

Abstract
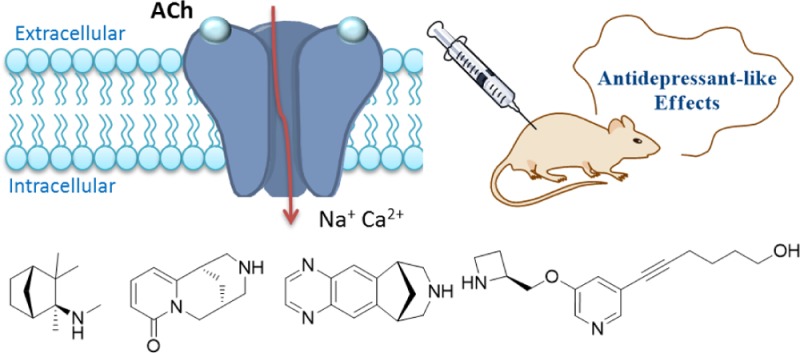
Nicotinic acetylcholine receptors (nAChRs) have been investigated for developing drugs that can potentially treat various central nervous system disorders. Considerable evidence supports the hypothesis that modulation of the cholinergic system through activation and/or desensitization/inactivation of nAChR holds promise for the development of new antidepressants. The introductory portion of this Miniperspective discusses the basic pharmacology that underpins the involvement of α4β2-nAChRs in depression, along with the structural features that are essential to ligand recognition by the α4β2-nAChRs. The remainder of this Miniperspective analyzes reported nicotinic ligands in terms of drug design considerations and their potency and selectivity, with a particular focus on compounds exhibiting antidepressant-like effects in preclinical or clinical studies. This Miniperspective aims to provide an in-depth analysis of the potential for using nicotinic ligands in the treatment of depression, which may hold some promise in addressing an unmet clinical need by providing relief from depressive symptoms in refractory patients.
Introduction
Depression is a common and frequently severe psychological condition with a distinct change of mood, characterized by sadness, loss of interest and pleasure, feelings of guilt or low self-worth, disturbed sleep or appetite, and feelings of tiredness and poor concentration, affecting approximately 120 million people worldwide.1 Numerous therapeutic agents exist for the treatment of depression that target monoamine transporters regulating the uptake of the neurotransmitters dopamine, serotonin, and norepinephrine.2 However, a considerable proportion of patients respond poorly to these drugs,3 as demonstrated by the NIMH-funded sequenced treatment alternatives to relieve depression (STAR*D) study conducted between 2001 and 2006, which highlighted the inadequacy of current medications for major depressive disorder (MDD).4 Therefore, there is still an urgent need for potent pharmacotherapies associated with novel biological mechanisms of action. In this Miniperspective, we review the association between depression and nicotinic acetylcholine receptors (nAChRs), especially the α4β2-nAChR subtype, from the perspective of clinical and preclinical findings. We highlight the most recently developed α4β2-nAChR agonists and antagonists that exhibit antidepressant-like effects in vivo.
The cholinergic hypothesis of depression proposes that hyperactivity of the cholinergic system over that of the adrenergic system leads to depression (Figure 1).5 Several lines of evidence from rodent and human studies support this hypothesis. Flinders sensitive rats, a line selectively bred for increased cholinergic sensitivity, were found to exhibit several depression-like behaviors,6,7 and increased acetylcholine (ACh) signaling in the hippocampus was found to promote behaviors in mice related to anxiety and depression.8 In humans, physostigmine, which potentiates cholinergic transmission by inhibiting acetylcholinesterase (AChE), the enzyme that breaks down ACh, produces depressive-like symptoms in individuals with and without a history of depression.5 Administration of the nonselective nicotinic antagonist mecamylamine (1) (Scheme 1),9−11 the partial agonist varenicline (2),12 or the muscarinic antagonist scopolamine (3)13,14 demonstrated putative antidepressant-like effects, especially in treatment-resistant patients receiving their regular psychotropic medications such as the selective serotonin reuptake inhibitor (SSRI) citalopram (4).9 Magnetic resonance imaging studies have shown that the levels of choline, the rate-limiting precursor to endogenous ACh, were elevated in the brains of patients with depression as well as in the frontal cortex of adolescents with depression.15,16 Additionally, a number of key antidepressants such as SSRIs (fluoxetine, sertraline (5), paroxetine, and citalopram 4), the norepinephrine reuptake inhibitor reboxetine (6), the norepinephrine dopamine reuptake inhibitor bupropion (7), and tricyclics (amitriptyline (8), imipramine (9), and nortriptyline)17 have all been shown to possess antagonistic activities at nAChRs,18−20 although, in most cases, drug concentrations achieved in the human brain would not be adequate to affect nAChR functions.21 Nicotine (10) itself, and some nicotinic agonists or antagonists, can potentiate the antidepressant-like effects of the SSRIs and SNRIs in rodent models,22,23 likely due to the common end point of reduced function due to receptor desensitization by agonists or antagonism. The development of nAChR ligands to attenuate cholinergic activity to treat depression could conceivably help to treat depressive symptoms in refractory patients.
Figure 1.

Role of the cholinergic system in depression. The cholinergic hypothesis of depression postulates a hyperactivity of the cholinergic system over that of the adrenergic system in the brain. Choline (the rate-limiting precursor to endogeneous ACh) crosses the blood–brain barrier to enter the brain and is actively transported into the cholinergic presynaptic terminals by an active uptake mechanism. The neurotransmitter ACh is synthesized from choline and acetyl coenzyme A, catalyzed by the enzyme choline acetyl transferase. ACh is sequestered into secretory vesicles by vesicular ACh transporters. Once released from the presynaptic terminals, ACh can interact with a variety of presynaptic and postsynaptic receptors. Two classes of the cholinergic ACh receptors are muscarinic (G protein-coupled) and nicotinic (ionotropic). Once activated, nAChRs form transient open cationic channels that allow the ions Na+, K+, and Ca2+ to flow across the plasma membrane and induce cellular responses. Prolonged exposure to ACh or nicotinic agonist causes a gradual decrease in the rate of this ionic response, leading to a high affinity, longer-lasting functionally inactive state, referred to as desensitization. ACh has its signal terminated primarily by the enzyme AChE, unlike many other monoaminergic neurotransmitters where reuptake mechanisms predominate.
Scheme 1. Compound Structures.

Nicotinic Acetylcholine Receptors and Depression
The two major types of cholinergic receptors are muscarinic ACh receptors (mAChRs) and nAChRs, both of which are widely distributed in the central and peripheral nervous systems.24 G protein-coupled mAChRs are believed to be involved in mood regulation and AChE-induced depressive behavior.5,13,14,25 Neuronal nAChRs belong to the ligand-gated ion channel superfamily of neurotransmitter receptors. Varying combinations of nAChR subunits (α1−α10, β1−β4, γ, δ, and ε; α2−α7 and β2−β4 are expressed in the brain) assemble into pentameric ion channels, allowing for diverse pharmacological properties.26 Each nAChR subunit consists of a large amino-terminal extracellular domain (ECD), a transmembrane domain comprising four α-helices (M1–M4), and a variable cytoplasmic domain between M3 and M4. ACh binding sites are thought to form between the subunit interfaces of the ECD bound by the C-loop containing the face of an α-type subunit and the face of an adjacent subunit. When acutely activated by endogenous ACh or exogenous nicotinic ligands, nAChRs form transient open cationic channels that allow the ions Na+, K+, and Ca2+ to flow across the plasma membrane and induce cellular responses.27 Prolonged exposure to ACh or nicotinic agonists causes a gradual decrease in the rate of this ionic response, leading to a longer-lasting functionally inactive state through a process referred to as desensitization. nAChRs have been found to contribute to mood control by regulating behavioral reinforcement in the mesolimbic dopamine system, corticotropin releasing factor and function in the hypothalamic–pituitary–adrenal (HPA) axis, circadian rhythms in the suprachiasmatic nucleus, and cytoplasticity in the hippocampus.11 Given nAChR subtype diversity and their involvement in the modulation of various key neurotransmitter systems, including dopamine, serotonin, norepinephrine, glutamate, and γ-aminobutyric acid (GABA), nicotinic ligands have the potential to treat a multitude of neurological and psychiatric disorders, including depression.9,28
The α4β2 heteropentameric and α7 homopentameric subtypes are the two major nAChR subtypes expressed in the brain (Figure 2).29 Both subtypes are implicated in the mediation of the pharmacological and behavioral effects of compound 10 and in nAChR-mediated modulation of monoamine release30,31 and are likely involved in the antidepressant effects of nicotinic ligands. Studies investigating the role of nAChRs in depression have focused primarily on the α4β2* (asterisk indicates possible assembly with other subunits) and α7-nAChR subtypes. α4β2*-nAChRs are widely distributed in the neuroanatomic regions implicated in depression, including the thalamus, basal ganglia, striatum, hypothalamus, amygdala, ventral tegmental area (VTA), locus coeruleus, and dorsal raphe nucleus,9 and α7-nAChRs are highly expressed in the hypothalamus, hippocampus, and cortex.32,33 α4β2*-nAChRs are thought to regulate the release of monoamine neurotransmitters through action in these areas.34,35 β2-knockout mice showed decreased immobility in the forced swim test (FST) compared to that of wild-type mice, indicating that the absence of β2-nAChRs-mediated signaling could manifest in an antidepressant-like phenotype in vivo.36 The antidepressant-like effect of the nAChR antagonist compound 1 was diminished when the β2- or α7-subunits were knocked out.37 Similarly, the antidepressant-like effects of the nAChR agonist sazetidine-A (11)38 and the tricyclic antidepressant compound 8(36) were absent in mice lacking the β2-subunit. Additionally, the efficacies of compounds 4 and 6 in the mouse FST were enhanced by agonists at either α4β2*- or α7-nAChRs.23 These findings suggest the involvement of β2- and α7-receptor subtypes in mediating the antidepressant-like effects of nicotinic ligands.
Figure 2.
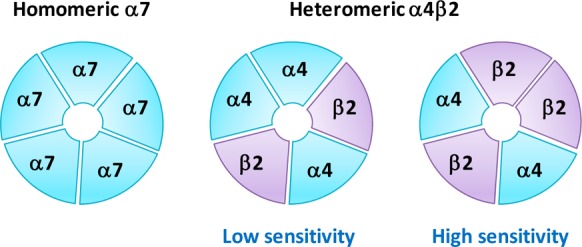
Selected nAChR subtypes. The high sensitivity (HS) α4β2-nAChR has a presumed α4/β2 subunit ratio of 2:3 and exhibits comparatively high sensitivity to nicotinic agonists, whereas the low sensitivity (LS) α4β2-nAChR, at which nicotinic agonists have lower observed potency, is composed presumably of α4 and β2 subunits in a 3:2 ratio.
Clinical studies provide additional evidence for a relationship between α4β2*-nAChRs and depression. Single photon emission computed tomography (SPECT) using the α4β2*-nAChR specific radioligand [123I]5-I-A-85380 ([123I]12) revealed that the β2*-nAChR availability across all brain regions in depressed patients was lower than that in healthy subjects.39 Additionally, positron emission tomography using 2-[18F]fluoro-3-(2[S]-2-azetidinylmethoxy)-pyridine showed reduced levels of ligand binding to α4β2*-nAChR in Parkinson’s patients with depressive symptoms.40 It was recently reported that the clinical action of compound 6 may be at least partially due to its inhibitory action on α4β2-nAChR.41
Designing a nicotinic ligand to provide maximal therapeutic efficacy and minimal side effects depends on a ligand’s ability to specifically target the desired combination of nAChR subtypes. Observations that nAChR α6 subunits are not widely distributed in the brain but are most prevalent in midbrain dopaminergic regions in the mammalian CNS suggest their potential involvement in mood control. As such, targeting α6*-nAChR may be indicated.11,42−44 In the basal ganglia, the VTA and substantia nigra, in particular, α6- and possibly nAChR β3 subunits, are included in α4β2*-nAChRs that appear to have high affinity for nicotinic agonists.34 α3β4*-nAChR subtypes are expressed at relatively low levels in the brain45 except for the interpeduncular nucleus, fasciculus retroflexus, and median habenula.46 However, activation or blockade of α3β4*-nAChR, which are also expressed in the peripheral nervous system, may result in side effects in vivo, including dysregulation of the autonomic nervous system.47,48 Selective and potent partial agonists of α4β2*-nAChRs, especially those with low affinity for α3β4*-nAChRs, are considered to have higher efficacy and likely fewer side effects in rodent behavioral models, although the involvement of α3β4*-nAChRs in mood control cannot be completely ruled out.49−51 We also point out that because of their roles as “accessory” subunits that are incapable of forming functional receptors alone or even in combination with nAChR α or β subunits (except, perhaps, as α7α5-nAChR), we have not delved deeply into the potential roles played by the α5 subunits. However, α5 subunits can integrate into α3β4*- and α4β2*-nAChRs and perhaps into α6*-nAChRs to further extend the diversity in receptor subtypes and isoforms, which may affect pharmacological profiles.52,53 Continuing studies will extend our knowledge of the role of α5 subunits in nAChRs.
The essential pharmacophore presented by nicotinic ligands consists of a cationic center (e.g., a quaternized or protonated nitrogen) and a hydrogen-bond acceptor (e.g., the pyridine nitrogen atom in the case of compound 10). The cationic nitrogen binds to a tryptophan residue of the principal α-subunit, forming a cation−π interaction.54−56 The hydrogen-bond acceptor has been shown to bind to the complementary β-subunit, likely mediated by a water molecule to the backbone NH of a Leu residue in the α4β2 receptor.57,58 Additionally, various studies support the idea that agonist interactions with the principal α-subunit are important for binding affinity, while interactions with key amino acids present in the complementary β-subunit affect agonist efficacy.59,60
High-resolution crystal structures of integral membrane protein mammalian nAChRs are not yet available. Invertebrate pentameric acetylcholine binding protein (AChBP) subunits share 20–24% sequence identity with that of the homologous ECDs of nAChR subunits. However, the putative binding site and some aromatic residues found in nAChR subunits are reported to be conserved. Therefore, AChBPs have been characterized and used to provide insights into ligand recognition sites and other elements of nAChR subunit ECDs and ligand binding site interfaces. The majority of ligand-bound structures have been obtained with AChBP from Aplysia californica (Ac) or Lymnaea stagnalis (Ls) (Figure 3). The Ls-AChBP was postulated to be a more suitable surrogate of α4β2-nAChR for a series of 1-(pyridin-3-yl)-1,4-diazepane analogues because the Trp 53 in Ls-AChBP subunits is Tyr in Ac-AChBP subunits, which interacts less favorably with the quaternized or protonated nitrogen of nicotinic ligands. Recently, the nonmolluscan acetylcholine binding protein from the marine annelid Capitella teleta (Ct), namely, Ct-AChBP, was identified61 and reported to more closely mimic α4β2-nAChR than does the Ac-AChBP based on comparisons of the ligand binding affinities.62 The X-ray crystal structure of Ct-AChBP with lobeline or compound 2 bound along with mutagenic studies highlight the importance of key interactions that are responsible for receptor activation or desensitization and for location of key residues in loops D (W57) and E (V111, F119, and L121) in the complementary subunit opposed to the α subunit at the ligand binding subunit interface.
Figure 3.
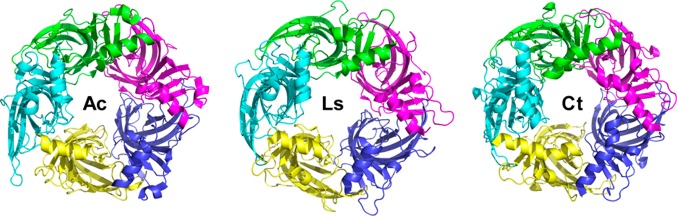
Top view of X-ray crystal structures of Ac-, Ls-, and Ct-AChBPs. The figure was generated using PDB files 2BR7, 1I9B, and 4B5D by PyMOL.
Because of the higher homology between Ct-AChBP and the human α4 and β2 subunits (as computed by ClustalX: α4/Ct-AChBP = 64.6% and β2/Ct-AChBP = 62.1%), especially for key residues involved in ligand recognition (Figure 4), a homology model was generated and refined based on a Ct-AChBP for the ligand binding domain of the human α4β2-nAChR. A ribbon structure for the ECD modeled α4β2-nAChR shows a 10-stranded β-sandwich capped by an N-terminal α-helix for each subunit (Figure 5A), for which the modeled β-sandwich can be nicely superimposed on that of Ct-AChBP (Figure 5B). This homology model may provide useful information for designing other selective α4β2-nAChR ligands.63
Figure 4.

Amino acid sequence alignment of the Ct-AChBP with extracellular domains of nAChR α4 or β2 subunits. Green boxes highlight positions of key residues of the α4 subunit, and blue boxes outline key residues of the β2 subunit. Residue numeration refers to that for the human α4 subunit. (This is for the mature α4 subunit, not including cleavage of the leaders sequence including the translational start methionine residue.).
Figure 5.

Homology model of the human α4β2-nAChR ECD including the ligand binding interface: (A) ribbon structure representation colored by subunit (yellow, α4; azure, β2) for the human α4β2-nAChR ECD and (B) superimposition of the modeled structure (in blue) with the experimental template from Ct-AChBP (in red).
nAChR Agonists and Antagonists Exhibiting Antidepressant-Like Effects
The diversity of nAChR subtypes provides an opportunity to generate subtype-specific ligands that could treat a variety of conditions, although the high sequence homology across individual subtypes of brain nAChRs poses a substantial challenge for the development of subtype-selective nicotinic drugs. Both academia and industry have contributed to a growing body of literature concerning the rational design of potential antidepressant ligands that more selectively target β2*-nAChR than does compound 10.
Rodent behavioral models have been used to assess antidepressant-like effects of these new chemical entities. The most widely used behavioral assay for antidepressant-like efficacy is the FST, in which a mouse or rat is placed in a beaker of water and the amount of time the animal spends passively floating is measured. Clinically therapeutic antidepressants typically reduce the time an animal will spend immobile. Similarly, the tail suspension test (TST) measures immobility time when the rodent is suspended by its tail. This test is relatively short in duration, and in the case where an antidepressant agent is given, the animal will struggle for longer periods of time compared to vehicle treated animals.64 The novelty-suppressed feeding (NSF) test is a behavioral paradigm originally utilized to measure anxiolytic-like effects of drugs, but more recently the NSF assay has been proposed as a behavioral platform sensitive to the chronic but not acute administration of antidepressants. In NSF, food-deprived mice experience a conflict between feeding and the fear of exploring the novel environment of a brightly lit open area or an unfamiliar cage containing food. Chronic treatment with antidepressants reduces latency to eat in the novel environment. Additionally, an alternative version of the NSF has been advanced that is called the novelty-induced hypophagia (NIH) test, but it utilizes a palatable food to eliminate the need for food restriction. In general, these models predict the onset of action of antidepressants consistent with the therapeutic time course found in humans and therefore validate the NSF/NIH tests as useful behavioral paradigms to gauge the antidepressant efficacy of compounds.65
nAChR Antagonists
(1R,2S,4S)-N,2,3,3-Tetramethylbicyclo[2.2.1]heptan-2-amine, 13

The nicotinic ligand that has received the most attention as a potential antidepressant is compound 1, a racemic noncompetitive and nonselective antagonist of nAChRs (IC50,α3β4 = 91–610 nM, IC50,α4β2 = 0.6–2.5 μM, and IC50,α7 = 1.6–6.9 μM). Originally developed as an antihypertensive agent, anecdotal reports of mood modulation and a hypothesis that traditional antidepressants might be acting in part through noncompetitive antagonism of nAChRs19 led to a preliminary controlled study that demonstrated therapeutic effects of compound 1 for mood disorders, including MDD, that were comorbid with Tourette’s disorder.10,50 However, the subgroup sizes were very small, and the comorbidity with Tourette’s disorder precluded any conclusions from being drawn about the antidepressant efficacy of compound 1 monotherapy in patients who do not have Tourette’s disorder.
In studies using receptor expression in Xenopus oocytes, the 2S-(+)-enantiomer of compound 1, TC-5214 (13),51 was found to dissociate more slowly than 2R-(−)-mecamylamine (14) from α4β2- and α3β4-nAChRs, as well as more slowly from α4β2- than α3β4-nAChRs. IC50 values for compound 13 at α3β4-, α4β2-, α7-, or α1β1γδ-nAChRs are similar to those of compound 14 (IC50,α3β4 = 0.2–0.6 and 0.05–0.4 μM, IC50,α4β2 = 0.5–3.2 and 0.5–1.7 μM, IC50,α7 = 1.2–4.6 and 2.2–5.8 μM, and IC50,α1β1γδ = 0.6–2.2 and 0.3–1.1 μM).51 Further pharmacological studies suggested that compound 13 is a more efficacious antagonist of LS α4β2-nAChRs than compound 1 and can act as a positive allosteric modulator at HS α4β2-nAChRs.66 This was hypothesized to be mechanistically important; however, the positive allosteric modulation at α4β2-nAChRs was inferred from a slight potentiation of the HS α4β2-selective agonist TC-2559 (15) in a mixed population of HS and LS α4β2-nAChRs. Potentiation of ACh was not demonstrated, and because the effect was not reproduced in a pure population of HS α4β2-nAChRs, activation of LS α4β2-nAChRs by the coapplication of compounds 15 and 13 was not excluded.
Compound 13 exhibited higher anxiolytic- and antidepressant-like effects in several animal models than racemic mecamylamine (forced swim and social interaction tests, light/dark assay). These behavioral activities were attributed to antagonist effects at α4β2*-nAChRs.67 Moreover, compound 13 showed a superior preclinical safety profile compared to that of either the racemic compound or the 2R-(−)-enantiomer. Compound 13 was found to be well-tolerated in acute and chronic toxicity studies in different animal models (mice, rats, and dogs) and showed acceptable pharmacologic, pharmacokinetic, and metabolic profiles for therapeutic development.
Targacept advanced compound 13 to a phase 2 study as an augmentation in patients who were inadequate responders to the SSRI compound 4. Initial results were very promising, with patients showing an average 6 point improvement on the primary end point, the Hamilton depression rating scale (HAM-D), as well as improvement on other secondary measures.68 A collaboration with AstraZeneca (Targacept and AstraZeneca’s RENAISSANCE program) followed, advancing compound 13 to phase 3 development as an adjunctive therapy in MDD patients who were inadequate responders to SSRI or SNRI monotherapy. Unfortunately, four phase 3 trials (two fixed and two flexible dose trials) failed to meet the primary end point of achieving a greater change in the Montgomery–Asberg depression rating scale total score for the experimental group receiving an adjunctive therapy of compound 13 combined with an SSRI or SNRI than for the placebo group receiving monotherapy with an SSRI or SNRI alone.69−72
There are several possible explanations for the failure of compound 13 in the phase 3 trials. One explanation is that the studies that utilized compound 13 as an adjunct therapy had an unusually high placebo response. Every dose group (compound 13 and placebo) showed at least a 40% improvement in the MADRS total score after 8 weeks of adjunct treatment. A contemporaneous phase 2b monotherapy study of compound 13, which had a lower placebo response, provided some indications of a dose-related antidepressant response, but the study was terminated early.73 It has been suggested that pursuing compound 13 as a monotherapy may have led to a better outcome than as a combination therapy with an SSRI or SNRI.73,74 Subjects in placebo groups in the combination study received SSRI or SNRI treatment, and manipulation of the serotonergic or noradrenergic system may have interfered with the antidepressant response of compound 13.73 Additionally, other authors have suggested that a combination study with a tricyclic antidepressant, which would have antimuscarinic effects, rather than an SSRI or SNRI devoid of antimuscarinic activity, may have been more effective. An antidepressant with antimuscarinic effects could have pro-nicotinic effects through a neuro-adaptive upregulation of cholinergic tone, which could be normalized by compound 1. Alternatively, combining an antinicotinic with an antimuscarinic may have produced a complementary effect in reducing a hypercholinergic state than either treatment alone. Papke and Picciotto74 also suggest that compound 1, in particular, the 2S-(+)-enantiomer 13, may not have been the best choice for the clinical trials. The rationale for using compound 13 was based on a study that suggested that compound 13 had both activating and inhibitory effects at α4β2-nAChRs, a better in vivo profile in animal studies, and a better safety profile than the 2R-(−)-enantiomer.67,75 However, more recent studies found little in vitro evidence to differentiate the pharmacological properties of compound 1 and the 2R- and 2S-enantiomers and only a modest difference in potency in vivo in the tail flick test between the stereoisomers.76 Furthermore, compound 1 is an antagonist that is more potent at inhibiting ganglionic α3β4-nAChRs than α4β2-nAChRs in the brain that are thought be the primary target of hypercholinergic activity associated with depression.25,77 A recent estimate of the free compound 1 concentration in the brain indicates that it is likely insufficient to be pharmacologically relevant, blocking approximately 20% of α4β2*-nAChR function.21 Further evidence that compound 13 did not provide sufficient inhibition on nAChRs comes from the failure of CP-601927 (16),78 an α4β2 partial agonist that was tested in a phase 2 clinical trial as an augmentation in treatment resistant subjects with major depression.79,80 Weber et al.21 propose that compound 16, which was predicted to inhibit only 23% of α4β2-nAChRs,81 would not translate into improved antidepressant efficacy.
Although it is possible that compound 13 could be an effective monotherapy, given its poor nAChR subtype selectivity and the failure in the phase 3 clinical trial, it seems unlikely that this ligand will have a future in the treatment of depressive disorders. The disappointing failure of compound 13 illustrates the need for a better understanding of the optimal pharmacological properties to maximize clinical antidepressant efficacy. More potent nAChR inhibitors that block >25% α4β2 nAChR would likely be more effective than compound 1 or compound 13. Inhibition may be achieved with more potent antagonists, partial agonists with very low intrinsic activity, or desensitizing agents.
2-(tert-Butylamino)-1-(3-chlorophenyl)propan-1-one, 7
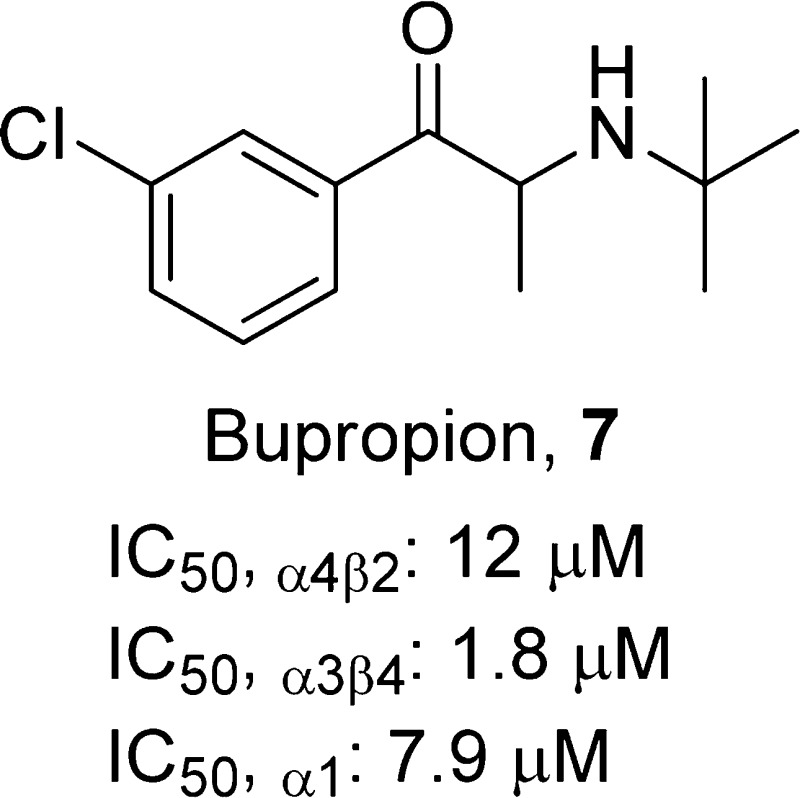
Compound 7(82) is an α-aminoketone believed to elicit antidepressant effects by acting as a dopamine and norepinephrine reuptake inhibitor. It was first reported to show efficacy as a smoking-cessation aid in nondepressed patients in 1994 and has subsequently proven to be a noncompetitive antagonist at a variety of nAChR subtypes.83,84 Compound 7 is most potent as a dopamine reuptake inhibitor, with an IC50 of 550 nM, while potencies at norepinephrine transporters and a number of nAChR subtypes fall in the low micromolar range. However, it has long been appreciated that clinical outcomes correlate poorly with plasma concentrations of compound 7(85) and that active metabolites reaching higher and longer-lasting concentrations in plasma likely play the leading role. The principal metabolite, 2S,3S-hydroxybupropion (17), has reduced potency at dopamine transporters and most nAChR subtypes, whereas its potency at norepinephrine transporters and α4β2-nAChRs is enhanced.76 It appears that a determining factor of compound 7’s efficacy is its metabolism to sufficiently high concentrations of compound 17, as differences in its plasma concentrations between responders and nonresponders was the most statistically significant difference in a 2006 study.86 Plasma concentrations reported for compound 17 were in the low micromolar range. Although compound 7 and another active metabolite, threohydrobupropion, were present at lower concentrations, they may have additive effects in combination with the principal metabolite that provide a significant contribution to a complex pharmacology mediated by norepinephrine transporters, dopamine transporters, and α4β2-nAChRs. Evaluation of compound 7 and its hydroxyl metabolites in the mouse FST showed that compound 17 was equally potent to that of compound 7, whereas the 2S,3R-isomer showed no effect on immobility.83 The 2S,3S-isomer was also a more potent inhibitor of [3H]norepinephrine uptake and [3H]dopamine update as well as a more potent antagonist of α4β2-nAChR function than that of the 2S,3R-isomer, suggesting that the 2S,3S-isomer may be a better candidate than compound 7 for antidepressant treatment.
(2S,13bS)-2-Methoxy-2,3,5,6,8,9,10,13-octahydro-1H,12H-pyrano[4′,3′:3,4]pyrido[2,1-i]indol-12-one, 18
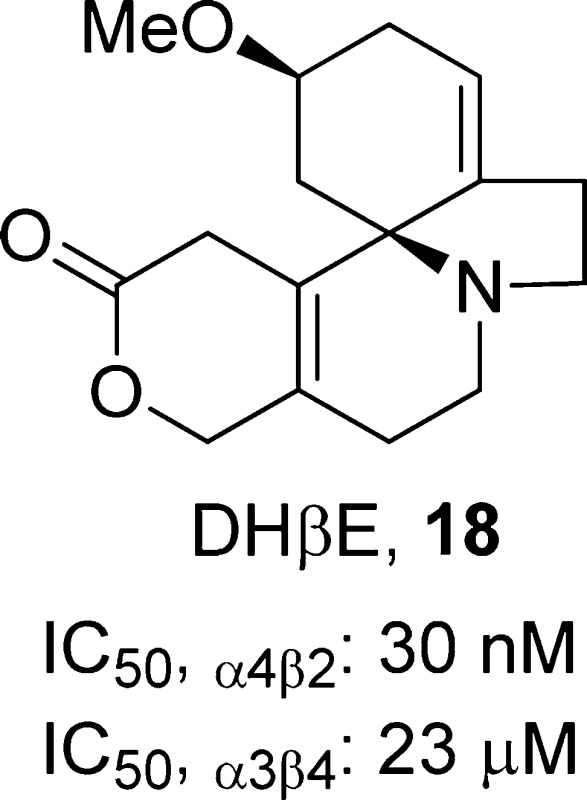
Dihydro-β-erythroidine (DHβE, 18)87 is an alkaloid that is isolated from the seeds of Erythrina L., a genus of trees and shrubs that is found in the tropics and subtropics around the globe. In Central America, Erythrina had an important role in folk medicine during precolonial times. Compound 18 acts as a competitive antagonist at α4β2-nAChRs (IC50,α4β2 = 30 nM and IC50,α3β4 = 23 μM)87 that blocks compound 10-induced dopamine release from rat striatal slices (IC50 = 30 nM).88 Compound 18 shows antidepressant-like effects in the mouse FST (3.0 mg/kg) and TST (1.0 and 3.0 mg/kg) without affecting locomotor activity.89 Additionally, compound 18 is able to potentiate the antidepressant-like effects of compound 9 (4 and 20 mg/kg) in the TST.22
((3S,6S,6aS,7R,7aR,8S,9R,10S,11aR,12S,12aS,13S)-1-Ethyl-11a,12-dihydroxy-6,8,10,13-tetramethoxydodecahydro-2H-3,6a,12-(epiethane[1,1,2]triyl)-7,9-methanonaphtho[2,3-b]azocin-3(4H)-yl)methyl 2-((S)-3-methyl-2,5-dioxopyrrolidin-1-yl)benzoate, 19

Methyllycaconitine (MLA, 19),90 a complex diterpenoid alkaloid isolated from the seeds of Delphinium brownii, is a competitive and selective α7-nAChR antagonist with an IC50 value in the subnanomolar range, as tested in Xenopus oocytes and α7-transfected SH-SY5Y cells.90,91 Compound 19 was found to partially inhibit anatoxin-evoked dopamine release from rat striatal slices92 and decrease the time spent immobile in NMRI female mice in the mouse FST (10 mg/kg) and TST (10 mg/kg) without affecting locomotor activity.89 Another study, however, found no effect of compound 19 (10 mg/kg) in the FST in BALB/c male mice.38 The discrepancies between these studies, which may be related to mouse strain, gender, or procedural differences, warrant further investigation into the role of α7 nAChR inhibition and antidepressant function.
7-(Pyridin-3-yl)-1,7-diazaspiro[4.4]nonane, 20
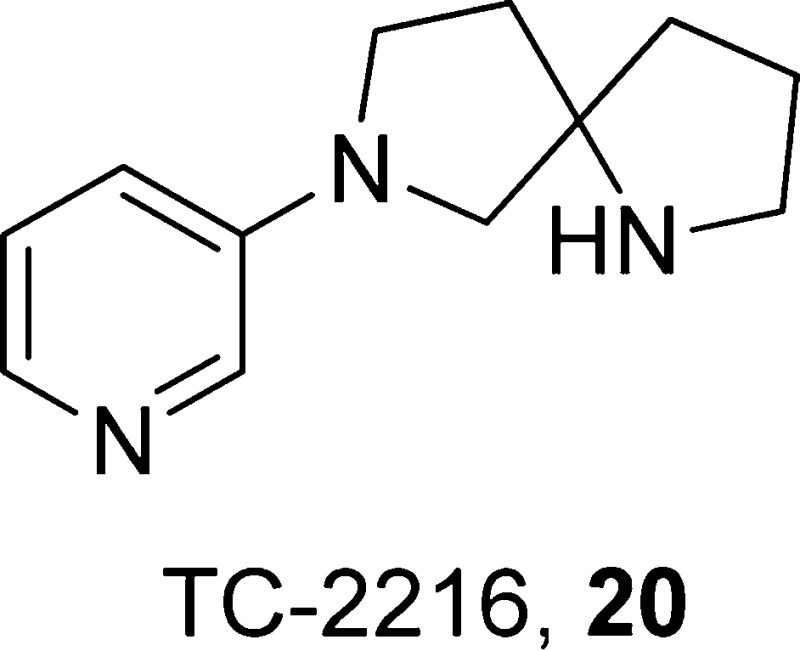
Another α4β2-nAChR antagonist is the Targacept compound TC-2216 (20).93 This compound was reported to show beneficial effects in preclinical studies, thereby promoting its further development for the treatment of depression and anxiety disorders.93 A phase 1 clinical trial of racemic compound 20 was initiated in 2008, but no further trials were conducted.
nAChR Agonists
Modulation of nAChRs with partial agonists is pharmacologically complex, as it involves potentially simultaneous and interacting effects. Under acute conditions, partial agonists elevate baseline cholinergic tone while simultaneously lowering the ceiling of nAChR-mediated signaling. Depending on potency, dose, and exposure, partial agonists could have an additive effect with endogenous ACh, thereby lowering the threshold for ACh signaling while simultaneously lowering the efficacy of that signal. Under conditions of chronic administration, partial agonists desensitize nAChR, and it is believed that the resulting attenuation of cholinergic signaling is the principal pharmacotherapeutic end point.
Cytisine and Derivatives
(1R,5S)-1,2,3,4,5,6-Hexahydro-8H-1,5-methanopyrido[1,2-a][1,5]diazocin-8-one, 21

(−)-Cytisine (21),94 a tricyclic quinolizidine alkaloid isolated from the seeds of Cytisus laburnum L., has been used to treat tobacco dependence in Eastern Europe since the 1960s. Compound 21 is an α4β2-nAChR partial agonist (Ki,α4β2 = 2 nM) and an α3β4 (Ki,α3β4 = 480 nM) and α7 (Ki,α7 = 5890 nM) full agonist that shows antidepressant-like activities in several rodent models,95 presumably mediated by a reduction of neuronal activity in the basolateral amygdala.96 Compound 21 (10 μM) was reported to exhibit 56% of the response relative to that of compound 10 and to inhibit 30% of the current evoked by compound 10 (10 μM) in Xenopus oocytes expressing human α4β2-nAChRs.97 However, it was reported to have unfavorable side effects, which include nausea, vertigo, abdominal pain, respiratory stimulation, and muscle weakness.98 Mineur et al. observed that compound 21 was not tolerated by mice at a dose of >1.5 mg/kg.95 Additionally, the poor absorption and brain penetration94,99−101 of compound 21 may also limit its application as a clinical antidepressant.
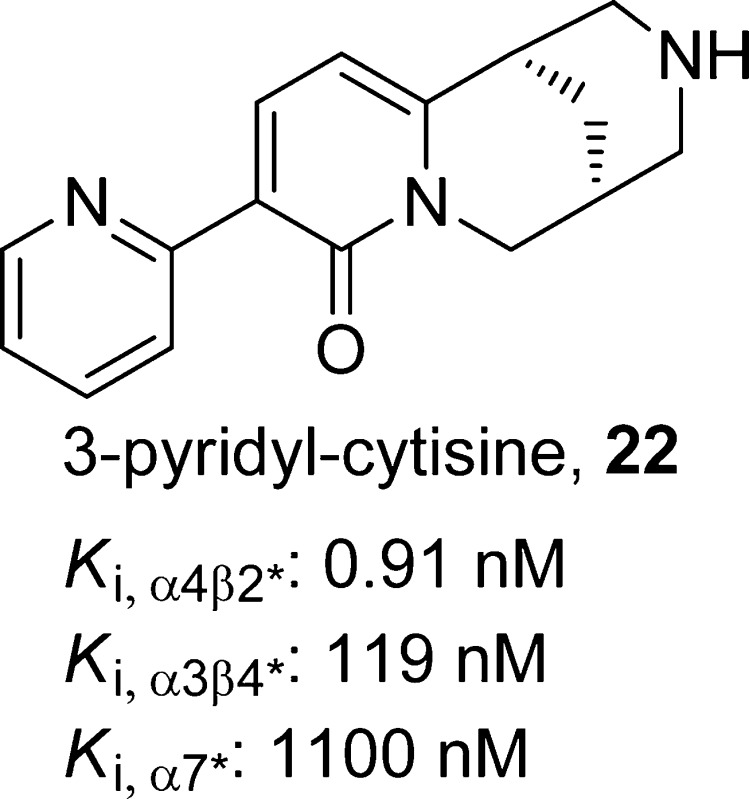
(1R,5S)-9-(Pyridin-2-yl)-1,2,3,4,5,6-hexahydro-8H-1,5-methanopyrido[1,2-a][1,5]diazocin-8-one, 22
(1R,5S)-11-Bromo-1,2,3,4,5,6-hexahydro-8H-1,5-methanopyrido[1,2-a][1,5]diazocin-8-one, 23

SAR studies of cytisine analogues revealed that phenyl ring replacements of the pyridone ring of compound 21 reduced the binding affinities and functional efficacies.97 Substitution of 21 at the C-3 (−CH=CH2,102 −NO2,103 or pyridinyl95), C-4 (−CH3),102 or C-5 (-Br)78 position could lead to the same or improved α4β2-nAChR binding affinity, whereas substitution at the piperidine nitrogen,97,103,104 the C-3 position (aromatic rings),102 or at the C-12 position (−CO2CH3 or −COCH2CH3)102 reduced affinity. Introduction of substituents on the piperidine ring nitrogen,97 C-4 (−CH3 or −CH2OH),102 or the C-5 (−NO2)103 position increases the selectivity for α4β2 receptor. Among these substituted cytisine analogues, 3-(2-pyridyl)cytisine (22)95 and 5-bromocytisine (23)95 were further tested in mouse antidepressant efficacy models. Both compounds displayed high affinity at α4β2*-nAChRs (Ki,α4β2* = 0.9 for 22 and 0.3 nM for 23), whereas compound 22 was found to show lower affinities at α3β4*-nAChRs and α7*-nAChRs (Ki,α3β4* = 119 nM and Ki,α7 = 1100 nM for 22 vs Ki,α3β4* = 3.8 nM and Ki,α7 = 28 nM for 23). In electrophysiology assays, compound 22 activates both HS and LS α4β2*-nAChRs (EC50,HS = 12 nM and EC50,LS = 31 nM) expressed in oocytes with low efficacies (less than 10% relative to the efficacy of ACh at both HS and LS receptors) while exhibiting little agonism at α3β4*.95 In the same assays, compound 23 activated both HS and LS α4β2*-nAChRs with similar potencies (EC50,HS = 13 nM and EC50,LS = 15 nM) and efficacies (17% relative to ACh). A comparison of binding and functional data for compound 21 and its selected analogues are presented in Table 1. The antidepressant-like effects of compound 22 were demonstrated in the TST (0.6 mg/kg, but not at 0.3 or 0.9 mg/kg), FST (0.3–0.9 mg/kg), and chronic NSF tests (15 days at 0.3 mg/kg, but not at 0.6 mg/kg) in C57BL/6 mice. On the other hand, compound 23 (0.3–1.2 mg/kg) failed to show any significant effects in the same tests 30 min postintraperitoneal injection.95 It has been suggested that the lack of efficacy of compound 23 was likely due to the low brain penetration, as 50 ng of the compound in 1 μL of artificial cerebrospinal fluid showed a significant antidepressant-like effect in the TST when administered centrally.
Table 1. Binding Affinities and Maximal Responses and Potencies of Compounds 2 and 21–23 with Respect to Activation of nAChRs Expressed in Oocytes95,101.
(1R,5S)-7-(Trifluoromethyl)-2,3,4,5-tetrahydro-1H-1,5-methanobenzo[d]azepine, 16
(1S,5R)-7-(Trifluoromethyl)-2,3,4,5-tetrahydro-1H-1,5-methanobenzo[d]azepine, 25
(6R,10S)-7,8,9,10-Tetrahydro-6H-6,10-methanoazepino[4,5-g]quinoxaline, 2
Inspired by morphine and its simplified analogues [3.3.1]- and [3.2.1]-bicyclic benzomorphans, Coe et al. identified the simplified cytisine analogue, benzazapine 24,105 as a nicotinic antagonist with a Ki value of 20 nM. Attachment of a trifluoromethyl group to the C-4 position of benzazapine 24 gives 16 and CP-601932 (25)78 (Figure 6). Compound 16 is a selective α4β2-nAChR partial agonist with weaker activity at the α3β4-nAChRs (Ki,α4β2 = 1.2 nM vs Ki,α3β4 = 102 nM),78 whereas its enantiomer, 25, shows similar affinities at both receptor subtypes, with Ki values of 21 nM and lower affinities for the α6- and α7-nAChR subtypes (Ki > 300 nM).106,107 Compound 16 decreased the time spent immobile in the FST at 0.75, 1, and 1.5 mg/kg, whereas no significant effect was found in the TST at the same doses.78 Moreover, compound 16 was found to be relatively safe and well-tolerated in phase 1 single- and multiple-dose studies, but it failed to show a statistically significant change on the primary efficacy scale in favor of the drug when used in the augmentation of antidepressant therapy in major depression in a phase 2 clinical study.108
Figure 6.

Selected nicotinic benzazapine analogues.
Nitration of benzazapine 24 in the presence of 2 equiv of nitronium triflate (CF3SO2O–NO2+) in dichloromethane afforded the 4,5-dinitrated benzazapine 26(105) with an unexpected regioselectivity (the meta-isomer was found to be less than 10% of the mixture). Reduction of 26 to the diamine, condensation with glyoxal, and deprotection followed by salt formation provided compound 2 (Figure 6).105 This achiral quinoxaline 2 is the most publicized cytisine analogue, which is known as varenicline. Its tartrate salt has been launched and marketed by Pfizer for smoking cessation in the U.S. (Chantix), Canada, Europe, and other countries (Champix). Compound 2 is a potent partial agonist at α4β2*-nAChRs, with a Ki value of 0.4 nM and an agonist efficacy of 40–60% of that of compound 10.109,110 In Xenopus laevis oocytes expressing α4β2-nAChRs, compound 2 was found to have an EC50 of 2.3 μM and an efficacy of 13.4% relative to that of ACh.109 It potently blocks compound 10 binding to α4β2*-nAChRs and produces a sustained increase in dopamine release to 60% of the maximal effect of compound 10 (188% at 0.32 mg/kg sc).110 In addition, 1 mg/kg po compound 2 reduced the dopamine-enhancing effects of a subsequent dose of 0.32 mg/kg sc compound 10 to the level of the effect of compound 2 alone.110 In a SPECT imaging study, compound 2 (0.5 mg) was found to completely saturate α4β2*-nAChRs in human brain.111 The dissociation half-life of compound 2 is about 5.4 h in a mouse ex vivo receptor occupancy study.38 Compound 2 alone has antidepressant-like effects in the FST and was able to augment the effects of compound 5 when administered in combination.112 It enhanced the mood and cognitive function associated with compound 10 withdrawal in patients in clinical trials for smoking cessation.113 In an 8 week open-label study, compound 2 augmented the effects of traditional antidepressants in depressed smokers who derived minimal benefit from standard antidepressant treatment.12 Recently, studies on a larger scale of community volunteers who wanted to quit smoking revealed that 12 week administration of compound 2 (0.5 mg/day for days 1–3 followed by 0.5 mg twice a day for days 4–7 and 1 mg twice a day thereafter) was coupled with a generalized suppression of depression compared with that of the placebo-controlled treatment.114
A-85380 and Analogues
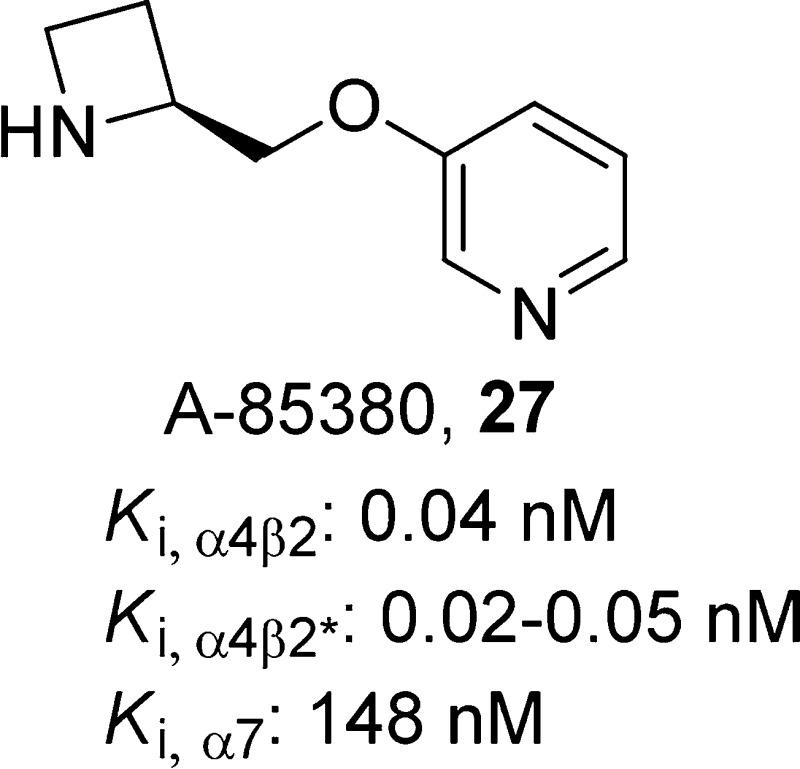
(S)-3-(Azetidin-2-ylmethoxy)pyridine, 27
(S)-3-(Azetidin-2-ylmethoxy)-5-iodopyridine, 12
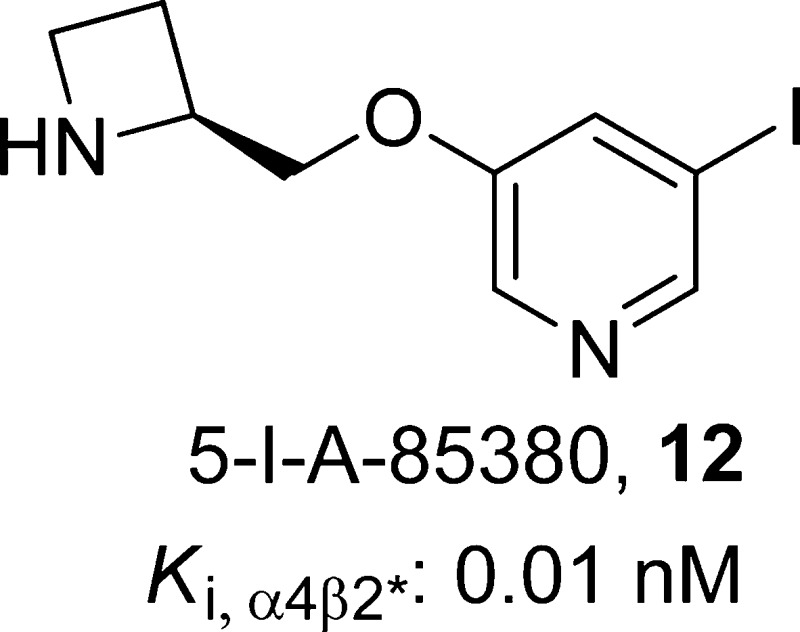
The pyridine ether-based ligands, in which a CH2O linker is inserted between the pyrrolidine ring (or in case of its analogues, an azetidine ring) of compound 10 and its pyridine ring have attracted considerable interest as α4β2-nAChR agonists due to their high potency.115−118 A-85380 (27)119 is a 3-pyridyl ether that exhibits both high potency and selectivity for β2-containing nAChRs, primarily α4β2* in brain (Ki,α4β2* = 0.02–0.05 nM for rat brain receptors and 0.04 nM for human brain receptors) relative to that of human α7-nAChRs (Ki,α7 = 148 nM) or muscle-type α1β1γδ-nAChRs (Ki,α1β1γδ = 314 nM).119,120 Functionally, compound 27 acts as a potent activator of both human α4β2-nAChRs (EC50 = 0.7 μM) and ganglionic α3β4*-nAChRs (EC50 = 0.8 μM), the effects of which are blocked by pretreatment with compound 1. In the FST, compound 27 was found to have antidepressant-like effects that could be blocked by pretreatment with the nAChR antagonists compounds 1 and 18, as well as by the nonselective serotonin receptor antagonist methiothepin, suggesting that compound 27 exerts its effects via neuronal nicotinic receptor activation of serotonergic pathways.119,121 As exemplified by compound 12(120) (Ki,α4β2* = 0.01 nM), introduction of a halogen substituent at the C-2 (fluoro only), C-5, or C-6 position of compound 27 leads to ligands that retain subnanomolar affinity for the α4β2*-nAChRs, as measured by radioligand binding assays utilizing rat brain membrane preparations.120 Similar to compound 27, 12 was also found to decrease immobility in the mouse FST.38 Recently, β2*-nAChR availability was found to be decreased across brain regions in depressed patients compared to that of healthy subjects using SPECT with the β2*-selective radioligand tracer [123I]12.39 A unifying explanation of these effects stems from the idea that desensitization and antagonism provide similar end points for agonists and antagonists. However, because agonists have an acute activation phase, behavioral assays may measure effects due to activation and/or inactivation of nAChRs. In this context, one would expect coadministration of an antagonist to reduce the behavioral signature of an agonist administered alone. More complicated explanations involving multiple nAChR subtypes also can be made, but they are highly speculative.
(S)-5-(Azetidin-2-ylmethoxy)-3-methylisoxazole, 28
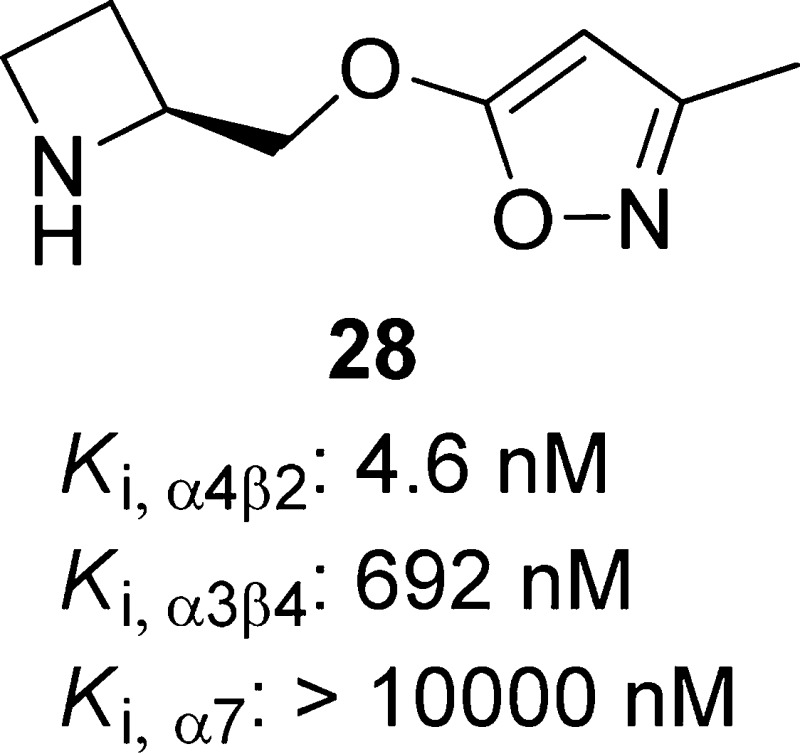
Replacement of the pyridine core of these ether-based ligands with a methylisoxazole group led to compound 28, which binds to α4β2-nAChRs with higher affinity than to the α3β4-nAChRs (Ki,α4β2 = 4.6 nM vs Ki,α3β4 = 692 nM).122 The functional potency of compound 28 (1.2 μM) at the α4β2-nAChR is similar to that of compound 2 (1.4 μM), whereas the efficacy of compound 28 is higher (110% vs 53%) in cells heterologously expressing a mixture of HS and LS α4β2-nAChRs (Table 2). Broad screening showed that this compound was highly selective for nAChRs and did not have significant binding affinity to the other 45 neurotransmitter receptors and transporters tested. In the mouse FST, compound 28 was found to decrease immobility at 1–5 mg/kg i.p. and 5 mg/kg po. This compound showed no significant hERG or CYP (1A2, 2B6, 2C9, 2C19, 2D6, and 3A4) inhibition at 10 μM. Between 53.4 and 73.7% of compound 28 was found to remain unchanged after 60 min incubation with mouse or human liver microsomes (1 and 10 μM).122
Table 2. Functional Potencies and Efficacies of Ligands 2, 10, 11, and 28–31: Agonism and Inactivation at Human α4β2-nAChRs122−125.
| agonism |
inactivation |
||||
|---|---|---|---|---|---|
| compound | EC50 (nM) | efficacy (%)a | efficacy HS (%)b | IC50 (nM) | efficacy (%) |
| 2 | 1400 | 53 | 110 | 85 | |
| 11 | 5.8 | 55 | 100 | 4.8 | 37 |
| 28 | 1200 | 110 | 92 | 169 | 78 |
| 29 | 10 | 21 | 92 | 9.4 | 63 |
| 30 | 42 | 22 | 61 | 31 | 68 |
| 31 | 8.4, 2300c | 21 | 45 | 58 | 85 |
| 10 | 290 | 88 | 110 | 430 | 93 |
The efficacies were measured in a mixture of HS and LS α4β2-nAChRs.
The efficacy values were extrapolated using compound 11 defined as a full agonist at the HS α4β2-nAChR.
Compound 31 activates both HS and LS α4β2-nAChRs with sufficient selectivity to distinguish activity at each subtype. Efficacy of 31 at LS α4β2-nAChRs is approximately 17%.
(S)-6-(5-(Azetidin-2-ylmethoxy)pyridin-3-yl)hex-5-yn-1-ol, 11

Compound 11, an analogue of compound 27 having an alkynyl substituent attached to the 5-position of the pyridine ring, was identified as a highly potent full agonist at HS α4β2-nAChR and a low efficacy agonist at LS nAChR (Table 2).126 Compared with compound 27, the selectivity of compound 11 at α4β2- relative to ganglionic α3β4*- and homomeric α7-nAChRs is greatly improved.127,128 Compound 11 potently binds to α4β2-nAChRs (Ki = 0.4 nM) but not to α3β4*- (Ki > 104 nM) or α7-nAChRs (Ki > 104 nM). Ex vivo receptor occupancy in the thalamus showed prolonged receptor occupancy. The dissociation half-life of compound 11 (8–24 h) was found to be significantly longer compared with that of compound 12 (3–5 h),38 which is likely due to the presence of the side chain in compound 11.
Robust antidepressant-like effects of compound 11 were observed in the FST, TST, and NIH assays, with no effect on baseline locomotor activity when given chronically.35,38,129 Compound 11 (1 mg/kg) significantly reduced immobility 2, 3, and 4 h but not 5 h after administration in the mouse FST. Repeated administration of compound 11 in the FST at 1 and 3 mg/kg doses for 2 weeks did not result in tolerance. Moreover, its activity in the FST was completely blocked by either compound 18 or compound 1,38 suggesting that activation of nAChRs may responsible for the activity of compound 11 in this behavioral model of antidepressant efficacy. In addition, β2 subunit knockout mice did not show any antidepressant response to compound 11 in the FST, suggesting that interaction with β2*-nAChRs plays a key role in the behavioral activity of compound 11.38 Stimulation of dopamine and noradrenaline release in rat striatal slices was observed with compound 11, an effect that was blocked by nicotinic antagonists 18 and compound 1.126 In addition, the α6*-selective antagonist α-conotoxin MII (100 nM) reduced the maximum effect of compound 11-induced dopamine release in rat by 48%, suggesting that α6*-nAChRs may be involved in the antidepressant-like effects of compound 11.126
Compound 11 has been reported to have other behavioral effects including increased hypothermia.130,131 It is unlikely that the hypothermic effects of compound 11 are related to its antidepressant-like effects observed in the FST. Although reduced, β2 knockout mice still showed a hypothermic response to compound 11,131 whereas the antidepressant-like effects of compound 11 was completely abolished in β2 knockout mice.38 The hypothermic effects of compound 11 (1 mg/kg) returned to baseline approximately 2 h postinjection,132 whereas the antidepressant-like effect persisted up to 4 h.38 Compound 11 has shown additional beneficial behavioral effects in animal models including analgesia,133 improving attentional function after disruption with compound 3 or MK-801,132,134 reducing anxiety following withdrawal from chronic compound 10,135 decreasing body weight gain following chronic treatment,136 and decreasing compound 10 self-administration and alcohol consumption.137−139 Interestingly, unlike compound 10 or compound 2, compound 11 did not upregulate nAChR in the brain after chronic administration140 and did not maintain receptor upregulation following chronic compound 10 administration,136 providing further evidence that compound 11 may exert its behavioral effects through a unique mechanism.
2-((1R,2S)-2-(5-(((S)-Azetidin-2-yl)methoxy)pyridin-3-yl)cyclopropyl)ethan-1-ol, 29

(S)-2-(5-(5-(Azetidin-2-ylmethoxy)pyridin-3-yl)isoxazol-3-yl)ethan-1-ol, 30

(S)-N-Phenyl-5-(pyrrolidin-2-ylmethoxy)pyridin-3-amine, 31

Replacing the acetylene bond of compound 11 with a substituted cyclopropane or an isoxazole ring lead to compounds 29 and 30, respectively.63,124,125,141 Introduction of different amino groups at the 5-position of the pyridine ring yielded a series of 3-alkoxy-5-aminopyridine derivatives exemplified by compound 31.123 These compound 11 analogues (29–31) act as α4β2-nAChRs partial agonists (Table 2) with low nanomolar binding affinities (Ki = 0.1–1.2 nM) and excellent subtype selectivity at β2*- over β4*-nAChRs (Table 3). Compound 29 has been cocrystallized with Ct-AChBP, and its binding mode was found to be similar to that reported for compound 2; both compounds rely on the presence of two cationic centers spaced ∼5.8 Å from each other for their binding interactions, including cation−π interactions with W153 (loop B) and Y201 (loop C) as well as a set of H-bonds with the backbone atoms of W153 (loop B), Q116 (loop E), and I128 (loop E). (Figure 7.) The hydroxyethyl group of 29 appears to be flexible and is able to adopt different orientations, thus highlighting the ability to carry out structural modifications of this appendage without causing significant alterations in activity. The antidepressant-like properties of compounds 29–31 were demonstrated in the classical mouse FST (1–30 mg/kg). The lack of significant interactions with other neurotransmitter receptors and transporters widely distributed throughout the CNS as well as the favorable preliminary absorption, distribution, metabolism, excretion, and toxicity (ADME-Tox) profiles place these specific α4β2-nAChR ligands in a favorable position to be further studied as new antidepressant drug candidates.
Table 3. Binding Affinities of Compounds 10, 11, and 27–31 at Seven nAChR Subtypes122−125.
|
Ki (nM)a |
||||||||
|---|---|---|---|---|---|---|---|---|
| compound | α2β2 | α2β4 | α3β2 | α3β4 | α4β2 | α4β4 | α4β2*b | selectivity α4β2/α3β4 |
| 11 | >10 000 | 0.4 | 0.9 | 24 000 | ||||
| 27 | 0.05 | 0.05 | ||||||
| 28 | 4.3 | 311 | 8.7 | 692 | 4.6 | 86.0 | 12.0 | 150 |
| 29 | 0.1 | 249 | 3.0 | 6520 | 0.1 | 82.6 | 0.5 | 65 200 |
| 30 | 1.0 | 935 | 15.4 | >10 000 | 0.6 | 1790 | 3.0 | >16 300 |
| 31 | 1.7 | 559 | 40.6 | 5640 | 1.2 | 16.9 | 1.4 | 4700 |
| 10 | 5.5 | 70 | 29 | 260 | 4.9 | 23 | 9.8 | 53 |
Ki values were determined by competition for [3H]epibatidine binding sites using radioligand binding.
α4β2*, prepared from rat forebrain.
Figure 7.
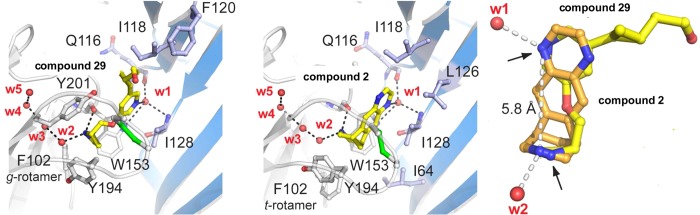
X-ray crystal structure of the Ct-AChBP in complex with compound 2 or 29.63
Other Ligands
(S,E)-5-(5-Isopropoxypyridin-3-yl)-N-methylpent-4-en-2-amine, 32

TC-1734 (32) is a potent α4β2-nAChR partial agonist (Ki = 11 nM) and is selective over α7-nAChRs (Ki > 50 000 nM).142 Compound 32 (10 μM) showed no significant inhibition when tested at 135 other receptor and enzyme systems. In functional studies, the agonist activity of compound 32 was measured using a 86Rb+ efflux assay in rat thalamic synaptosomes (α4β2 subtype, EC50 = 220 nM, efficacy = 57%) and the dopamine-release assay in striatal synaptosomes (α4β2/α6/α3β2, EC50 = 106 nM, efficacy = 55%). Compound 32 (up to 100 μM) did not show any detectable effects at either muscle-type (α1β1γδ) or ganglionic (α3β4) nAChRs. Compound 32 had favorable pharmacokinetic and metabolic profiles and was well-tolerated in acute and chronic oral toxicity studies in various animal species (mice, rats, and dogs), as well as in a human clinical studies using either single- or repeated-dose oral administration.143 Compound 32 exhibited antidepressant-like effects in the mouse FST when administered intraperitoneally, with a significant reduction in immobility at the minimal dose of 1 μmol/kg, and locomotor activity was not affected by repeated administration.142 In clinical trials, compound 32 has been studied in a variety of indications characterized by varying types and degrees of cognitive impairment, such as attention deficit/hyperactivity disorder, mild cognitive impairment, and age-associated memory impairment.144,145 Currently, Targacept is recruiting patients for a phase 2b study with this compound in order to assess efficacy, safety, and tolerability in patients with mild to moderate dementia due to Alzheimer’s disease.146,147
(S)-3-Ethynyl-5-(1-methylpyrrolidin-2-yl)pyridine, 33
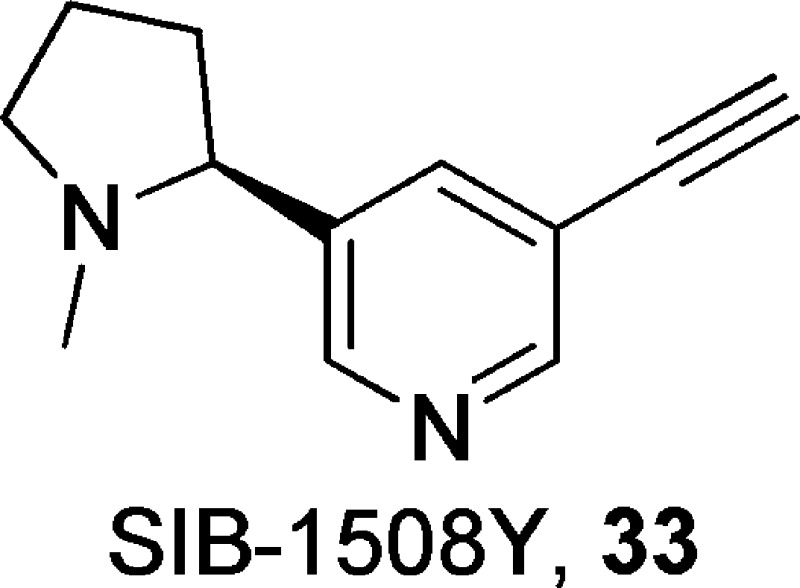
SIB-1508Y (33)148 is a selective α4β2-nAChR partial agonist (EC50 = 1.8 μM; efficacy = 49% relative to that of compound 10), having no activity at homomeric α7- or muscle-type α1β1γδ-nAChRs and weak activity at ganglionic α3β4-nAChRs (EC50 = 23 μM; efficacy = 52% relative to that of compound 10).148 In vitro, compound 33 stimulated dopamine release from slices of rat striatum prepared from various brain regions, and the effect was blocked by nAChR antagonists compound 1 or compound 18.149 On the other hand, compound 33 was found to show relatively weak effects on NE or 5-HT release. Subchronic or chronic administration of compound 33 demonstrated robust antidepressant-like effects in the learned helplessness model in rat, which were significantly blocked by nAChR antagonist compound 1 in the subchronic study.150 In addition, compound 33 was also found to improve cognitive and motor function in monkey models of Parkinson’s disease.151,152
1-(5-Chloropyridin-3-yl)-1,4-diazepane, 34
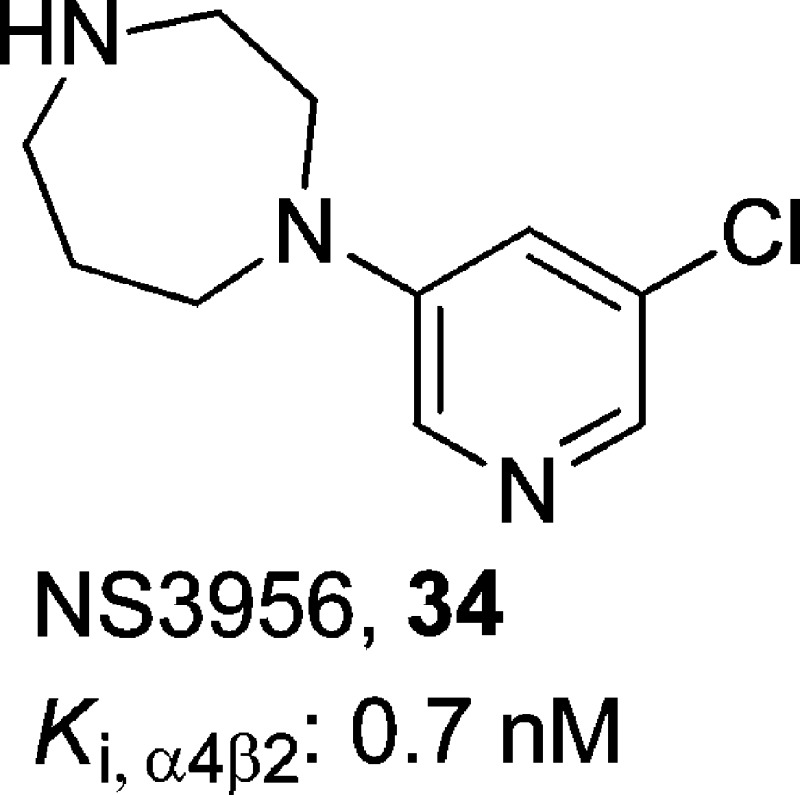
NS3956 (34)23 was found to be a partial agonist at human α4β2-nAChRs (Ki = 0.36 nM) with good selectivity (Ki,α7 = 399 nM and Ki,α1 = 944 nM). In the oocyte expression system, this compound was very potent at HS α4β2-nAChRs (EC50 = 11 nM), with an efficacy of 47% of the response to a maximally efficacious concentration of ACh.23 At LS α4β2-nAChRs, potency and efficacy were both reduced, with an EC50 of 604 nM and an efficacy of 38% relative to that of ACh. In reference to the subtype selectivity and off-target activity, actions at α7- or α3β4-nAChRs of compound 34 were weak or absent, and it did not show any significant interactions with the monoamine transporters, including 5-HT, DA, and NE. Moreover, compound 34 exhibited an ED50 value of 0.033 mg/kg in the [3H]epibatidine binding assay when tested in vivo, indicating that it was able to cross the BBB. Interestingly, compound 34 (0.3–3.0 mg/kg) was shown to be inactive in the mouse FST when tested alone, whereas when tested as a combination therapy, it significantly enhanced the antidepressant-like effects of the SSRI compound 4 and the SNRI compound 6 at 1.0 mg/kg.23 These results suggest the possibility of a synergistic interaction between nicotinic agonism and the action of antidepressant medications. In vitro binding studies along with the detailed interactions with Ls-AChBP revealed that a bromine substitution at the 6-position increased the efficacy at HS α4β2-nAChR, and a relatively small substitution, such as chloro, bromo, or ethoxy, at the 5-position of the pyridine ring in a 1-(pyridin-3-yl)-1,4-diazepane scaffold improved selectivity for α4β2- over α7-nAChRs.57
(R)-4-Chloro-N-(quinuclidin-3-yl)benzamide, 35
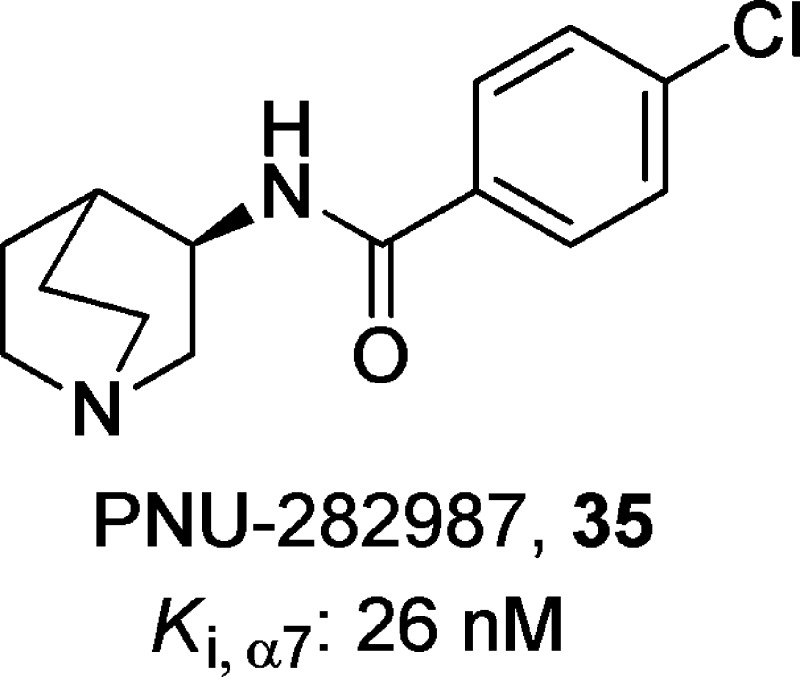
PNU-282987 (35)153 is a potent and selective α7-nAChR agonist (Ki = 26 nM) with good selectivity over α3β4- or α1β1γδ-nAChRs (≥60 μM). Compound 35 did not show significant interactions with monoamine, muscarine, glutamate, or GABA receptors when tested at concentrations of 1 μM, with the exception of the 5-HT3 receptor, where it showed a Ki value of 930 nM. Furthermore, compound 35 was able to evoke whole-cell currents from cultured rat hippocampal neurons and enhance GABAergic synaptic activity in hippocampal slices.153 Compound 35 (10–20 mg/kg) show no antidepressant-like activity when tested alone, but it significantly enhanced the antidepressant-like effect of the SSRI compound 4 or the SNRI compound 6.23
Conclusions
Considerable evidence supports the hypothesis that hyperactivity of the cholinergic system over that of the adrenergic system leads to depression. Clinical and preclinical studies provide evidence that modulating nAChRs may lead to antidepressant effects when given alone and in combination with traditional antidepressants (summarized in Table 4). Compound 10 has a clear and long-recognized activity as an antidepressant, and a childhood history of depression elevates the risk for those individuals becoming users of tobacco products, reflecting self-medication with compound 10 as a “street” antidepressant.154 It is difficult to determine the critical drug features that are responsible for compound 10’s antidepressant activity, given the pharmacodynamic and pharmacokinetic variables and the various effects of compound 10 on nAChRs in vitro. Our understanding is incomplete about where and which nAChR subtypes are involved in excitatory neurotransmission and/or in modulation of neurotransmission mediated by monoamines or other chemical messengers implicated in mood regulation. Pharmacokinetics bring additional uncertainties with regard to drug availability and efficacy in vivo and translation of ligand activities in vitro to those in vivo. For example, effects of several agonists in animal models are attenuated by coadministered antagonists, suggesting that nAChR activation contributes to some antidepressant-like drug effects. Antagonists alone also elicit antidepressant-like effects, suggesting that inhibition of nAChR function through processes such as desensitization also contribute to antidepressant efficacy, consistent with the cholinergic/adrenergic hypothesis. Moreover, compound 10 and many nicotinic ligands have limited selectivity across nAChR subtypes or subtype isoforms, meaning that they could have a matrix of effects across subtypes as well as on the activation–inactivation axis. There still is a need for reliable assays for several nAChR subtypes, in vitro, complicating the interpretation of results and strategies in identification of promising compounds. It is also important to be mindful that behavioral tests in animals measure changes in behavior that correlate with clinical efficacies of currently available antidepressants and do not necessarily predict effective antidepressant activity in humans, particularity in treatment-resistant patients.
Table 4. Clinical and Preclinical Evidence for the Viability of Targeting nAChRs in Depression.
| ID | receptor subtype selectivity | antidepressant/anxiolytic results | ref |
|---|---|---|---|
| 2 | α4β2 partial agonist, less potent α3β4 and α7 full agonist | Improved FST; augmented the antidepressant effects in depressed smokers | (12, 38, and 109−114) |
| 11 | α4β2 partial agonist | Improved FST, TST, and NIH | (35, 38, and 126−129) |
| 12 | α4β2 full agonist | Improved FST | (38, 39, and 120) |
| 13 | Nonselective noncompetitive antagonist | Improved FST, social interaction test, light/dark assay; failed phase 3 clinical study as add-on in treating resistant patientsdark assay | (19, 21, 50, 51, and 66−72) |
| 16 | α4β2 partial agonist | Improved FST Failed as augmentation of antidepressant therapy for major depression in phase 2 clinical study | (78 and 108) |
| 18 | α4β2 competitive antagonist | Improved FST and TST | (22 and 87−89) |
| 19 | α7 antagonist | Improved FST and TST | (89−92) |
| 20 | α4β2 antagonist | Antidepressant and anxiolytic like response in preclinical studies | (93) |
| 21 | α4β2 partial agonist, α3β4 and α7 full agonist | Antidepressant-like activities in several rodent models; safety issues, poor absorption and limited brain penetration | (94−101) |
| 22 | α4β2 partial agonist | Improved FST, TST, and chronic NSF | (95 and 97) |
| 27 | α4β2 partial agonist, α3β4 agonist | Improved FST | (119−121) |
| 28 | α4β2 partial agonist, less potent α3β4 agonist | Improved FST | (122) |
| 29 | α4β2 partial agonist | Improved FST | (63 and 125) |
| 30 | α4β2 partial agonist | Improved FST | (124 and 141) |
| 31 | α4β2 partial agonist | Improved FST | (123) |
| 32 | α4β2 partial agonist | Improved FST | (142−147) |
| 33 | α4β2 partial agonist | Improved learned helplessness | (148−152) |
| 34 | α4β2 partial agonist | No effect alone in FST; enhanced the antidepressant-like effect of SSRI (compound 4) and SNRI (compound 6) | (23 and 58) |
| 35 | α7 agonist | No effect alone at FST; enhanced the antidepressant-like effect of SSRI (compound 4) and SNRI (compound 6) | (23 and 153) |
Recent phase 3 findings indicated that the nAChR antagonist compound 13, the 2S-(+)-enantiomer of the broad-spectrum, noncompetitive nAChR antagonist, compound 1, did not show efficacy as an adjunct therapy for patients that were nonresponders to traditional antidepressant treatment. This suggests that simple antagonism of what is likely to be a number of nAChR subtypes has an insufficient antidepressant effect. However, the lack of nAChR subtype specificity and the marginal free concentration in brain are also significant issues for compound 13 that may have contributed to the negative outcomes in the phase 3 trials. Those outcomes are not likely to be accurate predictors of effects of nicotinic full or partial agonists with greater nAChR subtype selectivity.
Designing a compound that has actions more like those of compound 10 (short-lived nAChR agonism mediated by binding to the orthosteric sites followed by receptor desensitization) may produce a more efficacious antidepressant. Factors outlined above need to be addressed toward optimization of ligand action of the desired type and at the appropriate nAChR subtypes. These challenges aside, preclinical evidence shows potent antidepressant-like efficacy of compound 11 and some of its analogues in the mouse FST. These ligands have high selectivity at α4β2*-nAChR over other nAChR subtypes and act as partial agonists at the (α4)2(β2)3-nAChR isoform. These findings suggest and support the hypothesis that ligands selectively targeting the (α4)2(β2)3-nAChR isoform may hold significant promise as efficacious antidepressants. These ligands would offer advantages over nicotinic agents that are relatively nonselective and produce adverse side effects through actions at peripheral nAChRs or at central non-α4β2*-nAChRs.
Aside from their selectivity for α4β2*-nAChR, compound 11 and its analogues have favorable pharmacokinetic and toxicological profiles. Advancement of the best of these ligands to and through clinical trials still requires substantial investment of effort and capital. Our work funded under a National Cooperative Drug Discovery and Development program has provided important impetus to such advancement, but private sector engagement and commitment to treatment of depression or other psychiatric disorders will be necessary. This poses an additional challenge given the perhaps more proximal benefit of attention on new drugs to treat diseases of aging and metabolism and acute/chronic pain than to treat psychiatric disorders that are poorly understood, perhaps because of their etiopathogenic heterogeneity. If nAChR ligands are to be prescribed as antidepressants, then it may happen through the inverse route that compound 7 took to be prescribed as a smoking-cessation aid. α4β2*-selective agonists and partial agonists that are attractive as potential antidepressants may also be efficacious as smoking-cessation aids. It is entirely possible that this class of compounds will first reach the clinic as an improvement over compound 2 and that antidepressant efficacy is ascertained thereafter. Regardless, studies with α4β2*-nAChR-selective ligands have already contributed to an improved understanding about nAChR subtypes and isoforms, and elucidation of roles played by the nicotinic system in the effects of these ligands certainly have illuminated the neurobiology underlying depression.
Acknowledgments
We thank the National Institute of Mental Health (NIMH grant U19MH085193) for research support. The Phoenix research component was also supported in part by the Barrow Neurological Foundation, and work by this team was conducted in part in the Charlotte and Harold Simensky Neurochemistry of Alzheimer’s Disease Laboratory.
Glossary
Abbreviations Used
- STAR*D
sequenced treatment alternatives to relieve depression
- MDD
major depressive disorder
- nAChR(s)
nicotinic acetylcholine receptor(s)
- SSRI(s)
selective serotonin reuptake inhibitor(s)
- SNRI(s)
serotonin norepinephrine reuptake inhibitor(s)
- ACh
acetylcholine
- AChE
acetylcholinesterase
- mAChRs
muscarinic ACh receptors
- ECD
extracellular domain
- HPA
hypothalamic–pituitary–adrenal
- GABA
γ-aminobutyric acid
- LS
low sensitivity
- HS
high sensitivity
- VTA
ventral tegmental area
- SPECT
single photon emission computed tomography
- CNS
central nervous system
- AChBP(s)
acetylcholine binding protein(s)
- Ac
Aplysia californica
- Ls
Lymnaea stagnalis
- Ct
Capitella teleta
- FST
forced swim test
- TST
tail suspension test
- NSF
novelty-suppressed feeding
- NIH
novelty-induced hypophagia
- ADME-Tox
absorption, distribution, metabolism, excretion, and toxicity
Biographies
Li-Fang Yu received a Bachelor of Science degree in chemistry at Beijing Normal University (2004). She subsequently pursued graduate studies at Shanghai Institute of Materia Medica Chinese Academy of Sciences, where she obtained her Ph.D. in Medicinal Chemistry (2009) under the mentorship of Dr. Fa-Jun Nan. She completed her postdoctoral training at the University of Illinois at Chicago in the laboratory of Dr. Alan P. Kozikowski, where she worked on developing nicotinic ligands for use in depression. She is currently an associate professor of Medicinal Chemistry at East China Normal University. Her research interests are CNS drug design and development.
Han-Kun Zhang completed his Ph.D. in Medicinal Chemistry in 2009 at Shanghai Institute of Materia Medica, Chinese Academy of Sciences, under the mentorship of Dr. Li-Hong Hu. His dissertation research centered on the discovery of novel antidiabetic and antitumor agents derived from natural products. He began his postdoctoral research career at the University of Illinois at Chicago, under the direction of Dr. Alan P. Kozikowski, where his research focus was on the development of novel nAChR ligands and HDAC inhibitors. In 2013, he joined the School of Life Sciences, East China Normal University, as an Associate Professor, and his current research interests are focused on GPCR-based drug discovery.
Barbara J. Caldarone is the Director of the NeuroBehavior Laboratory at the Harvard NeuroDiscovery Center and an Instructor at Brigham and Women’s Hospital in Boston, Massachusetts. She completed her postdoctoral training at Yale University School of Medicine, where she began studying the relationship between nicotinic acetylcholine receptors and depression; she continued this work in industry at PsychoGenics, where she worked on characterizing novel nicotinic ligands for the treatment of depression. She currently works with investigators from the Harvard research community in utilizing mouse behavioral models to help understand and develop treatments for neurodegenerative and psychiatric disorders.
J. Brek Eaton received a Bachelor of Science degree in Microbiology from Arizona State University with an emphasis on Molecular Biology and Immunology in 1998 after studying engineering at Florida Institute of Technology. He is currently a research technician at Barrow Neurological Institute employed in the Neurochemistry Laboratory of Dr. Ronald J. Lukas. Current research is focused on the discovery of novel ligands targeting neuronal nicotinic acetylcholine receptors, both for use as therapeutics and as research tools to advance understanding of the biophysics underlying nicotinic acetylcholine receptor function.
Ronald J. Lukas received undergraduate and early graduate training in Physics at the State University of New York (SUNY) and Columbia University before earning a Ph.D. in Biophysics from the SUNY Health Sciences Center in Brooklyn and serving as a postdoctoral fellow in Chemical Biodynamics at the University of California, Berkeley, and in Neurobiology at Stanford University. He since has directed the Laboratory of Neurochemistry at the Barrow Neurological Institute, conducting multidisciplinary work concerning nicotinic acetylcholine receptor structure, function, and roles in neuropsychiatric disorders.
Alan P. Kozikowski is a Professor of Medicinal Chemistry and Pharmacognosy at the University of Illinois at Chicago. He has previously held positions at the University of Pittsburgh, the Mayo Clinic, and the Georgetown University Medical Center. He has made numerous contributions to the CNS and cancer fields and published over 500 research articles. His interest in epigenetics led to the identification of highly selective HDAC6 inhibitors for application to orphan indications such as Rett syndrome and Charcot–Marie–Tooth disease. Some of his designed PSMA inhibitors have been advanced to phase 2 clinical trials for prostate cancer imaging. His efforts in the GSK-3 area have recently led to a new startup company to advance novel therapeutics for pancreatic and brain cancers.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- World Health Organization. Mental health and development: targeting people with mental health conditions as a vulnerable group; WHO Press: Geneva, Switzerland, 2010.
- Berton O.; Nestler E. J. New approaches to antidepressant drug discovery: beyond monoamines. Nat. Rev. Neurosci. 2006, 7, 137–151. [DOI] [PubMed] [Google Scholar]
- Ruhe H. G.; Huyser J.; Swinkels J. A.; Schene A. H. Switching antidepressants after a first selective serotonin reuptake inhibitor in major depressive disorder: a systematic review. J. Clin. Psychiatry 2006, 67, 1836–1855. [DOI] [PubMed] [Google Scholar]
- Sinyor M.; Schaffer A.; Levitt A. The sequenced treatment alternatives to relieve depression (STAR*D) trial: a review. Can. J. Psychiatry 2010, 55, 126–135. [DOI] [PubMed] [Google Scholar]
- Janowsky D. S.; el-Yousef M. K.; Davis J. M.; Sekerke H. J. A cholinergic–adrenergic hypothesis of mania and depression. Lancet. 1972, 2, 632–635. [DOI] [PubMed] [Google Scholar]
- Overstreet D. H. Selective breeding for increased cholinergic function: development of a new animal model of depression. Biol. Psychiatry 1986, 21, 49–58. [DOI] [PubMed] [Google Scholar]
- Pucilowski O.; Overstreet D. H.; Rezvani A. H.; Janowsky D. S. Chronic mild stress-induced anhedonia: greater effect in a genetic rat model of depression. Physiol. Behav. 1993, 54, 1215–1220. [DOI] [PubMed] [Google Scholar]
- Mineur Y. S.; Obayemi A.; Wigestrand M. B.; Fote G. M.; Calarco C. A.; Li A. M.; Picciotto M. R. Cholinergic signaling in the hippocampus regulates social stress resilience and anxiety- and depression-like behavior. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 3573–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip N. S.; Carpenter L. L.; Tyrka A. R.; Price L. H. Nicotinic acetylcholine receptors and depression: a review of the preclinical and clinical literature. Psychopharmacology 2010, 212, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George T. P.; Sacco K. A.; Vessicchio J. C.; Weinberger A. H.; Shytle R. D. Nicotinic antagonist augmentation of selective serotonin reuptake inhibitor-refractory major depressive disorder: a preliminary study. J. Clin. Psychopharmacol. 2008, 28, 340–344. [DOI] [PubMed] [Google Scholar]
- Shytle R. D.; Silver A. A.; Lukas R. J.; Newman M. B.; Sheehan D. V.; Sanberg P. R. Nicotinic acetylcholine receptors as targets for antidepressants. Mol. Psychiatry 2002, 7, 525–535. [DOI] [PubMed] [Google Scholar]
- Philip N. S.; Carpenter L. L.; Tyrka A. R.; Whiteley L. B.; Price L. H. Varenicline augmentation in depressed smokers: an 8-week, open-label study. J. Clin. Psychiatry 2009, 70, 1026–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey M. L.; Drevets W. C. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch. Gen. Psychiatry 2006, 63, 1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets W. C.; Furey M. L. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol. Psychiatry 2010, 67, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles H. C.; Lazeyras F.; Krishnan K. R.; Boyko O. B.; Payne M.; Moore D. Brain choline in depression: in vivo detection of potential pharmacodynamic effects of antidepressant therapy using hydrogen localized spectroscopy. Prog. Neuropsychopharmacol. Biol. Psychiatry 1994, 18, 1121–1127. [DOI] [PubMed] [Google Scholar]
- Steingard R. J.; Yurgelun-Todd D. A.; Hennen J.; Moore J. C.; Moore C. M.; Vakili K.; Young A. D.; Katic A.; Beardslee W. R.; Renshaw P. F. Increased orbitofrontal cortex levels of choline in depressed adolescents as detected by in vivo proton magnetic resonance spectroscopy. Biol. Psychiatry 2000, 48, 1053–1061. [DOI] [PubMed] [Google Scholar]
- Arias H. R.; Rosenberg A.; Targowska-Duda K. M.; Feuerbach D.; Jozwiak K.; Moaddel R.; Wainer I. W. Tricyclic antidepressants and mecamylamine bind to different sites in the human alpha4beta2 nicotinic receptor ion channel. Int. J. Biochem. Cell Biol. 2010, 42, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennings E. C.; Kiss J. P.; Vizi E. S. Nicotinic acetylcholine receptor antagonist effect of fluoxetine in rat hippocampal slices. Brain Res. 1997, 759, 292–294. [DOI] [PubMed] [Google Scholar]
- Fryer J. D.; Lukas R. J. Antidepressants noncompetitively inhibit nicotinic acetylcholine receptor function. J. Neurochem. 1999, 72, 1117–1124. [DOI] [PubMed] [Google Scholar]
- Lopez-Valdes H. E.; Garcia-Colunga J. Antagonism of nicotinic acetylcholine receptors by inhibitors of monoamine uptake. Mol. Psychiatry 2001, 6, 511–519. [DOI] [PubMed] [Google Scholar]
- Weber M. L.; Hofland C. M.; Shaffer C. L.; Flik G.; Cremers T.; Hurst R. S.; Rollema H. Therapeutic doses of antidepressants are projected not to inhibit human alpha4beta2 nicotinic acetylcholine receptors. Neuropharmacology 2013, 72, 88–95. [DOI] [PubMed] [Google Scholar]
- Popik P.; Kozela E.; Krawczyk M. Nicotine and nicotinic receptor antagonists potentiate the antidepressant-like effects of imipramine and citalopram. Br. J. Pharmacol. 2003, 139, 1196–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen J. T.; Nielsen E. O.; Christensen J. K.; Olsen G. M.; Peters D.; Mirza N. R.; Redrobe J. P. Subtype-selective nicotinic acetylcholine receptor agonists enhance the responsiveness to citalopram and reboxetine in the mouse forced swim test. J. Psychopharmacol. 2011, 25, 1347–1356. [DOI] [PubMed] [Google Scholar]
- Eglen R. M. Muscarinic receptor subtypes in neuronal and non-neuronal cholinergic function. Auton. Autacoid Pharmacol. 2006, 26, 219–233. [DOI] [PubMed] [Google Scholar]
- Mineur Y. S.; Picciotto M. R. Nicotine receptors and depression: revisiting and revising the cholinergic hypothesis. Trends Pharmacol. Sci. 2010, 31, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas R. J.; Changeux J. P.; Le Novere N.; Albuquerque E. X.; Balfour D. J.; Berg D. K.; Bertrand D.; Chiappinelli V. A.; Clarke P. B.; Collins A. C.; Dani J. A.; Grady S. R.; Kellar K. J.; Lindstrom J. M.; Marks M. J.; Quik M.; Taylor P. W.; Wonnacott S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol. Rev. 1999, 51, 397–401. [PubMed] [Google Scholar]
- Changeux J. P. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat. Rev. Neurosci. 2010, 11, 389–401. [DOI] [PubMed] [Google Scholar]
- Arneric S. P.; Holladay M.; Williams M. Neuronal nicotinic receptors: a perspective on two decades of drug discovery research. Biochem. Pharmacol. 2007, 74, 1092–1101. [DOI] [PubMed] [Google Scholar]
- Picciotto M. R.; Zoli M.; Rimondini R.; Lena C.; Marubio L. M.; Pich E. M.; Fuxe K.; Changeux J. P. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 1998, 391, 173–177. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Strecker R. E.; McKenna J. T.; Thakkar M. M.; McCarley R. W.; Tao R. Effects on serotonin of (−)nicotine and dimethylphenylpiperazinium in the dorsal raphe and nucleus accumbens of freely behaving rats. Neuroscience 2005, 135, 949–958. [DOI] [PubMed] [Google Scholar]
- Tucci S. A.; Genn R. F.; File S. E. Methyllycaconitine (MLA) blocks the nicotine evoked anxiogenic effect and 5-HT release in the dorsal hippocampus: possible role of alpha7 receptors. Neuropharmacology 2003, 44, 367–373. [DOI] [PubMed] [Google Scholar]
- Dominguez del Toro E.; Juiz J. M.; Peng X.; Lindstrom J.; Criado M. Immunocytochemical localization of the alpha 7 subunit of the nicotinic acetylcholine receptor in the rat central nervous system. J. Comp. Neurol. 1994, 349, 325–342. [DOI] [PubMed] [Google Scholar]
- Seguela P.; Wadiche J.; Dineley-Miller K.; Dani J. A.; Patrick J. W. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J. Neurosci. 1993, 13, 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque E. X.; Pereira E. F.; Alkondon M.; Rogers S. W. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 2009, 89, 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C.; Riganti L.; Vailati S.; Clementi F. Brain neuronal nicotinic receptors as new targets for drug discovery. Curr. Pharm. Des. 2006, 12, 407–428. [DOI] [PubMed] [Google Scholar]
- Caldarone B. J.; Harrist A.; Cleary M. A.; Beech R. D.; King S. L.; Picciotto M. R. High-affinity nicotinic acetylcholine receptors are required for antidepressant effects of amitriptyline on behavior and hippocampal cell proliferation. Biol. Psychiatry 2004, 56, 657–664. [DOI] [PubMed] [Google Scholar]
- Rabenstein R. L.; Caldarone B. J.; Picciotto M. R. The nicotinic antagonist mecamylamine has antidepressant-like effects in wild-type but not beta2- or alpha7-nicotinic acetylcholine receptor subunit knockout mice. Psychopharmacology 2006, 189, 395–401. [DOI] [PubMed] [Google Scholar]
- Caldarone B. J.; Wang D.; Paterson N. E.; Manzano M.; Fedolak A.; Cavino K.; Kwan M.; Hanania T.; Chellappan S. K.; Kozikowski A. P.; Olivier B.; Picciotto M. R.; Ghavami A. Dissociation between duration of action in the forced swim test in mice and nicotinic acetylcholine receptor occupancy with sazetidine, varenicline, and 5-I-A85380. Psychopharmacology 2011, 217, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saricicek A.; Esterlis I.; Maloney K. H.; Mineur Y. S.; Ruf B. M.; Muralidharan A.; Chen J. I.; Cosgrove K. P.; Kerestes R.; Ghose S.; Tamminga C. A.; Pittman B.; Bois F.; Tamagnan G.; Seibyl J.; Picciotto M. R.; Staley J. K.; Bhagwagar Z. Persistent beta2*-nicotinic acetylcholinergic receptor dysfunction in major depressive disorder. Am. J. Psychiatry 2012, 169, 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer P. M.; Strecker K.; Kendziorra K.; Becker G.; Hesse S.; Woelpl D.; Hensel A.; Patt M.; Sorger D.; Wegner F.; Lobsien D.; Barthel H.; Brust P.; Gertz H. J.; Sabri O.; Schwarz J. Reduced alpha4beta2*-nicotinic acetylcholine receptor binding and its relationship to mild cognitive and depressive symptoms in Parkinson disease. Arch. Gen. Psychiatry 2009, 66, 866–877. [DOI] [PubMed] [Google Scholar]
- Arias H. R.; Fedorov N. B.; Benson L. C.; Lippiello P. M.; Gatto G. J.; Feuerbach D.; Ortells M. O. Functional and structural interaction of (−)-reboxetine with the human alpha4beta2 nicotinic acetylcholine receptor. J. Pharmacol. Exp. Ther. 2013, 344, 113–123. [DOI] [PubMed] [Google Scholar]
- Klink R.; de Kerchove d’Exaerde A.; Zoli M.; Changeux J. P. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J. Neurosci. 2001, 21, 1452–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Lukas R. J. Naturally-expressed nicotinic acetylcholine receptor subtypes. Biochem. Pharmacol. 2011, 82, 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M.; Perez X. A.; Grady S. R. Role of alpha6 nicotinic receptors in CNS dopaminergic function: relevance to addiction and neurological disorders. Biochem. Pharmacol. 2011, 82, 873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C.; Zoli M.; Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. [DOI] [PubMed] [Google Scholar]
- Perry D. C.; Xiao Y.; Nguyen H. N.; Musachio J. L.; Davila-Garcia M. I.; Kellar K. J. Measuring nicotinic receptors with characteristics of alpha4beta2, alpha3beta2 and alpha3beta4 subtypes in rat tissues by autoradiography. J. Neurochem. 2002, 82, 468–481. [DOI] [PubMed] [Google Scholar]
- Gotti C.; Clementi F.; Fornari A.; Gaimarri A.; Guiducci S.; Manfredi I.; Moretti M.; Pedrazzi P.; Pucci L.; Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009, 78, 703–711. [DOI] [PubMed] [Google Scholar]
- de Filippi G.; Mogg A. J.; Phillips K. G.; Zwart R.; Sher E.; Chen Y. The subtype-selective nicotinic acetylcholine receptor positive allosteric potentiator 2087101 differentially facilitates neurotransmission in the brain. Eur. J. Pharmacol. 2010, 643, 218–224. [DOI] [PubMed] [Google Scholar]
- Gong C. L.; Chiu Y. T.; Lin N. N.; Cheng C. C.; Lin S. Z.; Lee T. J.; Kuo J. S. Regulation of the common carotid arterial blood flow by nicotinic receptors in the medulla of cats. Br. J. Pharmacol. 2006, 149, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shytle R. D.; Silver A. A.; Sheehan K. H.; Sheehan D. V.; Sanberg P. R. Neuronal nicotinic receptor inhibition for treating mood disorders: preliminary controlled evidence with mecamylamine. Depression Anxiety 2002, 16, 89–92. [DOI] [PubMed] [Google Scholar]
- Papke R. L.; Sanberg P. R.; Shytle R. D. Analysis of mecamylamine stereoisomers on human nicotinic receptor subtypes. J. Pharmacol. Exp. Ther. 2001, 297, 646–656. [PubMed] [Google Scholar]
- George A. A.; Lucero L. M.; Damaj M. I.; Lukas R. J.; Chen X.; Whiteaker P. Function of human alpha3beta4alpha5 nicotinic acetylcholine receptors is reduced by the alpha5(D398N) variant. J. Biol. Chem. 2012, 287, 25151–25162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash B.; Chang Y.; Lukas R. J. Reporter mutation studies show that nicotinic acetylcholine receptor (nAChR) alpha5 subunits and/or variants modulate function of alpha6*-nAChR. J. Biol. Chem. 2011, 286, 37905–37918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W.; Gallivan J. P.; Zhang Y.; Li L.; Lester H. A.; Dougherty D. A. From ab initio quantum mechanics to molecular neurobiology: a cation-pi binding site in the nicotinic receptor. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 12088–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty D. A.; Stauffer D. A. Acetylcholine binding by a synthetic receptor: implications for biological recognition. Science 1990, 250, 1558–1560. [DOI] [PubMed] [Google Scholar]
- Xiu X.; Puskar N. L.; Shanata J. A.; Lester H. A.; Dougherty D. A. Nicotine binding to brain receptors requires a strong cation-pi interaction. Nature 2009, 458, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum A. P.; Lester H. A.; Dougherty D. A. Nicotinic pharmacophore: the pyridine N of nicotine and carbonyl of acetylcholine hydrogen bond across a subunit interface to a backbone NH. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13206–13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum A. P.; Van Arnam E. B.; German L. A.; Lester H. A.; Dougherty D. A. Binding interactions with the complementary subunit of nicotinic receptors. J. Biol. Chem. 2013, 288, 6991–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde L. A.; Ahring P. K.; Jensen M. L.; Nielsen E. O.; Peters D.; Helgstrand C.; Krintel C.; Harpsoe K.; Gajhede M.; Kastrup J. S.; Balle T. Intersubunit bridge formation governs agonist efficacy at nicotinic acetylcholine alpha4beta2 receptors: unique role of halogen bonding revealed. J. Biol. Chem. 2012, 287, 4248–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpsoe K.; Hald H.; Timmermann D. B.; Jensen M. L.; Dyhring T.; Nielsen E. O.; Peters D.; Balle T.; Gajhede M.; Kastrup J. S.; Ahring P. K. Molecular determinants of subtype-selective efficacies of cytisine and the novel compound NS3861 at heteromeric nicotinic acetylcholine receptors. J. Biol. Chem. 2013, 288, 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack T.; Petrovich R. M.; Mercier K. A.; DeRose E. F.; Cuneo M. J.; Williams J.; Johnson K. L.; Lamb P. W.; London R. E.; Yakel J. L. Identification and functional characterization of a novel acetylcholine-binding protein from the marine annelid Capitella teleta. Biochemistry 2010, 49, 2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billen B.; Spurny R.; Brams M.; van Elk R.; Valera-Kummer S.; Yakel J. L.; Voets T.; Bertrand D.; Smit A. B.; Ulens C. Molecular actions of smoking cessation drugs at alpha4beta2 nicotinic receptors defined in crystal structures of a homologous binding protein. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9173–9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. K.; Eaton J. B.; Yu L. F.; Nys M.; Mazzolari A.; van Elk R.; Smit A. B.; Alexandrov V.; Hanania T.; Sabath E.; Fedolak A.; Brunner D.; Lukas R. J.; Vistoli G.; Ulens C.; Kozikowski A. P. Insights into the structural determinants required for high-affinity binding of chiral cyclopropane-containing ligands to alpha4beta2-nicotinic acetylcholine receptors: an integrated approach to behaviorally active nicotinic ligands. J. Med. Chem. 2012, 55, 8028–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan J. F.; Mombereau C.; Vassout A. The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci. Biobehav. Rev. 2005, 29, 571–625. [DOI] [PubMed] [Google Scholar]
- Dulawa S. C.; Hen R. Recent advances in animal models of chronic antidepressant effects: the novelty-induced hypophagia test. Neurosci. Biobehav. Rev. 2005, 29, 771–783. [DOI] [PubMed] [Google Scholar]
- Fedorov N. B.; Benson L. C.; Graef J.; Lippiello P. M.; Bencherif M. Differential pharmacologies of mecamylamine enantiomers: positive allosteric modulation and noncompetitive inhibition. J. Pharmacol. Exp. Ther. 2009, 328, 525–532. [DOI] [PubMed] [Google Scholar]
- Lippiello P. M.; Beaver J. S.; Gatto G. J.; James J. W.; Jordan K. G.; Traina V. M.; Xie J.; Bencherif M. TC-5214 (S-(+)-mecamylamine): a neuronal nicotinic receptor modulator with antidepressant activity. CNS Neurosci. Ther. 2008, 14, 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targacept’s TC-5214 achieves all primary and secondary outcome measures in Phase 2b trial as augmentation treatment for major depressive disorder. Targacept, Inc., July 15, 2009; http://www.targacept.com/newsroom.
- A study to assess the efficacy and safety of TC-5214 as an adjunct therapy in patients with major depressive disorder (MDD). ClinicalTrials.gov, December 29, 2011; http://clinicaltrials.gov/ct2/show/NCT01157078?term=TC5214&rank=17.
- A study to assess the efficacy and safety of TC-5214 as an adjunct therapy in patients with major depressive disorder. ClinicalTrials.gov, November 19, 2012; http://www.clinicaltrials.gov/ct2/show/NCT01153347.
- A study to assess the efficacy and safety of TC-5214 as an adjunct therapy in patients with major depressive disorder. ClinicalTrials.gov, November 19, 2012; http://clinicaltrials.gov/ct2/show/results/NCT01180400.
- Vieta E.; Thase M. E.; Naber D.; D’Souza B.; Rancans E.; Lepola U.; Olausson B.; Szamosi J.; Wilson E.; Hosford D.; Dunbar G.; Tummala R.; Eriksson H. Efficacy and tolerability of flexibly-dosed adjunct TC-5214 (dexmecamylamine) in patients with major depressive disorder and inadequate response to prior antidepressant. Eur. Neuropsychopharmacol. 2014, 24, 564–574. [DOI] [PubMed] [Google Scholar]
- Nickell J. R.; Grinevich V. P.; Siripurapu K. B.; Smith A. M.; Dwoskin L. P. Potential therapeutic uses of mecamylamine and its stereoisomers. Pharmacol., Biochem. Behav. 2013, 108, 28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke R. L.; Picciotto M. R. Nicotine dependence and depression, what is the future for therapeutics?. J. Addict. Res. Ther. 2012, 3, e105. [Google Scholar]
- Fedorov N.; Moore L.; Gatto G.; Jordan K.; Bencherif M.. Differential effects of TC-5214 [S-(+)-mecamylamine] and TC-5213 [R-(−)-mecamylamine] at low and high sensitivity human alpha4beta2 nicotinic receptors and in animal models of depression and anxiety, 37th Annual Meeting of the Society for Neuroscience, San Diego, CA, Nov 3–7, 2007.
- Papke R. L.; Stokes C.; Muldoon P.; Imad Damaj M. Similar activity of mecamylamine stereoisomers in vitro and in vivo. Eur. J. Pharmacol. 2013, 720, 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip N. S.; Carpenter L. L.; Tyrka A. R.; Price L. H. The nicotinic acetylcholine receptor as a target for antidepressant drug development. Sci. World J. 2012, 2012, 104105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur Y. S.; Einstein E. B.; Seymour P. A.; Coe J. W.; O’Neill B. T.; Rollema H.; Picciotto M. R. alpha4beta2 nicotinic acetylcholine receptor partial agonists with low intrinsic efficacy have antidepressant-like properties. Behav. Pharmacol. 2011, 22, 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava M.; Ramey T.; Bell J.; Li X.; Boyer S.; Davidson W.; Billing B.; Arroyo S.. Augmenting SSRIs with an alpha4beta2 nAChR partial agonist: lack of efficacy in insufficient response major depressive disorder, ISCTM 8th Annual Scientific Meeting, Washington, DC, Feb 21–23, 2012.
- Ramey T.; Bell J.; Boyer S.. Pharmacological and clinical profile of CP- 601,927: evidence for activity at the nAChR site, 2012 Neuroscience Meeting, New Orleans, LA, Oct 13–17, 2012.
- Hurst R. S.; Rollema H.; Shaffer C. L.; Coe J. W.; Bertrand D.. Pharmacological profile of CP-601927 at neuronal nAChRs, 2012 Neuroscience Meeting, New Orleans, LA, Oct 13–17, 2012.
- Ferry L. H.; Burchette R. J.. Evaluation of bupropion versus placebo for treatment of nicotine dependence, 147th Annual Meeting of the American Psychiatric Association, Philadelphia, PA, May 21–26, 1994; pp 199–200.
- Damaj M. I.; Carroll F. I.; Eaton J. B.; Navarro H. A.; Blough B. E.; Mirza S.; Lukas R. J.; Martin B. R. Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol. Pharmacol. 2004, 66, 675–682. [DOI] [PubMed] [Google Scholar]
- Fryer J. D.; Lukas R. J. Noncompetitive functional inhibition at diverse, human nicotinic acetylcholine receptor subtypes by bupropion, phencyclidine, and ibogaine. J. Pharmacol. Exp. Ther. 1999, 288, 88–92. [PubMed] [Google Scholar]
- Golden R. N.; De Vane C. L.; Laizure S. C.; Rudorfer M. V.; Sherer M. A.; Potter W. Z. Bupropion in depression. II. The role of metabolites in clinical outcome. Arch. Gen. Psychiatry 1988, 45, 145–149. [DOI] [PubMed] [Google Scholar]
- Daviss W. B.; Perel J. M.; Brent D. A.; Axelson D. A.; Rudolph G. R.; Gilchrist R.; Nuss S.; Birmaher B. Acute antidepressant response and plasma levels of bupropion and metabolites in a pediatric-aged sample: an exploratory study. Ther. Drug Monit. 2006, 28, 190–198. [DOI] [PubMed] [Google Scholar]
- Harvey S. C.; Maddox F. N.; Luetje C. W. Multiple determinants of dihydro-beta-erythroidine sensitivity on rat neuronal nicotinic receptor alpha subunits. J. Neurochem. 1996, 67, 1953–1959. [DOI] [PubMed] [Google Scholar]
- Crooks P. A.; Ravard A.; Wilkins L. H.; Teng L.-H.; Buxton S. T.; Dwoskin L. P. Inhibition of nicotine-evoked [3H]dopamine release by pyridino N-substituted nicotine analogues: a new class of nicotinic antagonist. Drug Dev. Res. 1995, 36, 91–102. [Google Scholar]
- Andreasen J. T.; Olsen G. M.; Wiborg O.; Redrobe J. P. Antidepressant-like effects of nicotinic acetylcholine receptor antagonists, but not agonists, in the mouse forced swim and mouse tail suspension tests. J. Psychopharmacol. 2009, 23, 797–804. [DOI] [PubMed] [Google Scholar]
- Palma E.; Bertrand S.; Binzoni T.; Bertrand D. Neuronal nicotinic alpha 7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. J. Physiol. 1996, 491, 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchacz E.; Buisson B.; Bertrand D.; Lukas R. J. Functional expression of nicotinic acetylcholine receptors containing rat alpha 7 subunits in human SH-SY5Y neuroblastoma cells. FEBS Lett. 1994, 354, 155–159. [DOI] [PubMed] [Google Scholar]
- Kaiser S.; Wonnacott S. alpha-Bungarotoxin-sensitive nicotinic receptors indirectly modulate [3H]dopamine release in rat striatal slices via glutamate release. Mol. Pharmacol. 2000, 58, 312–318. [DOI] [PubMed] [Google Scholar]
- Romanelli M. N.; Gratteri P.; Guandalini L.; Martini E.; Bonaccini C.; Gualtieri F. Central nicotinic receptors: structure, function, ligands, and therapeutic potential. ChemMedChem. 2007, 2, 746–767. [DOI] [PubMed] [Google Scholar]
- Scharfenberg G.; Benndorf S.; Kempe G. [Cytisine (Tabex) as a pharmaceutical aid in stopping smoking]. Dtsch. Gesundheitsw. 1971, 26, 463–465. [PubMed] [Google Scholar]
- Mineur Y. S.; Eibl C.; Young G.; Kochevar C.; Papke R. L.; Gundisch D.; Picciotto M. R. Cytisine-based nicotinic partial agonists as novel antidepressant compounds. J. Pharmacol. Exp. Ther. 2009, 329, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur Y. S.; Somenzi O.; Picciotto M. R. Cytisine, a partial agonist of high-affinity nicotinic acetylcholine receptors, has antidepressant-like properties in male C57BL/6J mice. Neuropharmacology 2007, 52, 1256–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe J. W.; Vetelino M. G.; Bashore C. G.; Wirtz M. C.; Brooks P. R.; Arnold E. P.; Lebel L. A.; Fox C. B.; Sands S. B.; Davis T. I.; Schulz D. W.; Rollema H.; Tingley F. D. 3rd; O’Neill B. T. In pursuit of alpha4beta2 nicotinic receptor partial agonists for smoking cessation: carbon analogs of (−)-cytisine. Bioorg. Med. Chem. Lett. 2005, 15, 2974–2979. [DOI] [PubMed] [Google Scholar]
- Etter J. F. Cytisine for smoking cessation: a literature review and a meta-analysis. Arch. Int. Med. 2006, 166, 1553–1559. [DOI] [PubMed] [Google Scholar]
- Reavill C.; Walther B.; Stolerman I. P.; Testa B. Behavioural and pharmacokinetic studies on nicotine, cytisine and lobeline. Neuropharmacology 1990, 29, 619–624. [DOI] [PubMed] [Google Scholar]
- Barlow R. B.; McLeod L. J. Some studies on cytisine and its methylated derivatives. Br. J. Pharmacol. 1969, 35, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollema H.; Shrikhande A.; Ward K. M.; Tingley F. D. 3rd; Coe J. W.; O’Neill B. T.; Tseng E.; Wang E. Q.; Mather R. J.; Hurst R. S.; Williams K. E.; de Vries M.; Cremers T.; Bertrand S.; Bertrand D. Pre-clinical properties of the alpha4beta2 nicotinic acetylcholine receptor partial agonists varenicline, cytisine and dianicline translate to clinical efficacy for nicotine dependence. Br. J. Pharmacol. 2010, 160, 334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellappan S. K.; Xiao Y.; Tueckmantel W.; Kellar K. J.; Kozikowski A. P. Synthesis and pharmacological evaluation of novel 9- and 10-substituted cytisine derivatives. Nicotinic ligands of enhanced subtype selectivity. J. Med. Chem. 2006, 49, 2673–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boido C. C.; Tasso B.; Boido V.; Sparatore F. Cytisine derivatives as ligands for neuronal nicotine receptors and with various pharmacological activities. Farmaco 2003, 58, 265–277. [DOI] [PubMed] [Google Scholar]
- Nicolotti O.; Canu Boido C.; Sparatore F.; Carotti A. Cytisine derivatives as high affinity nAChR ligands: synthesis and comparative molecular field analysis. Farmaco 2002, 57, 469–478. [DOI] [PubMed] [Google Scholar]
- Coe J. W.; Brooks P. R.; Vetelino M. G.; Wirtz M. C.; Arnold E. P.; Huang J.; Sands S. B.; Davis T. I.; Lebel L. A.; Fox C. B.; Shrikhande A.; Heym J. H.; Schaeffer E.; Rollema H.; Lu Y.; Mansbach R. S.; Chambers L. K.; Rovetti C. C.; Schulz D. W.; Tingley F. D. 3rd; O’Neill B. T. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J. Med. Chem. 2005, 48, 3474–3477. [DOI] [PubMed] [Google Scholar]
- Campion S. N.; Hurtt M. E.; Chatman L. A.; Cappon G. D. Toxicity study in juvenile rats with the alpha(4) beta(2) nicotinic acetylcholine receptor partial agonist CP-601,927. Birth Defects Res., Part B 2011, 323–332. [DOI] [PubMed] [Google Scholar]
- Chatterjee S.; Steensland P.; Simms J. A.; Holgate J.; Coe J. W.; Hurst R. S.; Shaffer C. L.; Lowe J.; Rollema H.; Bartlett S. E. Partial agonists of the alpha3beta4* neuronal nicotinic acetylcholine receptor reduce ethanol consumption and seeking in rats. Neuropsychopharmacology 2011, 36, 603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A study of the efficacy and safety of CP-601,927 augmentation of antidepressant therapy in major depression. ClinicalTrials.gov, December 29, 2011; http://clinicaltrials.gov/ct2/show/NCT01098240.
- Mihalak K. B.; Carroll F. I.; Luetje C. W. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol. Pharmacol. 2006, 70, 801–805. [DOI] [PubMed] [Google Scholar]
- Rollema H.; Chambers L. K.; Coe J. W.; Glowa J.; Hurst R. S.; Lebel L. A.; Lu Y.; Mansbach R. S.; Mather R. J.; Rovetti C. C.; Sands S. B.; Schaeffer E.; Schulz D. W.; Tingley F. D. 3rd; Williams K. E. Pharmacological profile of the alpha4beta2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology 2007, 52, 985–994. [DOI] [PubMed] [Google Scholar]
- Lotfipour S.; Mandelkern M.; Alvarez-Estrada M.; Brody A. L. A single administration of low-dose varenicline saturates alpha4beta2* nicotinic acetylcholine receptors in the human brain. Neuropsychopharmacology 2012, 37, 1738–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollema H.; Guanowsky V.; Mineur Y. S.; Shrikhande A.; Coe J. W.; Seymour P. A.; Picciotto M. R. Varenicline has antidepressant-like activity in the forced swim test and augments sertraline’s effect. Eur. J. Pharmacol. 2009, 605, 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson F.; Jepson C.; Strasser A. A.; Loughead J.; Perkins K. A.; Gur R. C.; Frey J. M.; Siegel S.; Lerman C. Varenicline improves mood and cognition during smoking abstinence. Biol. Psychiatry 2009, 65, 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinciripini P. M.; Robinson J. D.; Karam-Hage M.; Minnix J. A.; Lam C.; Versace F.; Brown V. L.; Engelmann J. M.; Wetter D. W. Effects of varenicline and bupropion sustained-release use plus intensive smoking cessation counseling on prolonged abstinence from smoking and on depression, negative affect, and other symptoms of nicotine withdrawal. JAMA Psychiatry 2013, 70, 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abreo M. A.; Lin N. H.; Garvey D. S.; Gunn D. E.; Hettinger A. M.; Wasicak J. T.; Pavlik P. A.; Martin Y. C.; Donnelly-Roberts D. L.; Anderson D. J.; Sullivan J. P.; Williams M.; Arneric S. P.; Holladay M. W. Novel 3-pyridyl ethers with subnanomolar affinity for central neuronal nicotinic acetylcholine receptors. J. Med. Chem. 1996, 39, 817–825. [DOI] [PubMed] [Google Scholar]
- Decker M. W.; Meyer M. D.; Sullivan J. P. The therapeutic potential of nicotinic acetylcholine receptor agonists for pain control. Expert Opin. Invest. Drugs 2001, 10, 1819–1830. [DOI] [PubMed] [Google Scholar]
- Kondo S.; Marty A. Synaptic currents at individual connections among stellate cells in rat cerebellar slices. J. Physiol. 1998, 509, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks M. J.; Wageman C. R.; Grady S. R.; Gopalakrishnan M.; Briggs C. A. Selectivity of ABT-089 for alpha4beta2* and alpha6beta2* nicotinic acetylcholine receptors in brain. Biochem. Pharmacol. 2009, 78, 795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan J. P.; Donnelly-Roberts D.; Briggs C. A.; Anderson D. J.; Gopalakrishnan M.; Piattoni-Kaplan M.; Campbell J. E.; McKenna D. G.; Molinari E.; Hettinger A. M.; Garvey D. S.; Wasicak J. T.; Holladay M. W.; Williams M.; Arneric S. P. A-85380 [3-(2(S)-azetidinylmethoxy) pyridine]: in vitro pharmacological properties of a novel, high affinity alpha4beta2 nicotinic acetylcholine receptor ligand. Neuropharmacology 1996, 35, 725–734. [DOI] [PubMed] [Google Scholar]
- Koren A. O.; Horti A. G.; Mukhin A. G.; Gundisch D.; Kimes A. S.; Dannals R. F.; London E. D. 2-, 5-, and 6-Halo-3-(2(S)-azetidinylmethoxy)pyridines: synthesis, affinity for nicotinic acetylcholine receptors, and molecular modeling. J. Med. Chem. 1998, 41, 3690–3698. [DOI] [PubMed] [Google Scholar]
- Buckley M. J.; Surowy C.; Meyer M.; Curzon P. Mechanism of action of A-85380 in an animal model of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 723–730. [DOI] [PubMed] [Google Scholar]
- Yu L. F.; Tuckmantel W.; Eaton J. B.; Caldarone B.; Fedolak A.; Hanania T.; Brunner D.; Lukas R. J.; Kozikowski A. P. Identification of novel alpha4beta2-nicotinic acetylcholine receptor (nAChR) agonists based on an isoxazole ether scaffold that demonstrate antidepressant-like activity. J. Med. Chem. 2012, 55, 812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Eaton J. B.; Caldarone B.; Lukas R. J.; Kozikowski A. P. Chemistry and pharmacological characterization of novel nitrogen analogues of AMOP-H-OH (sazetidine-A, 6-[5-(azetidin-2-ylmethoxy)pyridin-3-yl]hex-5-yn-1-ol) as alpha4beta2-nicotinic acetylcholine receptor-selective partial agonists. J. Med. Chem. 2010, 53, 6973–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Yu L. F.; Eaton J. B.; Caldarone B.; Cavino K.; Ruiz C.; Terry M.; Fedolak A.; Wang D.; Ghavami A.; Lowe D. A.; Brunner D.; Lukas R. J.; Kozikowski A. P. Discovery of isoxazole analogues of sazetidine-A as selective alpha4beta2-nicotinic acetylcholine receptor partial agonists for the treatment of depression. J. Med. Chem. 2011, 54, 7280–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Tuckmantel W.; Eaton J. B.; Yuen P. W.; Yu L. F.; Bajjuri K. M.; Fedolak A.; Wang D.; Ghavami A.; Caldarone B.; Paterson N. E.; Lowe D. A.; Brunner D.; Lukas R. J.; Kozikowski A. P. Chemistry and behavioral studies identify chiral cyclopropanes as selective alpha4beta2-nicotinic acetylcholine receptor partial agonists exhibiting an antidepressant profile. J. Med. Chem. 2012, 55, 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R.; Carbone A. L.; Moroni M.; Bermudez I.; Mogg A. J.; Folly E. A.; Broad L. M.; Williams A. C.; Zhang D.; Ding C.; Heinz B. A.; Sher E. Sazetidine-A is a potent and selective agonist at native and recombinant alpha4beta2 nicotinic acetylcholine receptors. Mol. Pharmacol. 2008, 73, 1838–1843. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Fan H.; Musachio J. L.; Wei Z. L.; Chellappan S. K.; Kozikowski A. P.; Kellar K. J. Sazetidine-A, a novel ligand that desensitizes alpha4beta2 nicotinic acetylcholine receptors without activating them. Mol. Pharmacol. 2006, 70, 1454–1460. [DOI] [PubMed] [Google Scholar]
- Kozikowski A. P.; Eaton J. B.; Bajjuri K. M.; Chellappan S. K.; Chen Y.; Karadi S.; He R.; Caldarone B.; Manzano M.; Yuen P. W.; Lukas R. J. Chemistry and pharmacology of nicotinic ligands based on 6-[5-(azetidin-2-ylmethoxy)pyridin-3-yl]hex-5-yn-1-ol (AMOP-H-OH) for possible use in depression. ChemMedChem. 2009, 4, 1279–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J. R.; Castellano L. M.; Blendy J. A. Nicotinic partial agonists varenicline and sazetidine-A have differential effects on affective behavior. J. Pharmacol. Exp. Ther. 2010, 334, 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani A. H.; Timofeeva O.; Sexton H. G.; DeCuir D.; Xiao Y.; Gordon C. J.; Kellar K. J.; Levin E. D. Effects of sazetidine-A, a selective alpha4beta2* nicotinic receptor desensitizing agent, on body temperature regulation in mice and rats. Eur. J. Pharmacol. 2012, 682, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin E. D.; Sexton H. G.; Gordon K.; Gordon C. J.; Xiao Y.; Kellar K. J.; Yenugonda V. M.; Liu Y.; White M. P.; Paige M.; Brown M. L.; Rezvani A. H. Effects of the sazetidine-a family of compounds on the body temperature in wildtype, nicotinic receptor beta2–/– and alpha7–/– mice. Eur. J. Pharmacol. 2013, 718, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani A. H.; Cauley M.; Xiao Y.; Kellar K. J.; Levin E. D. Effects of chronic sazetidine-A, a selective alpha4beta2 neuronal nicotinic acetylcholine receptors desensitizing agent on pharmacologically-induced impaired attention in rats. Psychopharmacology 2013, 226, 35–43. [DOI] [PubMed] [Google Scholar]
- Cucchiaro G.; Xiao Y.; Gonzalez-Sulser A.; Kellar K. J. Analgesic effects of sazetidine-A, a new nicotinic cholinergic drug. Anesthesiology 2008, 109, 512–519. [DOI] [PubMed] [Google Scholar]
- Rezvani A. H.; Cauley M.; Sexton H.; Xiao Y.; Brown M. L.; Paige M. A.; McDowell B. E.; Kellar K. J.; Levin E. D. Sazetidine-A, a selective alpha4beta2 nicotinic acetylcholine receptor ligand: effects on dizocilpine and scopolamine-induced attentional impairments in female Sprague-Dawley rats. Psychopharmacology 2011, 215, 621–630. [DOI] [PubMed] [Google Scholar]
- Turner J. R.; Wilkinson D. S.; Poole R. L.; Gould T. J.; Carlson G. C.; Blendy J. A. Divergent functional effects of sazetidine-a and varenicline during nicotine withdrawal. Neuropsychopharmacology 2013, 38, 2035–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussmann G. P.; Dedominicis K. E.; Turner J. R.; Yasuda R. P.; Klehm J.; Forcelli P. A.; Xiao Y.; Richardson J. R.; Sahibzada N.; Wolfe B. B.; Lindstrom J.; Blendy J. A.; Kellar K. J. Chronic sazetidine-A maintains anxiolytic effects and slower weight gain following chronic nicotine without maintaining increased density of nicotinic receptors in rodent brain. J. Neurochem. 2014, 129, 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. E.; Slade S.; Wells C.; Petro A.; Sexton H.; Rezvani A. H.; Brown M. L.; Paige M. A.; McDowell B. E.; Xiao Y.; Kellar K. J.; Levin E. D. Assessing the effects of chronic sazetidine-A delivery on nicotine self-administration in both male and female rats. Psychopharmacology 2012, 222, 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin E. D.; Rezvani A. H.; Xiao Y.; Slade S.; Cauley M.; Wells C.; Hampton D.; Petro A.; Rose J. E.; Brown M. L.; Paige M. A.; McDowell B. E.; Kellar K. J. Sazetidine-A, a selective alpha4beta2 nicotinic receptor desensitizing agent and partial agonist, reduces nicotine self-administration in rats. J. Pharmacol. Exp. Ther. 2010, 332, 933–939. [DOI] [PubMed] [Google Scholar]
- Rezvani A. H.; Slade S.; Wells C.; Petro A.; Lumeng L.; Li T. K.; Xiao Y.; Brown M. L.; Paige M. A.; McDowell B. E.; Rose J. E.; Kellar K. J.; Levin E. D. Effects of sazetidine-A, a selective alpha4beta2 nicotinic acetylcholine receptor desensitizing agent on alcohol and nicotine self-administration in selectively bred alcohol-preferring (P) rats. Psychopharmacology 2010, 211, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussmann G. P.; Turner J. R.; Lomazzo E.; Venkatesh R.; Cousins V.; Xiao Y.; Yasuda R. P.; Wolfe B. B.; Perry D. C.; Rezvani A. H.; Levin E. D.; Blendy J. A.; Kellar K. J. Chronic sazetidine-A at behaviorally active doses does not increase nicotinic cholinergic receptors in rodent brain. J. Pharmacol. Exp. Ther. 2012, 343, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L. F.; Eaton J. B.; Fedolak A.; Zhang H. K.; Hanania T.; Brunner D.; Lukas R. J.; Kozikowski A. P. Discovery of highly potent and selective alpha4beta2-nicotinic acetylcholine receptor (nAChR) partial agonists containing an isoxazolylpyridine ether scaffold that demonstrate antidepressant-like activity. Part II. J. Med. Chem. 2012, 55, 9998–10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto G. J.; Bohme G. A.; Caldwell W. S.; Letchworth S. R.; Traina V. M.; Obinu M. C.; Laville M.; Reibaud M.; Pradier L.; Dunbar G.; Bencherif M. TC-1734: an orally active neuronal nicotinic acetylcholine receptor modulator with antidepressant, neuroprotective and long-lasting cognitive effects. CNS Drug Rev. 2004, 10, 147–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar G.; Demazieres A.; Monreal A.; Cisterni C.; Metzger D.; Kuchibhatla R.; Luthringer R. Pharmacokinetics and safety profile of ispronicline (TC-1734), a new brain nicotinic receptor partial agonist, in young healthy male volunteers. J. Clin. Pharmacol. 2006, 46, 715–726. [DOI] [PubMed] [Google Scholar]
- Dunbar G.; Boeijinga P. H.; Demazieres A.; Cisterni C.; Kuchibhatla R.; Wesnes K.; Luthringer R. Effects of TC-1734 (AZD3480), a selective neuronal nicotinic receptor agonist, on cognitive performance and the EEG of young healthy male volunteers. Psychopharmacology 2007, 191, 919–929. [DOI] [PubMed] [Google Scholar]
- Dunbar G. C.; Kuchibhatla R. V.; Lee G. A randomized double-blind study comparing 25 and 50 mg TC-1734 (AZD3480) with placebo, in older subjects with age-associated memory impairment. J. Psychopharmacol. 2011, 25, 1020–1029. [DOI] [PubMed] [Google Scholar]
- Frolich L.; Ashwood T.; Nilsson J.; Eckerwall G. Effects of AZD3480 on cognition in patients with mild-to-moderate Alzheimer’s disease: a phase IIb dose-finding study. J. Alzheimer’s Dis. 2011, 24, 363–374. [DOI] [PubMed] [Google Scholar]
- Efficacy, safety, & tolerability of AZD3480 patients with mild to moderate dementia of the Alzheimer’s Type (AD). TrialsUnited, April 25, 2013; http://www.trialsunited.com/studies/NCT01466088/.
- Cosford N. D.; Bleicher L.; Herbaut A.; McCallum J. S.; Vernier J. M.; Dawson H.; Whitten J. P.; Adams P.; Chavez-Noriega L.; Correa L. D.; Crona J. H.; Mahaffy L. S.; Menzaghi F.; Rao T. S.; Reid R.; Sacaan A. I.; Santori E.; Stauderman K. A.; Whelan K.; Lloyd G. K.; McDonald I. A. (S)-(−)-5-Ethynyl-3-(1-methyl-2-pyrrolidinyl)pyridine maleate (SIB-1508Y): a novel anti-parkinsonian agent with selectivity for neuronal nicotinic acetylcholine receptors. J. Med. Chem. 1996, 39, 3235–3237. [DOI] [PubMed] [Google Scholar]
- Rao T. S.; Adams P. B.; Correa L. D.; Santori E. M.; Sacaan A. I.; Reid R. T.; Cosford N. D. Pharmacological characterization of (S)-(2)-5-ethynyl-3-(1-methyl-2-pyrrolidinyl)pyridine HCl (SIB-1508Y, altinicline), a novel nicotinic acetylcholine receptor agonist. Brain Res. 2008, 1234, 16–24. [DOI] [PubMed] [Google Scholar]
- Ferguson S. M.; Brodkin J. D.; Lloyd G. K.; Menzaghi F. Antidepressant-like effects of the subtype-selective nicotinic acetylcholine receptor agonist, SIB-1508Y, in the learned helplessness rat model of depression. Psychopharmacology 2000, 152, 295–303. [DOI] [PubMed] [Google Scholar]
- Schneider J. S.; Pope-Coleman A.; Van Velson M.; Menzaghi F.; Lloyd G. K. Effects of SIB-1508Y, a novel neuronal nicotinic acetylcholine receptor agonist, on motor behavior in parkinsonian monkeys. Mov. Disord. 1998, 13, 637–642. [DOI] [PubMed] [Google Scholar]
- Schneider J. S.; Tinker J. P.; Van Velson M.; Menzaghi F.; Lloyd G. K. Nicotinic acetylcholine receptor agonist SIB-1508Y improves cognitive functioning in chronic low-dose MPTP-treated monkeys. J. Pharmacol. Exp. Ther. 1999, 290, 731–739. [PubMed] [Google Scholar]
- Hajos M.; Hurst R. S.; Hoffmann W. E.; Krause M.; Wall T. M.; Higdon N. R.; Groppi V. E. The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J. Pharmacol. Exp. Ther. 2005, 312, 1213–1222. [DOI] [PubMed] [Google Scholar]
- Tizabi Y.; Overstreet D. H.; Rezvani A. H.; Louis V. A.; Clark E. Jr.; Janowsky D. S.; Kling M. A. Antidepressant effects of nicotine in an animal model of depression. Psychopharmacology 1999, 142, 193–199. [DOI] [PubMed] [Google Scholar]