

Abstract

The G protein-coupled chemokine receptors CXCR1 and CXCR2 play key roles in inflammatory diseases and carcinogenesis. In inflammation, they activate and recruit polymorphonuclear cells (PMNs) through binding of the chemokines CXCL1 (CXCR1) and CXCL8 (CXCR1 and CXCR2). Structure–activity studies that examined the effect of a novel series of S-substituted 6-mercapto-N-phenyl-nicotinamides on CXCL1-stimulated Ca2+ flux in whole human PMNs led to the discovery of 2-[5-(4-fluorophenylcarbamoyl)pyridin-2-ylsulfanylmethyl]phenylboronic acid (SX-517), a potent noncompetitive boronic acid CXCR1/2 antagonist. SX-517 inhibited CXCL1-induced Ca2+ flux (IC50 = 38 nM) in human PMNs but had no effect on the Ca2+ flux induced by C5a, fMLF, or PAF. In recombinant HEK293 cells that stably expressed CXCR2, SX-517 antagonized CXCL8-induced [35S]GTPγS binding (IC50 = 60 nM) and ERK1/2 phosphorylation. Inhibition was noncompetitive, with SX-517 unable to compete the binding of [125I]-CXCL8 to CXCR2 membranes. SX-517 (0.2 mg/kg iv) significantly inhibited inflammation in an in vivo murine model. SX-517 is the first reported boronic acid chemokine antagonist and represents a novel pharmacophore for CXCR1/2 antagonism.
Introduction
The chemokine receptors CXCR1 and CXCR2 are closely related members of the class A (rhodopsin-like) family of seven transmembrane G-protein-coupled receptors.1 The chemokine CXCL8 (Interleukin-8, IL8) activates the receptors CXCR1 and CXCR2, whereas the chemokine CXCL1 (growth related oncogene α, GROα) is a selective agonist for CXCR2.2 CXCR1/2 signaling is sensitive to the pertussis toxin, indicating involvement of the Gαi subunit in the heterotrimeric G-protein.3,4 Agonist-induced changes in receptor conformation uncouple the Gβγ subunit from the heterotrimeric G-protein complex, activating signaling pathways that include phospholipase Cβ, phosphatidylinositol-3-kinase, and mitogen-activated protein kinases. Phospholipase Cβ in turn generates inositol-1,4,5-triphosphate, which binds to the endoplasmic reticulum and leads to a release of Ca2+ into the cytoplasm.5 CXCR1/2 signaling is involved in inflammation, wound healing, and angiogenesis, and their dysregulation has been implicated in a myriad of diseases involving acute and chronic inflammation,6−14 as well as tumorigenesis.15−20 In particular, CXCR1/2 signaling mediates agonist-induced neutrophil activation and recruitment to sites of inflammation (i.e., chemotaxis) and is therefore thought to play an important role in inflammatory diseases characterized by a significant neutrophil component.
Due to the involvement of these receptors in a wide range of inflammatory diseases and carcinogenesis, CXCR1 and CXCR2 have attracted attention as targets for small-molecule drug discovery (Figure 1).21 Reparixin 1 is a ketoprofen derivative being investigated in trials for the prevention and treatment of delayed graft function and pancreatic islet transplantation.22,23 Diarylureas exemplified by 2 (SB225002) have been disclosed as either selective CXCR2 antagonists24,25 or dual CXCR1/2 antagonists.26 The central urea motif in the diarylureas was later replaced with the cyclic urea bioisostere 3,4-diaminocyclobut-3-ene-1,2-dione to provide potent CXCR2-selective analogues as represented by SCH527123 (3).27 The diarylurea SB656933 has advanced into clinical trials for chronic obstructive pulmonary disease (COPD)28,29 and cystic fibrosis,30 and SCH527123 inhibited ozone inhalation-induced sputum neutrophil recruitment in healthy subjects.31 AZD-8309 (4) is representative of the bicyclic thiazolopyrimidine class of CXCR2 antagonists,32 and this antagonist effectively inhibited the increase of LPS-mediated neutrophil recruitment in the nasal lavage of healthy subjects.33
Figure 1.

CXCR1 and CXCR2 receptor antagonists.
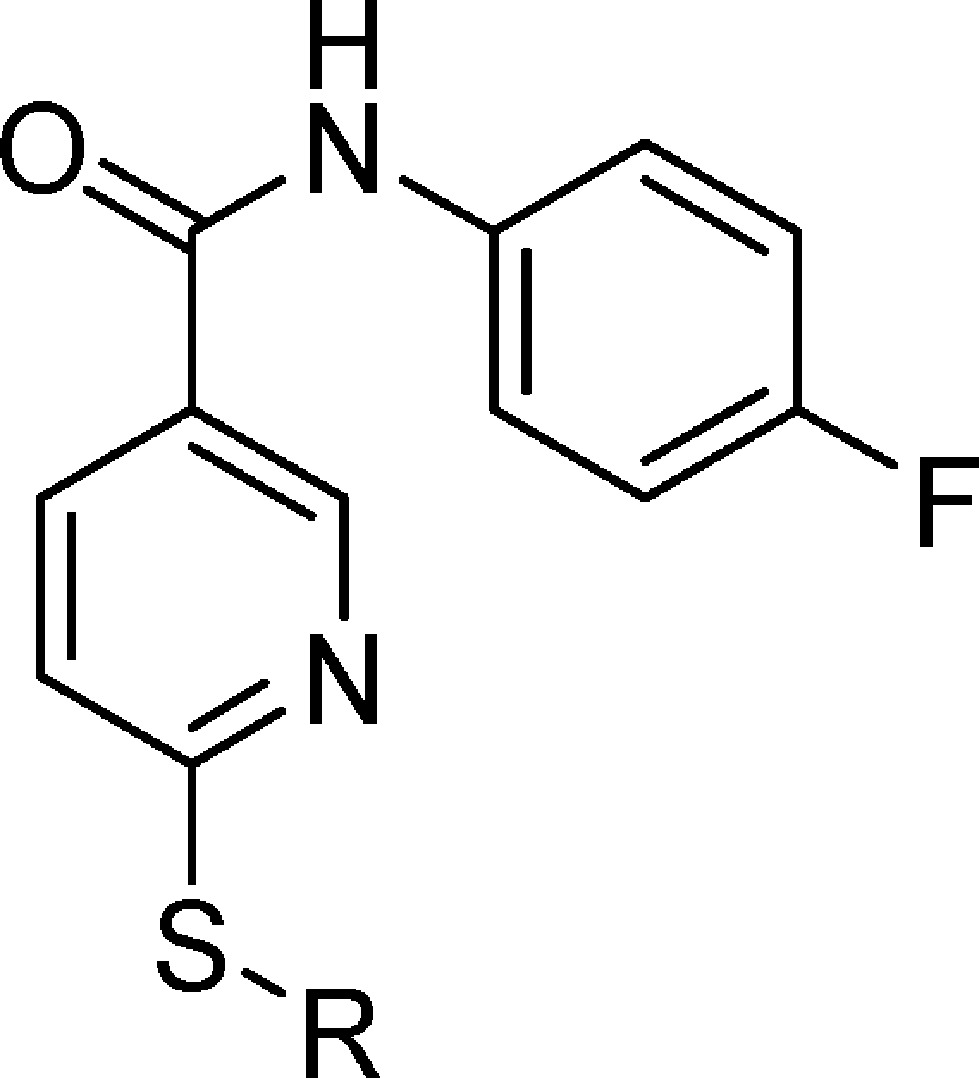
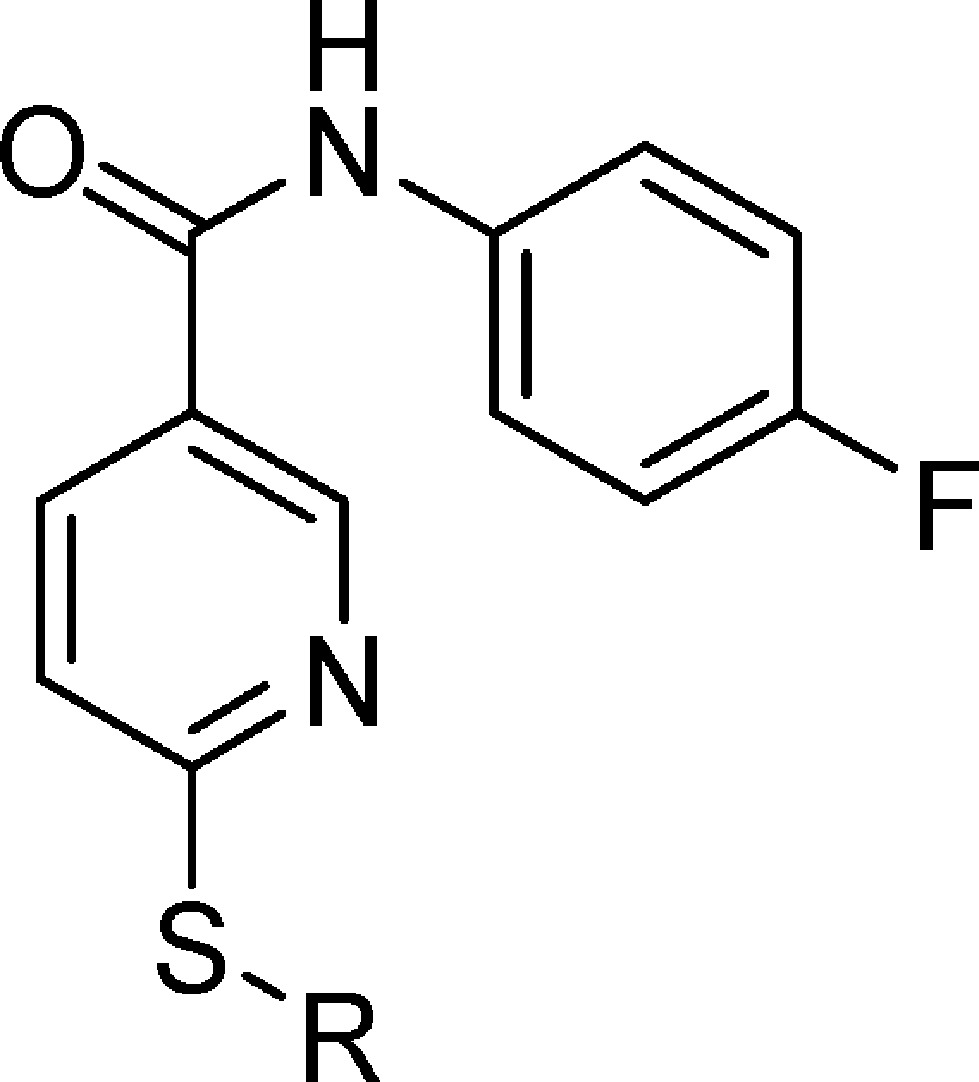
As disclosed by Cutshall and co-workers, nicotinamide glycolate esters exemplified by the methyl ester 5 (Figure 1) are antagonists of CXCR2-mediated human neutrophil chemotaxis and are distinct from the diarylurea and related cyclobutane classes.34 We have previously shown that CXCR2 antagonism by nicotinamide glycolate esters proceeded through a novel intracellular mechanism that required hydrolytic cleavage of the ester within the neutrophil for activity.35 However, the unique pharmacology of this class also led to rapid degradation in plasma, making it untenable as a therapeutic. The mechanistic insights gleaned from these studies inspired us to search for new nicotinamide templates that would directly antagonize CXCR2 without requiring intracellular hydrolytic activation for their activity. Herein, we report the discovery, structure–activity relationship (SAR), in vitro pharmacology, and in vivo biologic activity of 7 (SX-517). Compound 7 is a potent noncompetitive CXCR1/2 antagonist resulting from a novel series of S-substituted 6-mercapto-N-phenyl-nicotinamides active in their native form, and is the first reported boronic acid chemokine antagonist.
Results and Discussion
The compounds described in this study are shown in Tables 1–4, and their synthetic methods are outlined in Schemes 1–4.
Table 1. CXCL1-Inhibitory Activity of Carboxylate and Carboxylate Isostere Substituted Thionicotinamides.

Each IC50 determination was performed in triplicate with ≥6 concentrations, and reported as the mean ± standard error.
Table 4. Effect of N-Substituted 6-(Thiobenzyl-2-borono)-nicotinamides on CXCL1-Induced Ca2+ Flux in Human Neutrophils.
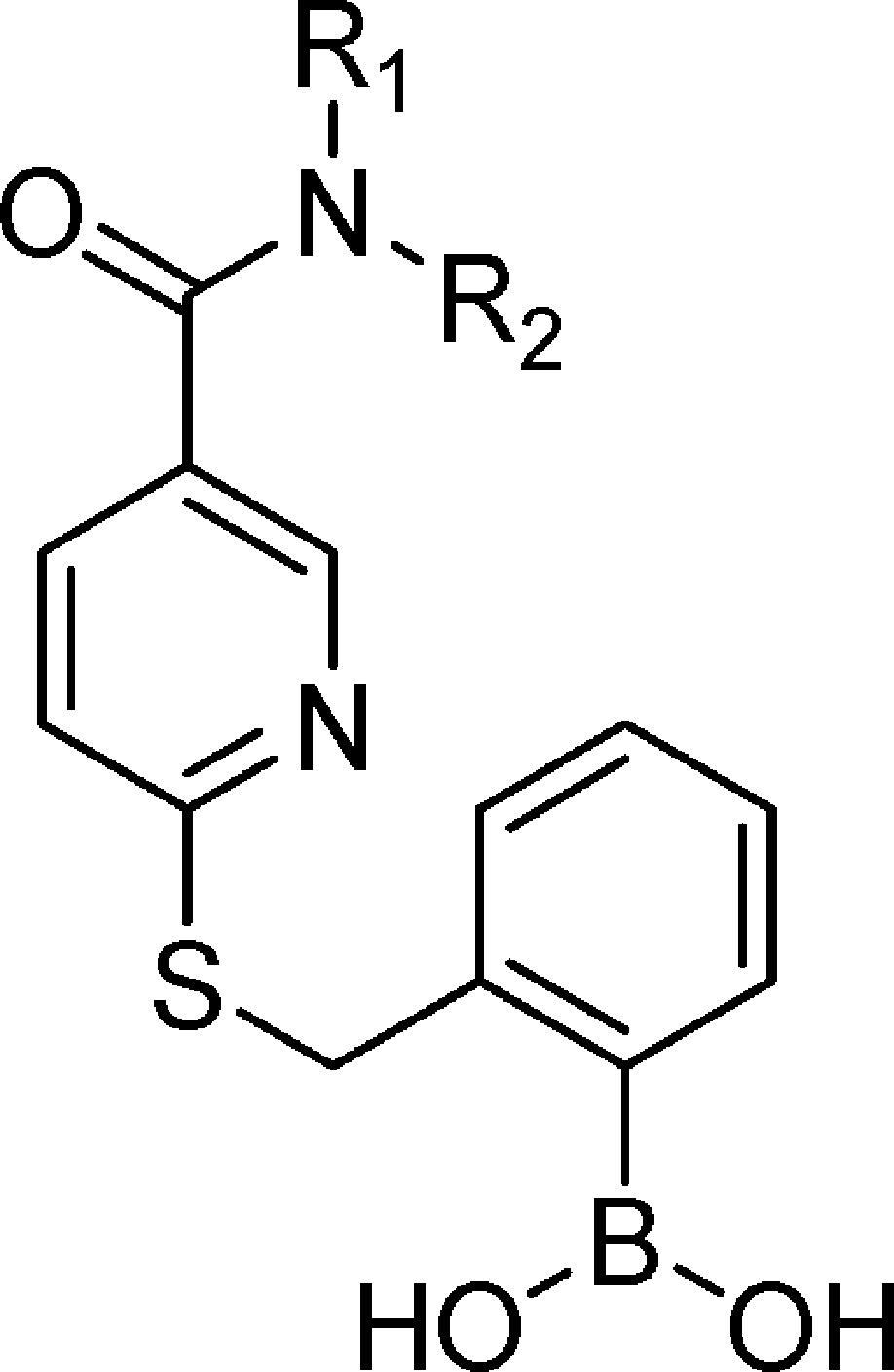
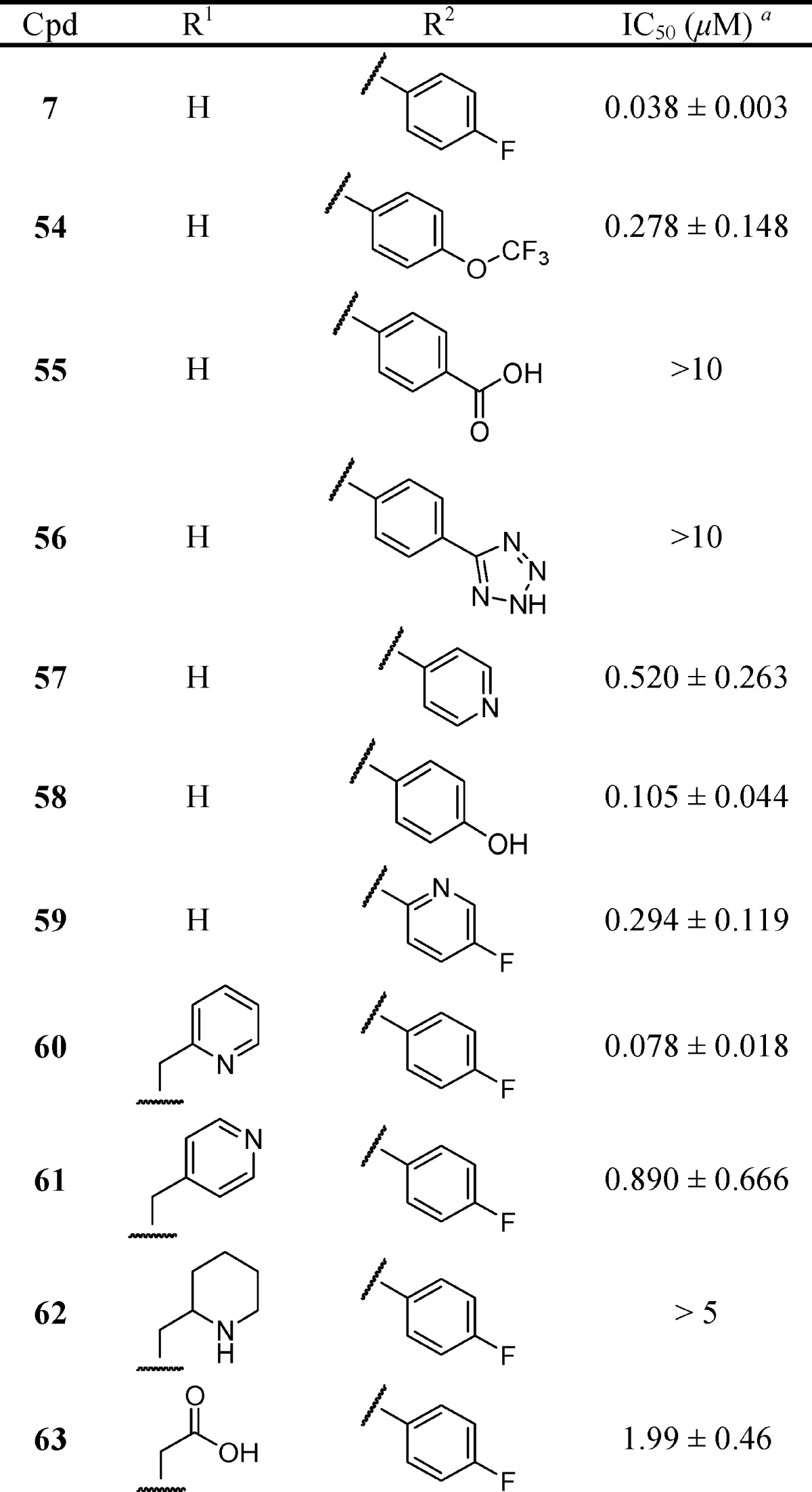
Each IC50 determination was performed in triplicate with ≥6 concentrations and reported as the mean ± standard error.
Scheme 1. Synthesis of S-Substituted N-(4-Fluoro-phenyl)-6-mercapto-nicotinamides 9–45, 47–48, 52, and 55.

Conditions: (i) 4-fluoroaniline, EEDQ, DMF, rt; (ii) method A: Br-CH2–R, solid-phase base (N-methylmorpholino-substituted resin), DMF, 60 °C; method B: Br-CH2–R or Cl-CH2–R, DMF, base (TEA, DIPEA, or), rt; method C: Br-CH2–R, EtOH, 1 N NaOH, reflux.
Scheme 4. Synthesis of N-Substituted 6-(Thiobenzyl-2-borono)-nicotinamides 60–63.

Conditions: (i) 4-fluoroaniline, K2CO3, THF, rt; (ii) N-Boc-2-piperidinecarbaldehyde, NaBH(OAc)3, DCE, glacial AcOH, rt; (iii) tert-butyl bromoacetate, DIPEA, DMF, 80 °C; (iv) phase-transfer reaction: bromomethylpyridine, toluene, 50% aq NaOH, TBAH, rt; (v) 6-chloronicotinoyl chloride, DBU, DMF, heat; (vi) NaHS, DMF, heat; (vii) 2-bromomethyl-phenylboronic acid, EtOH, 1 N aq NaOH, reflux; (viii) 2-bromomethyl-phenylboronic acid, DMF, TEA; (ix) 4 M HCl, dioxane, rt; and (x) 90% aq TFA, rt.
Synthetic Strategies and Focused Parallel Combinatorial Synthesis
The synthesis of S-substituted N-(4-fluorophenyl)-6-mercapto-nicotinamides (7–53) was carried out as described in Scheme 1 by first condensing 6-thio-nicotinic acid with 4-fluoroaniline using the coupling reagent 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ). The resulting N-(4-fluorophenyl)-6-mercapto-nicotinamide 64 was then S-alkylated with a commercially available alkyl halide, typically an alkyl bromide, using one of three methods that differed primarily in their solvent and base. Method A utilized the thio-nicotinamide intermediate 64 and bromomethyl building blocks (Maybridge Chemical Co., Cornwall, U.K.) dissolved in anhydrous DMF in the presence of resin-bound tertiary amine (4-methylmorpholino polystyrene resin, NovaBiochem, La Jolla, CA). The reaction mixture was heated at 60 °C for 1 h, followed by thiol scavenger resin (mercaptomethyl polystyrene resin, NovaBiochem, La Jolla, CA) addition. The suspension was heated to 60 °C for 2 h, and then the reaction mixture was filtered and the filtrate diluted with water. The resulting precipitate was collected by centrifugation. Purity of the synthesized compounds was assessed by HPLC, and the identification of compounds was done by electrospray ionization mass spectroscopy (ESI-MS). In all cases, the major reaction product was the derivatized thionicotinamide. Method A was used to initially synthesize compounds 13–43 in moderate to excellent purity by facile filtration. The synthesized compounds were screened at initial test concentrations of 5 and 10 μM for antagonism of CXCL1-mediated intracellular Ca2+ release in isolated human neutrophils (hPMNs). Compounds that exhibited greater than 50% inhibition at 5 μM were then resynthesized by either methods B or C and re-evaluated in the assay to obtain IC50 values of these selected compounds at a higher tested purity. Method B utilized an alkyl halide and a tertiary amine [typically triethylamine (TEA), diisopropylethylamine (DIPEA), or N-methylmorpholine (NMM)] to give the final compounds 8–13, 15, 17, 22, 43, 46, 48, and 50. Method C utilized an alkyl halide and aqueous NaOH in EtOH at reflux to give the final compounds 7, 30, and 53 in good yield.
Further chemical modification of intermediates to final test compounds is shown in Scheme 2. Treatment of 7 by potassium peroxymonosulfate (Oxone), according to the procedure of Webb and Levy for the hydroxylation of aryl boronic acids,36 afforded compound 44. Saponification of the methyl esters 46, 48, and 50 with NaOH afforded carboxylic acids 47, 49, and 51, respectively. The tetrazoles 8 and 52 were prepared via the cycloaddition of the cyano intermediates with trimethylsilyl azide and dibutyltin oxide37 in toluene under reflux.
Scheme 2. Synthesis of S-Substituted N-(4-Fluoro-phenyl)-6-mercapto-nicotinamides 8, 44, 47, 49, 51–52.

Conditions: (i) TMS-N3, Bu2SnO, toluene, reflux; (ii) 1 N NaOH, MeOH, rt; and (iii) oxone, aq NaHCO3, acetone, 4 °C
The synthesis of N-substituted 6-(thiobenzyl-2-borono)-nicotinamides 54–59 was performed as in Scheme 3. 6-Thio-nicotinic acid was coupled to 2-bromomethyl-phenylboronic acid using a modified method B employing TEA, DMF, and elevated temperature (60 °C) to give intermediate 67. Intermediate 67 was treated with neopentyl glycol in toluene under reflux to afford the protected neopentyl boronate ester. After cooling to 0 °C, pivaloyl chloride was added with triethylamine (TEA), and the reaction proceeded for an hour and then with gradual warming to room temperature for another hour to give the pivaloyl mixed anhydride intermediate 68, which was used without purification. Intermediate 68 was then coupled immediately to either 4-trifluoromethoxyaniline, 4-aminobenzoic acid, 5-(4-aminophenyl)-1H-tetrazole, 4-amino-pyridine, 4-hydroxy-aniline, or 2-amino-5-fluoropyridine under heating to give the corresponding 6-(thiobenzyl-2-neopentyl boronate ester)-nicotinamides. Exposure to water liberated the boronic acid moiety from the neopentyl boronate ester, which after purification by preparative HPLC afforded 54–59.
Scheme 3. Synthesis of Anilide Derivatives of 6-(Thiobenzyl-2-borono)-nicotinamides 54–59.

Conditions: (i) 2-bromomethyl-phenylboronic acid, DMF, TEA, 60 °C; (ii) neopentyl glycol, toluene, reflux; (iii) pivaloyl chloride, TEA, toluene, 0 °C to rt; and (iv) aniline derivative, TEA, DMF, 60 °C.
The synthesis of N,N-disubstituted 6-(thiobenzyl-2-borono)-nicotinamides 60–63 was performed through two alternative routes as described in Scheme 4. In one route, 6-chloronicotinoyl chloride was amidated with the secondary amines 70a and 70b, using DBU as the base to give the N,N-disubstituted 6-chloronicotinamides 71a and 71b, respectively. The secondary amine 70a was derived from reductive amination of N-Boc-2-piperidinecarbaldehyde (Chem-Impex, Wood Dale, IL) with 4-fluoroaniline and sodium triacetoxyborohydride. The secondary amine 70b was derived from the coupling of 4-fluoroaniline with tert-butyl bromoacetate in DMF with DIPEA as base. In the alternate route, 6-chloronicotinoyl chloride was amidated with 4-fluoroaniline in THF in the presence of potassium carbonate to form intermediate 69. Compounds 72a–72b were synthesized from intermediate 69 under phase transfer catalyzed conditions, utilizing either 2-bromomethylpyridine or 4-bromomethylpyridine, respectively. The tertiary amides 71a–71b and 72a–72b were then subjected to the same treatment with sodium hydrogen sulfide (NaHS) in DMF to displace the pyridinyl chloride and form the corresponding thio-nicotinamide intermediates 73a–73d, respectively. The resultant thio-nicotinamide intermediates 73a and 73b were alkylated with 2-bromomethyl-phenylboronic acid using method C to afford 60 and 61, respectively. Method C applied to 73c followed by treatment with 4 M HCl in dioxane deprotected the Boc-piperidyl moiety to yield 62. Method B applied to 73d followed by acid hydrolysis of the tert-butyl protected carboxyl group with 90% TFA yielded 63.
CXCL1-Stimulated Ca2+ Flux in Human Neutrophils
An increased flux of intracellular Ca2+ represents a key signaling event in CXCL1-induced neutrophil activation through CXCR2.2 Since nicotinamide-based chemokine antagonists act intracellularly,35 the activity of a compound will depend not only on its target affinity but also on its ability to transit into the intracellular compartment. To simultaneously capture both properties in initial SAR evaluations, all compounds prepared were evaluated for their ability to inhibit CXCL1-stimulated Ca2+ flux in whole hPMNs. Full dose–response curves were determined for select compounds using at least seven concentrations, and the resulting IC50 values listed in Tables 1–4 as part of these SAR studies are the mean of at least three determinations.
Structure–Activity Studies
The nicotinamide glycolate methyl ester 5 antagonized CXCL1-stimulated Ca2+ flux by an intracellular mechanism that required hydrolytic cleavage of the ester within the hPMN to liberate the active species.35 The required hydrolytic cleavage of the ester led to instability in plasma that was inseparable from the intrinsic activation mechanism of this pharmacophore class. Since in vivo stability of a potential therapeutic is essential, novel CXCR2 antagonists were sought that would directly antagonize the receptor without the need for hydrolytic activation and, thus, resist in vivo esterase activity. To this end and based on the above activation mechanism involving the glycolate ester moiety, new S-substituted nicotinamides and related congeners were evaluated for antagonism of CXCL1-stimulated Ca2+ flux in whole hPMNs, as summarized in Tables 1–4.
Our SAR efforts first involved the bioisosteric replacement of the ester/acid moiety of lead nicotinamides 5 and 6 (Table 1). Replacement of the carboxylate moiety with either a cyclic ester (9), a sulfone (10), a nitro (11), or a phosphite (12) resulted in loss of activity in our assay. We previously demonstrated that the negatively charged nicotinamide carboxylate 6 could not enter hPMNs, and its activity was only observed after hPMNs were electropermeabilized.35 On the basis of this observation, we hypothesized that the S-methyl-tetrazole 8 might serve as a more lipophilic bioisostere of the thioglycolate moiety that could passively enter hPMNs. Its activity (IC50 = 3900 nM) supported this hypothesis, but its potency was relatively weak in this capacity.
In order to widen our SAR efforts, a focused combinatorial library screen was implemented. As we discussed above, this successful parallel synthetic effort was made possible through the use of excess alkyl bromide reagent and heat to drive the alkylation to completion, as well as the use of solid phase scavenging resins to aid in product isolation. The compounds (13–43) were first screened for activity versus CXCL1-mediated calcium flux in hPMNs at test concentrations of 5 and 10 μM. In our initial screen, compounds 13, 15, 17, 20, 22, 30, and 43 exhibited greater than 50% inhibition of CXCL1-mediated intracellular Ca2+ release at a test concentration of 5 μM and were therefore identified as potential hits. These compounds were then resynthesized and retested, and the results are shown in Tables 2 and 3. Upon resynthesis and retesting, compounds 13, 15, 17, and 22 exhibited IC50 values greater than 5 μM. Inhibitory activity was confirmed for the tetrahydrofuran derivative 20 (IC50 = 540 nM), the S-benzyl derivative 30 (IC50 = 390 nM), and the S-dichlorobenzyl derivative 43 (IC50 = 1610 nM). The activity of S-benzyl derivative 30 was similar to the intrinsic activity of the de-esterified carboxylate species (IC50 = 100–500 nM) of nicotinamide methyl ester 5 reported previously in electropermeabilized hPMNs.35 Removal of the methylene between the sulfur and the aryl ring resulted in loss of activity (45). The results from this focused combinatorial chemistry effort indicated that the S-benzyl nicotinamide nucleus held potential as a new CXCR2 inhibitor template.
Table 2. CXCL1-Inhibitory Activity of Heterocyclyl- and Heteroalkyl-Substituted Thionicotinamides.
Select IC50 determinations were performed in triplicate with ≥6 concentrations, and reported as the mean ± standard error.
Table 3. CXCL1-Inhibitory Activity of Aryl-Substituted Thionicotinamides.

Select IC50 determinations were performed in triplicate with ≥6 concentrations and reported as the mean ± standard error.
Elaboration of the benzyl ring with a carboxyl group resulted in activity, with the 2-position (51, IC50 = 1480 nM) favored over both the 3-position (49, IC50 = 2120 nM) and the 4-position (47, IC50 > 10000 nM). We hypothesized that the reduced activity of 51 was the result of poor accumulation in the hPMN as a result of the negatively charged carboxylate adversely affecting uptake, efflux, or both. To examine this hypothesis further and because we have shown previously that the nicotinamide methyl ester 5 can be de-esterified within hPMNs to liberate high concentrations of the corresponding acid,35 we examined the uncharged S-methyl-benzoyl methyl esters 46, 48, and 50 as potential intracellular precursors to the corresponding acids. There was a 2-fold improvement in potency in the 2-methyl ester 50 relative to the corresponding free acid 51, but the 3- and 4-methyl esters 48 and 46 were inactive. It is possible that the limited improvement in potency arose because benzoyl methyl esters are poorer substrates for intracellular esterase activity than the thioglycolate methyl ester 5.
To explore bioisosteric replacements for the carboxylate in our intermediate lead compound, the tetrazole derivative was synthesized and evaluated for activity. Compound 52 (IC50 = 170 nM) exhibited an almost 10-fold increase in activity as compared to the 2-carboxylate derivative 51. Our SAR efforts then turned to carboxyl replacements at the 2-position. An important functionality being explored in recent pharmaceutical development is the boronic acid moiety.38,39 Inclusion of a boronic acid at the 2-postion of the S-benzyl nicotinamide scaffold resulted in compound 7 exhibiting potent inhibition of CXCR2 activation with an IC50 of 38 nM. Phenylboronic acid has a pKa of 8.9,40 and at neutral pH, compound 7 is expected to be mostly uncharged and readily transit into the cell interior. However, to test the possibility that greater lipophilicity would further increase potency, we prepared the corresponding pinacol ester 53. Although acyclic and unhindered cyclic esters of boronic acids are rapidly hydrolyzed in water, hydrolysis is slowed considerably for hindered cyclic aliphatic esters such as the pinacol ester.40 Remarkably, the pinacol ester derivative 53 retained the same order of activity as the parent boronic acid, with its activity modestly reduced by approximately 7-fold (IC50 = 275 nM). Whether the pinacol ester was active at the target or served to liberate the boronic acid inside the cell is unknown, but importantly the enhanced lipophilicity of the boronyl ester offered no advantage in potency over the underivatived boronyl group.
Concluding that the S-benzyl-2-borono scaffold in N-(4-fluorophenyl)-6-(thiobenzyl-2-borono)-nicotinamide 7 provided optimal nanomolar potency against CXCL1-induced Ca2+ flux, we undertook separate focused SAR studies of the apical 4-fluorophenyl-carboxamido domain (Table 4). The 4-fluoroanilide moiety was previously investigated and optimized for a related class of CXCR2 antagonists,41 but our focus was to introduce polar and/or ionizable substitutions that could potentially increase aqueous solubility while retaining potent chemokine antagonism. To accomplish this, the SAR of the apical 4-fluorophenyl-carboxamido- moiety in 7 was explored in compounds 54–59, a series of R2 N-monosubstituted 6-(thiobenzyl-2-borono)-nicotinamides (Table 4). The results clearly revealed the importance of the 4-fluoroanilide. Its replacement with carboxyl (55) or tetrazolyl (56) abrogated all activity. Its replacement with trifluoromethoxy (54), a pyridinyl nitrogen (57), hydroxyl (58), or inclusion of a ring nitrogen (59) resulted in a 7-, 14-, 3-, and 8-fold reduction in activity, respectively. The N,N-disubstituted 6-(thiobenzyl-2-borono)-nicotinamides 60–63 were explored by leaving the N-4-fluorophenyl R2 moiety constant and elaborating the R1 N-substitution. The N-methyl-2-pyridyl (60) was found to be well-tolerated (IC50 = 78 nM), yielding an activity nearly equivalent to 7 and providing evidence that the pyridyl group, which would be expected to be uncharged at neutral pH, does not preclude entry of the molecule into the cell. Surprisingly, the potency of the closely related congener 61 employing N-methyl-4-pyridyl was 11-fold lower than 60, suggesting a model where the pyridyl moieties are engaged in a highly structured and restricted environment. N-methyl-2-piperidyl 62 was explored as a nonplanar and nonaromatic analog of 60. It was completely devoid of activity, possibly because it was not accommodated at the target or because its positive charge prevented passive diffusion into the cell. A similar finding was obtained for the N-2-acetic acid analog 63. None of the compounds 60–63 exhibited significantly improved aqueous solubility relative to 7, despite attempts to prepare conjugate salts (data not shown).
On the basis of its optimal in vitro potency in inhibiting CXCL1-stimulated Ca2+ release in the above SAR studies, compound 7 was further evaluated with respect to its in vitro signaling pharmacology and in vivo biologic activity as described below.
G-Protein Coupling to CXCR2
CXC receptors transduce signals to the interior of the cell through activation of a coupled heterotrimeric G-protein, the most proximal signaling event after agonist binding to the receptor.3,4 CXCR2 is bound and activated by both CXCL1 and CXCL8.2 The effect of compound 7 on G-protein coupling to CXCR2 was evaluated by [35S]GTPγS binding in CXCR2 membranes prepared from HEK293 cells that stably expressed the human receptor (Figure 2). Compound 7 potently inhibited [35S]GTPγS binding in response to 10 nM CXCL8 with an IC50 of 60 ± 7 nM (mean + S.E.).
Figure 2.

Inhibition of chemokine-stimulated [35S]GTPγS binding by compound 7. HEK293 cells stably expressing CXCR2 were incubated with different concentrations of compound (from 10–10 to 10–4 M) at 37 °C for 60 min, and then the membranes were prepared by lysis and centrifugation. The membranes were then incubated in buffer containing 50 μM GDP, 8 nM [35S]GTPγS, and 10 nM CXCL8 at 30 °C for another 60 min. Membranes were harvested by rapid filtration and membrane-bound [35S]GTPγS quantitated. Data, expressed as a percentage of basal [35S]GTPγS bound in the absence of CXCL8, are the mean ± SE from three independent experiments, each done in triplicate.
CXCL8 Binding at CXCR2
We have shown that nicotinamide glycolates act away from the orthosteric chemokine binding site to antagonize CXCR2 through an intracellular mechanism.35 Maximal inhibition of radioligand binding by an allosteric antagonist can be observed in displacement experiments with radioligand concentrations much lower than the Kd value.42 We examined the ability of compound 7 to displace binding of [125I]-CXCL8 from CXCR2 membranes using a radioligand concentration (25 pM) that was >10-fold below the Kd for CXCL8 binding to CXCR2.43 Although compound 7 potently inhibited functional CXCR2 signaling by CXCL1 (IC50 = 38 nM, CXCL1-stimulated Ca2+ flux) and CXCL8 (IC50 = 60 nM, CXCL8-stimulated [35S]GTPγS binding), up to 10 μM failed to compete the binding of [125I]-CXCL8 to CXCR2 membranes (Figure 3). In parallel controls, unlabeled CXCL8 isopotently displaced its homologous radioligand (IC50 = 28 pM). Collectively, these data support a model where compound 7 acts as a noncompetitive, intracellular allosteric inhibitor of CXCR2. These findings mirrored those of the noncompetitive allosteric inhibitor reparixin 1, which inhibited CXCR1 responses with no effect on CXCL8 binding to CXCR1.22,23
Figure 3.

Competition binding assay with 7 and CXCL8 at human CXCR2. Membranes from recombinant HEK293-CXCR2 cells were incubated with the compound at the indicated concentrations and 25 pM [125I]-CXCL8. Radioligand binding to the membranes was measured by scintillation. Data show the mean ± SD (n = 3) radioligand binding, expressed as the percent of control specific radioligand binding with vehicle.
Cell Surface Expression of CXCR2
CXCR2 activation by chemokines is followed by receptor phosphorylation and subsequent down-regulation; events that are accompanied by receptor internalization.44 We considered the possibility that compound 7 may antagonize CXCR2 in whole cells at least partially through a similar mechanism of sequestering receptor away from the cell membrane signaling machinery. We therefore evaluated the effect of compound 7 on the CXCR2 surface expression in stably transfected HEK293 cells using a fluorescently labeled antibody to the receptor and fluorescence-activated cell sorting. As shown in Figure 4, 60 min exposure to 10 μM compound 7 did not significantly alter the cell surface expression of CXCR2. These data together with the data showing inhibition of CXCL8-stimulated [35S]GTPγS binding are most consistent with a mechanism of antagonism involving direct blockade of receptor activation.
Figure 4.

Effect of compound 7 on the cell surface expression of CXCR2. HEK293 cells stably expressing CXCR2 were pretreated with 1% DMSO (vehicle) or 10 μM compound (cpd. 7) for 60 min. HEK293 cells not expressing CXCR2 served as a negative isotype control (isotype). All cells were then incubated with R-phycoerythrin (PE)-conjugated antihuman CXCR2 mouse monoclonal antibody at 4 °C for 60 min. Cells were washed, fixed in 2% formaldehyde in PBS, and subjected to flow cytometric fluorescence-activated cell sorting (FACS) analysis of the PE signal. Results are representative of three independent experiments.
CXCR2 MAPK Signaling
CXCR1 and CXCR2 mediate downstream signaling in part through MAPK activation.4,45 To assess the ability of compound 7 to inhibit MAPK in HEK293 cells that stably expressed CXCR2, we measured ERK1/2 phosphorylation in response to CXCL8 in the presence of vehicle or 10 μM compound 7. As shown in the vehicle arm in Figure 5, CXCR2 induced a time-dependent phosphorylation of ERK1/2 upon activation by CXCL8. Maximum response was obtained at 15 min. In contrast, compound 7 completely blocked CXCR2-mediated phosphorylation of ERK1/2 by 30 min. There was no effect on the amount of ERK1/2 present in the lysates (Figure 5, lower panel) as assessed by an anti-ERK1/2 antibody recognizing ERK1/2 irrespective of its phosphorylation state. These data are consistent with receptor blockade by compound 7 as the common event that abrogates signaling pathways downstream of CXCR2 involving G-proteins, MAP kinases, and intracellular Ca2+.
Figure 5.
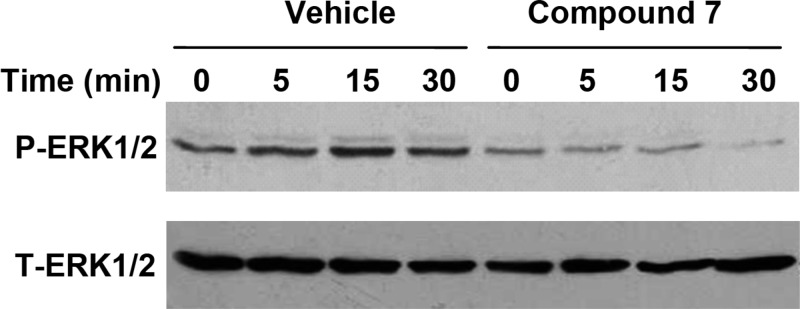
Effect of Compound 7 on CXCR2-induced ERK1/2 phosphorylation. HEK293 cells stably expressing CXCR2 were stimulated with CXCL8 (100 ng/mL) for 0–30 min in the presence of vehicle or 10 μM compound 7. Phosphorylated ERK1/2 and total ERK were determined by Western blotting using antiphospho-ERK1/2 (P-ERK1/2) and antitotal ERK1/2 (T-ERK1/2) antibodies, respectively. Data are representative of three independent experiments performed in triplicate.
Selectivity of Antagonism
CXCR1 and CXCR2 are expressed in equal numbers on the surface of hPMNs.46 Whereas CXCL8 activates both CXCR1 and CXCR2, CXCL1 is a selective agonist for CXCR2.2 Compound 7 inhibited both CXCL1- and CXCL8-stimulated Ca2+ flux in hPMNs with IC50 values of 38 and 36 nM, respectively (Table 5).
Table 5. Effect of 7 on Agonist-Induced Ca2+ Flux.
| IC50 (nM)a |
|||
|---|---|---|---|
| agonist | hPMN | HEK293-CXCR1 | HEK293-CXCR2 |
| CXCL1 | 38 ± 3 | NDb | ND |
| CXCL8 | 36 ± 11 | 880 ± 90 | 210 ± 40 |
| C5a | >5000 | ||
| PAFc | >5000 | ||
| fMLFd | >5000 | ||
Each IC50 determination was performed in triplicate with ≥6 concentrations and reported as the mean ± standard error.
ND = not determined.
Platelet activating factor.
Formyl-Met-Leu-Phe.
This inhibition was not partial, since a concentration of 1 μM compound 7 was sufficient to completely eliminate CXCL8-stimulated Ca2+ flux in hPMNs. The total antagonism of CXCL8-induced Ca2+ flux by compound 7 presumably reflected a dual blockade of both CXCR1 and CXCR2 signaling. The antagonism of CXCR1 and CXCR2 by compound 7 in hPMNs was found to be selective; however, as concentrations up to 5 μM failed to inhibit Ca2+ flux induced by optimal concentrations of the chemokines C5a, PAF, and fMLF (Table 5).
Although the inhibition of CXCL8-induced Ca2+ flux in hPMNs indicated dual antagonism of CXCR1 and CXCR2, the coexpression of these receptors in hPMNs precluded evaluating the relative inhibitory potency at each receptor. Additional experiments were therefore performed to separately evaluate the potency of compound 7 at CXCR1 and CXCR2. For these determinations, CXCL8-induced Ca2+ flux was measured in recombinant HEK293 cells that stably expressed CXCR1 (HEK293-CXCR1) or CXCR2 (HEK293-CXCR2) in the presence of vehicle and different concentrations of compound 7 (Table 5). For CXCL8-induced Ca2+ flux, the data showed that compound 7 exhibited a 4-fold selectivity in inhibition for CXCR2 (IC50 = 210 nM) over CXCR1 (IC50 = 880 nM) in these recombinant systems. As a positive control, the CXCR2-selective antagonist SB225002 inhibited CXCL8-induced Ca2+ flux in the HEK293-CXCR2 cells with an IC50 of 40 nM, a value consistent with its previously reported IC50 value of 30 nM for CXCL1-induced Ca2+ flux in hPMNs.24 The observed discrepancy in the potency of 7 when evaluated in isolated hPMNs versus HEK cells is not fully understood but may be attributed to intracellular differences between the native (hPMN) and artificially engineered (HEK) systems.
Effect on Inflammation in Vivo
To confirm the in vitro effects on the neutrophil function in vivo, compound 7 was evaluated in the murine air-pouch model of inflammation (Figure 6). An air-pouch was induced on the backs of male CD1 Swiss mice as described.47 Cohorts of five animals each were given vehicle (negative and positive control cohorts) or compound 7 dissolved in vehicle (0.02 mg/kg and 0.20 mg/kg test cohorts) by intravenous injection. Three hours afterward, the pouches in the negative control cohort were injected with sterile phosphate-buffered saline. Inflammation was induced in the pouches of the remaining three cohorts by injection with 2% carrageenan, which causes an inflammatory infiltrate consisting predominantly of neutrophils. The total cell count in the inflammatory infiltrate from each pouch was quantitated. At a dose of 0.2 mg/kg of compound 7, there was a significant reduction in cell count in the pouches of treated animals compared to the positive control cohort (**p < 0.01, Student’s t-test).
Figure 6.

An air-pouch was formed on the backs of 10–15 week old, male CD1 Swiss mice in four cohorts (n = 5 animals per cohort). Compound dissolved in vehicle (0.02 mg/kg and 0.20 mg/kg cohorts) or vehicle alone (positive and negative cohorts) was administered intravenously. After 3 h, each air pouch was injected with 1 mL of PBS (negative cohort) or 2% carrageenan in PBS (positive, 0.02 mg/kg and 0.20 mg/kg cohorts). After 4 h, the pouch fluid was collected and combined with an additional 2 mL PBS wash of the pouch. The cells in the combined fluid were stained with trypan blue and manually counted on a hemocytometer. Data show the mean ± SE of the absolute pouch cell count per cohort. Student’s t-test: **p < 0.01 vs positive cohort.
Conclusion
The results reported here describe SAR studies that examined the effect of a novel series of S-substituted 6-mercapto-N-phenyl-nicotinamides on CXCL1-stimulated Ca2+ flux in whole hPMNs. The SAR data established that an ortho-modified S-benzyl substituent played a critical role in determining activity, with an ortho-boronyl group being optimal. The fluorine in the 4-fluorophenyl-carboxamido- moiety was also an important requirement for optimal inhibitory activity. Among the derivatives exhibiting the most potent antagonism of CXCL1 described here, 7 was selected for further evaluation of its in vitro pharmacology and in vivo biologic activity.
Compound 7 was found to be a dual CXCR2/1 antagonist with a modest preference for CXCR2. It inhibited CXCL1- and CXCL8-induced Ca2+ flux (IC50 = 38 and 36 nM, respectively) in hPMNs. In response to CXCL8 stimulation, compound 7 directly antagonized [35S]GTPγS binding (IC50 = 60 nM) and ERK1/2 phosphorylation in HEK293 cells that stably expressed CXCR2. In an in vivo murine model of inflammation characterized by an inflammatory infiltrate predominated by neutrophils, compound 7 significantly reduced total cell count at an intravenous dose of 0.2 mg/kg.
Early work by others to develop small-molecule chemokine inhibitors focused on evolving compounds that were highly specific for either CXCR122,23 or CXCR2.24,25 These efforts were later followed by compounds with dual-activity at CXCR1 and CXCR226,48 in recognition that these homologous receptors mediate inflammation through unique and overlapping pathways. For example, whereas both CXCR1 and CXCR2 mediate CXCL8-induced chemotaxis,23,49 myeloperoxidase release from hPMNs occurs mainly through CXCR1 activation.50,51 CXCL8-mediated neutrophil chemotaxis is most effectively inhibited by dual CXCR1 and CXCR2 blockade.52 Compared to specific antagonists, dual CXCR2/1 inhibition by compound 7 may therefore offer a more complete therapeutic strategy in a number of inflammatory diseases where the CXCR2 pathway is involved specifically or in conjunction with CXCR1 signaling.
G-protein coupled receptors (GPCRs) are thought to exist in multiple conformational states, and activation by an agonist mediates signaling to the cell interior through poorly understood conformational changes in the receptor. It has been proposed that allosteric sites in the transmembrane domain of GPCRs may represent high-value targets for noncompetitive inhibitors that block agonist-induced G-protein activation.53 Indeed, studies have found that the antagonist reparixin (1),22,23 and the CXCR2-specific antagonists SB265610, SB332235, and SCH527123 (3), act as allosteric inhibitors.43,49,54−56 These inhibitors achieve receptor selectivity by exploiting amino acid differences between these homologous receptors, with reparixin (1) differentially binding the extracellular half of the seven-transmembrane core23 and SB332235 differentially binding an intracellular pocket made up of several transmembrane helices.55 We previously demonstrated that the nicotinamide glycolate methyl ester 5 inhibited CXCL1-induced effects in hPMNs through an intracellular mechanism.35 In the studies herein, potent antagonism of CXCL8-stimulated [35S]GTPγS binding by compound 7 localized inhibition to a proximal signaling event involving the CXCR2 receptor and its G-protein. Further data demonstrated compound 7 was CXCR2/1 specific, with no inhibition of fMLF-, C5a-, or PAF-induced Ca2+ flux through their corresponding GPCRs. We therefore speculate that compound 7 acts at an intracellular pocket, involving at least the CXCR2/1 receptor to lock the receptor in a conformation unable to activate downstream signaling. Studies to explore this postulated mechanism are underway in our group.
Consistent with being a noncompetitive inhibitor, compound 7 potently inhibited functional CXCR2 signaling by CXCL1 and CXCL8 but failed to compete with the binding of [125I]-CXCL8 to CXCR2 at concentrations ∼100-fold larger than its IC50 for inhibiting CXCR2 ligand-triggered Ca2+ flux. Others have earlier recognized the possibility of identifying allosteric small-molecule chemokine inhibitors that disrupt receptor function without displacing the chemokine from its orthosteric binding site.57 The SAR studies herein demonstrate that our whole-cell functional assay for CXCL1-induced Ca2+ flux in hPMNs beneficially allowed for the selection of a novel class of potent noncompetitive inhibitors that would have otherwise been missed by a competitive displacement screen. Our findings mirror those of the noncompetitive allosteric inhibitor reparixin, which likewise inhibited CXCR1 responses with no effect on CXCL8 binding to CXCR1.22,23
Compound 7 is notable for being the first reported boronic acid chemokine antagonist. Due to the unique chemical similarities and differences between boron and carbon, boronic acid based inhibitors have been the subject of considerable recent interest as a new source of small-molecule therapeutics.38 The clinical utility of the boronyl moiety has been validated with the U.S. and European approval of bortezomib (Velcade), and other boronic acids have been previously disclosed as inhibitors of serine proteases, proteasomes, arginase, nitric oxide synthase, and transpeptidases.39 The effectiveness of boronic acids as inhibitors of proteosomes58,59 and serine proteases60−63 has been ascribed to the ability of the boronyl group to form a reversible tetrahedral adduct with an active-site nucleophile that has most often been serine but has also included histidine.60 On the basis of crystallographic and NMR studies of boronic acid inhibitors in complex with their target enzymes, this adduct closely mimics the putative tetrahedral intermediate or transition-state formed between the enzyme and its substrates. Although CXCR1 and CXCR2 are not thought to possess intracellular enzymatic activity, we speculate that the marked increase in potency observed upon incorporation of the boronyl group at the 2-position of the S-benzyl scaffold in compound 7 may be due to an analogous reversible boronyl-mediated adduct with a receptor-based nucleophile in the putative intracellular binding pocket. The potential benefit of such a postulated adduct in prolonging pharmacodynamic effects in subjects remains to be investigated. However, like other CXCR2/1 inhibitors with a low off-rate (e.g., SCH527123),49 the benefit will likely be limited by the kinetics of new receptor biosynthesis and the 1–2 day tissue lifespan of the neutrophil itself.
Current efforts are underway in our laboratory to increase the oral bioavailability of compound 7 and to further investigate the interaction of this class of compounds with CXCR1 and CXCR2 receptors. Compound 7 (SX-517) represents a novel boronic acid containing pharmacophore for the antagonism of CXCR1/2 chemokine receptors and may prove useful in the treatment of inflammatory diseases with a significant neutrophil component.
Experimental Methods
A. Pharmacology and Biology
Materials and Reagents
Chemicals and carrageenan were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO). Chemokines were from PeproTech (Rocky Hill, NJ) or from R&D Systems (Minneapolis, MN). Control CXCR2 antagonists (SB265610 and SB225002) were from Tocris Biosciences (Ellisville, MO). The HEK293 cell line was from ATCC (Manassas, VA). Erk1/2 and phospho-Erk (p-Erk) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Cellulose nitrate membrane filters were from Whatman Inc. (Piscataway, NJ). [35S]GTPγS (1250 Ci/mmol), 125I-CXCL8 (2200 Ci/mmol), and Unifilter GF/C 96-well filter plates were from PerkinElmer Life and Analytical Sciences (Waltham, MA).
HEK293 Cell Stably Expressing Human CXCR1 and CXCR2
HEK293 cells were cultured in DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 50 units/mL penicillin, 50 μg/mL streptomycin, 3 mM glutamine, 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) at 37 °C, 5% CO2. Transfection with a human CXCR13 or CXCR2 plasmid44 was performed with Lipofectamine (Invitrogen Life Technology) based on the manufacturer’s protocol. For selection of stable polyclonal cell lines, 800 μg/mL G418 (Sigma, St Louis, MO) were added 24 h after transfection and cells were maintained in DMEM medium containing 800 μg/mL G418 through subculture procedures until a pooled, stable cell line was established. Surface expression of CXCR1 and CXCR2 was confirmed using R-phycoerythrin (PE)-conjugated antihuman CXCR1 or CXCR2 mouse monoclonal antibody (BD Pharmingen, San Diego, CA) and fluorescence-activated cell sorting as described below.
Isolation of Human Neutrophils
Blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at Montana State University. Human polymorphonuclear leukocytes (hPMNs) were purified from the blood using dextran sedimentation, followed by Histopaque 1077 gradient separation and hypotonic lysis of red blood cells, as described previously.64 hPMN preparations were routinely >95% pure, as determined by light microscopy, and >98% viable, as determined by trypan blue exclusion. Isolated hPMNs were washed twice and resuspended in RPMI containing 10% fetal bovine serum (FBS).
Animals
Male CD1 mice were obtained from Charles River Laboratories (Wilmington, MA). Animals were housed and acclimatized for 1 week under controlled temperature (20 ± 2 °C), humidity (55 ± 10%), and lighting (7 a.m. to 7 p.m.). Standard sterilized food and water were supplied ad libitum during acclimatization and experiments. All procedures and protocols were approved by the Institutional Animal Care and Use Committee and were carried out in accordance with NIH guidelines for the handling and use of laboratory animals.
Calcium Flux Assay
hPMNs (or cells expressing either CXCR1 or CXCR2) were suspended in HBSS– (Hank’s balanced salt solution without Ca2+ and Mg2+) containing 10 mM HEPES and FLIPR Calcium 3 dye (3.1 × 107 cells in total volume 1.7 mL). Cells were aliquoted (200 μL of the cell suspension per tube, 8 tubes total), and 2 μL of the designated compound (with appropriate dilutions) were added to each of 6 tubes. As controls, 2 μL of DMSO (1% final concentration) were added to two other tubes. Cells were incubated at 37 °C for 30 min. After dye loading, tubes were centrifuged at 6000 rpm for 1 min, supernatant was removed, and the cell pellet was resuspended in 200 μL of HBSS+ (with Ca2+ and Mg2+), containing 10 mM HEPES. The test compound or DMSO (control) were added again at the same concentrations that were used during cell loading. The cell suspension was aliquoted into a 96-well Reading Plate (Corning) in a volume of 90 μL (105 cells/well). The Compound Plate contained agonist in HBSS–) or HBSS– (control). After 15 s of reading the basal level of fluorescence by FlexStation II, 10 μL of agonist or HBSS– were automatically transferred from the compound plate into the reading plate. The agonists used and their final concentrations were 25 nM CXCL1, 1 nM CXCL8, 10 nM N-formyl-l-methionyl-l-leucyl-l-phenylalanine (fMLF), and 50 nM C5a. Changes in fluorescence were monitored (λex = 485 nm, λem = 525 nm) every 5 s for 240 to 500 s at room temperature. The maximum change in fluorescence, expressed in arbitrary units over baseline (max–min), was used to determine the agonist response. The effect of each compound on the agonist response was normalized and expressed as a percent of the DMSO control, which was designated as “100% response.” Curve fitting and calculation of the compound inhibitory concentration that reduced the level of the agonist response by 50% (IC50) was determined by nonlinear regression analysis of the dose–response curves generated using Prism 4 (GraphPad Software, Inc., San Diego, CA).
[35S]GTPγS Assay
[35S]GTPγS assays were performed as previously described65 with the following modifications: HEK293 cells stably expressing hCXCR2, pretreated with different concentrations of compound, were lysed in buffer containing 5 mM Tris-HCl, pH 7.5, 5 mM EDTA, and 5 mM EGTA, and the cell lysate was centrifuged at 30000g for 10 min. Protein concentration in membrane preparations was determined using the BioRad Protein Determination assay 18 from Bio-Rad (Hercules, CA). Membranes containing 50 μg of protein were incubated in 50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 1 mM EGTA, 100 mM NaCl, 50 μM GDP, 8 nM [35S]GTPγS, 10 nM CXCL8 in a total volume of 0.1 mL at 30 °C for 1 h. The reaction was terminated by dilution into phosphate-buffered saline and rapid filtration through Unifilter GF/C 96-well filter plates pretreated with 0.3% polyethylenimine and washed three times with ice-cold wash buffer (50 mM Na2HPO4 and 50 mM KH2PO4, pH 7.4). Bound radioactivity was determined using a MicroBeta counter (PerkinElmer Life and Analytical Sciences). Basal binding was assessed in the absence of CXCL8, and nonspecific binding was determined in the presence of 10 μM GTPγS. The percentage of CXCL8-stimulated [35S]GTPγS binding was calculated as [cpmCXCL8 – cpmnonspecific]/[cpmbasal – cpmnonspecific]. Curve fitting and calculation of the compound inhibitory concentration that reduced the percentage of CXCL8-stimulated [35S]GTPγS binding by 50% (IC50) was determined by nonlinear regression analysis of the dose–response curves generated using Prism 4 (GraphPad Software, Inc., San Diego, CA).
Competition 125I-CXCL8 Binding Assay
This was performed according to White et al. using HEK293-hCXCR2 membranes.24 Briefly, assays were performed in 96-well microtiter plates where the reaction mixture contained 1.0 μg/mL membrane protein in 20 mM Bis-trispropane, pH 8.0, with 1.2 mM MgSO4, 0.1 mM EDTA, 25 mM NaCl, and 0.03% CHAPS and compound (100 μM stock in DMSO) added at the indicated concentrations, the final DMSO concentration was <0.5% under standard binding conditions. Binding was initiated with 25 pM [125I]-CXCL8. Nonspecific binding was determined with 30 nM CXCL8. After 1 h incubation at room temperature, membranes were harvested by rapid filtration. The filter was dried and counted with a liquid scintillation counter. Specific CXCL8 binding to the receptors was defined as the difference between the total binding and the nonspecific binding determined in the presence of an excess of unlabeled CXCL8. The results were expressed as a percent of control specific binding: (specific binding with compound)/(control specific binding) × 100. IC50 for unlabeled CXCL8 was determined by nonlinear regression analysis of the concentration–response curve generated with mean replicate values fitted to the Hill equation.
CXCR2 Cell Surface Expression by Flow Cytometry
HEK293 cells stably expressing CXCR2 were pretreated with 1% DMSO (vehicle) or compound (10 μM) for 60 min. HEK293 cells not expressing CXCR2 served as a negative isotype control. All cells were then incubated with a 1:100 dilution of R-phycoerythrin (PE)-conjugated antihuman CXCR2 mouse monoclonal antibody (BD Pharmingen, San Diego, CA) at 4 °C for 60 min. Cells were washed, fixed in 2% formaldehyde in phosphate-buffered saline, and quantitated by fluorescence-activated cell sorting (FACS) using a FACScan flow cytometer equipped with CellQuest software (BD Biosciences, Mountain View, CA).
ERK1/2 Phosphorylation
HEK293 cells expressing CXCR2 were harvested and plated in equal number to 60 mm plates (5 × 106 cells/plate). The cells were then incubated with PBS containing 1% DMSO (vehicle) or 10 μM compound for 60 min at 37 °C, followed by the addition of 100 ng/mL CXCL8. All cells in a plate were then lysed at 0, 5, 15, and 30 min after agonist-treatment by adding lysis buffer: 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, 10 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, and 10 μg/mL leupeptin. Lysates containing equal amounts of protein (∼50 μg) were resolved by 10% SDS–polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and probed with antibody against phospho-ERK1/2 or ERK1/2. Visualization was carried out with a horseradish peroxidase-conjugated secondary antibody.
Murine Air-Pouch Model of Inflammation
An air-pouch was induced on the backs of 10–15 week old, male CD1 Swiss mice by subcutaneous injection (2 mL) of air as described.47 An additional subcutaneous injection of air (1.5 mL) was given to reinflate the pouch the following day. The compound was solubilized in vehicle consisting of a 20:20:80 mixture of PEG400:DMF:saline. Compound dissolved in vehicle, or vehicle alone was administered via iv tail vein injection at different doses according to body weight. After 3 h, either sterile phosphate-buffered saline (PBS, 1 mL) or 2% carrageenan in sterile PBS (1 mL) was injected into the air pouch. After 4 h, the mice were sedated with ketamine and sacrificed by cervical dislocation. The pouch fluid was then collected by syringe and combined with an additional 2 mL PBS wash of the pouch. The cells in the combined fluid were stained with trypan blue and manually counted on a hemocytometer under 20× magnification.
B. Chemistry
Materials and Reagents
General chemicals and reagents for synthesis were purchased from Sigma-Aldrich (Milwaukee, WI), and solvents were purchased from VWR International (West Chester, PA) and used without further purification. Commercial synthetic precursors and intermediates were from Acros Organics (Pittsburgh, PA), Sigma-Adrich Chemical (Milwaukee, WI), Avocado Research (Lancashire, U.K.), Bionet (Cornwall, U.K.), Boron Molecular (Research Triangle Park, NC), Combi-Blocks (San Diego, CA), Eastman Organic Chemicals, Eastman Kodak Company (Rochester, NY), Fisher Scientific Co. (Pittsburgh, PA), Frontier Scientific (Logan, UT), ICN Biomedicals, Inc. (Costa Mesa, CA), Lancaster Synthesis (Windham, NH), Maybridge Chemical Co. (Cornwall, U.K.), Pierce Chemical Co. (Rockford, IL), Riedel de Haen (Hannover, Germany), Santa Cruz Biotechnology (Dallas, TX), Spectrum Quality Product, Inc. (New Brunswick, NJ), TCI America (Portland, OR), and Wako Chemicals USA, Inc. (Richmond, VA). Solid phase scavenger resins were from NovaBiochem (La Jolla, CA).
General Analytical Procedures
Synthetic reaction progression was monitored by thin-layer chromatography (TLC) using precoated aluminum-backed plates with silica gel with fluorescent indicator (precoated F254 Macherey-Nagel plates, EMD Chemicals); the spots were examined with UV light. Chromatographic purification was performed with 230–400 mesh (32–63 μm) flash silica gel (Dynamic Adsorbents, Norcross, GA) or by preparative high-performance liquid chromatography (HPLC) using a Waters Delta Prep 4000 HPLC fitted with a Phenomenex Gemini 250 × 21 mm, 10 μm, C18 column and monitored at 254 nm. Retention time (RT) is reported in minutes (min), and purity as measured by UV absorbance is reported as a percentage of all peak areas. HPLC analyses were performed using the following gradients and systems:
Gradient A
Water:acetonitrile:formic acid (95:5:0.1) to water:acetonitrile:formic acid (5:95:0.1) at 35 °C over 12 min, on a Shimadzu HPLC system, with a Phenomenex Gemini 50 × 2 mm, 5 μm, C18 column, monitored at 254 nm.
Gradient B
Water:acetonitrile:formic acid (95:5:0.1) to water:acetonitrile:formic acid (5:95:0.1) at 30 °C over 30 min, on an Agilent 1100 Series HPLC system, with a Phenomenex Gemini 50 × 2 mm, 5 μm, C18 column, monitored at 254 nm.
Gradient C
Water:acetonitrile:trifluoroacetic acid (95:5:0.1) to water:acetonitrile:trifluoroacetic acid (5:95:0.1) at 35 °C over 0.5 min, held at 5:95:0.1 for 6.5 min, on an Agilent 1100 Series HPLC system, with Phenomenex Gemini 50 × 2 mm, 5 μm, C18 column, monitored at 254 nm.
Gradient D
Water:acetonitrile:formic acid (95:5:0.1) to water:acetonitrile:formic acid (5:95:0.1) at 35 °C over 4 min, held at 5:95:0.1 for 5 min, on a Shimadzu HPLC system, with Phenomenex Gemini 50 × 2 mm, 5 μm, C18 column, monitored at 254 nm.
Electrospray ionization mass spectrometric analysis (ESI-MS) was performed using a Micromass Quattro II mass spectrometer with MassLynx 4.0. 1H NMR spectra were obtained on a Bruker AVance (300 or 500 MHz, 1H) and are reported as parts per million downfield from tetramethylsilane with number of protons, multiplicities, and coupling constants in Hertz indicated parenthetically. Elemental analyses were performed by Atlantic Microlab (Norcross, GA).
General Procedures for the Synthesis of S-Substituted N-(4-Fluoro-phenyl)-6-mercapto-nicotinamides (Scheme 1)
Accomplished by one of the following three methods. If required, the crude was purified by either flash silica gel chromatography or preparative HPLC. Final product was characterized by HPLC, ESI-MS, and 1H NMR where indicated.
Method A
A solution of the bromomethyl derivative (2.5 equiv) in anhydrous DMF (5 mL/mmol) was added to thionicotinamide 64 (1 equiv) and N-methylmorpholino-substituted polystyrene resin (5 equiv) and heated at 60 °C for 2 h in a screw cap glass vial. Sulfhydryl-bearing scavenger resin (5 equiv) was then added to the reaction mixture and heated at 60 °C for a further 4 h. After cooling, the organic reaction solution was filtered, and then diluted into water (100 mL/mmol) to precipitate the product. The resulting suspension was then centrifuged at 5000 rpm for 15 min, the aqueous supernatant was decanted, and the product dried in a vacuum oven overnight at 50 °C. Compounds prepared by this method were used without further purification.
Method B
Thionicotinamide 64 (1 equiv) and the corresponding bromomethyl or chloromethyl derivative (1 equiv) was dissolved in anhydrous DMF (2 mL/mmol). To the solution, a tertiary amine base (diisopropylethylamine, triethylamine, or N-methylmorpholine, 1 equiv) was added. The reaction was allowed to proceed at room temperature and monitored by either TLC or LC–MS until complete (1–18 h). The crude product was then precipitated out of solution by the addition of water (5–50 mL/mmol).
Method C
Thionicotinamide 64 (1 equiv) and the corresponding bromomethyl derivative (1 equiv) were suspended in EtOH (5 mL/mmol). To the suspension, 1 N NaOH (1 equiv) was added, and the reaction mixture was brought to gentle reflux and monitored by either TLC or LC–MS until complete (0.5–2 h). The crude product was then precipitated out of solution by the addition of water (5–20 mL/mmol).
2-((5-(4-Fluorophenylcarbamoyl)pyridin-2-ylthio)methyl)phenylboronic acid (7, SX-517)
Prepared via method C using thionicotinamide 64 (1.27 g, 5.10 mmol) and 2-bromomethyl-phenylboronic acid (1.09 g, 5.10 mmol) suspended in EtOH (50 mL). To the suspension, 1 N NaOH (5.1 mL, 5.10 mmol) was added and the reaction mixture heated to gentle reflux for 2 h. Then water (50 mL) was added to the reaction mixture while still hot. Upon cooling, a white precipitate formed and this was filtered, washed with 50% aqueous EtOH, and then water and dried in an oven to yield 1.53 g (78%) of 7 (SX-517) as an off-white solid. ESI-MS m/z = 383.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.39 (s, 1H), 8.99 (s, 1H), 8.23 (s, 2H), 8.13–8.11 (m, 1H), 7.80–7.77 (m, 2H), 7.55 (d, J = 7.0 Hz, 1H), 7.45 (d, J = 8.0 Hz, 2H), 7.30 (t, J = 7.0 Hz, 1H), 7.22 (t, J = 8.8 Hz, 3H), 4.69 (s, 2H). 13C NMR (100.6 MHz, DMSO-d6): d 163.6, 162.7, 159.6, 157.2, 148.6, 141.5, 136.2, 135.5, 135.3, 134.0, 129.3, 129.0, 126.2, 122.2, 122.1, 120.7, 115.4, 115.1. Anal. Calcd for C19H16BFN2O3S: C, 59.71%; H, 4.22%; N, 7.33%; S, 8.39%. Found: C, 59.54%; H, 4.38%; N, 7.48%; S, 8.49%. HPLC (gradient B): RT = 18.45 min, purity 96.1%.
6-((2H-Tetrazol-5-yl)methylthio)-N-(4-fluorophenyl)nicotinamide (8)
The cyano intermediate was prepared via method B using thionicotinamide 64 and chloroacetonitrile. This intermediate was used without further purification. The cyano-intermediate (190 mg, 0.66 mmol), dibutyltin oxide (33 mg, 0.14 mmol) and trimethylsilyl azide (174 μL, 1.32 mmol) were suspended in toluene (50 mL) and refluxed for 24 h. The mixture was allowed to cool to room temperature, and the resulting precipitate was filtered and washed with toluene to yield 160 mg (73%) of 8 as a light yellow solid. TLC (1% AcOH/ethyl acetate): Rf = 0.32. ESI-MS m/z = 331.3 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.41 (s, 1H), 8.97 (s, 1H), 8.18 (d, J = 8.0 Hz, 1H), 7.77 (t, J = 6.0 Hz, 6.6 Hz, 2H), 7.58 (d, J = 8.2 Hz, 1H), 7.21 (t, J = 8.2 Hz, 2 H), 4.81 (s, 2H). HPLC (gradient B): RT = 14.52 min, purity 96.4%.
N-(4-Fluorophenyl)-6-(2-oxotetrahydrofuran-3-ylthio)nicotinamide (9)
Prepared via method B using thionicotinamide 64 and α-bromo-γ-butyrolactone and purified by flash silica gel chromatography (2:3 ethyl acetate:hexanes) to yield 135 mg (41%) of 9 as a white waxy solid. ESI-MS: m/z = 333.3 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.43 (s, 1H), 8.96 (s, 1H), 8.19 (dd, J = 8.3 Hz, 2.3 Hz, 1H), 7.80–7.77 (m, 2H), 7.57 (d, J = 8.4 Hz, 1H), 7.22 (t, J = 8.8 Hz, 2H), 4.77 (t, J = 9.7 Hz, 1H), 4.50–4.46 (m, 1H), 4.40–4.35 (m, 1H), 2.79–2.74 (m, 1H), 2.47–2.39 (m, 1H). Anal. Calcd for C16H13FN2O3S: C, 57.82%; H, 3.94%; N, 8.43%. Found: C, 57.61%; H, 4.04%; N, 8.14%. HPLC (gradient B): RT = 16.23 min, purity 99.8%.
N-(4-Fluorophenyl)-6-(phenylsulfonylmethylthio)nicotinamide (10)
Prepared via method B using thionicotinamide 64 and bromomethyl phenyl sulfone and purified by flash silica gel chromatography (step gradient of 1:2 to 2:3 ethyl acetate:hexanes) to yield 159 mg (40%) of 10 as a white waxy solid. ESI-MS: m/z = 403.3 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.41 (s, 1H), 8.86 (s, 1H), 8.15 (dd, J = 8.2 Hz, 2.0 Hz, 1H), 7.92 (s, 1H), 7.90 (d, J = 1.1 Hz, 1H), 7.80–7.77 (m, 2H), 7.69 (t, J = 7.3 Hz, 1H), 7.60 (t, J = 7.6 Hz, 2H), 7.57 (d, J = 8.6 Hz, 1H), 7.22 (t, J = 8.8 Hz, 2H), 5.33 (s, 2H). Anal. Calcd for C19H15FN2O3S2: C, 56.70%; H, 3.76%; N, 6.96%. Found: C, 56.20%; H, 3.97%; N, 6.51%. HPLC (gradient B): RT = 19.06 min, purity 97.9%.
N-(4-Fluorophenyl)-6-(nitromethylthio)nicotinamide (11)
Prepared via method B using thionicotinamide 64 and bromonitromethane to yield 197 mg (51%) of 11 as an off-white solid. ESI-MS: m/z = 308.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.44 (s, 1H), 8.97 (s, 1H), 8.24 (d, J = 8.3 Hz, 7.78 (t, J = 7.1 Hz, 5.3 Hz, 2H), 7.71 (d, J = 8.5 Hz, 1H), 7.22 (t, J = 8.0 Hz, 8.4 Hz, 2H), 6.32 (s, 2H). Purity determined by HPLC (gradient B): RT = 17.78 min, purity 88.4%.
Diethyl (5-(4-fluorophenylcarbamoyl)pyridin-2-ylthio)methylphosphonate (12)
The screened compound was prepared via method B using thionicotinamide 64 and diethyl-(bromomethyl)-phosphonate and purified by flash silica gel chromatography (step gradient of 1:1, 2:1, and 1:0 ethyl acetate:hexanes) to yield 138 mg (34.7%) of 12 as a white waxy solid. ESI-MS: m/z = 399.4 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.42 (s, 1H), 9.00 (s, 1H), 8.20 (dd, J = 8.4 Hz, 2.0 Hz, 1H), 7.80–7.77 (m, 2H), 7.60 (d, J = 8.3 Hz, 1H), 7.22 (t, J = 8.8 Hz, 2H), 4.08–4.03 (m, 4H), 3.78 (d, J = 13.1 Hz, 2H), 1.21 (t, J = 6.7 Hz, 7.2 Hz, 6H). Anal. Calcd for C17H20FN2O4PS: C, 51.25%; H, 5.06%; N, 7.03%. Found: C, 51.52%; H, 5.09%; N, 7.19%. HPLC (gradient B): RT = 17.56 min, purity 98.5%.
N-(4-Fluorophenyl)-6-((tetrahydro-2H-pyran-2-yl)methylthio)nicotinamide (13)
Prepared via method B using thionicotinamide 64 and 2-(bromomethyl)-tetrahydropyran to yield 143 mg (41%) of 13 as an off-white solid. ESI-MS: m/z = 346.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.37 (s, 1H), 8.97 (s, 1H), 8.13 (d, J = 8.2 Hz, 1H), 7.80–7.77 (m, 2H), 7.47 (d, J = 8.1 Hz, 1H), 7.21 (t, J = 7.8 Hz, 8.4 Hz, 2H), 3.90 (d, J = 11.1 Hz, 1H), 3.49–3.46 (m, 1H), 3.40–3.35 (m, 2H), 3.28–3.24 (m, 1H), 1.78–1.72 (m, 2H), 1.45 (broad m, 3H), 1.33–1.27 (m, 1H). Anal. Calcd for C18H19FN2O2S: C, 62.41%; H, 5.53%; N, 8.09%. Found: C, 62.37%; H, 5.49%; N, 8.20%. HPLC (gradient B): RT = 19.80 min, purity 95.5%.
N-(4-Fluorophenyl)-6-(4-(2-methylthiazol-4-yl)benzylthio)nicotinamide (14)
Prepared via method A using thionicotinamide 64 and 4-[4-(bromomethyl)phenyl]-2-methyl-1,3-thiazole. ESI-MS: m/z = 436.1 [M + H]+. HPLC (gradient C): RT = 6.66 min, purity 71.1%.
N-(4-Fluorophenyl)-6-(pyridin-4-ylmethylthio)nicotinamide (15)
Prepared via method B using thionicotinamide 64 and 4-(bromomethyl)pyridine to yield 146 mg (43%) of 15 as a white solid. ESI-MS: m/z = 340.4 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.39 (s, 1H), 8.98 (s, 1H), 8.53 (d, J = 3.9 Hz, 1H), 8.16 (d, J = 8.3 Hz, 1H), 7.79–7.75 (m, 3H), 7.54–7.53 (m, 2H), 7.29 (t, J = 2.9 Hz, 5.5 Hz, 1H), 7.21 (t, J = 8.2 Hz, 8.8 Hz, 2H), 4.61 (s, 2H). Anal. Calcd for C18H14FN3OS: C, 63.70%; H, 4.16%; N, 12.38%. Found: C, 63.28%; H, 3.90%; N, 12.11%. HPLC (gradient B): RT = 12.63 min, purity 93.7%.
Benzyl 4-((5-(4-Fluorophenylcarbamoyl)pyridin-2-ylthio)methyl)piperidine-1-carboxylate (16)
Prepared via method A using thionicotinamide 64 and benzyl 4-(bromomethyl)tetrahydro-1-(2H)-pyridinecarboxylate. ESI-MS: m/z = 495.0 [M + H]+. HPLC (gradient D): RT = 5.90 min, purity 89.1%.
N-(4-Fluorophenyl)-6-((tetrahydro-2H-pyran-4-yl)methylthio)nicotinamide (17)
Prepared via method B using thionicotinamide 64 and 4-(bromomethyl)-tetrahydropyran to yield 286 mg (83%) of 16 as a white solid. ESI-MS: m/z = 347.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.38 (s, 1H), 8.98 (s, 1H), 8.13 (d, J = 8.5 Hz, 1H), 7.80–7.77 (m, 2H), 7.48 (d, J = 8.3 Hz, 1H), 7.21 (t, J = 8.7 Hz, 9.1 Hz, 2H), 3.86–3.84 (m, 2H), 3.26 (t, J = 11.4 Hz, 2H), 3.20 (d, J = 6.7 Hz, 2H), 1.83–1.80 (m, 1H), 1.73 (d, J = 12.3 Hz, 2H), 1.33–1.23 (m, 2H). HPLC (gradient B): RT = 18.64 min, purity 89.1%.
N-(4-Fluorophenyl)-6-(4-(5-methyl-1,2,4-oxadiazol-3-yl)benzylthio)nicotinamide (18)
Prepared via method A using thionicotinamide 64 and 3-[3-(bromomethyl)phenyl]-5-methyl-1,2,4-oxadiazole. ESI-MS: m/z = 421.1 [M + H]+. HPLC (gradient C): RT = 6.63 min, purity 80.7%.
N-(4-Fluorophenyl)-6-((5-(trifluoromethyl)furan-2-yl)methylthio)nicotinamide (19)
Prepared via method A using thionicotinamide 64 and 2-(bromomethyl)-5-(trifluoromethyl)furan. ESI-MS: m/z = 397.1 [M + H]+. HPLC (gradient C): RT = 6.82 min, purity 82.0%.
N-(4-Fluorophenyl)-6-((tetrahydrofuran-2-yl)methylthio)nicotinamide (20)
Prepared via method C using thionicotinamide 64 and 2-(bromomethyl)tetrahydrofuran to yield 140 mg (41%) of 20 as a white solid. ESI-MS: m/z = 333.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.38 (s, 1H), 8.97 (s, 1H), 8.13 (d, J = 8.0 Hz, 1H), 7.78 (t, J = 6.1 Hz, 6.8 Hz, 2H), 7.49 (d, J = 8.3 Hz, 1H), 7.21 (t, J = 8.3 Hz, 8.6 Hz, 2H), 4.08–4.04 (m, 1H), 3.81 (q, J = 7.0 Hz, 1H), 3.66 (q, J = 7.0 Hz, 1H), 3.43–3.35 (m, 2H), 2.04–1.97 (m, 1H), 1.94–1.78 (m, 2H), 1.67–1.60 (m, 1H). Anal. Calcd for C17H17FN2O2S: C, 61.43%; H, 5.16%; 8.43%. Found: C, 61.34%; H, 5.07%; N, 8.47%. HPLC (gradient B): RT = 18.14 min, purity 95.6%.
6-(2-(3,5-Dimethyl-1H-pyrazol-4-yl)ethylthio)-N-(4-fluorophenyl)nicotinamide (21)
Prepared via method A using thionicotinamide 64 and 4-(2-bromoethyl)-3,5-dimethyl-1H-pyrazole. ESI-MS: m/z = 371.1 [M + H]+. HPLC (gradient D): RT = 5.47 min, purity 81.0%.
N-(4-Fluorophenyl)-6-((5-nitrofuran-2-yl)methylthio)nicotinamide (22)
The screened compound was prepared via method B using thionicotinamide 64 and 2-(bromomethyl)-5-(nitro)furan and purified by flash silica gel chromatography (1:2 ethyl acetate:hexanes) to yield 91 mg (24%) of 22 as an orange solid. ESI-MS: m/z = 374.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.42 (s, 1H), 9.02 (d, J = 2.0 Hz, 1H), 8.20 (dd, J = 8.3 Hz, 2.2 Hz, 1H), 7.80–7.77 (m, 2H), 7.64 (d, J = 3.6 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.23–7.20 (m, 2H), 6.79 (d, J = 3.8 Hz, 1H), 4.71 (s, 2H). Anal. Calcd for C17H12FN3O4S: C, 54.69%; H, 3.24%; N, 11.25%. Found: C, 55.20%; H, 3.24%; N, 11.01%. HPLC (gradient B): RT = 20.13 min, purity 91.1%.
N-(4-Fluorophenyl)-6-((5-methylisoxazol-3-yl)methylthio)nicotinamide (23)
Prepared via method A using thionicotinamide 64 and 3-(bromomethyl)-5-methylisoxazole. ESI-MS: m/z = 344.1 [M + H]+. HPLC (gradient C): RT = 6.21 min, purity 92.9%.
N-(4-Fluorophenyl)-6-((5-methyl-2-phenyl-2H-1,2,3-triazol-4-yl)methylthio)nicotinamide (24)
Prepared via method A using thionicotinamide 64 and 4-(bromomethyl)-5-methyl-2-phenyl-2H-1,2,3-triazole. ESI-MS: m/z = 420.1 [M + H]+. HPLC (gradient C): RT = 7.03 min, purity 91.7%.
N-(4-Fluorophenyl)-6-((3-methyl-5-phenylisoxazol-4-yl)methylthio)nicotinamide (25)
Prepared via method A using thionicotinamide 64 and 4-(bromomethyl)-3-methyl-5-phenylisoxazole. ESI-MS: m/z = 420.1 [M + H]+. HPLC (gradient C): RT = 6.81 min, purity 85.8%.
N-(4-Fluorophenyl)-6-((5-methyl-3-phenylisoxazol-4-yl)methylthio)nicotinamide (26)
Prepared via method A using thionicotinamide 64 and 4-(bromomethyl)-5-methyl-3-phenylisoxazole. ESI-MS: m/z = 420.1 [M + H]+. HPLC (gradient B): RT = 24.01 min, purity 72.0%.
N-(4-fluorophenyl)-6-((4-methyl-2-(4-(trifluoromethyl)phenyl)thiazol-5-yl)methylthio)nicotinamide (27)
Prepared via method A using thionicotinamide 64 and 5-(bromomethyl)-4-methyl-2-[4-(trifluoromethyl)phenyl]-1,3-thiazole. ESI-MS: m/z = 504.1 [M + H]+. HPLC (gradient D): RT = 6.41 min, purity 86.9%.
N-(4-Fluorophenyl)-6-((4-methyl-2-phenylthiazol-5-yl)methylthio)nicotinamide (28)
Prepared via method A using thionicotinamide 64 and 5-(bromomethyl)-4-methyl-2-[4-phenyl]-1,3-thiazole. ESI-MS: m/z = 436.1 [M + H]+. HPLC (gradient C): RT = 6.85 min, purity 70.7%.
6-(2-(3,5-Dimethyl-1H-pyrazol-4-yl)ethylthio)-N-(4-fluorophenyl)nicotinamide (29)
Prepared via method A using thionicotinamide 64 and 4-(2-bromoethyl)-3,5-dimethyl-1H-pyrazole. ESI-MS: m/z = 371.1 [M + H]+. HPLC (gradient D): RT = 5.47 min, purity 81.0%.
6-(Benzylthio)-N-(4-fluorophenyl)nicotinamide (30)
Prepared via method C using thionicotinamide 64 and benzyl bromide to yield 108 mg (64%) of 30 as an off-white powder. ESI-MS: m/z = 338.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.39 (s, 1H), 9.00 (s, 1H), 8.15 (d, J = 8.0 Hz, 1H), 7.80–7.77 (m, 2H), 7.49–7.45 (m, 3 H), 7.34 (t, J = 7.7 Hz, 6.9 Hz, 2H), 7.27–7.20 (m, 3H), 4.51 (s, 2H). Anal. Calcd for C19H15FN2OS: C, 67.44%; H, 4.47%; N, 8.28%. Found: C, 67.17%; H, 4.37%; N, 8.13%. HPLC (gradient B): RT = 21.66 min, purity 96.5%.
N-(4-Fluorophenyl)-6-(4-nitrobenzylthio)nicotinamide (31)
Prepared via method A using thionicotinamide 64 and 1-(bromomethyl)-4-nitrobenzene. ESI-MS: m/z = 384.1 [M + H]+. HPLC (gradient C): RT = 6.63 min, purity 66.3%.
N-(4-Fluorophenyl)-6-(4-(trifluoromethoxy)benzylthio)nicotinamide (32)
Prepared via method A using thionicotinamide 64 and 1-(bromomethyl)-4-(trifluoromethoxy)benzene. ESI-MS: m/z = 423.1 [M + H]+. HPLC (gradient C): RT = 7.12 min, purity 83.6%.
N-(4-Fluorophenyl)-6-(3-(trifluoromethoxy)benzylthio)nicotinamide (33)
Prepared via method A using thionicotinamide 64 and 1-(bromomethyl)-3-(trifluoromethoxy)benzene. ESI-MS: m/z = 423.1 [M + H]+. HPLC (gradient C): RT = 7.12 min, purity 86.3%.
N-(4-Fluorophenyl)-6-(2-(thiophen-2-yl)benzylthio)nicotinamide (34)
Prepared via method A using thionicotinamide 64 and 2-[2-(bromomethyl)phenyl]thiophene and screened without purification. ESI-MS: m/z = 421.1 [M + H]+. HPLC (gradient C): RT = 7.31 min, purity 73.7%.
N-(4-Fluorophenyl)-6-(4-(2-methylthiazol-4-yl)benzylthio)nicotinamide (35)
Prepared via method A using thionicotinamide 64 and 4-[4-(bromomethyl)phenyl]-2-methyl-1,3-thiazole. ESI-MS: m/z = 436.1 [M + H]+. HPLC (gradient C): RT = 6.66 min, purity 71.1%.
6-(4-(1H-1,2,4-Triazol-1-yl)benzylthio)-N-(4-fluorophenyl)nicotinamide (36)
Prepared via method A using thionicotinamide 64 and 1-[4-(bromomethyl)phenyl]-1H-1,2,4-triazole. ESI-MS: m/z = 406.1 [M + H]+. HPLC (gradient C): RT = 6.06 min, purity 85.3%.
N-(4-Fluorophenyl)-6-(3-(5-methyl-1,2,4-oxadiazol-3-yl)benzylthio)nicotinamide (37)
Prepared via method A using thionicotinamide 64 and 3-[3-(bromomethyl)phenyl]-5-methyl-1,2,4-oxadiazole. ESI-MS: m/z = 421.1 [M + H]+. HPLC (gradient C): RT = 6.63 min, purity 80.7%.
6-(4-(1H-Pyrazol-1-yl)benzylthio)-N-(4-fluorophenyl)nicotinamide (38)
Prepared via method A using thionicotinamide 64 and 1-[4-(bromomethyl)phenyl]-1H-pyrazole. ESI-MS: m/z = 405.1 [M + H]+. HPLC (gradient C): RT = 6.54 min, purity 92.6%.
N-(4-Fluorophenyl)-6-(3-(2-methylthiazol-4-yl)benzylthio)nicotinamide (39)
Prepared via method A using thionicotinamide 64 and 4-[3-(bromomethyl)phenyl]-2-methyl-1,3-thiazole. ESI-MS: m/z = 436.1 [M + H]+. HPLC (gradient C): RT = 6.68 min, purity 69.2%.
6-(4-(1H-Pyrrol-1-yl)benzylthio)-N-(4-fluorophenyl)nicotinamide (40)
Prepared via method A using thionicotinamide 64 and 1-[4-(bromomethyl)phenyl]-1H-pyrrole. ESI-MS: m/z = 404.1 [M + H]+. HPLC (gradient C): RT = 7.03 min, purity 70.7%.
6-(3-(1H-pyrrol-1-yl)benzylthio)-N-(4-fluorophenyl)nicotinamide (41)
Prepared via method A using thionicotinamide 64 and 1-[3-(bromomethyl)phenyl]-1H-pyrrole. ESI-MS: m/z = 404.1 [M + H]+. HPLC (gradient C): RT = 6.98 min, purity 91.8%.
6-(2-Fluoro-3-methylbenzylthio)-N-(4-fluorophenyl)nicotinamide (42)
Prepared via method A using thionicotinamide 64 and 1-(bromomethyl)-2-fluoro-3-methylbenzene. ESI-MS: m/z = 371.1 [M + H]+. HPLC (gradient C): RT = 6.97 min, purity 83.5%.
6-(3,4-Dichlorobenzylthio)-N-(4-fluorophenyl)nicotinamide (43)
Prepared via method B using thionicotinamide 64 and 1-(bromomethyl)-3,4-dichlorobenzene. The crude material was purified by flash chromatography using a gradient elution of 10% ethyl acetate/hexanes to 95% ethyl acetate/hexanes over 30 min to yield 355 mg (83%) of compound 43 as a white solid. TLC (ethyl acetate/hexane, 1:4): Rf = 0.27. ESI-MS: m/z = 407.0, 409.0 [M + H]+. 1H NMR (400 MHz, DMSO-d6): δ 10.35 (s, 1H), 8.96 (s, 1H), 8.12–8.10 (m, 1H), 7.75–7.72 (m, 2H), 7.69 (s, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 12.9 Hz, 1H), 7.41 (d, J = 8.5 Hz, 1H), 7.17 (t, J = 8.9 Hz, 2H), 4.47 (s, 2H). Purity determined by HPLC (gradient B): RT = 26.18 min, purity 71.2%.
N-(4-Fluorophenyl)-6-(2-hydroxybenzylthio)nicotinamide (44)
The boronic acid 6 (200 mg) was dissolved in 18 mL of THF/acetone/0.1 N NaOH (1:1:1) and cooled to −3 °C. OXONE (1.0 eq, 311 mg) dissolved in 0.4 mM EDTA (2.1 mL) was added, according to the procedure of Webb and Levy for the hydroxylation of aryl boronic esters or boronic acids.36 The reaction was then stirred for 1 h and quenched with sodium bisulfite (3 mL, 5 M aqueous), while being stirred for 10 min. The solvent was removed by reduced pressure rotary evaporation, leaving an aqueous suspension. The suspension was extracted 3× with ethyl acetate, and the combined ethyl acetate layers were washed with 0.01 N HCl, water, 0.01 N NaOH, and brine. They were dried over MgSO4, filtered, and dried under vacuum. The residue was dissolved in THF/MeOH and adhered to 2 g of silica gel. The adhered silica was loaded onto 20 g of silica gel, eluting the product with 3:1 hexanes:ethyl acetate, which was dried under vacuum to provide 67 mg of compound 44 (36% yield). ESI-MS: m/z = 355.4 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 9.75 (s, 1H), 9.01 (d, J = 2.1 Hz, 1H), 8.14 (dd, J = 8.7 Hz, 2.6 Hz, 1H), 7.80–7.77 (m, 3H), 7.48 (d, J = 8.3 Hz, 1H), 7.33 (dd, J = 7.4 Hz, 1.3 Hz, 1H), 7.23–7.20 (m, 2H), 7.11–7.07 (m, 1H), 6.85 (dd, J = 8.1 Hz, 0.8 Hz, 1H), 6.75–6.72 (m, 1H), 4.42 (s, 2H). Anal. Calcd for C19H15FN2O2S: C, 64.39%; H, 4.27%; N, 7.90%. Found: C, 64.50%; H, 4.47%; N, 7.72%. HRMS: calcd for C19H16FN2O2S [M + 1]+ 355.0911, found 355.0901; calcd for C19H15FN2NaO2S [M + Na]+ 377.0730, found 377.0718. HPLC (gradient B): RT = 20.18 min, purity 97.4%.
N-(4-Fluorophenyl)-6-(phenylthio)nicotinamide (45)
Chloronicotinamide 69 (200 mg, 0.75 mmol), thiophenol (77 μL, 0.75 mmol), potassium tert-butoxide (112 mg, 1.5 mmol) and 18-crown-6 (10 mg), was dissolved in NMP (3 mL) and heated to 160 °C for 3 h. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over Na2SO4 and purified using flash chromatography (7:3 hexanes/ethyl acetate) to yield 99 mg (31%) of 45 as an off-white powder. TLC (ethyl acetate/hexane, 1:1): Rf = 0.47. ESI-MS: m/z = 325.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.35 (s, 1H), 8.88 (d, J = 2.3 Hz, 1H), 8.08 (dd, J = 8.4 Hz, 2.3 Hz, 1H), 7.74–7.71 (m, 2H), 7.62–7.60 (m, 2H), 7.53–7.50 (m, 3H), 7.18 (t, J = 2.3 Hz, 2H), 7.02 (d, J = 8.5 Hz, 1H).
Methyl 4-((5-(4-Fluorophenylcarbamoyl)pyridin-2-ylthio)methyl)benzoate (46)
Prepared via method B using thionicotinamide 64 and methyl 4-bromomethylbenzoate to yield 343 mg (87%) of 46 as an off-white solid. ESI-MS: m/z = 397.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.38 (s, 1H), 9.00 (d, J = 1.6 Hz, 1H), 8.15 (dd, J = 8.6 Hz, 2.5 Hz, 1H), 7.92 (d, J = 8.3 Hz, 2H), 7.79–7.76 (m, 2H), 7.61 (d, J = 8.3 Hz, 2H), 7.50 (d, J = 8.2 Hz, 1H), 7.21 (t, J = 9.0 Hz, 2H), 4.59 (s, 2H), 3.84 (s, 3H). Anal. Calcd for C21H17FN2O3S: C, 63.63%; H, 4.32%; N, 7.07%. Found: C, 63.38%; H, 4.22%; N, 7.23%. Purity determined by HPLC (gradient B): RT = 21.59 min, purity 97.5%.
4-((5-(4-Fluorophenylcarbamoyl)pyridin-2-ylthio)methyl)benzoic acid (47)
The thionicotinamide methyl ester 46 (140 mg, 0.35 mmol) was saponified using 1 N NaOH (1.1 equiv) in MeOH (5 mL) at reflux for 1 h. The mixture was acidified using 1 N HCl and extracted into ethyl acetate. The organic layer was washed with saturated NaCl, dried over Na2SO4, filtered, and evaporated to yield 81 mg (83%) of compound 47 as a white solid. TLC (ethyl acetate/hexane 1:1): Rf = 0.23. ESI-MS: m/z = 383.0 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.55 (s, 1H), 9.01 (d, J = 1.6 Hz, 1H), 8.15 (dd, J = 8.6 Hz, 2.5 Hz, 1H), 7.92 (d, J = 8.3 Hz, 2H), 7.79–7.76 (m, 2H), 7.61 (d, J = 8.3 Hz, 2H), 7.50 (d, J = 8.2 Hz, 1H), 7.21 (t, J = 9.0 Hz, 2H), 4.59 (s, 2H), 3.84 (s, 3H).
Methyl 3-((5-(4-Fluorophenylcarbamoyl)pyridin-2-ylthio)methyl)benzoate (48)
Prepared via method B using thionicotinamide 64 and methyl 3-bromomethylbenzoate to yield 180 mg (45%) of 48 as a crystalline material. An analytical sample was recrystallized from hot ethyl acetate. ESI-MS: m/z = 397.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.40 (s, 1H), 8.99 (d, J = 1.4 Hz, 1H), 8.15 (dd, J = 8.6 Hz, 2.2 Hz, 1H), 8.07 (s, 1H), 7.85 (d, J = 8.2 Hz, 1H), 7.79–7.74 (m, 3H), 7.51–7.47 (m, 2H), 7.21 (t, J = 8.8 Hz, 2H), 4.59 (s, 2H), 3.85 (s, 3H). Anal. Calcd for C21H17FN2O3S: C, 63.63%; H, 4.32%; N, 7.07%. Found: C, 63.53%; H, 4.18%; N, 7.13%. Purity determined by HPLC (gradient B): RT = 21.57 min, purity 82.8%.
3-((5-(4-Fluorophenylcarbamoyl)pyridin-2-ylthio)methyl)benzoic acid (49)
The thionicotinamide methyl ester 48 (100 mg, 0.25 mmol) was saponified using 1 N NaOH (1.1 equiv) in MeOH at room temperature for 1 h. The mixture was acidified using 1 N HCl and extracted into ethyl acetate. The organic layer was washed with saturated NaCl, dried over Na2SO4, filtered and evaporated to yield 81 mg (83%) of compound 49 as a white solid. ESI-MS: m/z = 382.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 13.00 (br s, 1H), 10.39 (s, 1H), 9.00 (s, 1H), 8.16 (d, J = 8.6 Hz, 1H), 8.05 (s, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.78 (t, J = 7.3 Hz, 4.8 Hz, 2H), 7.71 (d, J = 7.5 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1 H), 7.46 (t, J = 7.7 Hz, 7.5 Hz, 1H), 7.21 (t, J = 7.5 Hz, 8.8 Hz, 2H), 4.59 (s, 2H). Anal. Calcd for C20H15FN2O3S: C, 62.82%; H, 3.95%; N, 7.33%. Found: C, 62.53%; H, 3.82%; N, 7.28%. Purity determined by HPLC (gradient B): RT = 19.11 min, purity 97.6%.
Methyl 2-((5-(4-Fluorophenylcarbamoyl)pyridin-2-ylthio)methyl)benzoate (50)
Prepared via method B using thionicotinamide 64 and methyl 2-bromomethylbenzoate to yield 300 mg (76%) of 50 as a white solid. ESI-MS: m/z = 397.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.40 (s, 1H), 9.00 (s, 1H), 8.13 (dd, J = 8.5 Hz, 1.8 Hz, 1H), 7.88 (d, J = 7.4 Hz, 1H), 7.80–7.77 (m, 2H), 7.66 (d, J = 7.9 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.46 (d, J = 8.6 Hz, 1H), 7.41 (t, J = 7.6 Hz, 7.1 Hz, 1H), 7.22 (t, J = 8.7 Hz, 9.0 Hz, 2H), 4.84 (s, 2H), 3.87 (s, 3H). HRMS: calcd for C21H18FN2O3S [M + 1]+ 397.1017, found 397.1006; calcd for C21H17FN2NaO3S [M + Na]+ 419.0836, found 419.0822. Purity determined by HPLC (gradient A): RT = 10.86 min, purity 95.2%.
2-((5-(4-Fluorophenylcarbamoyl)pyridin-2-ylthio)methyl)benzoic acid (51)
The thionicotinamide methyl ester 50 (100 mg, 0.25 mmol) was saponified using 1 N NaOH (1.55 mmol, 6.2 equiv) in MeOH at room temperature for 1 h. The mixture was acidified using 1 N HCl and extracted into ethyl acetate. The organic layer was washed with saturated NaCl, dried over Na2SO4, filtered and evaporated to yield 37.7 mg (40%) of compound 51 as a white solid. ESI-MS: m/z = 382.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 13.14 (s, 1H), 10.40 (s, 1H), 9.01 (s, 1H), 8.13 (dd, J = 8.7 Hz, 2.1 Hz, 1H), 7.90 (d, J = 7.5 Hz, 1H), 7.80–7.77 (m, 2H), 7.64 (d, J = 7.5 Hz, 1H), 7.50 (t, J = 6.9 Hz, 7.3 Hz, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.38 (t, J = 7.3 Hz, 1H), 7.22 (t, J = 8.8 Hz, 2H), 4.87 (s, 2H). HRMS: calcd for C20H16FN2O3S [M + 1]+ 383.0860, found 383.0852; calcd for C20H15FN2NaO3S [M + Na]+ 405.0680, found 405.0667. Purity determined by HPLC (gradient B): RT = 19.28 min, purity 98.3%.
6-(2-(2H-Tetrazol-5-yl)benzylthio)-N-(4-fluorophenyl)nicotinamide (52)
The cyanobenzyl intermediate 66 (363 mg, 1 mmol) was suspended in anhydrous toluene (25 mL). Dibutyltin oxide (25 mg, 0.1 mmol) was added, followed by trimethylsilyl azide (131 μL, 1 mmol). The mixture was then heated to reflux for 18 h. The dark yellow brown solution was allowed to cool to room temperature, at which time a brown precipitate was seen to form. The precipitate was filtered and washed with toluene to yield 190 mg (47%) of compound 52. ESI-MS: m/z = 407.2 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.39 (s, 1H), 8.93 (s, 1H), 8.11 (dd, J = 8.6 Hz, 1.6 Hz, 1H), 7.80–7.75 (m, 4H), 7.55–7.49 (m, 2H), 7.42 (d, J = 8.3 Hz, 1H), 7.22 (t, J = 8.6 Hz, 2H), 4.90 (s, 2H). HRMS: calcd for C20H16FN6OS [M + 1]+ 407.1085, found 407.1078; calcd for C20H15FN6NaOS [M + Na]+ 429.0904, found 429.0896. Purity determined by HPLC (gradient B): RT = 18.81 min, purity 94.6%.
N-(4-Fluorophenyl)-6-(2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzylthio)nicotinamide (53)
Prepared via method C using thionicotinamide 64 and 2-bromomethyl-phenylboronic acid pinacol ester, and purified by flash silica gel chromatography (step gradient of 1:6 and 1:3 ethyl acetate:hexanes) to yield 197 mg (42%) of 53 as an off-white hygroscopic solid. ESI-MS: m/z = 465.4 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.39 (s, 1H), 9.01 (s, 1H), 8.14 (d, J = 8.7 Hz, 1H), 7.80–7.77 (m, 2H), 7.68 (d, J = 7.2 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.46 (d, J = 8.3 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.26 (t, J = 7.1 Hz, 7.5 Hz, 1H), 7.21 (t, J = 8.8 Hz, 2H), 4.73 (s, 2H), 1.29 (s, 12H). Anal. Calcd for C25H26BFN2O3S: C, 64.66%; H, 5.64%; N, 6.03%. Found: C, 64.45%; H, 5.60%; N, 5.83%. Purity determined by HPLC (gradient B): RT = 24.93 min, purity 95.7%.
2-((5-(4-(Trifluoromethoxy)phenylcarbamoyl)pyridin-2-ylthio)methyl)phenylboronic acid (54)
Intermediate 68 (0.5 mmol) was reacted with 4-trifluoromethoxy aniline (177 mg, 0.5 mmol, 1 equiv) and TEA (70 mL, 0.5 mmol, 1 equiv) in anhydrous DMF. The reaction mixture was heated to 60 °C for 16 h. The crude material was diluted with ethyl acetate and the organic layer washed with water and saturated NaHCO3. The organic layer was dried over Na2SO4, filtered, and evaporated. The crude was purified using flash silica gel chromatography (ethyl acetate/hexanes 1:2 as eluent) to provide 53 mg (12%) of 54. ESI-MS: m/z = 449.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.52 (s, 1H), 9.00 (s, 1H), 8.23 (s, 2H), 8.14 (dd, J = 8.6 Hz, 1.8 Hz, 1H), 7.90–7.86 (m, 2H), 7.55 (d, J = 7.1 Hz, 1H), 7.46 (d, J = 3.8 Hz, 1H), 7.45 (d, J = 2.4 Hz, 1H), 7.40–7.37 (m, 2H), 7.30 (t, J = 7.1 Hz, 1H), 7.22 (t, J = 7.0 Hz, 1H), 4.69 (s, 2H). HRMS: calcd for C20H17BF3N2O4S [M + 1]+ 449.0949, found 449.0960. Purity determined by HPLC (gradient B): RT = 21.19 min, purity 98.3%.
4-(6-(2-Boronobenzylthio)nicotinamido)benzoic acid (55)
Following the same procedure described for 54 and starting from intermediate 68 (0.5 mmol) and 4-aminobenzoic acid (70 mg, 0.5 mmol, 1 equiv), and purified using preparative HPLC (60:40:0.1 water:acetonitrile:formic acid isocratic) to yield 23 mg (11%) of compound 55 as a white solid. ESI-MS: m/z = 409.2 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 12.79 (s, 1H), 10.61 (s, 1H), 9.00 (s, 1H), 8.23 (s, 2H), 8.15 (dd, J = 8.2 Hz, 1.7 Hz, 1H), 7.97–7.90 (m, 4H), 7.54 (d, J = 6.8 Hz, 1H), 7.47–7.44 (m, 2H), 7.30 (t, J = 6.9 Hz, 7.2 Hz, 1H), 7.22 (t, J = 8.3 Hz, 7.1 Hz, 1H), 4.69 (s, 2H). HRMS: calcd for C20H18BN2O5S [M + 1]+ 409.1024, found 409.1018; calcd for C20H17BN2NaO5S [M + Na]+ 431.0843, found 431.0837. Purity determined by HPLC (gradient B): RT = 16.61 min, purity 98.4%.
2-((5-(4-(2H-Tetrazol-5-yl)phenylcarbamoyl)pyridin-2-ylthio)methyl)phenylboronic acid (56)
Following the same procedure described for 54 and starting from intermediate 68 (0.5 mmol) and the 4-tetrazole-aniline (80 mg, 0.5 mmol, Santa Cruz Biotechnology, Dallas, TX), the crude material after workup was further purified using preparative HPLC (60:40:0.1 water:acetonitrile:formic acid isocratic) to yield 38 mg (18%) of compound 56 as a white solid. ESI-MS: m/z = 433.2 [M + H]+. 1H NMR (500 MHz, DMSO-d6) 10.63 (s, 1H), 9.02 (s, 1H), 8.23 (s, 1H), 8.16 (dd, J = 8.6 Hz, 1.8 Hz, 1H), 8.06–8.00 (m, 4H), 7.55 (d, J = 6.9 Hz, 1H), 7.46 (t, J = 8.5 Hz, 7.7 Hz, 2H), 7.31 (t, J = 7.3 Hz, 1H), 7.22 (t, J = 7.2 Hz, 1H), 4.70 (s, 2H). HRMS: calcd for C20H18BN6O3S [M + 1]+ 433.1249, found 433.1239. HPLC (gradient B): RT = 16.29 min, purity 100.0%.
2-((5-(Pyridin-4-ylcarbamoyl)pyridin-2-ylthio)methyl)phenylboronic acid (57)
Following the same procedure described for 54 and starting from intermediate 68 (0.33 mmol) and 4-amino-pyridine (31 mg, 0.33 mmol), and purified by preparative HPLC (95:5:0.1 water:acetonitrile:formic acid to 5:95:0.1 water:acetonitrile:formic acid over 30 min) to yield 18 mg (21%) of compound 57 as a white solid. ESI-MS: m/z = 366.2 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.68 (s, 1H), 9.01 (d, J = 1.7 Hz, 1H), 8.51 (d, J = 5.9 Hz, 2H), 8.24 (s, 2H), 8.15 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 8.01–7.95 (m, 1H), 7.79 (dd, J = 5.0 Hz, 1.3 Hz, 2H), 7.48–7.43 (m, 2H), 7.32–7.23 (m, 2H), 4.70 (s, 2H). HPLC (gradient B): RT = 10.79 min, purity 95.7%.
2-((5-(4-Hydroxyphenylcarbamoyl)pyridin-2-ylthio)methyl)phenylboronic acid (58)
Following the same procedure described for 54 and starting from intermediate 68 (0.33 mmol) and 4-hydroxy-aniline (36 mg, 0.33 mmol), the crude material after workup was further purified using preparative HPLC (60:40:0.1 water:acetonitrile:formic acid isocratic) to yield 21 mg (17%) of compound 58 as a white solid. ESI-MS: m/z = 381.3 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.11 (s, 1H), 9.30 (s, 1H), 8.96 (s, 1H), 8.23 (s, 1H), 8.11 (dd, J = 8.2 Hz, 1.6 Hz, 1H), 7.54–7.52 (m, 3H), 7.45–7.39 (m, 2H), 7.30 (t, J = 6.0 Hz, 7.5 Hz, 1H), 7.22 (t, J = 7.2 Hz, 1H), 6.76 (d, J = 8.9 Hz, 2H), 4.68 (s, 2H). HRMS: calcd for C19H18BN2O4S [M + 1]+ 381.1075, found 381.1093; calcd for C19H17BN2NaO4S [M + Na]+ 403.0894, found 403.0910. HPLC (gradient B): RT = 15.40 min, purity 98.8%.
2-((5-(5-Fluoropyridin-2-ylcarbamoyl)pyridin-2-ylthio)methyl)phenylboronic acid (59)
Following the same procedure described for 54 and starting from intermediate 68 (0.22 mmol) and 2-amino-5-fluoropyridine (25 mg, 0.22 mmol), the crude material after workup was further purified using preparative HPLC (60:40:0.1 water:acetonitrile:formic acid isocratic) to yield 17 mg (20%) of compound 59 as a white solid. ESI-MS: m/z = 383.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 11.08 (s, 1H), 9.02 (s, 1H), 8.42 (s, 1H), 8.23–8.16 (m, 4H), 7.82 (t, J = 8.8 Hz, 8.6 Hz, 1H), 7.54 (d, J = 7.3 Hz, 1H), 7.43 (t, J = 9.3 Hz, 10.5 Hz, 2H), 7.30 (t, J = 7.0 Hz, 7.8 Hz, 1H), 7.22 (t, J = 7.4 Hz, 7.2 Hz, 1H), 4.68 (s, 2H). HPLC (gradient B): RT = 17.24 min, purity 97.8%.
2-((5-((4-Fluorophenyl)(pyridin-2-ylmethyl)carbamoyl)pyridin-2-ylthio)methyl)phenylboronic acid (60)
According to method C, N-(4-fluorophenyl)-N-pyridin-2-yl-methyl-6-mercapto-nicotinamide intermediate 73a (2.9 g, 8.7 mmol) and 2-bromomethyl-phenylboronic acid (2.9 g, 8.7 mmol) were suspended in EtOH (100 mL). One N NaOH (8.7 mL, 8.7 mmol) was added and the suspension brought to reflux. After 2 h, the mixture was concentrated in vacuo and partioned between ethyl acetate and water. The ethyl acetate was extracted 3× with water and evaporated to yield 2.9 g (70%) of compound 60 as a white solid. ESI-MS: m/z = 473.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 8.52 (d, J = 4.4 Hz, 1H), 8.37 (s, 1H), 8.17 (s, 2H), 7.77 (t, J = 8.0 Hz, 7.4 Hz, 1H), 7.51–7.47 (m, 3H), 7.32–7.23 (m, 5H), 7.21–7.16 (m, 2H), 7.11 (t, J = 8.6 Hz, 2H), 5.15 (s, 2H), 4.56 (s, 2H). HPLC (gradient B): RT = 16.08 min, purity 95.9%.
2-((5-((4-Fluorophenyl)(pyridin-4-ylmethyl)carbamoyl)pyridin-2-ylthio)methyl)phenylboronic acid (61)
Following the same procedure described for 60 and starting from the N-(4-fluorophenyl)-N-pyridin-4-yl-methyl-6-mercapto-nicotinamide intermediate 73b (2.8 g, 8.0 mmol) and 2-bromomethyl-phenylboronic acid (1.72 g, 8.0 mmol), workup yielded 2.88 g (76%) of compound 61 as a light yellow foam. An analytical sample was purified by flash silica gel chromatography (97:3 ethyl acetate:MeOH). ESI-MS: m/z = 473.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 8.52–8.49 (m, 2H), 8.40–8.37 (m, 1H), 8.18 (s, 2H), 7.52–7.51 (m, 2H), 7.37 (d, J = 4.1 Hz, 2H), 7.33–7.17 (m, 5H), 7.14–7.10 (m, 2H), 7.06–7.03 (m, 1H), 5.12 (s, 2H), 4.56 (s, 2H). Anal. Calcd for C25H21BFN3O3S: C, 63.44%; H, 4.47%; N, 8.88%. Found: C, 63.70%; H, 4.55%; N, 8.80%. HPLC (gradient B): RT = 13.13 min, purity 96.8%.
2-((5-((4-Fluorophenyl)(piperidin-2-ylmethyl)carbamoyl)pyridin-2-ylthio)methyl)phenylboronic acid (62)
Thiol alkylation was achieved according to method C by suspending the thionicotinamide intermediate 73c (90 mg, 1 equiv) and 2-bromomethylphenyl boronic acid (50 mg, 1.1 equiv) in EtOH (1 mL). Then 1 N NaOH (0.2 mL, 1 equiv) was added and the solution gently refluxed for 2 h. Alkylated product 74a was extracted with ethyl acetate (3 × 5 mL), washed with water (3 × 5 mL), brine (3 × 5 mL), and dried over MgSO4. The organic layer was filtered to remove MgSO4, and then removed by rotary evaporation and dried in vacuo to afford 90 mg of the Boc-protected piperidine boronic acid. The Boc group was then removed by adding 4 M HCl in dioxane (1 mL) to the Boc-protected piperidine boronic acid (20 mg). The resulting piperidine compound was then purified using preparative HPLC (80:20:0.1 water:acetonitrile:formic acid to 70:30:0.1 water:acetonitrile:formic acid over 30 min) to yield 5 mg (29%) of 62 as a white solid. ESI-MS: m/z = 480.1 [M + H]+. 1H NMR (500 MHz, D2O): δ 8.35 (s, 1H), 8.08 (s, 1H), 7.41–7.37 (m, 2H), 7.28–7.15 (m, 4H), 7.05 (d, J = 7.6 Hz, 1H), 6.96–6.92 (m, 2H), 4.30 (s, 3H), 3.87 (dd, J = 14.8 Hz, 3.7 Hz, 1H), 3.38 (d, J = 10.8 Hz, 1H), 3.28 (br s, 1H), 2.88–2.84 (m, 1H), 1.78 (d, J = 7.7 Hz, 3H), 1.61–1.56 (m, 1H), 1.48–1.43 (m, 1H), 1.39–1.36 (m, 1H). HPLC (gradient B): RT = 19.29 min, purity 90.1%.
2-(6-(2-Boronobenzylthio)-N-(4-fluorophenyl)nicotinamido)acetic acid (63)
The thionicotinamide 73d (110 mg, 0.3 mmol) and 2-bromomethyl-phenylboronic acid were coupled using method B to yield the tert-butyl ester thionicotinamide intermediate 74b (138 mg, 93%). TLC (ethyl acetate): Rf = 0.6; ESI-MS: m/z = 496.9 [M + H]+. The intermediate was dissolved in 90% aq TFA and incubated at room temperature for 2 h. The TFA was removed by rotary evaporation to and the crude material purified by preparative HPLC (65:35:01 water:acetonitrile:formic acid isocratic) to yield 74 mg (61%) of compound 63 as a white hygroscopic solid. ESI-MS: m/z = 440.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 12.95 (br s, 1H), 8.37 (s, 1H), 8.30 (s, 1H), 7.53 (d, J = 5.9 Hz, 1H), 7.41–6.92 (br m, 10H), 4.57 (s, 2H), 4.46 (s, 2H). HPLC (gradient B): RT = 16.75 min, purity 96.6%.
N-(4-Fluoro-phenyl)-6-mercaptonicotinamide (64)
To 150 mL DMF was added and stirred 6-mercapto-nicotinic acid (50 mmol, 7.76 g). To the stirred solution, 4-fluoroaniline (50 mmol, 4.8 mL) was added followed by the addition of 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ, 50 mmol, 12.36 g). The dark brown mixture was stirred for 12 h. The mixture was then diluted with water (500 mL), and the precipitate collected by filtration and washed repeatedly with water. The off-white solid was dried in an oven (50 °C) for 72 h to afford 6.42 g (52%) of the thionicotinamide product. ESI-MS: m/z = 248.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 13.87 (s, 1H), 10.23 (s, 1H), 8.32 (s, 1H), 7.89 (dd, J = 8.8 Hz, 2.2 Hz, 1H), 7.74–7.71 (m, 2H), 7.38 (d, J = 9.3 Hz, 1H), 7.19 (t, J = 8.7 Hz, 2H). TLC (ethyl acetate/hexanes/MeOH 1:1:0.1): Rf = 0.7.
6-(Cyanomethylthio)-N-(4-fluorophenyl)nicotinamide (65)
Thionicotinamide 64 (248 mg. 1.00 mmol) dissolved in anhydrous DMF (3 mL) with chloroacetonitrile (63 μL, 1.0 mmol) was then added, followed by triethylamine (140 μL, 1.00 mmol). The reaction was heated to 100 °C for 30 min. After cooling to room temperature, the solution was diluted into water (60 mL), and the resulting precipitate was filtered and washed with water to yield the cyano-intermediate 65 as a white solid (259 mg, 90%). ESI-MS: m/z = 288.2 [M + H]+. 1H NMR (300 MHz, DMSO-d6): δ 10.47 (s, 1H), 9.06 (dd, J = 2.4 Hz, 0.9 Hz, 1H), 8.22 (dd, J = 8.4, 2.4 Hz, Hz, 1H), 7.78 (dd, J = 9.0 Hz, 4.8 Hz, 2H), 7.64 (dd, J = 8.4 Hz, 0.6 Hz, 1H), 7.22 (t, J = 8.7 Hz, 2 H), 4.36 (s, 2H).
6-(2-Cyanobenzylthio)-N-(4-fluorophenyl)nicotinamide (66)
Prepared via method B using thionicotinamide 64 (0.5 mmol) and α-bromo-o-tolunitrile (0.5 mmol) to yield 137 mg (75%) of 66 as an off-white solid. TLC (ethyl acetate/hexane, 1:1): Rf = 0.44. ESI-MS: m/z = 364.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.38 (s, 1H), 8.97 (d, J = 1.6 Hz, 1H), 8.11 (dd, J = 9.5 Hz, 2.1 Hz, 1H), 7.81 (d, J = 7.5 Hz, 1H), 7.75–7.68 (m, 3H), 7.62 (t, J = 7.3 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.42 (t, J = 8.3 Hz, 1H), 7.17 (t, J = 9.0 Hz, 2H), 4.64 (s, 2H).
6-(2-Boronobenzylthio)nicotinic acid (67)
6-Mercaptonicotinic acid (1.55 g, 10 mmol) and 2-bromomethyl-phenylboronic acid (2.14 g, 10 mmol) were dissolved in anhydrous DMF (20 mL), and then TEA (2.78 mL, 20 mmol) was added. The mixture was warmed to 60 °C for 1 h, and then removed from heat and let cool to room temperature. The solution was acidified with 1 N HCl and extracted with ethyl acetate. The ethyl acetate layer was washed with water, saturated NaCl, dried over Na2SO4, and evaporated to yield intermediate 67 as a yellow solid (1.54 g, 53%). TLC (AcOH/ethyl acetate/EtOH, 0.1:80:20): Rf = 0.52; ESI-MS: m/z = 290.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 13.23 (br s, 1H), 8.92 (s, 1H), 8.19 (br s, 2H), 8.05 (dd, J = 8.4 Hz, 2.0 Hz, 1H), 7.53 (d, J = 7.3 Hz, 1H), 7.43–7.36 (m, 2H), 7.29 (t, J = 7.3 Hz, 1H), 7.21 (t, J = 7.4 Hz, 1H), 4.67 (s, 2H). HPLC (gradient B): RT = 14.03 min, purity 83.4%.
6-(2-(5,5-Dimethyl-1,3,2-dioxaborinan-2-yl)benzylthio)nicotinic pivalic anhydride (68)
Intermediate 67 (290 mg, 1 mmol) and neopentyl glycol (1 mmol) were suspended in anhydrous toluene (5 mL) and set to reflux over 24 h. Without isolation, the resulting mixture containing the boronic ester was cooled in an ice bath under argon. Pivaloyl chloride (120 μL, 1 mmol) and triethylamine (140 μL, 1 mmol) were then added to the cooled solution. The reaction was allowed to proceed at the lowered temperature for 1 h, and then warmed to room temperature for an additional hour. The resulting crystalline triethylammonium salt was filtered away, and the reaction mixture concentrated by rotary evaporation. The resulting oil containing 70 was diluted with anhydrous DMF (3 mL). This solution was used without further isolation.
N-(4-Fluoro-phenyl)-6-chloro-nicotinamide (69)
4-Fluoroaniline (5.8 mL, 60 mmol) was stirred in THF (300 mL) and potassium carbonate (16.6 g, 120 mmol) was added, followed by 6-chloronicotinoyl chloride (10.6 g, 60 mmol) and stirred overnight. The salts were removed by filtration, and the organic solution was cooled in an ice bath and diluted with water while stirring. The resulting white precipitate was filtered to yield 8.3 g (55%) of 69. TLC (ethyl acetate/hexanes 1:1) Rf = 0.51. ESI-MS: m/z = 251.0 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 10.53 (s, 1H), 8.95 (d, J = 2.5 Hz, 1H), 8.37 (dd, J = 8.4 Hz, 2.5 Hz, 1H), 7.81–7.70 (m, 3H), 7.26–7.18 (m, 2H). HPLC (gradient B): RT = 13.8 min, purity 94.3%.
tert-Butyl 2-((4-fluorophenylamino)methyl)piperidine-1-carboxylate (70a)
N-Boc-2-piperidinecarbaldehyde (1.1 g, 1 eq., Combi-Blocks, San Diego, CA) and 4-fluoroaniline (481 μL, 1 equiv) were dissolved in DCE (20 mL). Under an inert atmosphere, NaBH(OAc)3 (1.5 g, 1.4 equiv) and glacial acetic acid (294 μL, 1 equiv) were added, and the reaction was monitored by TLC and LC–MS. The mixture was diluted with ethyl acetate and washed with 10% citric acid (×3), brine (×1) and dried over MgSO4. The mixture was filtered and the solvent removed by rotary evaporation and dried in vacuo to afford 1.4 g of the secondary aniline 70a (90% yield). The intermediate was carried forward without further characterization. ESI-MS: m/z = 309.1 [M + H]+.
tert-Butyl 2-(4-fluorophenylamino)acetate (70b)
A solution of 4-fluoroaniline (1.00 mL, 10 mmol) and DIPEA (1.74 mL, 10 mmol) in anhydrous DMF (10 mL) was warmed to 80 °C, and then a solution of tert-butyl bromoacetate (1.47 mL, 10 mmol) in anhydrous DMF (10 mL) was added dropwise over 1 h. After addition, the mixture was kept at 80 °C for 4 h. The mixture was then concentrated by rotary evaporation and then partitioned between ethyl acetate and water. The organic layer was washed with water and then evaporated to yield 70b as a dark brown liquid (1.96 g, 87%). A sample was purified by flash silica gel chromatography (1:10 ethyl acetate/hexanes). TLC (ethyl acetate/hexanes, 1:4) Rf = 0.31. ESI-MS: m/z = 225.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 6.94–6.90 (m, 2H), 6.55–6.52 (m, 2H), 5.85 (s, 1H), 3.75 (d, J = 4.3 Hz, 2H), 1.41 (s, 9H). HPLC (gradient B): RT = 18.78 min, purity 86.5%.
tert-Butyl 2-((6-chloro-N-(4-fluorophenyl)nicotinamido)methyl)piperidine-1-carboxylate (71a)
The secondary aniline intermediate 70a (0.5 g, 1 equiv) was stirred in DMF (5 mL) at 80 °C followed by the addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 240 μL, 1 equiv) and 6-chloronicotinoyl chloride (0.3 g, 1 equiv). The reaction was monitored by TLC and LC–MS and was stirred overnight at 80 °C. The mixture was then diluted with ethyl acetate (25 mL), washed with water, saturated NaHCO3, water, 10% citric acid, water, saturated NaCl, dried over Na2SO4, and filtered. The organic solvent was then removed by rotary evaporation and dried in vacuo to afford 0.56 g of the N-substituted N-(4-fluorophenyl)-6-chloro-nicotinamide 71a (78% yield). The intermediate was carried forward without further purification. ESI-MS: m/z = 448.2 [M + H]+.
tert-Butyl 2-(6-chloro-N-(4-fluorophenyl)nicotinamido)acetate (71b)
The disubstituted aniline 70b (0.41 g, 2.31 mmol) was coupled to 6-chloronicotinoyl chloride (0.52 g, 2.31 mmol) in anhydrous DMF (2 mL) using DBU (344 mL, 2.31 mmol). The mixture was heated to 65 °C for 48 h, diluted with ethyl acetate, and then the organic layer washed with water, saturated NaHCO3, water, 10% citric acid, water, saturated NaCl, dried over Na2SO4, and filtered. The crude material was concentrated by rotary evaporation, and purified by flash chromatography (ethyl acetate/hex 1:2) to yield 387 mg (46%) of 6-chloronicotinamide intermediate 71b as a clear oil. TLC (ethyl acetate/hexanes, 1:2) Rf = 0.28; ESI-MS: m/z = 364.9 [M + H]+. 1H NMR (300 MHz, DMSO-d6): δ 8.25 (s, 1H), 7.69 (d, J = 7.0 Hz, 1H), 7.46 (d, J = 8.7 Hz, 1H), 7.30–7.26 (m, 2H), 7.17 (t, J = 8.8 Hz, 2H), 4.48 (s, 2H), 1.42 (s, 9H). HPLC (gradient B): RT = 19.88 min, purity 83.5%.
6-Chloro-N-(4-fluorophenyl)-N-(pyridin-2-ylmethyl)nicotinamide (72a)
N-(4-fluorophenyl)-6-chloro-nicotinamide 69 (1.0 g, 4.0 mmol) and 2-bromomethyl-pyridine hydrobromide (1.01 g, 4.0 mmol) were suspended in toluene (5 mL). 50% aqueous NaOH (3.0 mL) was added to the mixture, followed by tetra-n-butyl-ammonium hydroxide (TBAH, 100 μL). The biphasic reaction mixture was vigorously stirred overnight and the aqueous layer removed by pipet. The organic layer was diluted with ethyl acetate and washed with water (3 × 25 mL) and saturated aqueous NaCl (3 × 25 mL). The water washes were back-extracted once with ethyl acetate (25 mL). The combined organic layers were dried over Na2SO4, filtered, and dried by rotary evaporation to yield 1.3 g (96%) of N-(4-fluorophenyl)-N-pyridin-2-ylmethyl-6-chloro-nicotinamide intermediate 72a as an off-white solid. ESI-MS: m/z = 341.9, 343.9 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 8.52 (d, J = 4.3 Hz, 1H), 8.35 (d, J = 2.3 Hz, 1H), 7.79–7.73 (m, 2H), 7.50 (d, J = 5.4 Hz, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.33–7.27 (m, 3H), 7.10 (t, J = 8.6 Hz, 2H), 5.16 (s, 2H).
6-Chloro-N-(4-fluorophenyl)-N-(pyridin-4-ylmethyl)nicotinamide (72b)
Following the same procedure described for 72a and starting from the N-(4-fluorophenyl)-6-chloro-nicotinamide 69 (3.0 g, 12 mmol, 1.2 equiv) and 4-bromomethyl-pyridine hydrobromide (2.5 g, 10 mmol, 1.0 equiv), workup yielded 2.5 g (74%) of the N-(4-fluorophenyl)-N-pyridin-4-ylmethyl-6-chloro-nicotinamide intermediate 72b. ESI-MS: m/z = 342.1, 344.1 [M + H]+. 1H NMR (500 MHz, DMSO-d6): δ 8.52 (s, 1H), 8.35 (s, 1H), 7.79–7.76 (m, 2H), 7.49 (br s, 1H), 7.47 (d, J = 7.9 Hz, 1H), 7.33–7.31 (m, 2H), 7.28 (t, J = 6.1 Hz, 1H), 7.10 (t, J = 8.1 Hz, 2H), 5.16 (s, 2H). HPLC (gradient B): RT = 13.45 min, purity 99.1%.
N-(4-Fluorophenyl)-6-mercapto-N-(pyridin-2-ylmethyl)nicotinamide (73a)
N-(4-fluorophenyl)-N-pyridin-2-ylmethyl-6-chloro-nicotinamide intermediate 72a (3.4 g, 10 mmol) and anhydrous sodium hydrogen sulfide (1.1 g, 20 mmol) were suspended in anhydrous DMF (30 mL). The suspension was heated to reflux, and the mixture turned a deep green color. After 2 h, the mixture was diluted with ethyl acetate (150 mL) and extracted with water. The aqueous layer was acidified with glacial AcOH to pH 6–7 and back extracted with ethyl acetate (4 × 50 mL), and the organic layers were combined, dried over Na2SO4, filtered, and evaporated by rotary evaporation to yield 2.9 g (87%) of N-(4-fluorophenyl)-N-pyridin-2-ylmethyl-6-mercapto-nicotinamide intermediate 73a as a dark yellow oil. TLC (ethyl acetate/hexanes, 2:1): Rf = 0.1; ESI-MS: m/z = 339.9 [M + H]+.
N-(4-Fluorophenyl)-6-mercapto-N-(pyridin-4-ylmethyl)nicotinamide (73b)
Following the same procedure described for 73a and starting from the N-(4-fluorophenyl)-N-pyridin-4-ylmethyl-6-chloro-nicotinamide intermediate 72b (2.5 g, 7.4 mmol), workup yielded 2.8 g (quant.) of compound crude 72b, which was carried forward without further purification. TLC (ethyl acetate/hexanes, 2:1): Rf = 0.1; ESI-MS: m/z = 339.9 [M + H]+.
tert-Butyl 2-((N-(4-fluorophenyl)-6-mercaptonicotinamido)methyl)piperidine-1-carboxylate (73c)
Thiolation of 71a was achieved by suspending the tertiary amide (0.56 g, 1 equiv) and sodium hydrogen sulfide (0.14 g, 2 equiv) in anhydrous DMF (2 mL) under an inert atmosphere. The reaction was gently refluxed for 2 h and then diluted with ethyl acetate (30 mL) and extracted with water (3 × 20 mL). The aqueous layer was then acidified with 10% citric acid and stored at 2–8 °C. The fine white precipitate was then collected by filtration to afford 90 mg of thionicotinamide intermediate 73c (16% yield). The intermediate was carried forward without further purification.
tert-Butyl 2-(N-(4-fluorophenyl)-6-mercaptonicotinamido)acetate (73d)
The 6-chloronicotinamide 71b (211 mg, 0.58 mmol) and sodium hydrogen sulfide (74 mg) were dissolved in DMF (1 mL). The mixture was heated to 85 °C for 15 min, and then allowed to cool to room temperature. The mixture was diluted in ethyl acetate, washed with water, saturated NaHCO3, water, 10% citric acid, water, saturated NaCl, and dried over Na2SO4. The crude material was filtered and evaporated to yield 123 mg (58%) of 73d as a yellow oil, which was carried forward without further purification. TLC (ethyl acetate): Rf = 0.53; ESI-MS: m/z = 362.9 [M + H]+.
Acknowledgments
This work was supported by National Institutes of Health Grant R44HL072614 (D.Y.M.) from the National Heart Lung and Blood Institute. We thank both Dr. Joel Morgan and David Nguyen (Syntrix Biosystems, Inc.) for careful review of the manuscript.
Glossary
Abbreviations Used
- CXCR1
CXC receptor 1
- CXCR2
CXC receptor 2
- CXCL1
CXC ligand 1
- CXCL8
CXC ligand 8
- DIPEA
diisopropylethylamine
- FACS
fluorescence-activated cell sorting
- C5a
complement component 5a
- fMLF
formyl-methionyl-leucyl-phenylalanine
- PAF
platelet-activating factor
- iv
intravenous
- NMM
N-methylmorpholine
- PMN
polymorphonuclear cells
- hPMN
human PMN
- RT
retention time
- SAR
structure activity relationship
- TEA
triethylamine
Author Present Address
G.F.: Neuroinflammation Discovery Performance Unit, GlaxoSmithKline R&D, Shanghai 201203, China.
Author Contributions
A.M.P. and A.D.S. contributed equally to this manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Murphy P. M.; Baggiolini M.; Charo I. F.; Hebert C. A.; Horuk R.; Matsushima K.; Miller L. H.; Oppenheim J. J.; Power C. A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 2000, 52, 145–176. [PubMed] [Google Scholar]
- Petersen F.; Flad H. D.; Brandt E. Neutrophil-activating peptides NAP-2 and IL-8 bind to the same sites on neutrophils but interact in different ways: Discrepancies in binding affinities, receptor densities, and biologic effects. J. Immunol. 1994, 152, 2467–2478. [PubMed] [Google Scholar]
- Neptune E. R.; Iiri T.; Bourne H. R. G alpha(i) is not required for chemotaxis mediated by G(i)-coupled receptors. J. Biol. Chem. 1999, 274, 2824–2828. [DOI] [PubMed] [Google Scholar]
- Jones S. A.; Moser B.; Thelen M. A Comparison of postreceptor signal-transduction events in Jurkat cells transfected with either IL-8R1 or IL-8R2 - chemokine mediated activation of P42/P44 MAP-Kinase (ERK-2). Febs Lett. 1995, 364, 211–214. [DOI] [PubMed] [Google Scholar]
- Schorr W.; Swandulla D.; Zeilhofer H. U. Mechanisms of IL-8-induced Ca2+ signaling in human neutrophil granulocytes. Eur. J. Immunol. 1999, 29, 897–904. [DOI] [PubMed] [Google Scholar]
- Kurdowska A.; Noble J. M.; Grant I. S.; Robertson C. R.; Haslett C.; Donnelly S. C. Anti-interleukin-8 autoantibodies in patients at risk for acute respiratory distress syndrome. Crit. Care Med. 2002, 30, 2335–2337. [DOI] [PubMed] [Google Scholar]
- Erdem H.; Pay S.; Serdar M.; Simsek I.; Dinc A.; Musabak U.; Pekel A.; Turan M. Different ELR(+) angiogenic CXC chemokine profiles in synovial fluid of patients with Behcet’s disease, familial Mediterranean fever, rheumatoid arthritis, and osteoarthritis. Rheumatol. Int. 2005, 26, 162–167. [DOI] [PubMed] [Google Scholar]
- Lally F.; Smith E.; Filer A.; Stone M. A.; Shaw J. S.; Nash G. B.; Buckley C. D.; Rainger G. E. A novel mechanism of neutrophil recruitment in a coculture model of the rheumatoid synovium. Arthritis Rheum. 2005, 52, 3460–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich K.; Garbe C.; Blaschke V.; Maurer C.; Middel P.; Westphal G.; Lippert U.; Neumann C. Response of psoriasis to interleukin-10 is associated with suppression of cutaneous type 1 inflammation, downregulation of the epidermal interleukin-8/CXCR2 pathway and normalization of keratinocyte maturation. J. Invest. Dermatol. 2001, 116, 319–329. [DOI] [PubMed] [Google Scholar]
- Banks C.; Bateman A.; Payne R.; Johnson P.; Sheron N. Chemokine expression in IBD. Mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn’s disease. J. Pathol. 2003, 199, 28–35. [DOI] [PubMed] [Google Scholar]
- Beeh K. M.; Kornmann O.; Buhl R.; Culpitt S. V.; Giembycz M. A.; Barnes P. J. Neutrophil chemotactic activity of sputum from patients with COPD: Role of interleukin 8 and leukotriene B-4. Chest 2003, 123, 1240–1247. [DOI] [PubMed] [Google Scholar]
- Keatings V. M.; Collins P. D.; Scott D. M.; Barnes P. J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 530–534. [DOI] [PubMed] [Google Scholar]
- Chapman R. W.; Phillips J. E.; Hipkin R. W.; Curran A. K.; Lundell D.; Fine J. S. CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol. Ther. 2009, 121, 55–68. [DOI] [PubMed] [Google Scholar]
- Donnelly L. E.; Barnes P. J. Chemokine receptor CXCR2 antagonism to prevent airways inflammation. Drugs Future 2011, 36, 465–472. [Google Scholar]
- Clarke M.; Jamieson T.; Steele C.; Olson M.; Samuel M.; Das S.; Sansom O.; Nibbs R. Inhibition of neutrophil chemokine receptor CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. Immunology 2012, 137, 190–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L.; Cumpian A. M.; Caetano M. S.; Ochoa C. E.; De la Garza M. M.; Lapid D. J.; Mirabolfathinejad S. G.; Dickey B. F.; Zhou Q. H.; Moghaddam S. J. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol. Cancer 2013, 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L.; Jiang B.; Wu H.; Wang X. D.; Tang X. J.; Huang J. F.; Zhu J. High expression of CXCR2 is associated with tumorigenesis, progression, and prognosis of laryngeal squamous cell carcinoma. Med. Oncol. 2012, 29, 2466–2472. [DOI] [PubMed] [Google Scholar]
- Jamieson T.; Clarke M.; Steele C. W.; Samuel M. S.; Neumann J.; Jung A.; Huels D.; Olson M. F.; Das S.; Nibbs R. J. B.; Sansom O. J. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Invest. 2012, 122, 3127–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh H.; Wang D. Z.; Daikoku T.; Sun H. Y.; Dey S. K.; DuBois R. N. CXCR2-Expressing Myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157. [DOI] [PubMed] [Google Scholar]
- Busch-Petersen J. Small molecule antagonists of the CXCR2 and CXCR1 chemokine receptors as therapeutic agents for the treatment of inflammatory diseases. Curr. Top. Med. Chem. 2006, 6, 1345–1352. [DOI] [PubMed] [Google Scholar]
- Allegretti M.; Bertini R.; Cesta M. C.; Bizzarri C.; Di Bitondo R.; Di Cioccio V.; Galliera E.; Berdini V.; Topai A.; Zampella G.; Russo V.; Di Bello N.; Nano G.; Nicolini L.; Locati M.; Fantucci P.; Florio S.; Colotta F. 2-Arylpropionic CXC chemokine receptor 1 (CXCR1) ligands as novel noncompetitive CXCL8 inhibitors. J. Med. Chem. 2005, 48, 4312–4331. [DOI] [PubMed] [Google Scholar]
- Bertini R.; Allegretti M.; Bizzarri C.; Moriconi A.; Locati M.; Zampella G.; Cervellera M. N.; Di Cioccio V.; Cesta M. C.; Galliera E.; Martinez F. O.; Di Bitondo R.; Troiani G.; Sabbatini V.; D’Anniballe G.; Anacardio R.; Cutrin J. C.; Cavalieri B.; Mainiero F.; Strippoli R.; Villa P.; Di Girolamo M.; Martin F.; Gentile M.; Santoni A.; Corda D.; Poli G.; Mantovani A.; Ghezzi P.; Colotta F. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: Prevention of reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 11791–11796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J. R.; Lee J. M.; Young P. R.; Hertzberg R. P.; Jurewicz A. J.; Chaikin M. A.; Widdowson K.; Foley J. J.; Martin L. D.; Griswold D. E.; Sarau H. M. Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J. Biol. Chem. 1998, 273, 10095–10098. [DOI] [PubMed] [Google Scholar]
- Podolin P. L.; Bolognese B. J.; Foley J. J.; Schmidt D. B.; Buckley P. T.; Widdowson K. L.; Jin Q.; White J. R.; Lee J. M.; Goodman R. B.; Hagen T. R.; Kajikawa O.; Marshall L. A.; Hay D. W.; Sarau H. M. A potent and selective nonpeptide antagonist of CXCR2 inhibits acute and chronic models of arthritis in the rabbit. J. Immunol. 2002, 169, 6435–6444. [DOI] [PubMed] [Google Scholar]
- Traves S. L.; Smith S. J.; Barnes P. J.; Donnelly L. E. Specific CXC but not CC chemokines cause elevated monocyte migration in COPD: A role for CXCR2. J. Leukocyte Biol. 2004, 76, 441–450. [DOI] [PubMed] [Google Scholar]
- Dwyer M. P.; Yu Y.; Chao J.; Aki C.; Biju P.; Girijavallabhan V.; Rindgen D.; Bond R.; Mayer-Ezel R.; Jakway J.; Hipkin R. W.; Fossetta J.; Gonsiorek W.; Bian H.; Fan X.; Terminelli C.; Fine J.; Lundell D.; Merritt J. R.; Rokosz L. L.; Kaiser B.; Li G.; Wang W.; Stauffer T.; Ozgur L.; Baldwin J.; Taveras A. G. Discovery of 2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methylfuran-2-yl)propyl]amino]-3,4-dioxocyclobut-1-enylamino}benzamide (SCH 527123): A potent, orally bioavailable CXCR2/CXCR1 receptor antagonist. J. Med. Chem. 2006, 49, 7603–7606. [DOI] [PubMed] [Google Scholar]
- Aul R.; Patel S.; Summerhill S.; Kilty I.; Plumb J.; Singh D. LPS challenge in healthy subjects: An investigation of neutrophil chemotaxis mechanisms involving CXCR1 and CXCR2. Int. Immunopharmacol. 2012, 13, 225–231. [DOI] [PubMed] [Google Scholar]
- Lazaar A. L.; Sweeney L. E.; MacDonald A. J.; Alexis N. E.; Chen C.; Tal-Singer R. SB-656933, a novel CXCR2 selective antagonist, inhibits ex vivo neutrophil activation and ozone-induced airway inflammation in humans. Br. J. Clin. Pharmacol. 2011, 72, 282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss R. B.; Mistry S. J.; Konstan M. W.; Pilewski J. M.; Kerem E.; Tal-Singer R.; Lazaar A. L.; Investigators C. F. Safety and early treatment effects of the CXCR2 antagonist SB-656933 in patients with cystic fibrosis. J. Cystic Fibrosis 2013, 12, 241–248. [DOI] [PubMed] [Google Scholar]
- Holz O.; Khalilieh S.; Ludwig-Sengpiel A.; Watz H.; Stryszak P.; Soni P.; Tsai M.; Sadeh J.; Magnussen H. SCH527123, a novel CXCR2 antagonist, inhibits ozone-induced neutrophilia in healthy subjects. Eur. Respir. J. 2010, 35, 564–570. [DOI] [PubMed] [Google Scholar]
- Dwyer M. P.; Yu Y. N. CXCR2 modulators: a patent review (2009–2013). Expert Opin. Ther. Pat. 2014, 24, 519–534. [DOI] [PubMed] [Google Scholar]
- Virtala R.; Ekman A. K.; Jansson L.; Westin U.; Cardell L. O. Airway inflammation evaluated in a human nasal lipopolysaccharide challenge model by investigating the effect of a CXCR2 inhibitor. Clin. Exp. Allergy 2012, 42, 590–596. [DOI] [PubMed] [Google Scholar]
- Cutshall N. S.; Kucera K. A.; Ursino R.; Latham J.; Ihle N. C. Nicotinanilides as inhibitors of neutrophil chemotaxis. Bioorg. Med. Chem. Lett. 2002, 12, 1517–1520. [DOI] [PubMed] [Google Scholar]
- Maeda D. Y.; Quinn M. T.; Schepetkin I. A.; Kirpotina L. N.; Zebala J. A. Nicotinamide glycolates antagonize CXCR2 activity through an intracellular mechanism. J. Pharmacol. Exp. Ther. 2010, 332, 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb K. S.; Levy D. A facile oxidation of boronic acids and boronic esters. Tetrahedron Lett. 1995, 36, 5117–5118. [Google Scholar]
- Wittenberger S. J.; Donner B. G. Dialkyltin oxide mediated addition of trimethylsilyl azide to nitriles. A novel preparation of 5-substituted tetrazoles. J. Org. Chem. 1993, 58, 4139–4141. [Google Scholar]
- Hunter P. Not boring at all Boron is the new carbon in the quest for novel drug candidates. EMBO Rep. 2009, 10, 125–U110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. Q.; Gao X. M.; Wang B. H. Boronic acid compounds as potential pharmaceutical agents. Med. Res. Rev. 2003, 23, 346–368. [DOI] [PubMed] [Google Scholar]
- Hall D. G.Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; WILEY-VCH: Weinheim, 2005. [Google Scholar]
- Cutshall N. S.; Ursino R.; Kucera K. A.; Latham J.; Ihle N. C. Nicotinamide N-oxides as CXCR2 antagonists. Bioorg. Med. Chem. Lett. 2001, 11, 1951–1954. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nat. Rev. Drug Discovery 2002, 1, 198–210. [Google Scholar]
- de Kruijf P.; van Heteren J.; Lim H. D.; Conti P. G.; van der Lee M. M.; Bosch L.; Ho K. K.; Auld D.; Ohlmeyer M.; Smit M. J.; Wijkmans J. C.; Zaman G. J.; Smit M. J.; Leurs R. Nonpeptidergic allosteric antagonists differentially bind to the CXCR2 chemokine receptor. J. Pharmacol. Exp. Ther. 2009, 329, 783–790. [DOI] [PubMed] [Google Scholar]
- Mueller S. G.; White J. R.; Schraw W. P.; Lam V.; Richmond A. Ligand-induced desensitization of the human CXC chemokine receptor-2 is modulated by multiple serine residues in the carboxyl-terminal domain of the receptor. J. Biol. Chem. 1997, 272, 8207–8214. [DOI] [PubMed] [Google Scholar]
- Shyamala V.; Khoja H. Interleukin-8 receptors R1 and R2 activate mitogen-activated protein kinases and induce c-fos, independent of Ras and Raf-1 in Chinese hamster ovary cells. Biochemistry 1998, 37, 15918–15924. [DOI] [PubMed] [Google Scholar]
- Hammond M. E. W.; Lapointe G. R.; Feucht P. H.; Hilt S.; Gallegos C. A.; Gordon C. A.; Giedlin M. A.; Mullenbach G.; Tekampolson P. IL-8 induces neutrophil chemotaxis predominantly via type-I IL-8 receptors. J. Immunol. 1995, 155, 1428–1433. [PubMed] [Google Scholar]
- Posadas I.; Terencio M. C.; Guillen I.; Ferrandiz M. L.; Coloma J.; Paya M.; Alcaraz M. J. Co-regulation between cyclo-oxygenase-2 and inducible nitric oxide synthase expression in the time-course of murine inflammation. N-S Arch. Pharmacol. 2000, 361, 98–106. [DOI] [PubMed] [Google Scholar]
- Moriconi A.; Cesta M. C.; Cervellera M. N.; Aramini A.; Coniglio S.; Colagioia S.; Beccari A. R.; Bizzarri C.; Cavicchia M. R.; Locati M.; Galliera E.; Di Benedetto P. Vigilante, P.; Bertini, R.; Allegretti, M. Design of noncompetitive interleukin-8 inhibitors acting on CXCR1 and CXCR2. J. Med. Chem. 2007, 50, 3984–4002. [DOI] [PubMed] [Google Scholar]
- Gonsiorek W.; Fan X.; Hesk D.; Fossetta J.; Qiu H.; Jakway J.; Billah M.; Dwyer M.; Chao J.; Deno G.; Taveras A.; Lundell D. J.; Hipkin R. W. Pharmacological characterization of Sch527123, a potent allosteric CXCR1/CXCR2 antagonist. J. Pharmacol. Exp. Ther. 2007, 322, 477–485. [DOI] [PubMed] [Google Scholar]
- Jones S. A.; Wolf M.; Qin S.; Mackay C. R.; Baggiolini M. Different functions for the interleukin 8 receptors (IL-8R) of human neutrophil leukocytes: NADPH oxidase and phospholipase D are activated through IL-8R1 but not IL-8R2. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6682–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser T.; Dewald B.; Ehrengruber M. U.; Clarklewis I.; Baggiolini M. The interleukin-8-related chemotactic cytokines GRO-alpha, GRO-beta, and GRO-gamma activate human neutrophil and basophil leukocytes. J. Biol. Chem. 1993, 268, 15419–15424. [PubMed] [Google Scholar]
- Kaur M.; Singh D. Neutrophil chemotaxis caused by chronic obstructive pulmonary disease alveolar macrophages: The role of CXCL8 and the receptors CXCR1/CXCR2. J. Pharmacol. Exp. Ther. 2013, 347, 173–180. [DOI] [PubMed] [Google Scholar]
- May L. T.; Avlani V. A.; Sexton P. M.; Christopoulos A. Allosteric modulation of G protein-coupled receptors. Curr. Pharm. Design 2004, 10, 2003–2013. [DOI] [PubMed] [Google Scholar]
- Bradley M. E.; Bond M. E.; Manini J.; Brown Z.; Charlton S. J. SB265610 is an allosteric, inverse agonist at the human CXCR2 receptor. Br. J. Pharmacol. 2009, 158, 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D. J.; Tomkinson N. P.; Wiley K. E.; Brammall A.; Bowers L.; Grahames C.; Gaw A.; Meghani P.; Shelton P.; Wright T. J.; Mallinder P. R. Identification of a putative intracellular allosteric antagonist binding-site in the CXC chemokine receptors 1 and 2. Mol. Pharmacol. 2008, 74, 1193–1202. [DOI] [PubMed] [Google Scholar]
- de Kruijf P.; Lim H. D.; Roumen L.; Renjaan V. A.; Zhao J.; Webb M. L.; Auld D. S.; Wijkmans J. C.; Zaman G. J.; Smit M. J.; de Graaf C.; Leurs R. Identification of a novel allosteric binding site in the CXCR2 chemokine receptor. Mol. Pharmacol. 2011, 80, 1108–1118. [DOI] [PubMed] [Google Scholar]
- Onuffer J. J.; Horuk R. Chemokines, chemokine receptors and small-molecule antagonists: Recent developments. Trends Pharmacol. Sci. 2002, 23, 459–467. [DOI] [PubMed] [Google Scholar]
- Groll M.; Berkers C. R.; Ploegh H. L.; Ovaa H. Crystal Structure of the Boronic Acid-Based Proteasome Inhibitor Bortezomib in Complex with the Yeast 20S Proteasome. Structure 2006, 14, 451–456. [DOI] [PubMed] [Google Scholar]
- Hu G. Q.; Lin G.; Wang M.; Dick L.; Xu R. M.; Nathan C.; Li H. L. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol. Microbiol. 2006, 59, 1417–1428. [DOI] [PubMed] [Google Scholar]
- Tsilikounas E.; Kettner C. A.; Bachovchin W. W. B-11 NMR-spectroscopy of peptide boronic acid inhibitor complexes of alpha-lytic protease: Direct evidence for tetrahedral boron in both boron histidine and boron serine adduct complexes. Biochemistry 1993, 32, 12651–12655. [DOI] [PubMed] [Google Scholar]
- Farrjones S.; Smith S. O.; Kettner C. A.; Griffin R. G.; Bachovchin W. W. Crystal versus solution structure of enzymes: NMR-spectroscopy of a peptide boronic acid-serine protease complex in the crystalline state. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 6922–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin W. W.; Wong W. Y. L.; Farrjones S.; Shenvi A. B.; Kettner C. A. N-15 NMR-spectroscopy of the catalytic-triad histidine of a serine protease in peptide boronic acid inhibitor complexes. Biochemistry 1988, 27, 7689–7697. [DOI] [PubMed] [Google Scholar]
- Bone R.; Shenvi A. B.; Kettner C. A.; Agard D. A. Serine protease mechanism: Structure of an inhibitory complex of alpha-lytic protease and a tightly bound peptide boronic acid. Biochemistry 1987, 26, 7609–7614. [DOI] [PubMed] [Google Scholar]
- Siemsen D. W.; Schepetkin I. A.; Kirpotina L. N.; Lei B.; Quinn M. T. Neutrophil isolation from nonhuman species. Methods Mol. Biol. 2007, 412, 21–34. [DOI] [PubMed] [Google Scholar]
- Hall D. A.; Beresford I. J.; Browning C.; Giles H. Signalling by CXC-chemokine receptors 1 and 2 expressed in CHO cells: A comparison of calcium mobilization, inhibition of adenylyl cyclase and stimulation of GTPgammaS binding induced by IL-8 and GROalpha. Br. J. Pharmacol. 1999, 126, 810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]