)
Method C: 7 (0.030 g, 0.11 mmol), 4-fluoromethylbenzyl bromide 10cc (0.017 mL, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4p (0.028 g, 0.075 mmol, 67% yield). 1H NMR (400 MHz, CDCl3): δ 7.51–7.40 (m, 4H), 7.32–7.22 (m, 6H), 7.20–7.13 (m, 2H), 7.02–6.92 (m, 2H), 3.47 (s, 2H), 2.98–2.85 (m, 2H), 2.49–2.37 (m, 1H), 2.29 (br s, 1H), 2.05–1.93 (m, 2H), 1.49 (td, J = 8.8, 7.8, 3.7 Hz, 4H). 13C NMR (125 MHz, CDCl3): δ 162.0 (d, J = 243.2 Hz), 145.9, 130.7 (d, J = 7.8 Hz), 128.1, 126.5, 125.8, 114.9 (d, J = 21.0 Hz), 79.5, 62.2, 53.7, 44.1, 26.3. LCMS retention time: 3.734 min. LCMS purity 96.5%. HRMS (ESI): m/z calcd for C25H26FNO [M + H]+ 376.2070, found 376.2081.
(1-(3-Fluorobenzyl)piperidin-4-yl)diphenylmethanol (4q)
Method C: 7 (0.030 g, 0.11 mmol), 3-fluorobenzyl bromide 10dd (0.017 mL, 0.135 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4q (0.028 g, 0.074 mmol, 66% yield). 1H NMR (400 MHz, CDCl3): δ 7.52–7.39 (m, 4H), 7.34–7.26 (m, 4H), 7.25–7.21 (m, 1H), 7.20–7.14 (m, 2H), 7.09–6.97 (m, 2H), 6.96–6.85 (m, 1H), 3.48 (s, 2H), 2.96–2.85 (m, 2H), 2.49–2.37 (m, 1H), 2.27 (s, 1H), 2.06–1.95 (m, 2H), 1.54–1.42 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 162.9 (d, J = 244.1 Hz), 145.9, 141.2 (d, J = 6.8 Hz), 129.5 (d, J = 8.1 Hz), 128.1, 126.5, 125.8, 124.5 (d, J = 2.7 Hz), 115.7 (d, J = 21.1 Hz), 113.7 (d, J = 21.1 Hz), 79.5, 62.6, 62.5, 53.9, 44.1, 26.5. LCMS retention time: 2.41 min. LCMS purity 99%. HRMS (ESI): m/z calcd for C25H26FNO [M + H]+ 376.2070, found 376.2069.
(1-(2-Fluorobenzyl)piperidin-4-yl)diphenylmethanol (4r)
Method C: 7 (0.030 g, 0.11 mmol), 2-fluorobenzyl bromide 10ee (0.016 mL, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4r (0.038 g, 0.10 mmol, 91% yield). 1H NMR (400 MHz, CDCl3): δ 7.49–7.44 (m, 4H), 7.35 (td, J = 7.5, 1.9 Hz, 1H), 7.31–7.26 (m, 4H), 7.26–7.20 (m, 1H), 7.20–7.14 (m, 2H), 7.09 (td, J = 7.5, 1.2 Hz, 1H), 7.06–6.96 (m, 1H), 3.60 (d, J = 1.5 Hz, 2H), 3.01–2.89 (m, 2H), 2.50–2.17 (m, 2H), 2.14–2.06 (m, 2H), 1.50 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 161.4 (d, J = 244.1 Hz), 145.9, 131.7 (d, J = 4.5 Hz), 128.7 (d, J = 8.2 Hz), 128.1, 126.5, 125.8, 124.5 (d, J = 14.8 Hz), 123.7 (d, J = 3.5 Hz), 115.2, 115.0, 79.5, 55.2, 55.2, 53.5, 44.0, 26.4. LCMS retention time: 3.733 min. LCMS purity 98.6%. HRMS (ESI): m/z calcd for C25H26FNO [M + H]+ 376.2070, found 376.2074.
(1-(4-Methoxybenzyl)piperidin-4-yl)diphenylmethanol (4s)
To a vial was added the 7 (0.030 g, 0.11 mmol) and 4-methoxybenzaldehyde 11a (0.015 g, 0.013 mL, 0.11 mmol) in THF (0.5 mL) to form a solution. The reaction began to stir at rt, and after 30 min the sodium triacetoxyborohydride (0.026 g, 0.12 mmol) was added and the reaction continued to stir at rt. The reaction was then quenched with water (1.5 mL) and extracted with CH2Cl2 (1.5 mL). The organic layer was dried (MgSO4), concentrated, and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4s (0.007 g, 0.018 mmol, 16% yield). 1H NMR (400 MHz, CDCl3): δ 7.51–7.40 (m, 4H), 7.33–7.23 (m, 4H), 7.21–7.10 (m, 4H), 6.88–6.76 (m, 2H), 3.78 (s, 3H), 3.44 (s, 2H), 2.97–2.84 (m, 2H), 2.50–2.33 (m, 1H), 2.07–1.89 (m, 3H), 1.54–1.39 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 158.6, 146.0, 130.4, 130.0, 128.1, 126.4, 125.8, 113.4, 79.5, 62.5, 55.2, 53.7, 44.1, 26.4. LCMS retention time: 2.35 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C26H29NO2 [M + H]+ 388.2272, found 388.2295.
(1-(3-Methoxybenzyl)piperidin-4-yl)diphenylmethanol (4t)
Method C: 7 (0.030 g, 0.11 mmol), 3-methoxybenzyl bromide 10ff (0.016 mL, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4t (0.034 g, 0.088 mmol, 79% yield). 1H NMR (400 MHz, CDCl3): δ 7.53–7.38 (m, 4H), 7.35–7.24 (m, 4H), 7.24–7.11 (m, 3H), 6.91–6.84 (m, 2H), 6.82–6.73 (m, 1H), 3.79 (s, 3H), 3.49 (s, 2H), 3.04–2.83 (m, 2H), 2.51–2.23 (m, 2H), 2.05–1.96 (m, 2H), 1.56–1.38 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 159.5, 146.0, 139.8, 129.0, 128.1, 126.5, 125.8, 121.5, 114.6, 112.4, 79.5, 63.1, 55.2, 53.8, 44.1, 26.4. LCMS retention time: 3.733 min. LCMS purity 97.3%. HRMS (ESI): m/z calcd for C26H29NO2 [M + H]+ 388.2272, found 388.2291.
(1-(2-Methoxybenzyl)piperidin-4-yl)diphenylmethanol (4u)
To a vial was added the diphenyl(piperidin-4-yl)methanol (0.030 g, 0.11 mmol) and 2-methoxybenzaldehyde 11b (0.015 g, 0.013 mL, 0.11 mmol) in THF (0.5 mL) to form a solution. The reaction began to stir at rt, and after 30 min the sodium triacetoxyborohydride (0.026 g, 0.12 mmol) was added and the reaction continued to stir at rt. The reaction was then quenched with water (1.5 mL) and extracted with CH2Cl2 (1.5 mL). The organic layer was dried (MgSO4), concentrated, and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4u (0.011 g, 0.028 mmol, 25% yield). 1H NMR (400 MHz, CDCl3): δ 7.53–7.40 (m, 4H), 7.34–7.25 (m, 5H), 7.22 (td, J = 8.1, 1.8 Hz, 1H), 7.19–7.13 (m, 2H), 6.91 (td, J = 7.4, 1.1 Hz, 1H), 6.85 (dd, J = 8.2, 1.1 Hz, 1H), 3.80 (s, 3H), 3.58 (s, 2H), 3.04–2.90 (m, 2H), 2.49–2.35 (m, 1H), 2.17 (s, 1H), 2.08 (td, J = 11.3, 3.6 Hz, 2H), 1.60–1.38 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 157.8, 146.0, 130.7, 128.1, 128.0, 126.4, 125.8, 120.2, 110.3, 79.5, 56.0, 55.4, 53.8, 44.1, 26.4. LCMS retention time: 2.40 min. LCMS purity 97.7%. HRMS (ESI): m/z calcd for C26H29NO2 [M + H]+ 388.2270, found 388.2242.
(1-(4-Hydroxybenzyl)piperidin-4-yl)diphenylmethanol (4v)
To a vial was added the 7 (0.030 g, 0.11 mmol) and 4-hydroxybenzaldehyde 11c (0.013 g, 0.11 mmol) in THF (0.5 mL) to form a solution. The reaction began to stir at rt, and after 30 min the sodium triacetoxyborohydride (0.026 g, 0.12 mmol) was added and the reaction continued to stir at rt. The reaction was then quenched with water (1.5 mL) and extracted with CH2Cl2 (1.5 mL). The organic layer was dried with MgSO4, filtered, concentrated, and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce (4v) (0.012 g, 0.032 mmol, 29% yield). 1H NMR (400 MHz, CDCl3): δ 7.50–7.40 (m, 4H), 7.32–7.21 (m, 4H), 7.20–7.11 (m, 2H), 7.11–7.01 (m, 2H), 6.73–6.65 (m, 2H), 3.45 (s, 2H), 2.98 (d, J = 11.4 Hz, 2H), 2.51–2.35 (m, 2H), 2.04 (m, 2H), 1.51 (m, 5H). 13C NMR (125 MHz, CDCl3): δ 155.9, 145.9, 130.9, 128.1, 126.5, 125.7, 125.7, 115.4, 79.4, 62.3, 53.4, 43.9, 25.8. LCMS retention time: 1.35 min. LCMS purity 95.8%. HRMS (ESI): m/z calcd for C25H27NO2 [M + H]+ 374.2114, found 374.2107.
(1-(3-Hydroxybenzyl)piperidin-4-yl)diphenylmethanol (4w)
Method C: 7 (0.030 g, 0.11 mmol), 3-hydroxybenzyl bromide 10gg (0.025 g, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4w (0.029 g, 0.077 mmol, 69% yield). 1H NMR (400 MHz, CDCl3): δ 7.48–7.40 (m, 4H), 7.31–7.22 (m, 4H), 7.18–7.11 (m, 2H), 7.08 (t, J = 7.8 Hz, 1H), 6.78–6.69 (m, 2H), 6.69–6.64 (m, 1H), 3.43 (s, 2H), 2.98–2.87 (m, 2H), 2.47–2.33 (m, 1H), 2.05–1.94 (m, 4H), 1.59–1.37 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 156.3, 145.9, 138.8, 129.2, 128.1, 126.4, 125.8, 125.7, 121.3, 116.8, 114.8, 79.4, 62.9, 53.7, 43.9, 25.9. LCMS retention time: 1.28 min. LCMS purity 97.7%. HRMS (ESI): m/z calcd for C25H27NO2 [M + H]+ 374.2114, found 374.2129.
(1-(2-Hydroxybenzyl)piperidin-4-yl)diphenylmethanol (4x)
To a vial was added the 7 (0.030 g, 0.11 mmol) and 2-hydroxybenzaldehyde 11d (0.013 g, 0.011 mL, 0.11 mmol) in THF (0.5 mL) to form a solution. The reaction began to stir at rt, and after 30 min the sodium triacetoxyborohydride (0.026 g, 0.12 mmol) was added and the reaction continued to stir at rt. The reaction was then quenched with water (1.5 mL) and extracted with CH2Cl2 (1.5 mL). The organic layer was dried with MgSO4, filtered, concentrated, and purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4x (0.009 g, 0.024 mmol, 21% yield). 1H NMR (400 MHz, CDCl3): δ 7.50–7.43 (m, 4H), 7.34–7.27 (m, 4H), 7.22–7.11 (m, 3H), 6.95 (dd, J = 7.5, 1.7 Hz, 1H), 6.79 (dd, J = 8.1, 1.2 Hz, 1H), 6.75 (td, J = 7.4, 1.2 Hz, 1H), 3.69 (s, 2H), 3.12–2.97 (m, 2H), 2.57–2.40 (m, 1H), 2.22–2.06 (m, 3H), 1.63–1.46 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 157.8, 145.6, 128.8, 128.6, 128.2, 126.7, 125.7, 119.0, 116.2, 79.3, 53.4, 43.8, 26.2. LCMS retention time: 1.41 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C25H27NO2 [M + H]+ 374.2114, found 374.2108.
Methyl 4-((4-(Hydroxydiphenylmethyl)piperidin-1-yl)methyl)benzoate (4y)
Method C: 7 (0.15 g, 0.56 mmol), methyl 4-(bromomethyl)benzoate 10hh (0.15 g, 0.67 mmol), and triethylamine (0.12 mL, 0.84 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4y (0.21 g, 0.52 mmol, 92% yield). 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 8.2 Hz, 2H), 7.51–7.42 (m, 4H), 7.37 (d, J = 8.1 Hz, 2H), 7.33–7.26 (m, 4H), 7.23–7.12 (m, 2H), 3.90 (s, 3H), 3.54 (s, 2H), 2.90 (d, J = 10.4 Hz, 2H), 2.43 (p, J = 8.6, 7.8 Hz, 1H), 2.14 (s, 1H), 2.04 (s, 2H), 1.55–1.42 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 167.0, 145.9, 129.5, 128.9, 128.1, 126.5, 125.8, 79.5, 62.8, 54.0, 52.0, 44.1, 26.4. LCMS retention time: 3.697 min. LCMS purity 99.6%. HRMS (ESI): m/z calcd for C27H29NO3 [M + H]+ 416.2219, found 416.2225.
Methyl 3-((4-(Hydroxydiphenylmethyl)piperidin-1-yl)methyl)benzoate (4z)
Method C: 7 (0.030 g, 0.11 mmol), methyl 3-(bromomethyl)benzoate 10ii (0.031 g, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4z (0.028 g, 0.067 mmol, 60% yield). 1H NMR (400 MHz, CDCl3): δ 7.97–7.87 (m, 2H), 7.53–7.43 (m, 5H), 7.40–7.33 (m, 1H), 7.31–7.24 (m, 4H), 7.20–7.14 (m, 2H), 3.90 (s, 3H), 3.54 (s, 2H), 2.97–2.83 (m, 2H), 2.50–2.37 (m, 1H), 2.29 (s, 1H), 2.07–1.95 (m, 2H), 1.49 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 167.2, 145.9, 138.7, 133.7, 130.2, 130.0, 128.3, 128.2, 128.1, 126.5, 125.8, 79.5, 62.7, 53.8, 52.1, 44.1, 26.4. LCMS retention time: 3.695 min. LCMS purity 98.5%. HRMS (ESI): m/z calcd for C27H29NO3 [M + H]+ 416.2219, found 416.2227.
Methyl 2-((4-(Hydroxydiphenylmethyl)piperidin-1-yl)methyl)benzoate (4aa)
Method C: 7 (0.030 g, 0.11 mmol), methyl 2-(bromomethyl)benzoate 10jj (0.031 g, 0.14 mmol), and triethylamine (0.023 mL, 0.17 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4aa (0.044 g, 0.10 mmol, 95% yield). 1H NMR (400 MHz, CDCl3): δ 7.97–7.87 (m, 2H), 7.53–7.43 (m, 5H), 7.40–7.33 (m, 1H), 7.31–7.24 (m, 4H), 7.20–7.14 (m, 2H), 3.90 (s, 3H), 3.54 (s, 2H), 2.97–2.83 (m, 2H), 2.50–2.37 (m, 1H), 2.29 (s, 1H), 2.07–1.95 (m, 2H), 1.49 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 167.2, 145.9, 138.7, 133.7, 130.2, 130.0, 128.3, 128.2, 128.1, 126.5, 125.8, 79.5, 62.7, 53.8, 52.1, 44.1, 26.4. LCMS retention time: 3.695 min. LCMS purity 97.4%. HRMS (ESI): m/z calcd for C27H29NO3 [M + H]+ 416.2219, found 416.2248.
(1-(4-Iodobenzyl)piperidin-4-yl)diphenylmethanol (4bb)
Method C: 7 (0.300 g, 1.12 mmol), 4-iodobenzyl bromide 10kk (0.40 g, 1.35 mmol), and triethylamine (0.24 mL, 1.68 mmol) in acetonitrile (2 mL). The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4bb (0.51 g, 1.06 mmol, 94% yield). 1H NMR (400 MHz, CDCl3): δ 7.65–7.57 (m, 2H), 7.49–7.40 (m, 4H), 7.33–7.26 (m, 4H), 7.22–7.11 (m, 2H), 7.06 (d, J = 7.9 Hz, 2H), 3.46 (s, 2H), 2.91 (d, J = 11.2 Hz, 2H), 2.50–2.34 (m, 1H), 2.13 (s, 1H), 2.05 (s, 2H), 1.50 (s, 4H).
4-((4-(Hydroxydiphenylmethyl)piperidin-1-yl)methyl)benzoic Acid (4cc)
To a vial was added the 4y (0.18 g, 0.42 mmol) and THF (1 mL). The 2.0 M LiOH (1.48 mL, 2.96 mmol) in water was added and the reaction stirred at rt for 24 h. The reaction was acidified with 1.0 M HCl to pH 2–3 and then extracted with CH2Cl2 (3 × 15 mL). The organic layers were combined and dried with MgSO4, filtered, and concentrated then purified by reverse-phase MPLC (10–100% CH3CN:water) to produce (4cc) (0.011 g, 0.027 mmol, 6% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 7.99 (d, J = 8.2 Hz, 2H), 7.67 (d, J = 7.9 Hz, 2H), 7.58–7.42 (m, 4H), 7.28 (t, J = 7.8 Hz, 4H), 7.20–7.07 (m, 2H), 4.28 (s, 2H), 3.82–3.52 (m, 2H), 3.17 (s, 1H), 2.85 (d, J = 52.0 Hz, 3H), 1.88–1.69 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ 167.4, 147.3, 143.7, 129.2, 128.6, 127.8, 125.8, 125.7, 78.5, 62.0, 53.5, 43.3, 26.0. LCMS retention time: 1.250 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C26H27NO3 [M + H]+ 402.2064, found 402.2062.
3-((4-(Hydroxydiphenylmethyl)piperidin-1-yl)methyl)benzoic Acid (4dd)
Same procedure as 4cc using 4z (0.012 g, 0.030 mmol), 2.0 M LiOH (0.10 mL, 0.21 mmol) in water, and THF (1 mL). Purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4dd (0.005 g, 0.012 mmol, 42% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 8.11 (s, 1H), 8.00 (d, J = 7.7 Hz, 1H), 7.82–7.70 (m, 1H), 7.57 (t, J = 7.7 Hz, 1H), 7.49 (d, J = 7.8 Hz, 4H), 7.28 (t, J = 7.7 Hz, 4H), 7.18–7.10 (m, 2H), 4.37–4.12 (m, 2H), 3.76–3.53 (m, 2H), 3.21–3.14 (m, 1H), 3.03–2.68 (m, 3H), 1.91–1.56 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 167.9, 147.3, 138.7, 132.1, 129.5, 127.9, 127.8, 125.8, 125.7, 78.5, 62.1, 53.4, 43.3, 26.0. LCMS retention time: 1.250 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C26H27NO3 [M + H]+ 402.2064, found 402.2092.
2-((4-(Hydroxydiphenylmethyl)piperidin-1-yl)methyl)benzoic Acid (4ee)
Same procedure as 4cc using 4ee (0.043 g, 0.10 mmol), 2.0 M LiOH (0.36 mL, 0.72 mmol) in water, and THF (1 mL). Purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4ee (0.007 g, 0.018 mmol, 17% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.05 (br s, 1H), 7.87–7.77 (m, 1H), 7.55–7.46 (m, 4H), 7.44–7.37 (m, 2H), 7.36–7.23 (m, 5H), 7.20–7.10 (m, 2H), 3.98 (s, 2H), 3.02 (d, J = 11.9 Hz, 2H), 2.83–2.72 (m, 1H), 2.72–2.58 (m, 2H), 2.47 (br s, 1H), 1.68–1.47 (m, 2H), 1.47–1.30 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.7, 146.7, 138.4, 131.8, 131.4, 131.3, 130.1, 128.7, 127.9, 126.1, 125.6, 78.2, 59.3, 50.2, 41.6, 24.4. LCMS retention time: 1.50 min. LCMS purity 99%. HRMS (ESI): m/z calcd for C26H27NO3 [M + H]+ 402.2064, found 401.2059.
1-(4-(Thiophen-3-yl)benzyl)piperidin-4-yl)diphenylmethanol (4ff)
Same procedure as 3l using thiophen-3-ylboronic acid (0.011 g, 0.087 mmol), 4bb (0.035 g, 0.072 mmol), 1,1′-bis(di-tert-butylphosphino)ferrocene palladium dichloride (2 mg, 3.6 μmol) and potassium carbonate (0.015 g, 0.11 mmol), acetonitrile (1 mL), and water (1 mL). Purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4ff (0.027 g, 0.062 mmol, 85% yield) as an oil. 1H NMR (400 MHz, CDCl3): δ 7.56–7.51 (m, 2H), 7.51–7.45 (m, 4H), 7.45–7.42 (m, 1H), 7.40–7.35 (m, 2H), 7.35–7.26 (m, 6H), 7.21–7.13 (m, 2H), 3.53 (s, 2H), 3.02–2.87 (m, 2H), 2.52–2.37 (m, 1H), 2.18 (s, 1H), 2.10–1.95 (m, 2H), 1.62–1.41 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 146.0, 142.1, 137.0, 134.6, 129.7, 128.1, 126.5, 126.3, 126.2, 126.1, 125.8, 120.0, 79.5, 62.8, 53.9, 44.1, 26.4. LCMS retention time: 2.65 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C29H29NOS [M + H]+ 440.2042, found 440.2033.
(1-(4-(Thiophen-2-yl)benzyl)piperidin-4-yl)diphenylmethanol (4gg)
To a microwave vial was added the 4bb (0.035 g, 0.073 mmol), RuPhos (4 mg, 8.8 μmol), palladium(II) acetate (1.0 mg, 4.4 μmol), potassium trifluoro(thiophen-2-yl)borate (0.015 g, 0.077 mmol), and sodium bicarbonate (0.015 g, 0.15 mmol). The vial was evacuated with argon three times, and then degassed ethanol (0.5 mL) was added. The reaction then stirred at 100 °C for 60 min in the microwave. The reaction was cooled to rt and diluted with saturated NaHCO3 (10 mL) and extracted with EtOAc (3 × 15 mL). The organic layers were combined, dried with MgSO4, filtered, and concentrated. The reaction was purified by reverse-phase MPLC (10–100% CH3CN:water) to produce (4gg) (0.022 g, 0.050 mmol, 69% yield). 1H NMR (400 MHz, CDCl3): δ 7.59–7.51 (m, 2H), 7.50–7.44 (m, 4H), 7.32–7.22 (m, 8H), 7.21–7.15 (m, 2H), 7.11–7.05 (m, 1H), 3.48 (s, 2H), 3.00–2.87 (m, 2H), 2.50–2.37 (m, 1H), 2.17 (s, 1H), 2.10–1.95 (m, 2H), 1.56–1.38 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 144.3, 133.2, 129.7, 128.1, 127.9, 126.5, 125.8, 125.7, 124.6, 122.9, 79.5, 62.8, 53.8, 44.1, 26.4. LCMS retention time: 2.71 min. LCMS purity 99.3%. HRMS (ESI): m/z calcd for C29H29NOS [M + H]+ 440.2042, found 440.2056.
(1-(4-(Furan-3-yl)benzyl)piperidin-4-yl)diphenylmethanol (4hh)
Same procedure as 3l using furan-3-ylboronic acid (0.010 g, 0.091 mmol), 4bb (0.037 g, 0.076 mmol), 1,1′-bis(di-tert-butylphosphino)ferrocene palladium dichloride (3 mg, 4 μmol), and potassium carbonate (0.016 g, 0.11 mmol). Purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4hh (0.028 g, 0.067 mmol, 89% yield) as an oil. 1H NMR (400 MHz, CDCl3): δ 7.71 (s, 1H), 7.51–7.45 (m, 5H), 7.45–7.37 (m, 2H), 7.34–7.24 (m, 6H), 7.23–7.09 (m, 2H), 6.69 (dd, J = 1.9, 0.9 Hz, 1H), 3.48 (s, 2H), 2.99–2.86 (m, 2H), 2.49–2.36 (m, 1H), 2.18 (s, 1H), 2.08–1.93 (m, 2H), 1.55–1.42 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 146.0, 143.6, 138.3, 136.9, 131.1, 129.7, 128.1, 126.5, 126.2, 125.8, 125.6, 108.8, 79.5, 62.9, 53.8, 44.1, 26.4. LCMS retention time: 2.52 min. LCMS purity 99.3%. HRMS (ESI): m/z calcd for C29H29NO2 [M + H]+ 424.2270, found 424.2250.
(1-(4-(Furan-2-yl)benzyl)piperidin-4-yl)diphenylmethanol (4ii)
Same procedure as 4gg using 4bb (0.036 g, 0.073 mmol), RuPhos (4 mg, 8.8 μmol), palladium(II) acetate (1 mg, 4.4 μmol), potassium trifluoro(furan-2-yl)borate (0.013 g, 0.077 mmol), and sodium carbonate (0.016 g, 0.15 mmol) and degassed ethanol (0.5 mL). Purified by reverse-phase MPLC (10–100% CH3CN:water) to produce 4ii (0.022 g, 0.050 mmol, 68% yield). 1H NMR (400 MHz, CDCl3): δ 7.63–7.54 (m, 2H), 7.52–7.42 (m, 5H), 7.29 (m, 6H), 7.22–7.07 (m, 2H), 6.62 (dd, J = 3.4, 0.8 Hz, 1H), 6.46 (dd, J = 3.4, 1.8 Hz, 1H), 3.48 (s, 2H), 2.99–2.86 (m, 2H), 2.51–2.36 (m, 1H), 2.17 (s, 1H), 2.08–1.95 (m, 2H), 1.56–1.42 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 154.0, 146.0, 141.9, 137.3, 129.7, 129.5, 128.1, 126.5, 125.8, 123.6, 111.6, 104.7, 79.5, 62.9, 53.8, 44.1, 26.4. LCMS retention time: 2.60 min. LCMS purity 97.4%. HRMS (ESI): m/z calcd for C29H29NO2 [M + H]+ 424.2270, found 424.2271.
(1-(4-(1H-Pyrrol-1-yl)benzyl)piperidin-4-yl)diphenylmethanol (4jj)
To a vial was added the 4bb (0.030 g, 0.062 mmol), copper powder (0.8 mg, 0.012 mmol), pyrrole (7 μL, 0.093 mmol), and cesium carbonate (0.071 g, 0.22 mmol) in acetonitrile (1 mL). The vial was then purged with argon three times and then stirred at 80 °C for 21 h. The reaction was then diluted with EtOAc (10 mL) and filtered through Celite. The filtrate was concentrated then purified with reverse-phase MPLC (10–100% CH3CN:water) to produce 4jj (0.012 g, 0.029 mmol, 47% yield). 1H NMR (400 MHz, CDCl3): δ 7.52–7.44 (m, 4H), 7.36–7.26 (m, 8H), 7.22–7.12 (m, 2H), 7.07 (t, J = 2.4 Hz, 2H), 6.33 (t, J = 2.2 Hz, 2H), 3.51 (s, 2H), 3.00–2.88 (m, 2H), 2.51–2.37 (m, 1H), 2.13 (s, 1H), 2.08–1.94 (m, 2H), 1.57–1.44 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 145.9, 139.7, 130.3, 128.1, 126.5, 125.8, 120.2, 119.3, 110.2, 79.5, 62.4, 53.8, 44.1, 26.4. LCMS retention time: 2.60 min. LCMS purity 97.4%. HRMS (ESI): m/z calcd for C29H30N2O [M + H]+ 423.2430, found 423.2441.
1-(4-(tert-Butyl)phenyl)-4-(4-(hydroxy(phenyl)methyl)piperidin-1-yl)butan-1-ol (5a)
Method B: 35 (0.026 g, 0.066 mmol) and MeOH (2 mL) and sodium borohydride (10 mg, 0.27 mmol) to produce 5a (0.019 g, 0.048 mmol, 72% yield). 1H NMR (400 MHz, CDCl3): δ 7.36–7.24 (m, 9H), 4.63–4.57 (m, 1H), 4.33 (d, J = 7.6 Hz, 1H), 3.23–2.83 (m, 2H), 2.42–2.35 (m, 2H), 2.13–1.89 (m, 3H), 1.86–1.52 (m, 6H), 1.46–1.16 (m, 4H), 1.31 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 149.4, 143.4, 142.8, 128.3, 127.63, 127.61, 126.6, 125.4, 125.0, 78.74, 78.67, 73.4, 58.9, 54.30, 54.26, 52.8, 52.7, 43.21, 43.2, 39.9, 34.4, 31.4, 28.4, 28.3, 28.2, 24.2, 24.1. LCMS retention time: 3.707 min. LCMS purity 97.1%. HRMS (ESI): m/z calcd for C26H37NO2 [M + H]+ 396.2896, found 396.2891.
4-(4-Benzylpiperidin-1-yl)-1-(4-(tert-butyl)phenyl)butan-1-ol (5b)
Method B: 36 (0.29 g, 0.77 mmol), MeOH (5 mL), and sodium borohydride (0.12 g, 3.08 mmol) to produce 5b (0.23 g, 0.61 mmol, 79% yield). 1H NMR (400 MHz, CDCl3): δ 7.35–7.25 (m, 6H), 7.21–7.12 (m, 3H), 4.64–4.60 (m, 1H), 3.13–3.07 (m, 1H), 2.94–2.88 (m, 1H), 2.54 (d, J = 7.0 Hz, 2H), 2.44–2.34 (m, 2H), 2.03–1.93 (m, 2H), 1.90–1.38 (m, 10H), 1.31 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 149.4, 142.9, 140.7, 129.1, 128.2, 125.8, 125.4, 125.0, 73.5, 59.0, 54.6, 52.9, 42.9, 40.1, 38.0, 34.4, 31.8, 31.6, 31.4, 24.3. LCMS retention time: 4.330 min. LCMS purity 99.1%. HRMS (ESI): m/z calcd for C26H37NO [M + H]+ 380.2948, found 380.2974.
4-(4-Benzhydrylpiperazin-1-yl)-1-(4-(tert-butyl)phenyl)butan-1-ol (5c)
Method B: 38 (0.086 g, 0.19 mmol), MeOH (3 mL), and sodium borohydride (0.014 g, 0.39 mmol) to produce 5c (0.232 g, 0.61 mmol, 79% yield). 1H NMR (400 MHz, CDCl3): δ 7.44–7.37 (m, 4H), 7.35–7.29 (m, 2H), 7.29–7.22 (m, 6H), 7.20–7.13 (m, 2H), 4.60 (dd, J = 8.3, 2.7 Hz, 1H), 4.22 (s, 1H), 2.86–2.16 (m, 10H), 1.94 (m, 1H), 1.87–1.75 (m, 1H), 1.72–1.55 (m, 2H), 1.29 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 149.5, 142.9, 142.8, 128.5, 128.3, 128.0, 127.9, 127.8, 126.9, 126.8, 125.3, 125.0, 77.3, 76.2, 73.4, 58.7, 53.3, 51.6, 39.7, 34.4, 31.4, 23.9. LCMS retention time: 4.270 min. LCMS purity 97.3%. HRMS (ESI): m/z calcd for C31H40N2O [M + H]+ 457.3213, found 457.3207.
1-(4-(tert-Butyl)phenyl)-4-(4-(diphenylmethylene)piperidin-1-yl)butan-1-ol (5d)
Method B: 40 (0.019 g, 0.042 mmol), MeOH (3 mL), and sodium borohydride (6 mg, 0.17 mmol) to produce 5d (0.010 g, 0.022 mmol, 52% yield). 1H NMR (400 MHz, CDCl3): δ 7.35–7.26 (m, 8H), 7.23–7.18 (m, 2H), 7.13–7.10 (m, 4H), 4.68–4.65 (m, 1H), 2.66–2.60 (m, 2H), 2.55–2.42 (m, 8H), 2.00–1.94 (m, 1H), 1.90–1.81 (m, 1H), 1.73–1.63 (m, 3H), 1.30 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 149.5, 142.7, 142.3, 136.4, 134.6, 129.7, 128.0, 126.4, 125.3, 125.0, 73.3, 58.7, 55.0, 39.6, 34.4, 31.6, 31.4, 31.1, 24.0, 22.6. LCMS retention time: 4.454 min. LCMS purity 97.1%. HRMS (ESI): m/z calcd for C32H39NO [M + H]+ 454.3104, found 454.3130.
4-(4-Benzhydrylpiperidin-1-yl)-1-(4-(tert-butyl)phenyl)butan-1-ol (5e)
Method B: 42 (0.025 g, 0.056 mmol), sodium borohydride (4 mg, 0.11 mmol), and MeOH (2 mL) to produce 5e (0.017 g, 0.036 mmol, 66% yield) as an oil. 1H NMR (400 MHz, chloroform-d) δ 7.27–7.21 (m, 2H), 7.21–7.13 (m, 10H), 7.05 (m, 2H), 4.51 (dd, J = 8.4, 2.5 Hz, 1H), 3.44 (d, J = 11.1 Hz, 1H), 3.00 (d, J = 11.4 Hz, 1H), 2.82 (d, J = 11.3 Hz, 1H), 2.32 (s, 2H), 2.13–1.77 (m, 4H), 1.76–1.64 (m, 1H), 1.59 (m, 2H), 1.53–1.44 (m, 2H), 1.38–1.24 (m, 3H), 1.21 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 149.5, 143.7, 143.7, 142.9, 128.5, 128.5, 128.0, 128.0, 126.2, 125.4, 125.0, 73.5, 58.6, 53.0, 39.6, 34.4, 31.4, 29.7. LCMS retention time: 1.76 min. LCMS purity 98.6%. HRMS (ESI): m/z calcd for C32H41NO [M + H]+ 456.3260, found 456.3245.
(1-(3-([1,1′-Biphenyl]-4-yloxy)propyl)piperidin-4-yl)diphenylmethanol (6)
To a vial was added the 12 (0.050 g, 0.15 mmol) and THF (3 mL). The reaction was cooled to 0 °C, and triphenylphosphine (0.060 g, 0.23 mmol), DIAD (0.045 mL, 0.23 mmol), and 4-phenylphenol (0.033 g, 0.19 mmol) were added and the reaction was allowed to warm to rt and stirred for 18 h. The reaction was then adsorbed to silica and purified by MPLC (0–10% MeOH:CH2Cl2) to produce the desired product 6 (0.057 g, 0.12 mmol, 77% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.57–7.44 (m, 8H), 7.44–7.37 (m, 2H), 7.30 (dd, J = 8.4, 7.1 Hz, 5H), 7.21–7.15 (m, 2H), 6.98–6.92 (m, 2H), 4.04 (t, J = 6.3 Hz, 2H), 3.09–2.95 (m, 2H), 2.57–2.51 (m, 2H), 2.51–2.41 (m, 1H), 2.17 (s, 1H), 2.08–1.93 (m, 4H), 1.59–1.45 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 158.5, 145.9, 140.8, 133.7, 128.7, 128.2, 128.1, 126.7, 126.6, 126.5, 125.8, 114.7, 79.5, 66.4, 55.4, 54.1, 44.1, 26.9, 26.3. LCMS retention time: 4.155 min. LCMS purity 100%. HRMS (ESI): m/z calcd for C33H35NO2 [M + H]+ 478.2740, found 478.2737.
Strains and Conditions
S. aureus strain UAMS-1,14 a methicillin susceptible clinical osteomyelitis isolate, was used to evaluate the antimicrobial activity of terfenadine and corresponding structural derivatives. Where indicated, antimicrobial activity measures were expanded to include: CRC61 and CRC118, which are spontaneous ciprofloxacin-resistant derivatives of UAMS-1 that were selected by growth on Mueller–Hinton agar (MHA) (Becton, Dickinson & Company, Franklin Lakes, NJ) at 1.5× MIC ciprofloxacin (0.75 μg/mL, Sigma-Aldrich, Saint Louis, MO); S. aureus strains USA 300–0114100 (methicillin resistant), Mu50200 (vancomycin intermediate resistant), and VRSA-1300 (vancomycin resistant); E. faecium 824–05;24E. faecalis OG1RF;400A. baumannii 983709;500K. pneumoniae CKP4;600E. coli 8314 (URMC Clinical Isolate); M. tuberculosis strain mc26020,700 and E. colitolC/imp−.800 Terfenadine was purchased (Sigma-Aldrich), and its analogues were synthesized and provided by the Specialized Chemistry Center at the University of Kansas, Lawrence.
Minimum Inhibitory Concentration (MIC) Testing
Minimum inhibitory concentration (MIC) testing was performed to determine the minimum concentration of the indicated agent necessary to inhibit visible growth of bacteria according to Clinical and Laboratory Standards (CLSI) guidelines.28 Briefly, with the exception of M. tuberculosis, overnight cultures of the indicated bacterial species/strain were subcultured 1:100 in fresh Mueller–Hinton broth (MHB) to early exponential phase (OD600 nm between 0.180 and 0.2). Then, 1 × 105 colony forming units (CFU)/mL bacteria were inoculated into the individual wells of a 96-well round-bottom microtiter plate containing 88 μL of MHB. To the first column, 2 μL of the test compound’s corresponding solvent were also added to each well (negative control). To the next 10 columns, 2 μL of the test compound (dissolved in DMSO for terfenadine and its derivatives or sterile water for ciprofloxacin) were added in increasing 2-fold increments of 0.5–256 μg/mL (final concentration) to each successive well; each compound was evaluated in duplicate. Plates were incubated at 37 °C incubator for 16 h, at which point the minimum inhibitory concentration was determined to be the lowest concentration of test compound that inhibited bacterial growth, as judged by the unaided eye. For M. tuberculosis, MIC measures of the attenuated Biosafety Level 2 approved strain mc26020 was performed using Alamar Blue staining, as previously described.900
S. aureus DNA Gyrase Supercoiling Assay
S. aureus gyrase supercoiling assays were performed in the absence or presence of the indicated concentration of terfenadine and corresponding analogues using the S. aureus Gyrase Supercoiling Assay Kit according to the manufacturer’s recommendations (Inspiralis, Norwich, UK). Briefly, assays (30 μL) were performed in gyrase buffer (40 mM HEPES·KOH pH 7.6, 10 mM magnesium acetate, 10 mM DTT, 2 mM ATP, 500 mM potassium glutamate, 0.5 mg/mL albumin), containing relaxed plasmid pBR322 DNA (250 ng/μL), using 0.5 units (U) of S. aureus DNA gyrase. Where indicated, reactions were carried out in the presence of the indicated concentration (2 μL) of ciprofloxacin or test compounds; their vehicles, water, and DMSO served as negative controls, respectively. The reactions were incubated at 37 °C for 30 min, followed by the addition of 30 μL of STEB (40% w/v sucrose, 100 mM pH 8 Tris-HCl, 1 mM EDTA, 0.5 mg/mL bromophenol blue) stop buffer and 30 μL of 24:1 chloroform:isoamyl alcohol (Amresco, Solon, OH) for a total volume of 90 μL. Reaction products were then loaded on 1% agarose TAE gels and allowed to sit for 40 min prior to electrophoresis, allowing for the salts to diffuse. Gels were stained with 0.5 μg/mL ethidium bromide, and images were analyzed using densitometry (ImageJ, NIH). The IC50 values for each test compound were determined to be the compound concentration that inhibited S. aureus DNA gyrase activity by 50% (determined from densitometry values relative to positive and negative controls of reactions either with or without the enzyme, respectively).
S. aureus Topoisomerase IV Decatenation Assay
An S. aureus topoisomerase IV activity assay was performed according to the manufacturer’s recommendations (Inspiralis) on terfenadine and corresponding analogues to determine if they inhibited the ability of S. aureus topoisomerase IV to decatenate kDNA. The assay was performed by incubating 0.25 U per reaction S. aureus topoisomerase IV enzyme with 200 ng kDNA (kinetoplast DNA isolated from Crithidia fasciculate) in kit-supplied assay buffer (50 mM Tris·HCl pH 7.5, 5 mM MgCl2, 5 mM DTT, 1.5 mM ATP, 350 mM potassium glutamate, and 0.05 mg/mL albumin) with varying amounts of water, DMSO, or test compounds (2 μL total at the desired concentrations) for a total volume of 30 μL at 37 °C for 30 min. Reactions were stopped by the addition of 30 μL of STEB buffer, followed by the addition of 30 μL of 24:1 chloroform:isoamyl alcohol (total volume 90 μL). Reaction products were then loaded on 1% agarose TAE gels and allowed to sit for 40 min prior to electrophoresis, allowing for the salts to diffuse. Gels were stained with 0.5 μg/mL ethidium bromide, and images were analyzed using densitometry (ImageJ, NIH). The IC50 values for each test compound were determined to be the compound concentration that inhibited S. aureus topoisomerase IV activity by 50% (determined from densitometry values relative to positive and negative controls of reactions either with or without the enzyme, respectively).
Transcription Profiling
To study the gene expression profile in response to treatment with terfenadine and ciprofloxacin, GeneChip studies were performed with S. aureus UAMS-1, as previously described.48,49 Briefly, bacterial cells were grown in TSB to early exponential phase (OD600 = 0.250) and were treated with DMSO or 0.5× MIC of ciprofloxacin or terfenadine for 30 min, at which point RNA was extracted. Then, Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA) was used to reverse transcribe 7 μg of each of the three bacterial RNA samples. This was then purified with the QIAquick PCR Purification Kit (Qiagen, Gaithersburg, MD) and subsequently partially digested with DNase I (Ambion, Austin, TX). The Enzo Bioarray Terminal Labeling Kit (Enzo Life Sciences, Farmingdale, NY) was used to 3′ biotinylate the fragmented DNA. This labeled cDNA (2 μg) was hybridized to an S. aureus Affymetrix GeneChip. The manufacturer’s recommendations for the processing of prokaryotic arrays were then followed (Affymetrix; Santa Clara, CA). This was done in biological replicates a minimum of two times. In the data analysis, the control poly(A) cDNA signal intensity or the signals from the entire GeneChip were used to normalize each of the RNA species. These were then averaged with GeneSpring 7.2 software (Agilent Technologies, Redwood City, CA). Those isolates designated as differentially expressed were defined as the RNA species, which had at least a 2-fold difference in signal compared to the mock-treated sample (t-test, P = 0.05).
Docking Studies
Using the Surflex module of the SYBYL 8.0 software package (Certara, St Louis, Mo), the receptor proteins (PDB IDs 2XCS, 2XCT and 3U2K) were prepared by removing the bound solved ligand then defining a pocket as a 20 Å sphere around the original ligand site. The corresponding terfenadine-based ligands were then docked into the original ligand site. The compounds were sketched and protonated in SYBYL, and Gasteiger–Marsili charges were assigned. Default Surflex settings were utilized, and the generated 30 docking poses per compound were visually inspected. The best pose was selected on the basis of Combined CScore (ChemScore, G_Score, D_Score, and PMF_Score) from the SYBYL software.
hERG Inhibition Assay
This assay was performed off-site at Eurofins Panlabs (Taiwan) and was adapted from a previous procedure.50 Human recombinant potassium channel hERG is expressed in human HEK-293 cells in HEPES buffer at pH 7.4, 0.1% BSA, 5 mM KCl, 0.8 mM MgCl2, 130 mM NaCl, 1 mM EGTA, and 10 mM glucose. A 10 μg aliquot was incubated with [3H] Astemizole, and either compound 4g or 6 in assay buffer containing 1% DMSO with 10 increasing doses (0.3 nM to 10 μM). Binding was terminated by filtration onto glass filters, followed by three washes with assay buffer. Radioactivity was determined by scintillation counting on each filter, and specifically bound [3H] astemizole was determined. A dose–response curve was generated to provide the Ki and IC50 for the two compounds tested.
Acknowledgments
We acknowledge Prof. Jeffrey Aubé for encouragement and support. We also thank Sarah Flaherty for creating artwork. We also thank Patrick Porubsky, Benjamin Neuenswander, and David Hill for lab and purification support and Jennifer Golden for scientific guidance. Funding for analogue synthesis was provided by National Institutes of Health for the University of Kansas Chemical Methodologies and Library Development Center (CMLD) Grant 5P50GM069663 assigned to Prof. Jeffrey Aubé (PI). Support for the University of Kansas NMR instrumentation was provided by NIH Shared Instrumentation Grant number S10RR024664 and NSF Major Research Instrumentation Grant number 0320648. Funding for microbiology and enzymology studies was, in part, provided by National Institutes of Health grant 5R01AI103507 awarded to Prof. Paul Dunman and Prof. Damian Krysan. Jessamyn I. Perlmutter was supported by a University of Rochester Take-Five award.
Glossary
Abbreviations Used
- ESKAPE
Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species
- NBTI
novel bacterial topoisomerase inhibitor
- MPLC
medium performance liquid chromatography
- HAI
hospital-acquired infection
- MIC
minimum inhibitory concentration
- SAR
structure–activity relationship
- HTS
high-throughput screen
- IC50
inhibitory concentration of 50%
- TSB
tryptic soy broth
- TSA
tryptic soy agar
- MHB
Mueller–Hinton broth
- MHA
Mueller–Hinton agar
- VISA
vancomycin-intermediate Staphylococcus aureus
- VRSA
vancomycin-resistant Staphylococcus aureus
- MRSA
methicillin-resistant Staphylococcus aureus
- hERG
human ether-à-go-go-related gene
Supporting Information Available
Full synthetic experimentals for intermediates not shown in Schemes 1–8. Microarray data for terfenadine inhibition, log P vs MIC data, and hERG inhibition data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hidron A. I.; Edwards J. R.; Patel J.; Horan T. C.; Sievert D. M.; Pollock D. A.; Fridkin S. K. Antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 2008, 29, 996–1011. [DOI] [PubMed] [Google Scholar]
- Klevens R.; Morrison M. A.; Nadle J.; Petit S.; Gershman K.; Ray S.; Harrison L. H.; Lynfield R.; Dumyati G.; Townes J. M.; Craig A. S.; Zell E. R.; Fosheim G. E.; McDougal L. K.; Carey R. B.; Fridken S. K. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA, J. Am. Med. Assoc. 2007, 298, 1763–1771. [DOI] [PubMed] [Google Scholar]
- Kim J.; Lee D.; Park C.; So W.; Jo M.; Ok T.; Kwon J.; Kong S.; Jo S.; Kim Y.; Choi J.; Kim H. C.; Ko Y.; Choi I.; Park Y.; Yoon J.; Ju M. K.; Kim J.; Han S.-J.; Kim T.-H.; Cechetto J.; Nam J.; Sommer P.; Liuzzi M.; Lee J.; No Z. Discovery of phenylaminopyridine derivatives as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. ACS Med. Chem. Lett. 2012, 3, 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalit I.; Berger S.; Gorea A.; Frimerman H. Widespread quinolone resistance among methicillin-resistant Staphylococcus aureus isolates in a general hospital. Antimicrob. Agents Chemother. 1989, 33, 593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [DOI] [PubMed] [Google Scholar]
- Scheld W. M. Maintaining fluoroquinolone class efficacy: review of influencing factors. Emerg. Infect. Dis. 2003, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby G. A. Mechanisms of resistance to quinolones. Clin. Infect. Dis. 2005, 41, S120–S126. [DOI] [PubMed] [Google Scholar]
- Mitscher L. A. Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chem. Rev. 2005, 105, 559–592. [DOI] [PubMed] [Google Scholar]
- Harnett N.; Brown S.; Krishnan C. Emergence of quinolone resistance among clinical isolates of methicillin-resistant Staphylococcus aureus in Ontario, Canada. Antimicrob. Agents Chemother. 1991, 35, 1911–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black M. T.; Stachyra T.; Platel D.; Girard A.-M.; Claudon M.; Bruneau J.-M.; Miossec C. Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 2008, 52, 3339–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basarab G. S.; Manchester J. I.; Bist S.; Boriack-Sjodin P. A.; Dangel B.; Illingworth R.; Sherer B. A.; Sriram S.; Uria-Nickelsen M.; Eakin A. E. Fragment-to-hit-to-lead discovery of a novel pyridylurea scaffold of ATP competitive dual targeting type II topoisomerase inhibiting antibacterial agents. J. Med. Chem. 2013, 56, 8712–8735. [DOI] [PubMed] [Google Scholar]
- Brvar M.; Perdih A.; Renko M.; Anderluh G.; Turk D.; Solmajer T. Structure-based discovery of substituted 4,5′-bithiazoles as novel DNA gyrase inhibitors. J. Med. Chem. 2012, 55, 6413–6426. [DOI] [PubMed] [Google Scholar]
- Reck F.; Alm R. A.; Brassil P.; Newman J. V.; Ciaccio P.; McNulty J.; Barthlow H.; Goteti K.; Breen J.; Comita-Prevoir J.; Cronin M.; Ehmann D. E.; Geng B.; Godfrey A. A.; Fisher S. L. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II with reduced pKa: antibacterial agents with an improved safety profile. J. Med. Chem. 2012, 55, 6916–6933. [DOI] [PubMed] [Google Scholar]
- Gillaspy A. F.; Hickmon S. G.; Skinner R. A.; Thomas J. R.; Nelson C. L.; Smeltzer M. S. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect. Immun. 1995, 63, 3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck F.; Alm R.; Brassil P.; Newman J.; DeJonge B.; Eyermann C. J.; Breault G.; Breen J.; Comita-Prevoir J.; Cronin M. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: broad-spectrum antibacterial agents with reduced hERG activity. J. Med. Chem. 2011, 54, 7834–7847. [DOI] [PubMed] [Google Scholar]
- Bax B. D.; Chan P. F.; Eggleston D. S.; Fosberry A.; Gentry D. R.; Gorrec F.; Giordano I.; Hann M. M.; Hennessy A.; Hibbs M.; Huang J.; Jones E.; Jones J.; Brown K. K.; Lewis C. J.; May E. W.; Saunders M. R.; Singh O.; Spitzfaden C. E.; Shen C.; Shillings A.; Theobald A. J.; Wohlkonig A.; Pearson N. D.; Gwynn M. N. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [DOI] [PubMed] [Google Scholar]
- Coates A. M.; Halls G.. Antibiotics in Phase II and II clinical trials. In Antibiotic Resistance; Coates A. R. M., Ed.; Springer: Berlin, Heidelberg, 2012; Vol. 211, pp 167–183. [DOI] [PubMed] [Google Scholar]
- Projan S. J. Why is big pharma getting out of antibacterial drug discovery. Curr. Opin. Microbiol. 2003, 6, 427–430. [DOI] [PubMed] [Google Scholar]
- White A. R.; Blaser M.; Carrs O.; Cassell G.; Fishman N.; Guidos R.; Levy S.; Powers J.; Norrby R.; Tillotson G. Effective antibacterials: at what cost? The economics of antibacterial resistance and it’s control. J. Antimicrob. Chemother. 2011, 66, 1948–1953. [DOI] [PubMed] [Google Scholar]
- Aubé J. Drug repurposing and the medicinal chemist. ACS Med. Chem. Lett. 2012, 3, 442–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I.; Bauman J. E.; Bologa C. G.; Buranda T.; Chigaev A.; Edwards B. S.; Jarvik J. W.; Gresham H. D.; Haynes M. K.; Hjelle B.; Hromas R.; Hudson L.; Mackenzie D. A.; Muller C. Y.; Reed J. C.; Simons P. C.; Smagley Y.; Strouse J.; Surviladze Z.; Thompson T.; Ursu O.; Waller A.; Wandinger-Ness A.; Winter S. S.; Wu Y.; Young S. M.; Larson R. S.; Willman C.; Sklar L. A. Drug repurposing from an academic perspective. Drug Discovery Today: Ther. Strat. 2011, 8, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburn T. T.; Thor K. B. Drug repositioning: identifying and developing new uses for existing drugs. Nature Rev. Drug Discovery 2004, 3, 673–683. [DOI] [PubMed] [Google Scholar]
- Huang R.; Southall N.; Wang Y.; Yasgar A.; Shinn P.; Jadhav A.; Nguyen D.-T.; Austin C. P. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med. 2011, 3, 80ps16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. C.; DiDone L.; Jobson J.; Sofia M. K.; Krysan D.; Dunman P. M. Adenylate kinase release as a high-throughput-screening-compatible reporter of bacterial lysis for identification of antibacterial agents. Antimicrob. Agents Chemother. 2013, 57, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woosley R. L.; Chen Y.; Freiman J. P.; Gillis R. A. Mechanism of the cardiotoxic actions of terfenadine. JAMA, J. Am. Med. Assoc. 1993, 269, 1532–1536. [PubMed] [Google Scholar]
- Monahan B. P.; Ferguson C. L.; Killeavy E. S.; Lloyd B. K.; Troy J.; Cantilena L. R. Torsades de pointes occurring in association with terfenadine use. JAMA, J. Am. Med. Assoc. 1990, 264, 2788–2790. [PubMed] [Google Scholar]
- Roy M.-L.; Dumaine R.; Brown A. M. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation 1996, 94, 817–823. [DOI] [PubMed] [Google Scholar]
- CLSI Performance Standards for Antimicrobial Susceptibility Testing: 23rd Informational Supplement, M100; Clinical Laboratory Standards Institute (CLSI): Wayne, PA, 2013.
- DeMartino J. K.; Hwang I.; Connelly S.; Wilson I. A.; Boger D. L. Asymmetric synthesis of inhibitors of glycinamide ribonucleotide transformylase. J. Med. Chem. 2008, 51, 5441–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley J. D.; Murray P. M.; Turp E. R.; Tyler S. N.; Burn R. T. A mild robust generic protocol for the Suzuki reaction using an air stable catalyst. Tetrahedron 2012, 68, 6010–6017. [Google Scholar]
- Honig P. K.; Woosley R. L.; Zamani K.; Conner D. P.; Cantilena L. R. Changes in the pharmacokinetics and electrocardiographic pharmacodynamics of terfenadine with concomitant administration of erythromycin. Clin. Pharmacol. Ther. 1992, 52, 231–238. [DOI] [PubMed] [Google Scholar]
- Kawai S. H.; Hambalek R. J.; Just G. A facile synthesis of an oxidation product of terfenadine. J. Org. Chem. 1994, 59, 2620–2622. [Google Scholar]
- Zhu R.; Xing L.; Wang X.; Cheng C.; Su D.; Hu Y. Highly Practical “Ligand-Free-Like” Copper-Catalyzed N-Arylation of Azoles in Lower Nitrile Solvents. Adv. Synth. Catal. 2008, 350, 1253–1257. [Google Scholar]
- Zhang M. Q.; ter Laak A. M.; Timmerman H. Structure–activity relationships within a series of analogues of the histamine H1-antagonist terfenadine. Eur. J. Med. Chem. 1993, 28, 165–173. [Google Scholar]
- Barnes-Seeman D.; Jain M.; Bell L.; Ferreira S.; Cohen S.; Chen X.-H.; Amin J.; Snodgrass B.; Hatsis P. Metabolically stable tert-butyl replacement. ACS Med. Chem. Lett. 2013, 4, 514–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuitschik G.; Carreira E. M.; Wagner B. r.; Fischer H.; Parrilla I.; Schuler F.; Rogers-Evans M.; Müller K. Oxetanes in drug discovery: structural and synthetic insights. J. Med. Chem. 2010, 53, 3227–3246. [DOI] [PubMed] [Google Scholar]
- Lee J.; Kang S.-U.; Lim J.-O.; Choi H.-K.; Jin M.-k.; Toth A.; Pearce L. V.; Tran R.; Wang Y.; Szabo T.; Blumberg P. M. N-[4-(Methylsulfonylamino)benzyl]thiourea analogues as vanilloid receptor antagonists: analysis of structure–activity relationships for the C-region. Bioorg. Med. Chem. 2004, 12, 371–385. [DOI] [PubMed] [Google Scholar]
- Edgar D. M.; Hangauer D. G.; Leighton H. J.; Mignot E. J. M.; Methods of treating sleep disorders. PCT patent WO2003032912A2, 2003.
- Wu Y.; Tai H.-H.; Cho H. Synthesis and SAR of thiazolidinedione derivatives as 15-PGDH inhibitors. Bioorg. Med. Chem. 2010, 18, 1428–1433. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Johnstone C. A quantitative assessment of hERG liability as a function of lipophilicity. Bioorg. Med. Chem. Lett. 2007, 17, 1759–1764. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nature Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- Tetko I. V. Computing chemistry on the web. Drug Discovery Today 2005, 10, 1497–1500. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Gasteiger J.; Todeschini R.; Mauri A.; Livingstone D.; Ertl P.; Palyulin V. A.; Radchenko E. V.; Zefirov N. S.; Makarenko A. S. Virtual computational chemistry laboratory—design and description. J. Comput.-Aided Mol. Des. 2005, 19, 453–463. [DOI] [PubMed] [Google Scholar]
- Schröder W.; Goerke C.; Wolz C. Opposing effects of aminocoumarins and fluoroquinolones on the SOS response and adaptability in Staphylococcus aureus. J. Antimicrob. Chemother. 2013, 68, 529–538. [DOI] [PubMed] [Google Scholar]
- Peng Q.; Zhou S.; Yao F.; Hou B.; Huang Y.; Hua D.; Zheng Y.; Qian Y. Baicalein suppresses the SOS response system of Staphylococcus aureus induced by ciprofloxacin. Cell. Physiol. Biochem. 2011, 28, 1045–1050. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- Eakin A. E.; Green O.; Hales N.; Walkup G. K.; Bist S.; Singh A.; Mullen G.; Bryant J.; Embrey K.; Gao N. Pyrrolamide DNA gyrase inhibitors: fragment-based nuclear magnetic resonance screening to identify antibacterial agents. Antimicrob. Agents Chemother. 2012, 56, 1240–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazakova S.; Hageman J.; Matava M.; Srinivasan A.; Phelan L.; Garfinkel B.; Boo T.; McAllister S.; Anderson J.; Jensen B.; Dodson D.; Lonsway D.; McDougal L.; Arduino M.; Fraser V.; Killgore G.; Tenover F. C.; Cody S.; Jernigan D. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N. Engl. J. Med. 2005, 352, 468–475. [DOI] [PubMed] [Google Scholar]
- Hiramatsu K.; Hanaki H.; Ino T.; Yabuto K.; Oguri T.; Tenover F. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. Antimicrob. Chemother. 1997, 40, 135–136. [DOI] [PubMed] [Google Scholar]
- Sievert D.; Boulton M.; Stolman G.; Johnson D.; Stobierski M.; Downes F.; Somsel P.; Rudrik J.; Brown W.; Hafeez W.; Lundstrom T.; Flanagan E.; Johnson R.; Mitchell J.; Chang S. Staphylococcus aureus resistant to vancomycin. MMWR Morb. Mortal. Wkly. Rep. 2002, 51, 565–567.12139181 [Google Scholar]
- Murray B. E.; Singh K. V.; Ross R. P.; Heath J. D.; Dunny G. M.; Weinstock G. M. Generation of restriction map of Enterococcus faecalis strain OG1 and investigation of growth requirements and regions encoding biosynthetic function. J. Bacteriol. 1993, 175, 5216–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. C.; Hood I.; Boyd K. L.; Olson P. D.; Morrison J. M.; Carson S.; Sayood K.; Iwen P. C.; Skaar E. R.; Dunman P. M. Inactivation of phospholipase D diminishes Acinetobacter baumannii pathogenesis. Infect. Immun. 2010, 78, 1952–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomakova D. K.; Hsiao C. B.; Beanan J. M.; Olson R.; MacDonald U.; Keynan Y.; Russo T. A. Clinical and phenotypic differences between classis and hypervirulent Klebsiella pneumoniae: an emerging and under-recognized pathogenic variant. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 981–989. [DOI] [PubMed] [Google Scholar]
- Sambandamurthy V. K.; Wang X.; Chen B.; Russell R. G.; Derrick S.; Collins F. M.; Morris S. L.; Jacobs W. R. Jr. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 2002, 8, 1171–1174. [DOI] [PubMed] [Google Scholar]
- Sumandeep K. G.; Garcia G. A. Rifamycin inhibition of WT and Rifresistant Mycobacterium tuberculosis and Escherichia coli RNA polymerases in vitro. Tuberculosis 2011, 91, 361–369. [DOI] [PubMed] [Google Scholar]
- Franzblau S. G.; Witzig R. S.; McLaughlin J. C.; Torres P.; Madico G.; Hernandez A.; Degnan M. T.; Cook M. B.; Quenzer V. K. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. L.; Roberts C.; Disz T.; Vonstein V.; Hwang K.; Overbeek R.; Olsen P. D.; Projan S. J.; Dunman P. M. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J. Bacteriol. 2006, 188196739–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utaida S.; Dunman P. M.; Macapagal D.; Murphy E.; Projan S. J.; Singh V. K.; Jayaswal R. K.; Wilkinson B. J. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 2003, 149102719–2732. [DOI] [PubMed] [Google Scholar]
- Finlayson K.; Turnbull L.; January C. T.; Sharkey J.; Kelly J. S. [3H]Dofetilide binding to HERG transfected membranes: a potential high throughput preclinical screen. Eur. J. Pharmacol. 2001, 430, 147–148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.