

Synovial sarcoma is an aggressive sarcoma subtype characterized by a recurrent translocation t(X;18). We report here that the genomic complexity assessed by comparative genomic hybridization is strongly associated with outcome independently of response to neoadjuvant chemotherapy.
Keywords: synovial sarcoma, genomic index, prognosis, chemotherapy
Abstract
Background
Synovial sarcoma (SS) is an aggressive soft-tissue tumor. Despite being considered as a chemosensitive disease, the real impact of perioperative chemotherapy on metastasis-free survival (MFS) is controversial. We have shown that metastatic relapse of SS is strongly associated with genomic complexity. There are no data regarding the potential correlation between genomic complexity and response to chemotherapy.
Patients and methods
The study population included 65 SS patients diagnosed between 1991 and 2013 and with available tissue material. Genomic profiling was carried out by using array-CGH. Forty-five SS out of the 65 patients were treated with neoadjuvant anthracycline/ifosfamide-based chemotherapy. Radiological response was assessed according to RECIST criteria. Histological response was defined by the percentage of recognizable tumor cells on the surgical specimen.
Results
Genomic complexity was significantly associated with MFS. However, there was no statistically significant association between radiological or histological response and genomic complexity.
Conclusion
The absence of significant association between response to chemotherapy and genomic complexity suggests that the prognostic value of chromosome instability in SS is independent of response to chemotherapy; mechanisms leading to metastatic relapse of SS are intrinsic to the biology of the tumor and current cytotoxic drugs are only poorly efficient to prevent it.
introduction
Synovial sarcoma (SS) is an aggressive soft-tissue sarcoma (STS), accounting for 6%–9% [1] of STS.
Surgery plus radiotherapy remains the cornerstone of treatment of localized SS [2]. The use of perioperative chemotherapy is controversial in this setting with several studies showing contradictory results in terms of survival benefit [3–7].
SS is characterized by a specific translocation t(X;18) (p11.2;q11.2) that occurs in more than 95% of patients and leads to two main chimeric fusion genes, SYT-SSX1 or SYT-SSX2 [8]. We have recently reported that a genomic biomarker—the Genomic Index (GI)—that is based on the number and type of chromosomal aberrations was a prognostic factor for metastasis-free survival (MFS) in pediatric and adult SS [9]. In that series, 82% of children and 42% of adults received perioperative chemotherapy in order to decrease their risk of metastatic relapse. The main issue with perioperative chemotherapy of SS patients is the identification of predictive factors of response to chemotherapy that could avoid for a majority of them ineffective and potentially toxic treatment with impaired quality of life. We hypothesized that GI was predictive of response to chemotherapy and represented a potential biomarker to select patients who were likely to benefit from perioperative chemotherapy. To address this point, we decided to analyze a new series of SS with a majority of them treated in the neoadjuvant setting. Indeed, compared with adjuvant chemotherapy, neoadjuvant chemotherapy allows assessment of tumor response and identification of in vivo tumor sensitivity and represents therefore an ideal setting to identify molecular predictive factors of sensitivity to chemotherapy by correlating tumor response with genetic profile.
patients and methods
tumor samples
Sixty-five patients (25 children and 40 adults) were included in this study. Inclusion criteria were as follows: (i) diagnosis of localized SS according to the World Health Organization Classification of Tumors between 1987 and 2013 [1]; (ii) availability of primary tumor material for molecular analysis obtained before any treatment procedure; (iii) availability of follow-up data for statistical correlations. Patients were from eight European institutions that agreed to participate in this study. Clinical, pathologic and radiological data were collected by reviewing medical records at each institution and were then entered into a comprehensive database. The histological diagnosis was reviewed for all specimens by two experts. The specific translocation t(X;18) was present in all cases (confirmed either by FISH or by RT-qPCR). The histological grade was determined as previously described according to the Fédération Nationale des Centres de Lutte Contre le Cancer grading system [10].
Among the previous 65 cases, a subset of 43 adult and pediatric patients with localized SS treated with neoadjuvant anthracycline/ifosfamide-based chemotherapy was tested for response to neoadjuvant chemotherapy. Radiological response to neoadjuvant chemotherapy was assessed by RECIST 1.0 criteria [11]. To these 43 patients, we added another 2 patients from the previous series [9] who received anthracyclines/ifosfamide-based chemotherapy and whose response was assessable according to RECIST 1.0 criteria.
Patients and tumor characteristics of all patients are described in Table 1.
Table 1.
Patients characteristics
| N | % | |
|---|---|---|
| (a) Characteristics of overall population n = 65 | ||
| Age (overall population), years | ||
| Median | 37 | |
| Range | 5–73 | |
| Age (pediatric population), years (n = 24) | ||
| Median | 14 | |
| Range | 4–18 | |
| Gender | ||
| Male | 37 | 56.9 |
| Female | 28 | 43.1 |
| Location | ||
| Extremities | 38 | 58.5 |
| Others | 27 | 41.5 |
| Grade | ||
| 2 | 28 | 43.0 |
| 3 | 31 | 47.5 |
| Unknown | 6 | 9.5 |
| Tumor size, cm | ||
| >5 | 43 | 66.0 |
| ≤5 | 18 | 27.5 |
| Unknown | 4 | 6.5 |
| Adjuvant radiotherapy | ||
| Yes | 41 | 63.0 |
| No | 14 | 37.0 |
| (b) Characteristics of patients treated with neoadjuvant chemotherapy (n = 45) | ||
| Age (overall population), years | ||
| Median | 27 | |
| Range | 4–64 | |
| Age (pediatric population), years (n = 14) | ||
| Median | 14 | |
| Range | 4–18 | |
| Gender | ||
| Male | 27 | 60.0 |
| Female | 18 | 40.0 |
| Location | ||
| Extremities | 24 | 53.5 |
| Others | 21 | 46.5 |
| Grade | ||
| 2 | 17 | 38.0 |
| 3 | 23 | 51.0 |
| Unknown | 5 | 11.0 |
| Tumor size, cm | ||
| >5 | 30 | 66.5 |
| ≤5 | 11 | 24.5 |
| Unknown | 4 | 9.0 |
| Number of cycles of chemotherapy | ||
| Median | 4 | |
| Range | 2–7 | |
Ethical approval from the board of Institut Bergonié was obtained. The tumor samples were anonymized and centralized in the Biological Resources Center of Institut Bergonié (CRB-IB), which received the agreement from the French authorities to deliver samples for scientific research (number AC-2008-812).
array-CGH analysis
Genomic DNA was extracted from paraffin-embedded tumors according to Agilent protocol for DNA isolation on formalin-fixed, paraffin-embedded (FFPE) tissues (http://www.chem.agilent.com/cag/prod/dn/G4410-90020_CGH_Protocol_FFPE1_0.pdf) (Agilent Technologies).
DNA was then treated using a DNase as previously described [12] and hybridized to 8 × 60 K whole-genome Agilent arrays (G4450A) as previously described [13]. Microarray slides were scanned using an Agilent DNA microarray scanner; images were analyzed by Feature Extraction V10.1.1.1 and then analyzed by Agilent Genomic Workbench Lite Edition 6.5.0.18 (Agilent). The ADM-2 algorithm was used to identify DNA copy-number anomalies at the probe level. A low-level copy-number gain was defined as a log2 ratio >0.25 and a copy-number loss was defined as a log2 ratio <−0.25.
The GI was calculated and applied for each profile as previously described [13]: GI = A2/C, where A is the total number of alterations (segmental gains and losses) and C is the number of involved chromosomes. Profiles were sorted into two different groups: cases where no alteration was present (even CGH profile) corresponded to the GI1 group, cases presenting one alteration or more (rearranged CGH profile) formed the GI2 group.
evaluation of response to neoadjuvant chemotherapy
Radiological response to chemotherapy was assessed according to RECIST 1.0 criteria [11] based on central review of imaging or radiological reports when imaging data were no longer available (n = 12). Histological response was arbitrarily defined by the percentage of recognizable tumor cells on the surgical specimen after central review by two experts: good histological response was defined as a percentage of tumor cells remaining after neoadjuvant chemotherapy ≤10%, intermediate histological response was 10%–50% of residual tumor cells and poor histological response was defined when there were 50% or more recognizable tumor cells after chemotherapy.
We defined overall response a composite criteria taking into account radiological and histological responses. Good overall response was defined as partial response (PR) or stable disease (SD) according to RECIST 1.0 and ≤10% tumor cells. Poor response was defined as SD or progressive disease according to RECIST 1.0 and ≥50% of tumor cells. All the other cases were defined as intermediate response. The cutoff for proportions of tumor cells to define response to chemotherapy were chosen based on the grading of Salzer-Kuntschik for bone sarcomas [14].
statistical analysis
The statistical analysis of baseline demographics and clinical outcome are based on all data available up to the cutoff date of 30 March 2013. MFS was defined as the interval between diagnosis and the time of distant recurrence or the last follow-up. Survival rates were estimated with the use of the Kaplan–Meier method and compared using the log-rank test. Descriptive statistics were used to show the distribution of variables in the population. All statistical tests were two sided, and P < 0.05 indicated statistical significance. All statistical analyses were carried out using the R statistical environment (R version 2.14.1).
results
prognostic value of GI
Among the 65 CGH profiles obtained, 18 (27.7%) were even and 47 (72.3%) were rearranged. We found a significant difference in terms of proportion of rearranged profiles between adult and pediatric patients: 14 children (56%) and 33 adults (82.5%) presented a rearranged profile [odds ratio 3.6227; 95% confidence interval (CI) 1.0362–13.6058; P = 0.03]. We identified several recurrent genomic aberrations (occurring in >20% of cases of rearranged profiles): gains in chromosome 12 (region 12q13.12–12q14.1 in 35% of cases), chromosome 8 (28%), chromosome 9qter (26%), chromosome 17 (22%) and chromosome 19 (22%), and losses in chromosome 3 (24%), chromosome 10 (22%) and chromosome 11 (22%). There were no significant difference in terms of grade, localization, cumulative dose of anthracyclines and ifosfamide between patients with high GI and low GI.
To take into account the number and the type of changes in rearranged profiles, we applied the previously described GI with scores ranging from 0 to 127 across the entire series. Stratification by GI at a cutoff of 1 splits the tumors into two groups with a significant different outcome in terms of MFS (hazard ratio 3.79; 95% CI 1.13–12.76; P = 0.02) (Figure 1) .
Figure 1.
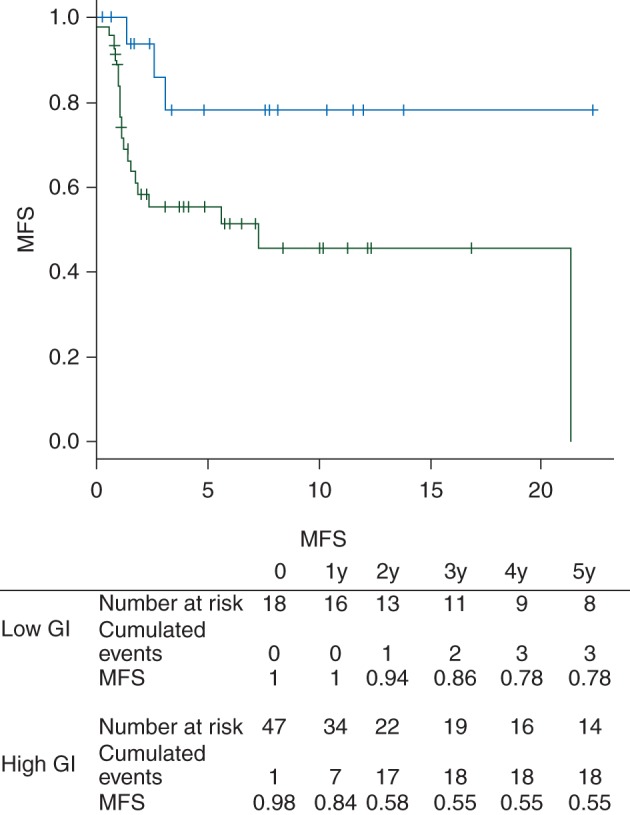
Metastasis-free survival according to Genomic Index (blue curve: low genomic index, green curve: high genomic index).
is genomic complexity associated to response to chemotherapy?
Forty-five patients had been treated with neoadjuvant chemotherapy. Twelve (26.7%) had an even genomic profile whereas 33 (73.3%) had a rearranged profile.
All the patients had evaluable radiological response. According to RECIST 1.0, 19 patients (43%) experienced PR and 26 (57%) had SD (Table 2). Their respective proportions according to GI were not significantly different (P = 0.49) (Figure 2).
Table 2.
Patterns of response to neoadjuvant chemotherapy (n = 45)
| Radiological response (RECIST 1.0) | |||
|---|---|---|---|
| Partial response | Stable disease | Total | |
| Histological response (% recognizable tumor cells) | |||
| <10 | 2 | 2 | 4 |
| 10–50 | 5 | 3 | 8 |
| >50 | 8 | 19 | 27 |
| N/A | 4 | 2 | 6 |
| Total | 19 | 26 | 45 |
N/A, not assessable.
Figure 2.
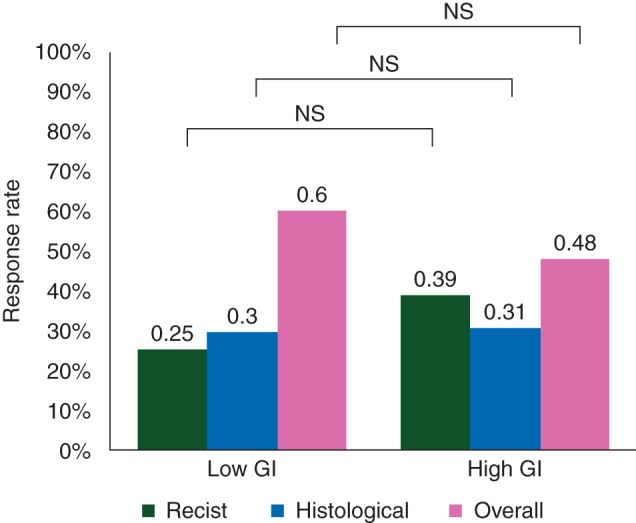
Radiological, histological and overall response to chemotherapy according to Genomic Index (GI).
Histological response was assessable for 39 patients. Four patients (10.2%) experienced good response, 8 (20.5%) intermediate response and 27 (69.3%) poor response (69.3%) (Table 2). As observed for radiological response, the proportions of responders and nonresponders were not statistically different according to GI (P = 1) (Figure 2).
Altogether, 4 patients of 39 (10.2%) had a good overall response (histological and radiological, see materials and methods), 16 (41%) had an intermediate overall response and 19 (48.8%) had a poor overall response (Table 2) without any correlation with GI (P = 0.72) (Figure 2).
Response to chemotherapy whatever the criteria used (radiological or overall) was not correlated with MFS. MFS was not different between patients with high GI having objective response according to RECIST versus SD (P = 0.77) as well as between patients having good or intermediate overall response versus poor overall response (P = 0.96).
Our data showing no association between GI and overall response to neoadjuvant chemotherapy, we reanalyzed the series for recurrent genomic aberrations that could be associated with nonresponse or response to chemotherapy (Figure 3).
Figure 3.
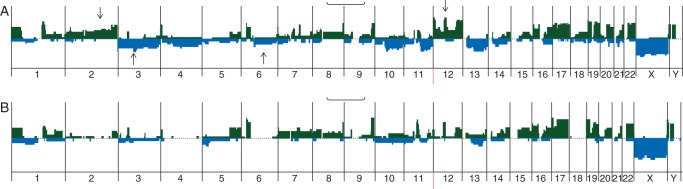
Recurrent genomic aberrations identified by comparative genomic hybridization in synovial sarcoma patients treated with neoadjuvant chemotherapy. (A) Patients with good and intermediate overall response to chemotherapy. (B) Patients with poor overall response to chemotherapy. Arrows indicate aberrations present in more than 20% of cases.
We found a trend for a higher incidence for gain in chromosomes 2 (found in 50% of patients with good or intermediate overall response versus 7% of poor responders and 12 (found in 50% of patients with good or intermediate overall response versus 7% of poor responders and deletion of chromosomes 3 (found in 50% of patients with good or intermediate overall response versus 13% of poor responders and 6 (found in 29% of patients with good or intermediate overall response versus 0% of poor responders).
discussion
We confirmed here the prognostic impact of genomic complexity in SS and the association between age and genomic complexity as we have previously reported [9].
Despite the fact that SS is often considered as a chemosensitive disease [15–20], the radiological objective response rate we observed was similar to that previously reported in miscellaneous soft-tissue sarcomas (STSs) [21]. Previous studies evaluating radiological response of SS to neoadjuvant chemotherapy were mainly limited pediatric series and showed objective response rate ranging between 20% and 65% [22–24]. None of them have assessed histological response, the prognostic value of which being still a matter of debate for STS.
Whatever the criteria used (radiological or histological), response to chemotherapy was not associated with outcome. This confirms the data we have reported recently showing no prognostic impact of response to neoadjuvant chemotherapy in a series of 93 SS patients [25]. Moreover, we did not found any correlation between genomic complexity and response to neoadjuvant chemotherapy whatever the criteria used. This demonstrates that GI can be considered as a prognostic biomarker rather than a predictive marker of response to chemotherapy. This also suggests that metastatic relapse of SS is more influenced by the biology of the tumor than by response to the current drugs used in the perioperative setting. Therefore, the real benefit of perioperative chemotherapy is questionable particularly in patients with a low GI such as the majority of children with SS. A histology-specific randomized trial with a stratification based on GI would be the best way to clarify the role of chemotherapy in this setting. However, SS sarcoma is a rare disease representing 80–100 new cases diagnosed yearly in a country of 65 million inhabitants such as France. Therefore, the feasibility of such a trial is clearly compromised by the difficulties to reach a reasonable accruement goal.
Although the GI was not predictive of chemotherapy efficacy, our study highlighted some genomic alterations that were significantly associated with overall response to chemotherapy, i.e. gains in chromosomes 2 and 12 and losses in chromosomes 3 and 6 are overrepresented in the group of patients with good and intermediate overall response. Even and rearranged profiles being found in both the group of patients with good or intermediate overall response and the group with poor overall response; we can conclude that factors predicting response to chemotherapy are beyond CGH resolution. Nevertheless, the overrepresentation of distinct rearrangements in both groups might imply that these regions or chromosomes include genes that could be involved in response to chemotherapy. Expression profiling using RNAseq and/or exome sequencing of these tumors could allow targeting these genes and by the way setting up a signature predictive of response to ifosfamide–adriamycin chemotherapy.
Our study confirmed that good-quality CGH profiles could be obtained by using FFPE samples and that this technique is therefore applicable to daily practice. We are therefore setting up a prospective European cohort of all newly diagnosed cases of pediatric SSs and performing systematic genomic profiles as well as centralizing clinical data and information on response to chemotherapy in order to confirm our results on a larger cohort of patients, in which Exome sequencing will also be carried out to look for genomic deregulations undetectable with CGH.
This prospective clinical validation of the GI would allow making it a molecular prognostic marker that could then help for therapeutic management.
funding
French National Cancer Institute (Grant ‘INCa-DGOS-Inserm 6046’), European Connective Tissue Cancer Network, French Ligue Regionale Contre le Cancer, Sarcoma Patients Euronet and association ‘Le fil d'Oriane’ (no grant number).
disclosure
The authors have declared no conflicts of interest.
Supplementary Material
acknowledgements
We thank Christophe Perrin, Emmanuelle Bompas, Anne Moreau, Christine Chevreau, Sophie Le Guellec, Natacha Entz-Werle, Jean-Emmanuel Kurtz and Pascaline Boudou-Rouquette for providing additional histologic material and clinical information for cases in this study.
references
- 1.Suurmeijer AJH, de Bruijn D, Geurts van Kessel A, Miettinen MM. Synovial sarcoma. In: Fletcher CD, Bridge JA, Hogendoorn PCM, Mertens F, editors. WHO Classification of Tumours of Soft Tissue and Bone. Lyon: IARC; 2013. pp. 213–215. [Google Scholar]
- 2.Herzog CE. Overview of sarcomas in the adolescent and young adult population. J Pediatr Hematol Oncol. 2005;27:215–218. doi: 10.1097/01.mph.0000161762.53175.e4. [DOI] [PubMed] [Google Scholar]
- 3.Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23:92–99. doi: 10.1093/annonc/mds253. [DOI] [PubMed] [Google Scholar]
- 4.Eilber FC, Brennan MF, Eilber FR, et al. Chemotherapy is associated with improved survival in adult patients with primary extremity synovial sarcoma. Ann Surg. 2007;246:105–113. doi: 10.1097/01.sla.0000262787.88639.2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Italiano A, Penel N, Robin YM, et al. Neo/adjuvant chemotherapy does not improve outcome in resected primary synovial sarcoma: a study of the French Sarcoma Group. Ann Oncol. 2008;20:425–430. doi: 10.1093/annonc/mdn678. [DOI] [PubMed] [Google Scholar]
- 6.Palmerini E, Staals EL, Alberghini M, et al. Synovial sarcoma: retrospective analysis of 250 patients treated at a single institution. Cancer. 2009;115:2988–2998. doi: 10.1002/cncr.24370. [DOI] [PubMed] [Google Scholar]
- 7.Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101:627–634. doi: 10.1002/cncr.20386. [DOI] [PubMed] [Google Scholar]
- 8.Clark J, Rocques P, Crew A, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet. 1994;7:502–508. doi: 10.1038/ng0894-502. [DOI] [PubMed] [Google Scholar]
- 9.Lagarde P, Przybyl J, Brulard C, et al. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol. 2013;31:608–615. doi: 10.1200/JCO.2012.46.0147. [DOI] [PubMed] [Google Scholar]
- 10.Guillou L. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol. 2004;22:4040–4050. doi: 10.1200/JCO.2004.11.093. [DOI] [PubMed] [Google Scholar]
- 11.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 12.Hostetter G, Kim SY, Savage S, et al. Random DNA fragmentation allows detection of single-copy, single-exon alterations of copy number by oligonucleotide array CGH in clinical FFPE samples. Nucleic Acids Res. 2010;38:e9. doi: 10.1093/nar/gkp881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagarde P, Perot G, Kauffmann A, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:826–838. doi: 10.1158/1078-0432.CCR-11-1610. [DOI] [PubMed] [Google Scholar]
- 14.Salzer-Kuntschik M, Delling G, Beron G, Sigmund R. Morphological grades of regression in osteosarcoma after polychemotherapy—study COSS 80. J Cancer Res Clin Oncol. 1983;106(Suppl):21–24. doi: 10.1007/BF00625047. [DOI] [PubMed] [Google Scholar]
- 15.Pappo AS, Devidas M, Jenkins M, et al. Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol. 2005;23:4031–4038. doi: 10.1200/JCO.2005.03.209. [DOI] [PubMed] [Google Scholar]
- 16.Karavasilis V, Seddon BM, Ashley S, et al. Significant clinical benefit of first-line palliative chemotherapy in advanced soft-tissue sarcoma: retrospective analysis and identification of prognostic factors in 488 patients. Cancer. 2008;112:1585–1591. doi: 10.1002/cncr.23332. [DOI] [PubMed] [Google Scholar]
- 17.Kampe CE, Rosen G, Eilber F, et al. Synovial sarcoma. A study of intensive chemotherapy in 14 patients with localized disease. Cancer. 1993;72:2161–2169. doi: 10.1002/1097-0142(19931001)72:7<2161::aid-cncr2820720716>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen OS, Judson I, van Hoesel Q, et al. Effect of high-dose ifosfamide in advanced soft tissue sarcomas. A multicentre phase II study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2000;36:61–67. doi: 10.1016/s0959-8049(99)00240-3. [DOI] [PubMed] [Google Scholar]
- 19.Okcu MF, Munsell M, Treuner J, et al. Synovial sarcoma of childhood and adolescence: a multicenter, multivariate analysis of outcome. J Clin Oncol. 2003;21:1602–1611. doi: 10.1200/JCO.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Ladenstein R, Treuner J, Koscielniak E, et al. Synovial sarcoma of childhood and adolescence. Report of the German CWS-81 study. Cancer. 1993;71:3647–3655. doi: 10.1002/1097-0142(19930601)71:11<3647::aid-cncr2820711129>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 21.Gortzak E, Azzarelli A, Buesa J, et al. A randomised phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer. 2001;37:1096–1103. doi: 10.1016/s0959-8049(01)00083-1. [DOI] [PubMed] [Google Scholar]
- 22.Orbach D, Mc Dowell H, Rey A, et al. Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors (SIOP-MMT) Working Group. Pediatr Blood Cancer. 2011;57:1130–1136. doi: 10.1002/pbc.23138. [DOI] [PubMed] [Google Scholar]
- 23.Al-Hussaini H, Hogg D, Blackstein ME, et al. Clinical features, treatment, and outcome in 102 adult and pediatric patients with localized high-grade synovial sarcoma. Sarcoma. 2011;2011:231789. doi: 10.1155/2011/231789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brennan B, Stevens M, Kelsey A, et al. Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children's Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer. 2010;55:85–90. doi: 10.1002/pbc.22453. [DOI] [PubMed] [Google Scholar]
- 25.Chakiba C, Terrier P, Ranchere-Vince D, et al. Prognostic significance of radiologic and histologic response to neoadjuvant chemotherapy in localized synovial sarcoma. J Clin Oncol. 2013;31(Suppl) abstract 10578. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.