

Abstract
Improved understanding of soft-tissue sarcoma (STS) biology has led to better distinction and subtyping of these diseases with the hope of exploiting the molecular characteristics of each subtype to develop appropriately targeted treatment regimens. In the care of patients with extremity STS, adjunctive radiation therapy (RT) is used to facilitate limb and function, preserving surgeries while maintaining five-year local control above 85%. In contrast, for STS originating from nonextremity anatomical sites, the rate of local recurrence is much higher (five-year local control is approximately 50%) and a major cause of death and morbidity in these patients. Incorporating novel technological advancements to administer accurate RT in combination with novel radiosensitizing agents could potentially improve local control and overall survival. RT efficacy in STS can be increased by modulating biological pathways such as angiogenesis, cell cycle regulation, cell survival signaling, and cancer-host immune interactions. Previous experiences, advancements, ongoing research, and current clinical trials combining RT with agents modulating one or more of the above pathways are reviewed. The standard clinical management of patients with STS with pretreatment biopsy, neoadjuvant treatment, and primary surgery provides an opportune disease model for interrogating translational hypotheses. The purpose of this review is to outline a strategic vision for clinical translation of preclinical findings and to identify appropriate targeted agents to combine with radiotherapy in the treatment of STS from different sites and/or different histology subtypes.
Over the last decade, advancements in genomics and molecular biology have led to an increasing number of molecular targets and agents to be tested and used clinically in different cancers. While the combination of these targeted agents with chemotherapy has been actively explored, research on the complementarity and combination of different molecularly targeted therapies with radiotherapy is lagging (1). In order to promote research in this area, the National Cancer Institute (NCI) held the first workshop on developing of radiosensitizers in August 2012, from which a set of recommendations was recently published (1). In concordance with the NCI’s efforts, the NCI-Radiation Therapy Oncology Group (RTOG) translational program also published their strategic guidelines to foster multi-institutional efforts to accelerate the development of radiosensitizers for different cancers, including soft-tissue sarcomas (STSs) (2).
The management of STS is challenging because of the rarity of the cancer, the wide variety of sites of origins, and subtypes with differing clinical, phenotypical, and genomic characteristics that may alter their sensitivity to chemotherapy and radiotherapy. A recent major advancement in STS came with the publication of the World Health Organization (WHO) 2002 pathology guidelines, which was a result of improved understanding in the molecular biology of STS. This publication has, for example, abolished the diagnosis of malignant fibrous histiocytomas (MFH) (3), which was once the most common STS diagnosis. Many previously diagnosed MFH are now reclassified as other STS subtypes using more sophisticated methods such as immunohistochemistry and fluorescent in-situ hybridization analysis (3–7). Furthermore, newfound molecular and genomic understanding of each STS subtype has led to the identification of subtype-specific genomic aberrations that may be sarcomagenic and are currently being investigated as potential targets for molecular agents used as monotherapies or in combination with chemotherapy and/or radiotherapy (7,8).
The primary modality in the management of patients with STS remains surgical, with radiotherapy used adjunctively to reduce the surgical extent and preserve patient function (9,10). Efficacious chemotherapy that improves patient survival remains elusive (11–15), hence opportunities exist for examining molecular pathways to discover and develop novel systemic agents against metastasis, the main cause of death in STS originating from the extremities. While the five-year local control of the disease ranges from 80% to 95% in patients with STS of the extremities treated with surgery and/or radiotherapy (9,16–18), local relapse is more prevalent in STS originating from other sites (head and neck, trunk, retroperitoneum, intra-abdomen and pelvis). In these body regions, the five-year local relapse rate is approximately 50%, and a majority of mortality is secondary to the complications related to local tumor progression (19–22). The inferior local control at these sites may be secondary to differences in tumor biology and/or the challenging anatomy, because adjacent critical structures and organs may limit the ability to obtain wide surgical margins and to deliver a sufficiently high dose of radiation (22). Incorporating novel technological advancements to administer accurate radiation therapy in combination with novel radiosensitizing agents could potentially improve local control and overall survival in STS from nonextremity sites.
Although radiation-induced DNA damage is postulated to be the main source of cell death, the toxicity of radiotherapy is also modulated by molecular pathways and tumor microenvironmental factors such as (23): angiogenesis, cell cycle regulation, cell survival signaling, and cancer-host immune interaction (Figure 1). Therefore, molecular alteration of one or more of these pathways has the potential to improve the efficacy of radiotherapy. This review aims to describe recent advancements in the modulation of these pathways using molecular agents in combination with radiotherapy for the treatment of patients with STS.
Figure 1.

Radiation (RT)-induced cellular toxicity occurs through the production of double-stranded DNA break, which promptly activates a series of DNA damage response (DDR) that may repair the damage and rescue the cell from death through apoptosis, senescence, necrosis, mitotic catastrophe, and autophagy. Beyond DDR, the cell’s ability to survive from RT is also modulated by other biological pathways related to A) angiogenesis, B) cell cycle regulation, C) survival signaling, and D) cancer-host immune interaction. The activity of these pathways may be modulated by different targeted agents that in combination with RT may enhance the cytotoxicity of RT in soft-tissue sarcomas. DDR = DNA damage response; RT = radiotherapy.
Angiogenesis Inhibitors
Since the original description of angiogenesis and its acceptance as a critical component in tumorgenesis and progression (24,25), multiple inhibitors of varied components in angiogenesis have been developed and are in clinical use for multiple cancers. Several antiangiogenics agents, including sorafenib, sunitinib, pazopanib, and bevacizumab, have been investigated for use in STS.
Sorafenib
Sorafenib is a multikinase inhibitor with antiproliferative and antiangiogenic effects with a wide range of targets, including RAF, vascular endothelial growth factor receptors (VEGFR), and platelet-derived growth factor receptor (PDGFR), amongst others. The safety and clinical activity of sorafenib monotherapy was established through phase I studies in patients with solid tumors (26,27). Its efficacy in renal cell cancer and hepatocellular carcinoma was subsequently defined by Phase III clinical trials (28–30). Phase II studies in STS suggested that sorafenib is most active against angiosarcomas and leiomyosarcomas (31,32) in comparison with other STS histologies. To date, two published clinical trials have evaluated the efficacy and safety of combining radiotherapy and sorafenib in STS (33,34) and determined the maximum tolerated dose of sorafenib to be 400mg daily when combined with chemoradiation (epirubicin/ifosfamide and 28Gy/8 fractions) or radiotherapy (50Gy/25 fractions). In the former trial, when preoperative sorafenib was combined with chemoradiation therapy, 44% of case patients had more than 95% histopathologic tumor necrosis. However, 15 of the 16 patients developed grade 3–4 toxicity (primarily hematopoietic), and six patients (38%) had major wound complications (34). Comparatively, a similar trial that also combined sorafenib and preoperative radiotherapy, but excluded systemic chemotherapy, reported less grade 3–4 toxicity (50%), considerably fewer wound complications (12.5%), and similar but slightly lower rate of more than 95% tumor necrosis (38%) (33). Taken together, these studies suggest the relative safety and potential efficacy of combining neoadjuvant radiotherapy and sorafenib in the management of STS.
Sunitinib
Sunitinib is a receptor tyrosine kinase inhibitor with activity against multiple receptors, including VEGFR, PDGFR, c-KIT, FMS-like tyrosine kinase 3 (Flt3), CSF-1 receptor, and RET (35). Although preclinical studies evaluating the activity of sunitinib showed promising results in leukemia and some neuroblastoma models (36,37), sunitinib was not efficacious when used alone or in combination with other systemic therapies in clinical trials involving breast, colorectal, prostate, and lung cancers (38–43) (clinicaltrials.gov NCT00676650). In STS, however, a phase III randomized clinical study demonstrated that sunitinib prolonged the time to progression from 6.4 weeks (placebo) to 27.3 weeks in patients with gastrointestinal stromal tumors (GIST) that were resistant or intolerant to imatinib (44). Therefore, sunitinib may not only target c-KIT, but may also act by targeting angiogenesis. Two phase II studies further suggested sunitinib’s efficacy in various mesenchymal tumors, such as liposarcoma (LPS), leiomyosarcoma (LMS), MFH, desmoplastic round cell tumors, and solitary fibrous tumors, by delaying disease progression when used as monotherapy in heavily pretreated patients (45,46). A pediatric (age = 2–21 years) phase I trial determined the maximum tolerated dose (MTD) for this population to be 15mg/m2/day, which is lower than the approved adult dose of 50mg/day (35). Despite the lower dose of sunitinib administered, one of the eight STS (four nonspecified, two Ewing’s sarcoma, one desmoplastic small round cell tumor, one gastrointestinal stromal tumor) patients from the trials had stable disease for nine cycles (35).
Theoretically, angiogenesis inhibitors may improve the efficacy of radiotherapy either by normalizing tumor blood vessels to improve tumor oxygenation and reduce intratumoral pressure or by increasing the rate of tumor apoptosis through direct inhibition of cellular survival signals (Figure 2) (47). Akin to other cancers, STSs are also known to contain hypoxic regions, as well as abnormal tumor vascularization (48); hence, the concurrent use of this class of agents may sensitize STS to the effects of radiotherapy. The combinatorial effect of sunitinib and radiotherapy was previously characterized by Yoon et al. through their murine STS in vivo model (49). In their experiments, tumor growth was most delayed in mice treated with concurrent sunitinib and radiotherapy (10Gy x2) (T/C 71%) in comparison with those that received sunitinib (T/C 56%) only or radiotherapy (T/C 41%) only. Tumors that were treated with sunitinib (+/- radiotherapy) showed reduced microvessel densities and endothelial cell apoptosis, which were not observed in tumors that received radiotherapy alone (49). Finally, this preclinical study also validated previously described biomarkers of sunitinib activity in which sunitinib treatment increased plasma VEGF, PGF, and endoglin while reducing VEGFR2 and monocyte counts (49). These biomarkers of response and prognosis could possibly be examined and validated in future early clinical trials.
Figure 2.
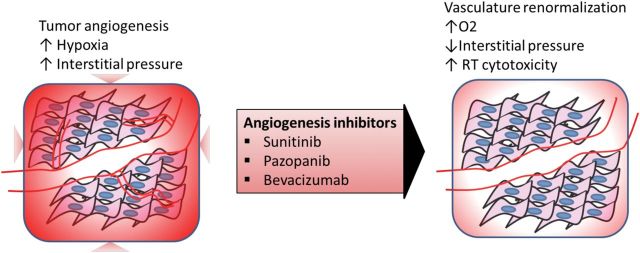
Tumor angiogenesis is frequently found in soft-tissue sarcomas (STSs), leading to high levels of aberrant vasculatures, acute and chronic hypoxia, and increased interstitial pressures within the STS microenvironment. Treatment of STS with angiogenesis inhibitors may renormalize the tumor vasculature. This would in turn increase tumor tissue oxygenation and reduce the interstitial pressure, thus increasing the sensitivity of the cells to radiotherapy. RT = radiotherapy.
Several groups have published small clinical trials and case reports that described the efficacy and toxicities in combining sunitinib with radiotherapy. In these trials, radiotherapy was often given in slightly hypofractionated forms (range = 2-5Gy/fraction) together with a range of sunitinib doses (25mg - 50mg daily) (50–55). According to three phase I adult trials, the maximum tolerated dose for the concurrent use of sunitinib ranges from 25mg to 37.5mg daily (54–56). Two case reports suggested potential recall effect (pneumonitis and cerebral perimetastasis edema increase) occurring in patients who received sequential sunitinib one week following the completion of radiotherapy (57,58). These initial experiences in the concurrent use of sunitinib and radiotherapy suggested that a tolerable regimen exists that should be tested in a controlled research setting to ensure meticulous monitoring for previously known as well as unexpected toxicities from either therapy during and following the completion of the treatments. To our knowledge, there are two ongoing trials that are testing for the safety of combining radiotherapy and sunitinib in the treatment of STS (NCT00753727 and NCT01498835).
Pazopanib
Pazopanib is another antiangiogenic agent that targets VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-a/β, and c-kit (59). Given the similarity in molecular targets between sunitinib and pazopanib, pazopanib was examined for its use in renal cell cancer (RCC) and was subsequently approved for use in the treatment of RCC (60). This was followed by a direct comparison between sunitinib and pazopanib in a recently published phase III trial for their efficacy (PFS), safety and quality-of-life (QOL) profiles in patients with metastatic RCC. Results from this trial demonstrated that pazopanib was not inferior to sunitinib and had better patient safety and QOL profile than sunitinib (61). For STS, a phase III trial had previously shown that pazopanib was active in the treatment of patients with metastatic nonadipocytic STS previously treated with chemotherapy (62,63). This trial specifically took into account a prior Phase II study that suggested a lack of response of pazopanib in adipocytic STS (64) and hence included only nonadipocytic tumors (synovial sarcomas, LMS and “other”) into the trial. In these STS subtypes, statistically significantly prolonged PFS (4.6 months vs 1.6 months, P < .0001) was observed in patients receiving pazopanib vs placebo, despite a modest 6% objective response rate and a lack of overall survival improvement (survival 12.5 months vs 10.7 months, P = .25) in pazopanib-treated patients (62), suggesting a cytostatic mechanism of action by pazopanib in STS, consistent with an antiangiogenic activity.
The combination of radiotherapy with pazopanib had been studied in the adjuvant treatment setting of breast cancer patients (65), who unexpectedly developed fewer dermatological toxicities than matched control patients treated with radiotherapy only. Two trials are currently ongoing to assess the addition of pazopanib to radiotherapy in the neoadjuvant treatment of STS. In the German phase II study (NCT01543802), the investigators’ primary objective is to determine whether the addition of pazopanib reduces the metabolic response on PET-CT one month after the completion of neoadjuvant therapy, with other biological, radiological, and clinical parameters as secondary endpoints.
Similarly, the Children’s Oncology Group (COG) and RTOG have jointly developed a trial to study this concept, which will open in early 2014. In this trial, titled “Pazopanib Neoadjuvant Trial in Non-Rhabdomyosarcoma Soft Tissue Sarcomas (PAZNTIS): A Phase II/III Randomized Trial of Preoperative Chemoradiation or Preoperative Radiation Plus or Minus Pazopanib” (ARST1321), patients are divided into two cohorts based on their STS’s chemosensitivity, which was determined from the assessment of pathological necrosis of STS treated with neoadjuvant chemoradiotherapy from prior COG (ARST0332) and RTOG (RTOG 9415) studies. The first cohort consist of patients with large (>5cm), high-grade (grade III/III) “chemosensitive” STSs who will be randomly assigned to neoadjuvant chemoradiotherapy (adriamycin and ifosfamide based regimen and 50Gy in 25 fractions) +/-pazopanib. Patients who will receive neoadjuvant chemoradiotherapy and pazopanib will also receive adjuvant chemotherapy and pazopanib for six months, while patients treated with neoadjuvant chemoradiotherapy alone will receive adjuvant chemotherapy only for six months. The second cohort will comprise patients with intermediate-to-high-grade (grade II-III) “chemoresistant” STS and some “chemosensitive” STS patients that are not eligible or refuse chemotherapy. They will be randomly assigned to receive neoadjuvant radiotherapy with or without pazopanib. Postoperatively, patients treated with neoadjuvant radiotherapy and pazopanib will receive adjuvant pazopanib for six months, while the patients treated with neoadjuvant radiotherapy alone will be observed. The primary endpoints of this study are: 1) more than 90% pathologic necrosis in the resected STS after neoadjuvant treatment (Phase II component) and 2) event-free survival (Phase III component) (Figure 3). Translational research components will include tumor response assessed by MR and PET images, determination of actionable mutations through whole genome sequencing, correlation of tumor microvessel density, and circulating tumor DNA with the patterns of failure and survival of patients.
Figure 3.

A schema of the “Pazopanib Neoadjuvant Trial in Non-Rhabdomyosarcoma Soft Tissue Sarcomas” (PAZNTIS) (ARST1321). All patients will receive radiotherapy (50 Gy in 25 fractions) preoperatively. Neoadjuvant chemotherapy, which consists of four cycles of ifosfamide and two cycles of doxorubicin, will be offered to those who have soft-tissue sarcomas (STSs) that are sensitive to chemotherapy. Following trial entry, patients in each cohort will be randomly assigned to receive preoperative daily (QD) pazopanib in addition to their neoadjuvant treatments. After their definitive surgery, patients who received systemic preoperative treatments will receive six months of postoperative systemic therapy. STS = soft-tissue sarcoma.
Bevacizumab
In addition to tyrosine kinase inhibitors, bevacizumab, a humanized monoclonal antibody that binds to and neutralizes VEGF-A, has been investigated for use in STS (66,67). In a preclinical study, Myers et al. explored the potential of combining bevacizumab with radiotherapy using a rhabdomyosarcoma xenograft murine model and observed normalization of tumor vasculature, improved oxygenation, and increased tumor radiosensitivity when tumors were radiated two to five days after bevacizumab (68). With 80% of angiosarcomas overexpressing VEGF (69), Agulnik et al. tested the efficacy of bevacizumab monotherapy through a phase II trial involving unresectable angiosarcomas and epithelioid hemangioendotheliomas (70). Of the 32 patients, 13% and 50% obtained partial response and stable disease, respectively, as best response, with a 26-week median time to progression (70). In light of recent case reports describing patients achieving complete response following concurrent bevacizumab and radiotherapy (50-60Gy) (71,72), Agulnik et al. suggested for future efforts to explore combining bevacizumab with chemotherapy and radiotherapy to improve outcomes (70).
To our knowledge, a phase II STS trial combining bevacizumab with gemcitabine/docetaxel has completed accrual (NCT00887809), but the results are not yet published. In assessing the combination of bevacizumab with radiotherapy, results from a phase II trial in the preoperative treatment of “high-risk” STSs demonstrated that, of the 20 tumors treated, nine tumors had more than 80% pathological necrosis, including three complete responses (73). Despite these responses, four patients had microscopically positive margins and one patient had gross residual disease, suggesting that surgical feasibility was not improved (9,74) by the additional response from bevacizumab as compared with historical results from patients who received neoadjuvant radiotherapy only. Twenty-five percent of the patients developed major wound complication, which is comparable with rates published in other studies utilizing preoperative radiotherapy (73). Finally, the authors described several candidate biomarkers of response (VEGF, PIGF, and sVEGFR3), as well as a 24-gene predictive signature for future validation. Three large ongoing clinical trials (NCT00643565, NCT01746238, NCT01871766) with differing timing and use of bevacizumab with chemotherapy or radiotherapy will help characterize the combinatorial effects and candidate response and prognostic biomarkers. As the phase II trial by Yoon et al. failed to demonstrate a definitive benefit from the addition of bevacizumab to radiotherapy, results from ongoing trials will be needed to determine the merits and risks of the approach.
Cell Cycle Regulators
MDM2 Inhibitors
The tumor suppressor p53 is activated in response to a variety of cellular perturbations, in particular genotoxic stress, and functions to prevent fixation of DNA damage and proliferation of cells with genetic alterations (75). Activation of p53 in response to genotoxic stress leads to induction or repression of p53-responsive genes at the transcriptional level, resulting in cell cycle arrest, senescence, or apoptosis (76). The activity of p53 is tightly regulated by post-translational modifications that affect its cellular localization and ability to bind DNA consensus sequences (77,78). However, p53 is principally regulated by its high turnover rate and degradation mediated by the E3 ubiquitin ligase, MDM2 (mouse double minute 2). MDM2 catalyzes p53 ubiquitination and subsequent degradation by the proteasome (Figure 4) (78–81).
Figure 4.
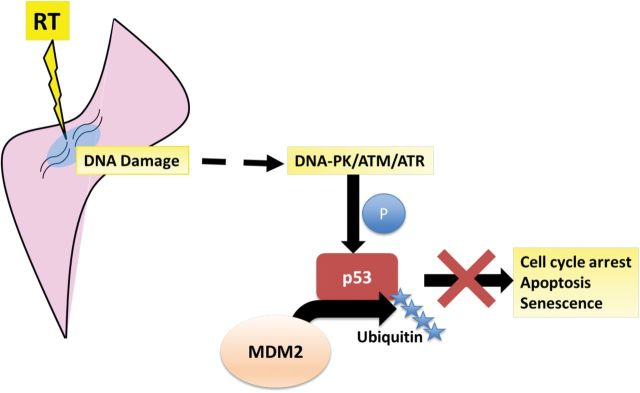
DNA damage secondary to radiotherapy induces the activation of DNA-protein kinases (DNA-PK), ataxia telangiectasia mutated, and ataxia telangiectasia and Rad3-related proteins, which subsequently induce p53 via phosphorylation of various substrates. In sarcomas overexpressing murine double minute 2 homolog (MDM2), p53 is targeted by MDM2 for ubiquitination and degradation, thereby inhibiting p53’s ability to induce cell cycle arrest, apoptosis, and senescence. ATM = ataxia telangiectasia mutated; ATR = ataxia telangiectasia and Rad3-related proteins; RT = radiotherapy.
An analysis of 4000 human tumor samples found that MDM2 is overexpressed in 7% of cancers and its overexpression is mutually exclusive from p53 mutations (82). It has long been observed that 90% of well-differentiated (WD) and dedifferentiated (DD) LPSs contain supernumerary rings and/or giant rod chromosomes (83) that contain amplified segments of the 12q13-15 region (84,85). Of the genes residing in this amplified chromosomal region, MDM2 (murine double minute) and CDK4 (cyclin-dependent kinase 4) amplifications are used in pathology to help differentiate WD and DD LPSs from other STS subtypes and lipomas (5,83,85–88).
Multiple competitive inhibitors (eg, nutlin, MI-219, RG7112) of the MDM2-p53 interaction have been developed to counter the effect of MDM2 and restore the activity of cellular p53 (81,89–91). These MDM2 antagonists seem to function only in tumors with wild-type p53 (90,92,93). Preclinical studies that evaluated these inhibitors in the treatment of osteosarcoma, and LPS cell lines overexpressing MDM2 demonstrated that these MDM2 antagonists were able to reactivate p53 activity, induce G2 cell cycle arrest and apoptosis in vitro (88,90,92). Correspondingly, Tovar et al. observed that the cytotoxicity of nutlin-3 in an array of cancer cell lines was associated with the level of MDM2 amplification (94). Xenografts from human sarcoma cell lines demonstrated tumor regression (90,92) following MDM2 inhibitor monotherapy (90,92,94), thus supporting further exploration of this class of agents for STS therapy (95). Ray-Coquard et al. recently published a proof-of-mechanism study that evaluated biological markers of RG7112 activity following neoadjuvant treatments of patients with WD or DDLPS using 1440mg/m2 of RG7112 daily for 10 days on 28-day cycles, a regimen derived from an initial phase 1 trial (91). In this trial of 20 patients, although p53 and p21 levels were statistically significantly increased from baseline following RG7112 treatment, their magnitude of change did not correlate with drug exposure (91). Furthermore, following the initial dose of MDM2 antagonist, the authors observed increased macrophage inhibitory cytokine-1 secretion (MIC-1) that correlated with drug exposure and tumor apoptosis. While the study has potentially identified an important candidate biomarker for RG7112 activity, it also demonstrated the toxicity of the monotherapy with eight (of 20) patients developing serious adverse events. In addition, interpretation of the results is hindered by the inclusion of patients with tumors that had p53 mutations (two of 20 analyzed) or normal MDM2 copies (three of 17 analyzed) (91).
In a prior evaluation of RG7112 against pediatric cancer cell lines, and a variety of p53 wild-type xenografts in immune-deficient mice (96), cell sensitivity in vitro differed based on p53 mutation status. It was observed that p53-mutant cell lines were approximately 10-fold less sensitive to this MDM2 inhibitor than cells with wild-type p53. In contrast, RG7112 induced regressions in only five of 26 solid tumor models that had wild-type p53 by sequencing (96). However, despite the relatively modest antitumor activity, RG7112 did induce p53 and downstream targets (p21, PUMA), hence clearly inhibited the MDM2:p53 interaction by stabilizing p53. In the “nonresponsive” tumors, the increase in p53 was inadequate to induce apoptosis, or cell cycle arrest, but the combination of an MDM2 inhibitor in the context of DNA damage may be potentially synergistic in stabilizing p53 and inducing cell death. This is the rationale for combining a second generation MDM2 inhibitor, RG7338, with ionizing radiation. In vivo studies combining RG7338, using either daily or weekly schedules of administration, with daily fractionated radiation (2Gy fractions) to a total of 10, 20, or 30Gy are ongoing, using childhood sarcoma models transplanted into immune-deficient mice (Peter Houghton, personal communication).
CDK4 Inhibitors
Similarly, CDK4, which resides within the amplified chromosomal region found in WD- and DD-LPS, is integral to the functioning of cellular checkpoints. CDK4 functions by forming a complex with cyclin D to phosphorylate and inactivate RB, thus releasing the E2F transcription factor to promote cell cycle progression through the G1-S checkpoint (97). Inhibition of CDK4 activity by the CDK4/6 inhibitor PD033991 prevents RB phosphorylation, resulting in cytostasis and tumor regression in RB-positive carcinoma xenografts (98).
Hagen et al. observed that knock-down of CDK4 radiosensitized breast cancer cells and cancer stem cells in vitro by increasing the rate of apoptosis following irradiation (99). However, the increased radiosensitivity in CDK4 knock-down cells was not correlated with cell cycle alterations or impaired DNA repair (99), suggesting that CDK4 inhibition may confer its effects through inhibition of cell survival pathway (AKT/CyclinD/CDK) signal transduction (100,101). The effect of CDK4/6 inhibition by PD033991 was similarly observed in LPS cell lines (102), which provided the basis for the translation of CDK4 inhibition to STS patients first achieved through a phase I trial by Schwartz et al. (103). In this study, four out of seven patients with LPS obtained disease stabilization and two had prolonged stabilization (11 and ≥23 cycles) on PD033991 monotherapy (103). These results hinted at the potential efficacy of the agent as monotherapy for LPS, which was further explored in a phase II trial that enrolled patients with CDK4-amplified LPS-expressing RB (104). PD033991 monotherapy resulted in 66% of the patients being free of disease progression at 12 weeks, with a median PFS of 18 weeks. Grade 3 or higher toxicities (mostly haematological) were, however, observed in 50% of patients (104). Given the theoretical and preclinical association between CDK4 modulation and radiosensitization, combining radiotherapy with the inhibition of CDK4 or the survival pathway could yield both spatial and in-field cooperation between the modalities.
Inhibition of Cell Survival Pathways
As mentioned above, the radiosensitizing mechanism of CDK4 inhibition may be because of reduced survival signalling through the AKT/CyclinD/CDK pathway. As many growth factor–signalling pathways converge to activate PI3K, followed by downstream stimulation of AKT and mTOR activity, inhibition of PI3K and mTOR may potentially lead to radiosensitization effects.
Toulany et al. recently reviewed preclinical studies that support the role of the PI3K-AKT pathway in directly mediating DNA repair following radiation (105). In vitro experiments from multiple carcinoma models have demonstrated that activity of PI3K/AKT is correlated with radioresistance independent of p53 status (106–109). Furthermore, Toulany et al. demonstrated that AKT promotes DNA-PKcs accumulation at DNA-DSB and colocalizes with yH2AX following radiotherapy (110), thus suggesting a potential role of combining radiotherapy with molecular inhibitors of this pathway. In conjunction, cell with constitutively active AKT are more resistant, while the inhibition of upstream PI3K using LY294002 induced radiosensitization in multiple in vitro cancer models. (111–114). Interestingly, some PI3K inhibitors with broad specificity, such as the dual PI3K/mTOR inhibitor NVP-BEZ235, also inhibit the PI3K-related protein kinases ATM and DNA-PKCs (115). Because ATM and DNA-PKCs are activated by DNA damage to regulate the cellular response to radiation and because inhibition of these enzymes increases radiosensitivity (116), the ability of some PI3K inhibitors to radiosensitize tumors may be, in part, because of inhibition of ATM and/or DNA-PKCs.
In a zebrafish model of LPS that expresses constitutively active AKT2, the dual inhibition of PI3K and mTOR by BEZ235 induces cytostasis and apoptosis in a dose-dependent manner (117). Moreover, Dodd and colleagues observed sensitization of a primary mouse model of undifferentiated pleomorphic sarcoma to the combination of BEZ235 and doxorubicin (118). Furthermore, Marklein et al. observed that pretreatment or concurrent treatment with two PI3K/mTOR inhibitors, PI103 and GDC-0941, augmented doxorubicin cytotoxicity by inducing apoptosis in several sarcoma cell lines and xenografts (119). Using synovial sarcoma cell lines, Hosaka et al. observed that PI3K-AKT suppression following the administration of pazopanib was correlated with cellular sensitivity to cytostasis and tumor growth delay in xenografts (120). These results, taken together with the convergence of multiple growth factor signalling through the PI3K-AKT pathway, suggest that regulation of the PI3K-AKT pathway may be another potential mechanism by which pazopanib and other antiangiogenic agents induce their effects in STS beyond modulating the tumor vasculature.
Cancer-Host Immune Modulation
Recent developments in immunotherapy in cancer treatments have yielded clinical success in the treatment of prostate cancer and melanoma through the respective use of Sipuleucel-T, an autologous dentritic cell (DC) vaccine, and ipilimumab, an antibody specific for cytotoxic T lymphocyte-associated antigen 4 (CTLA4) (121). The potential of immunotherapy may be further boosted by combining it with targeted therapies and cytotoxic agents to prime the specificity of T cells and to trigger the release of antigenic debris from tumor cell death (121). Many cancers, including sarcoma, reportedly have upregulated expression of PDL1 (122,123), which inhibits the activation of T cells. Further research using newly developed molecular agents that modulate PD1 and PDL1 activities may therefore yield promising results in the treatment of STS. Furthermore, preclinical in vivo and clinical data suggest that localized radiotherapy can have abscopal effects on nonirradiated tumor sites through immunostimulation (124,125), which could be exploited and combined with cancer immunotherapy agents. In a retrospective review of 37 paired samples of sarcoma before and after radiotherapy, Sharma et al. observed changes in the expression of various immune-related transcripts and cancer-testis antigens that implicate the possibility that radiotherapy stimulates immune reactions and, hence, support the potential combination of immunotherapy with radiotherapy in STS (126).
Several groups have reported successful results in syngeneic tumor models by combining an apoptosis-inducing treatment with intratumoral injection of dentritic cells (DCs) (127). Candido et al. demonstrated that injections of DCs into tumors resulted in a high rate of spontaneous apoptosis and in an antitumor effect (128), and potential generation of systemic immunity. Combination of radiotherapy with autologous DC administration eliminates the cumbersome requirement for patient selection based on MHC class I type in future clinical development. Similarly, use of autologous DCs eliminates subject selection criteria based on expression of defined tumor-associated antigens (TAAs) and reduces the problem of TAA loss during tumor progression. This would reduce time and cost of development compared with immune-based treatment directed at specific TAAs.
Using a mouse sarcoma tumor model, it was demonstrated that the combination of radiation with local DC administration into the tumor site resulted in the induction of a tumor-specific immune response and a statistically significant antitumor effect (129). Teitz-Tennenbaum and colleagues confirmed that radiotherapy potentiates the efficacy of intratumoral DC administration (130). To test the safety and immunological efficacy of this approach, Finkelstein et al. conducted a pilot clinical trial of sarcoma patients with high-risk of recurrence (High grade STSs larger than 5cm) (131). In this study, patients were treated with neoadjuvant radiotherapy with three weekly intratumoral injections of DC. Induction of an immune response in peripheral blood mononuclear cells to tumor cell lysates was observed in nine out of 17 patients, at one or more times point following treatment. Wound complication rates and Grade 2 or greater toxicities were in line with those observed in the previous NCI-Canada SR2 preoperative vs postoperative radiotherapy in extremity sarcoma trial (9). Future investigation will be needed to compare and validate the efficacy of cancer-host immunotherapy agents such as nivolumab and ipilumimab in addition to radiotherapy in stimulating an antitumoral immune response and in inducing abscopal effects against distant metastases, translating into improved patient survival.
Translation to the Clinic
Extremity and Superficial Trunk Primary Sarcomas
Results of recent trials have demonstrated that preoperative modern radiotherapy, especially image-guided intensity-modulated radiotherapy, is associated with a statistically significant reduction of chronic side effects compared with preoperative conventional radiotherapy (132–134). However, preoperative modern radiotherapy is still associated with an increased rate of wound complication (>30%) (9,132,135). Improving the efficacy of radiotherapy through the use of molecular agents may reduce the dose of radiation needed in the adjunctive setting and, when combined with precise radiotherapy, may widen the therapeutic window between tumor and normal tissue to reduce the rate of radiation related side effects. Volumetric reduction of tumors from neoadjuvant radiotherapy of myxoid liposarcomas was associated with reduced field of surgery and local recurrences (136). The addition of STS-targeted radiosensitizing agents to preoperative radiotherapy may similarly lead to increased volumetric and pathological responses, which may enhance surgical resectability and local control. As each of the molecular pathways discussed may potentiate the effects of radiotherapy, the application of targeted agents in extremity STS should be explored. Several such clinical trials are underway (NCT00753727, NCT01498835, NCT01543802).
Retroperitoneal, Visceral, Thoracic, and Head and Neck Primary Sarcomas
For STSs originating from the retroperitoneum, viscera, thorax, head and neck regions, surgery alone results in suboptimal local control (30% to 50%) (22,74,137), and loco-regional recurrences are the leading cause of death and morbidity in these patients. To improve local control, the addition of preoperative radiotherapy (50.4Gy in 28 fractions) to surgery in the management retroperitoneal STS is being investigated through an ongoing phase III randomized trial (EORTC-62092-22092) (NCT01344018). Even with the addition of radiotherapy, prior clinical series suggest that long-term local control is achieved in about 70% of the patients (22) who have resectable primary diseases. As administration of radiation doses beyond 45-50Gy with brachytherapy did not improve local control and frequently induced Grade 3–5 toxicities in the surrounding radiosensitive normal organs (138,139), combining targeted radiotherapy with STS-targeted agents may be an alternate avenue to improve the efficacy of radiotherapy, resectability, and local control. Consistent with the above observation, also because 50% of retroperitoneal STSs are LPSs (140), agents (RG7112 and PD033991) targeting MDM2 and CDK4, which are frequently amplified in LPSs, have been tested mainly in LPSs originating from the retroperitoneum (85% to 97% of the study patients) (91,104). Thus, the combination of radiotherapy with these agents in tumors with wild-type p53, or immunotherapy agents such as nivolumab or antiangiogenesis inhibitors such as pazopanib in p53 mutant tumors, could be the next steps for clinical and translational investigations in nonextremity STS. As the normal tissues in these regions are highly radiosensitive, preclinical data on normal tissue effects will be needed to select for the optimal agents and dose regimens (for the drugs and radiotherapy) to proceed to clinical trials.
Oligometastatic Disease
Eighty to ninety percent of STSs metastasize to the lungs. Selected patients who undergo metastatectomies have five-year overall survival of 13% to 44% (141). However, not all patients are amenable to surgery, and, in the absence of efficacious chemotherapy, stereotactic body radiotherapy (SBRT) of STS lung metastasis may provide an alternative treatment. Multiple prospective studies have demonstrated that SBRT for inoperable primary lung cancers yields a local control rate approximating 90% (142–145). Although evidence supporting the use of SBRT in the management of oligometastatic lung disease is less robust, published retrospective studies of its use in inoperable STS pulmonary metastases suggest that this treatment is both tolerable (no Grade 3 acute toxicities) and efficacious in controlling lung disease (two-year local control: 88% to 90%) (146–148). The NRG oncology sarcoma working group is currently designing a prospective randomized trial to evaluate this highly precise radiation technique in combination with different targeted agents for different STS subtypes in the treatment of inoperable pulmonary sarcoma oligometastasis. It is anticipated that important knowledge on each agent’s effect and interaction with radiotherapy through these proposed trials will help identify an effective method of long-term maintenance therapy in patients with limited metastasis after effective local radiotherapy.
STSs With Pathognomonic Mutations
Inflammatory myofibroblastic tumors (IMTs) are rare entities with a wide range of local aggressiveness and low (<5%) metastatic potential. Following complete excision, local recurrence rates are high, especially in IMTs expressing anaplastic lymphoma kinase (ALK) from rearrangements at 2p23, which occurs in 50% of IMTs (149). Prior case series described the potential activity of crizotinib, an ALK inhibitor in IMTs overexpressing ALK (150,151). The combination of crizotinib with radiation was investigated preclinically using non–small cell lung cancer cells and suggested that ALK inhibition might radiosensitize cells harboring specific EML4-ALK fusions (152).
Synovial sarcoma represent approximately 10% of STSs. The majority carry the pathognomonic t (X;18) reciprocal translocation that results in the SS18-SSX fusion protein that deregulates the SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeling complex (153). The associations between SS18-SSX, SWI/SNF, and chromatin structure suggest that molecules altering chromatin structures, such as histone deacetylase (HDAC) inhibitors, could be exploited as a therapeutic strategy in this disease. As HDAC inhibitors may also inhibit DNA double strand break repairs, combining HDAC inhibitors with radiotherapy may improve the efficacy of radiation therapy, a strategy that showed promise in osteosarcoma and rhabdomyosarcoma cell lines (154), and could be evaluated for synovial sarcoma.
Translocations involving the EWSR1 gene are found the Ewing STS family, including Ewing’s sarcoma, clear cell sarcoma, and primitive neuroectodermal tumors. The resulting fusion products interact with PARP-1 or predict for the tumor’s sensitivity to PARP inhibitors (155,156). A preclinical study demonstrated that the combination of PARP inhibition synergistically increased the cytotoxicity of radiotherapy in Ewing sarcoma cells and xenografts (157). There are six trials involving PARP inhibitors and Ewing’s sarcomas registered on ClinicalTrials.gov; none of these trials are evaluating the combination of PARP inhibition with radiotherapy.
PDGF/PDGFR pathways are implicated in multiple subtypes of sarcomas, among which dermatofibrosarcoma protruberans (DFSP) are known for t (17;22) translocations that result in a COL1A1-PDGFB fusion protein that stimulates the cell’s PDGFR through an autocrine signaling mechanism (158). Logically, PDGFR inhibition using Imatinib was explored and is now approved for the treatment of DFSP (159). Akin to the inhibition of cell survival pathways by EGFR or VEGFR inhibition, PDGFR inhibition using imatinib increased radiosensitivity in glioma models (160). Preclinical evaluation of the effects of PDGFR inhibition on radiation sensitivity in sarcoma and DFSP is lacking.
Risk of Surgical Complications
If targeted agents are tested in combination with neoadjuvant radiotherapy in clinical trials, then the rate of surgical complication should be assessed. In carcinomas, prospective trials suggest that the addition of bevacizumab to preoperative chemoradiotherapy or radiotherapy does not increase wound complications in rectal (161) or liver surgeries (162); the rate of postoperative complications was not reduced in patients who stopped bevacizumab two months vs one month before surgery (163). However, other feasibility trials observed unacceptable rates of wound complications from the addition of bevacizumab to neoadjuvant oxaliplatin, 5-FU and radiotherapy (rectal cancer) (164) or cisplatin (breast cancer) (165). For tyrosine kinase inhibitors, complication rates from nephrectomies of renal cancers were not increased by the preoperative use of sorafenib (166) or sunitinib (167,168), which were stopped zero to five days prior to surgery. Wound complication rates will be estimated in the COG-NRG trial (ARST1321), in which preoperative chemoradiotherapy or radiotherapy with or without pazopanib is used to treat extremity STS.
Developing New Therapies
Sarcomas, like childhood cancers, are rare diseases with approximately 12000 new cases per year in the United States (Figure 5). However, whereas the COG enters approximately 90% of patients on protocol (169), there is no cooperative group in the United States focused to a similar extent on adult sarcoma patients. Organizations such as the Sarcoma Alliance for Research through Collaboration (SARC) partially fill this void. One approach developed for identifying new therapeutics to treat pediatric cancers is the Pediatric Preclinical Testing Program (PPTP) (170). The PPTP developed panels of molecularly characterized pediatric cancer xenograft and cell lines with the primary objective of identifying novel drugs in early clinical development or drug combinations that show broad spectrum or histotype-selective efficacy. Based on PPTP testing, several novel agents and combinations have entered phase I trials and are progressing to phase II trials in pediatric cancers. Development of a similar program for adult sarcomas that includes combinations with radiation therapy would facilitate more rapid development of new and more effective therapies for adult sarcoma patients.
Figure 5.
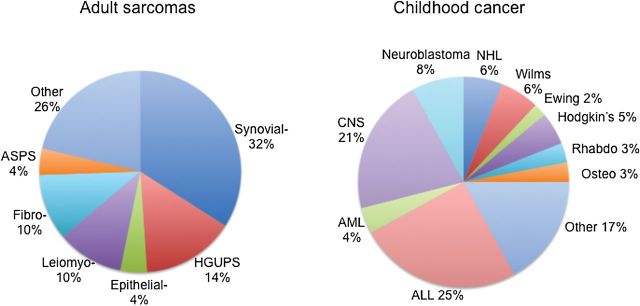
Distribution of adult sarcomas and childhood cancers. ASPS = alveolar soft part sarcoma (ASPS); ALL = acute lymphoblastic leukemia; AML = acute myeloid leukemia; CNS = central nervous system; Epithelial- = epithelial sarcoma; Fibro- = fibrosarcoma; HGUPS = high-grade undifferentiated pleomorphic sarcoma; Leiomyo-= leiomyosarcoma; NHL = non-Hodgkin’s lymphoma; Osteo = osteosarcoma; Rhabdo = rhabdomyosarcoma; Synovial- = synovial sarcoma.
Although investigations in genomics in STS is less advanced than in carcinomas, it has helped define pathognomonic changes for certain STS subtypes (171), as well as potential therapeutic targets in others. Few prognostic biomarkers have been identified and validated in STS (172–175). Furthermore, the impact of prognostic markers is untested in the clinical setting, as no effective systemic drugs are available. Thus, predictive biomarkers for different agents in combination with traditional cytotoxic treatments are being sought, which would complement ongoing histology and genomic-driven systemic therapies (176). The upcoming COG-NRG Pazopanib STS trial (ARST1321) will pursue the correlations between the patterns of failure and patient survival with actionable mutations found via whole genome sequencing and circulating tumor DNA. The clinical genomic-outcome data will be tested in preclinical models to increase understanding of STS biology and aid development of new translational hypotheses (Figure 6).
Figure 6.

A roadmap for the advancement of radiotherapy research in sarcoma. The NRG Oncology sarcoma group can serve to coordinate with other NRG Oncology site groups and collaborative groups (Sarcoma Alliance for Research through Collaboration [SARC], Children’s Oncology Group (COG), European Organisation for Research and Treatment of Cancer [EORTC], others) to initiate and solve pertinent clinical and translational questions in sarcoma. Radiotherapy-related issues can initially be tested via the NRG Oncology sarcoma group resources using the proposed sarcoma preclinical testing program (SPTP) in collaboration with the National Cancer Institute (NCI) Radiation Research Program and Molecular Pharmacology Branch. The SPTP would provide a platform to test novel combinations between radiotherapy and molecular agents across multiple histotype specific in vitro/in vivo models with different genomic alterations. These experiments may subsequently lead to important prognostic and predictive markers to better segregate patients to different treatment regimens. Complementarily, sarcoma biopsy specimens may be tested for their response to combination treatments using microfluidic chips. Available tissue microarrays (TMAs) and tissue banks from prior RTOG trials can be used to confirm laboratory findings or serve as feedback to generate new hypotheses for further elucidation at the laboratory level. With ongoing NRG Oncology trials integrating tissue collection and translational questions, these studies would further expand the NRG Oncology sarcoma group’s resources and capacity to lead future translational research and trials. RTOG = Radiation Therapy Oncology Group.
Conclusions
In summary, novel molecular agents are seldom examined in combination with radiotherapy, and hence their combined toxicities and efficacies are understudied, even though biologically sound hypotheses for synergisms for their combined use in STS exist. A number of possibilities, many of which could be utilized in the near future, have been described in this report, and there are other potential targets that could be developed. With the routine use of preoperative radiotherapy in the management of STS, these diseases present an ideal clinical setting for the evaluation of translational questions, as pretreatment core biopsies and postneoadjuvant treatment surgical specimens are regularly obtained. Paired comparisons of pre- and post-treatment specimens can be made to determine the tumor biological response and normal tissue toxicities from different neoadjuvant combined regimens, thus allowing for basket trial approaches to accelerate the implementation of targeted agents into radiotherapy regimens (177). Further success in advancing the field of radiotherapy in STS will be determined by the upcoming clinical trials that will demand strong support and joint efforts to translate the fundamental science to clinical use by defining the following: Who requires more aggressive local and/or systemic treatments? Which combinations of STS subtype, molecular agents, and radiotherapy timing are safe and efficacious? Can immunotherapy and radiotherapy be combined to improve local control and mount a systemic immune effect? The answers to these questions are the basis to precision oncology and personalized medicine, which will in effect lead to optimal evaluation of new agents and ideas in these rare diseases through international and/or national sarcoma collaborations.
To encourage and accelerate preclinical studies, the NCI-NRG Oncology sarcoma working group has amalgamated in vitro and in vivo models among its members to encourage collaborative research through shared resources. The sarcoma group has and is developing additional proposals for clinical trials that integrate preclinical data from both tumors and normal tissues. The preclinical barrier can be bridged with appropriate models and timely development of data. In addition, research identifying new targeted therapy for metastatic STS from SARC via industry-sponsored research or the Specialized Program of Research Excellence (SPORE) grant awarded to SARC may be very helpful in selecting potential agents to combine with radiotherapy in localized STS in the future. In order for the NCI to achieve its important goal of launching trials for less common diseases such as sarcoma (The Cancer Letter, April 16, 2010) the NRG sarcoma working group and collaborating scientists have developed a research plan for which support for the essential preclinical studies and clinical trials is necessary. This effort addresses the critically important challenge presented by Dr. James Doroshow (Director of the NCI Division of Cancer Treatment and Diagnosis), “This new [cooperative group clinical trials] network system will not be effective if we cannot launch trials in some less common cancers such as head and neck and sarcomas…” And we agree with the important goal of this new structure in this transformational period for clinical trials: “If we have a new system three years from now and we still can’t do trials in sarcoma or head and neck cancer, we haven’t really done much…” (The Cancer Letter, December 17, 2010). NRG sarcoma working group, SARC, and other sarcoma groups will be able to complement each other to identify targeted therapies and new management strategies to improve the outcome for patients with STS.
Funding
This work was supported by the Radiation Therapy Oncology Group (National Cancer Institute grant number U10 CA21661-36), the Radiation Therapy Oncology Foundation, and NRG Oncology (U10 CA180868).
The study funders had no role in the writing of the manuscript or the decision to submit the manuscript for publication.
References
- 1. Lin SH, George TJ, Ben-Josef E, et al. Opportunities and challenges in the era of molecularly targeted agents and radiation therapy. J Natl Cancer Inst. 2013;105(10):686–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lawrence YR, Vikram B, Dignam JJ, et al. NCI-RTOG translational program strategic guidelines for the early-stage development of radiosensitizers. J Natl Cancer Inst. 2013;105(1):11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fletcher CDM, Unni KK, Mertens F, et al. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002 [Google Scholar]
- 4. Fletcher CD, Gustafson P, Rydholm A, et al. Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: prognostic relevance of subclassification. J Clin Oncol. 2001;19(12):3045–3050 [DOI] [PubMed] [Google Scholar]
- 5. Dei Tos AP, Doglioni C, Piccinin S, et al. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumours. J Pathol. 2000;190(5):531–536 [DOI] [PubMed] [Google Scholar]
- 6. Folpe AL, Inwards CY. Bone and soft tissue pathology. Philadelphia, PA: Saunders Elsevier; 2010 [Google Scholar]
- 7. Demicco EG. Sarcoma diagnosis in the age of molecular pathology. Adv Anat Pathol. 2013;20(4):264–274 [DOI] [PubMed] [Google Scholar]
- 8. Demicco EG, Maki RG, Lev DC, et al. New therapeutic targets in soft tissue sarcoma. Adv Anat Pathol. 2012;19(3):170–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet. 2002;359(9325):2235–2241 [DOI] [PubMed] [Google Scholar]
- 10. Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg. 1982;196(3):305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Italiano A, Delva F, Mathoulin-Pelissier S, et al. Effect of adjuvant chemotherapy on survival in FNCLCC grade 3 soft tissue sarcomas: a multivariate analysis of the French Sarcoma Group Database. Ann Oncol. 2010;21(12):2436–2441 [DOI] [PubMed] [Google Scholar]
- 12. Verma S, Younus J, Stys-Norman D, et al. Meta-analysis of ifosfamide-based combination chemotherapy in advanced soft tissue sarcoma. Cancer Treat Rev. 2008;34(4):339–347 [DOI] [PubMed] [Google Scholar]
- 13. Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008;113(3):573–581 [DOI] [PubMed] [Google Scholar]
- 14. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet. 1997;350(9092):1647–1654 [PubMed] [Google Scholar]
- 15. Le Cesne A, Van Glabbeke M, Woll PJ, et al. The end of adjuvant chemotherapy (adCT) era with doxorubicin-based regimen in resected high-grade soft tissue sarcoma (STS): Pooled analysis of the two STBSG-EORTC phase III clinical trials. In. 2008 ASCO Annual Meeting. J Clin Oncol; 2008; Abstract 10525.
- 16. Alektiar KM, Leung D, Zelefsky MJ, et al. Adjuvant radiation for stage II-B soft tissue sarcoma of the extremity. J Clin Oncol. 2002;20(6):1643–1650 [DOI] [PubMed] [Google Scholar]
- 17. Yang JC, Chang AE, Baker AR, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol. 1998;16(1):197–203 [DOI] [PubMed] [Google Scholar]
- 18. Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol. 1996;14(3):859–868 [DOI] [PubMed] [Google Scholar]
- 19. Stojadinovic A, Leung DH, Allen P, et al. Primary adult soft tissue sarcoma: time-dependent influence of prognostic variables. J Clin Oncol. 2002;20(21):4344–4352 [DOI] [PubMed] [Google Scholar]
- 20. LeVay J, O’Sullivan B, Catton C, et al. Outcome and prognostic factors in soft tissue sarcoma in the adult. Int J Radiat Oncol Biol Phys. 1993;27(5):1091–1099 [DOI] [PubMed] [Google Scholar]
- 21. Lewis JJ, Leung D, Woodruff JM, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg. 1998;228(3):355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swallow CJ, Catton CN. Improving outcomes for retroperitoneal sarcomas: a work in progress. Surg Oncol Clin N Am. 2012;21(2):317–331 [DOI] [PubMed] [Google Scholar]
- 23. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11(4):239–253 [DOI] [PubMed] [Google Scholar]
- 24. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186 [DOI] [PubMed] [Google Scholar]
- 25. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674 [DOI] [PubMed] [Google Scholar]
- 26. Awada A, Hendlisz A, Gil T, et al. Phase I safety and pharmacokinetics of BAY 43–9006 administered for 21 days on/7 days off in patients with advanced, refractory solid tumours. Br J Cancer. 2005;92(10):1855–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Strumberg D, Richly H, Hilger RA, et al. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43–9006 in patients with advanced refractory solid tumors. J Clin Oncol. 2005;23(5):965–972 [DOI] [PubMed] [Google Scholar]
- 28. Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34 [DOI] [PubMed] [Google Scholar]
- 29. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390 [DOI] [PubMed] [Google Scholar]
- 30. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–134 [DOI] [PubMed] [Google Scholar]
- 31. Santoro A, Comandone A, Basso U, et al. Phase II prospective study with sorafenib in advanced soft tissue sarcomas after anthracycline-based therapy. Ann Oncol. 2013;24(4):1093–1098 [DOI] [PubMed] [Google Scholar]
- 32. Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27(19):3133–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Canter RJ, Borys D, Olusanya A, et al. Phase I Trial of Neoadjuvant Conformal Radiotherapy Plus Sorafenib for Patients with Locally Advanced Soft Tissue Sarcoma of the Extremity. Ann Surg Oncol. 2014;21(5):1616–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meyer JM, Perlewitz KS, Hayden JB, et al. Phase I trial of preoperative chemoradiation plus sorafenib for high-risk extremity soft tissue sarcomas with dynamic contrast-enhanced MRI correlates. Clin Cancer Res. 2013;19(24):6902–6911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dubois SG, Shusterman S, Ingle AM, et al. Phase I and pharmacokinetic study of sunitinib in pediatric patients with refractory solid tumors: a children’s oncology group study. Clin Cancer Res. 2011;17(15):5113–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maris JM, Courtright J, Houghton PJ, et al. Initial testing (stage 1) of sunitinib by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;51(1):42–48 [DOI] [PubMed] [Google Scholar]
- 37. Zhang L, Smith KM, Chong AL, et al. In vivo antitumor and antimetastatic activity of sunitinib in preclinical neuroblastoma mouse model. Neoplasia. 2009;11(5):426–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barrios CH, Liu MC, Lee SC, et al. Phase III randomized trial of sunitinib versus capecitabine in patients with previously treated HER2-negative advanced breast cancer. Breast Cancer Res Treat. 2010;121(1):121–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bergh J, Bondarenko IM, Lichinitser MR, et al. First-line treatment of advanced breast cancer with sunitinib in combination with docetaxel versus docetaxel alone: results of a prospective, randomized phase III study. J Clin Oncol. 2012;30(9):921–929 [DOI] [PubMed] [Google Scholar]
- 40. Saltz LB, Rosen LS, Marshall JL, et al. Phase II trial of sunitinib in patients with metastatic colorectal cancer after failure of standard therapy. J Clin Oncol. 2007;25(30):4793–4799 [DOI] [PubMed] [Google Scholar]
- 41. Carrato A, Swieboda-Sadlej A, Staszewska-Skurczynska M, et al. Fluorouracil, leucovorin, and irinotecan plus either sunitinib or placebo in metastatic colorectal cancer: a randomized, phase III trial. J Clin Oncol. 2013;31(10):1341–1347 [DOI] [PubMed] [Google Scholar]
- 42. Han JY, Kim HY, Lim KY, et al. A phase II study of sunitinib in patients with relapsed or refractory small cell lung cancer. Lung Cancer. 2013;79(2):137–142 [DOI] [PubMed] [Google Scholar]
- 43. Scagliotti GV, Krzakowski M, Szczesna A, et al. Sunitinib plus erlotinib versus placebo plus erlotinib in patients with previously treated advanced non-small-cell lung cancer: a phase III trial. J Clin Oncol. 2012;30(17):2070–2078 [DOI] [PubMed] [Google Scholar]
- 44. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–1338 [DOI] [PubMed] [Google Scholar]
- 45. Mahmood ST, Agresta S, Vigil CE, et al. Phase II study of sunitinib malate, a multitargeted tyrosine kinase inhibitor in patients with relapsed or refractory soft tissue sarcomas. Focus on three prevalent histologies: leiomyosarcoma, liposarcoma and malignant fibrous histiocytoma. Int J Cancer. 2011;129(8):1963–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. George S, Merriam P, Maki RG, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J Clin Oncol. 2009;27(19):3154–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wachsberger P, Burd R, Dicker AP. Tumor response to ionizing radiation combined with antiangiogenesis or vascular targeting agents: exploring mechanisms of interaction. Clin Cancer Res. 2003;9(6):1957–1971 [PubMed] [Google Scholar]
- 48. Rajendran JG, Wilson DC, Conrad EU, et al. [(18)F]FMISO and [(18)F]FDG PET imaging in soft tissue sarcomas: correlation of hypoxia, metabolism and VEGF expression. Eur J Nucl Med Mol Imaging. 2003;30(5):695–704 [DOI] [PubMed] [Google Scholar]
- 49. Yoon SS, Stangenberg L, Lee YJ, et al. Efficacy of sunitinib and radiotherapy in genetically engineered mouse model of soft-tissue sarcoma. Int J Radiat Oncol Biol Phys. 2009;74(4):1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hui EP, Ma BB, King AD, et al. Hemorrhagic complications in a phase II study of sunitinib in patients of nasopharyngeal carcinoma who has previously received high-dose radiation. Ann Oncol. 2011;22(6):1280–1287 [DOI] [PubMed] [Google Scholar]
- 51. Chi KH, Liao CS, Chang CC, et al. Angiogenic blockade and radiotherapy in hepatocellular carcinoma. Int J Radiat Oncol Biol Phys. 2010;78(1):188–193 [DOI] [PubMed] [Google Scholar]
- 52. Kao J, Chen CT, Tong CC, et al. Concurrent sunitinib and stereotactic body radiotherapy for patients with oligometastases: Final report of a prospective clinical trial. Target Oncol. 2013;9(2):145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Staehler M, Haseke N, Stadler T, et al. Feasibility and effects of high-dose hypofractionated radiation therapy and simultaneous multi-kinase inhibition with sunitinib in progressive metastatic renal cell cancer. Urol Oncol. 2012;30(3):290–293 [DOI] [PubMed] [Google Scholar]
- 54. Corn PG, Song DY, Heath E, et al. Sunitinib plus androgen deprivation and radiation therapy for patients with localized high-risk prostate cancer: results from a multi-institutional phase 1 study. Int J Radiat Oncol Biol Phys. 2013;86(3):540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wuthrick EJ, Kamrava M, Curran WJ, Jr, et al. A phase 1b trial of the combination of the antiangiogenic agent sunitinib and radiation therapy for patients with primary and metastatic central nervous system malignancies. Cancer. 2011;117(24):5548–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kao J, Packer S, Vu HL, et al. Phase 1 study of concurrent sunitinib and image-guided radiotherapy followed by maintenance sunitinib for patients with oligometastases: acute toxicity and preliminary response. Cancer. 2009;115(15):3571–3580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yuasa T, Kitsukawa S, Sukegawa G, et al. Early onset recall pneumonitis during targeted therapy with sunitinib. BMC Cancer. 2013;13:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kelly PJ, Weiss SE, Sher DJ, et al. Sunitinib-induced pseudoprogression after whole-brain radiotherapy for metastatic renal cell carcinoma. J Clin Oncol. 2010;28(25):e433–e435 [DOI] [PubMed] [Google Scholar]
- 59. Verweij J, Sleijfer S. Pazopanib, a new therapy for metastatic soft tissue sarcoma. Expert Opin Pharmacother. 2013;14(7):929–935 [DOI] [PubMed] [Google Scholar]
- 60. van Geel RM, Beijnen JH, Schellens JH. Concise drug review: pazopanib and axitinib. Oncologist. 2012;17(8):1081–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369(8):722–731 [DOI] [PubMed] [Google Scholar]
- 62. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–1886 [DOI] [PubMed] [Google Scholar]
- 63. Bramwell VH. Pazopanib and the treatment palette for soft-tissue sarcoma. Lancet. 2012;379(9829):1854–1856 [DOI] [PubMed] [Google Scholar]
- 64. Sleijfer S, Ray-Coquard I, Papai Z, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). J Clin Oncol. 2009;27(19):3126–3132 [DOI] [PubMed] [Google Scholar]
- 65. Goyal S, Shah S, Khan AJ, et al. Evaluation of acute locoregional toxicity in patients with breast cancer treated with adjuvant radiotherapy in combination with pazopanib. ISRN Oncology. 2012;2012:896202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Presta LG, Chen H, O’Connor SJ, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57(20):4593–4599 [PubMed] [Google Scholar]
- 67. Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333(2):328–335 [DOI] [PubMed] [Google Scholar]
- 68. Myers AL, Williams RF, Ng CY, et al. Bevacizumab-induced tumor vessel remodeling in rhabdomyosarcoma xenografts increases the effectiveness of adjuvant ionizing radiation. J Pediatr Surg. 2010;45(6):1080–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zietz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Pathol. 1998;153(5):1425–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24(1):257–263 [DOI] [PubMed] [Google Scholar]
- 71. Koontz BF, Miles EF, Rubio MA, et al. Preoperative radiotherapy and bevacizumab for angiosarcoma of the head and neck: two case studies. Head Neck. 2008;30(2):262–266 [DOI] [PubMed] [Google Scholar]
- 72. De Yao JT, Sun D, Powell AT, et al. Scalp Angiosarcoma Remission with Bevacizumab and Radiotherapy without Surgery: A Case Report and Review of the Literature. Sarcoma. 2011;2011:160369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yoon SS, Duda DG, Karl DL, et al. Phase II study of neoadjuvant bevacizumab and radiotherapy for resectable soft tissue sarcomas. Int J Radiat Oncol Biol Phys. 2011;81(4):1081–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stojadinovic A, Leung DH, Hoos A, et al. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tissue sarcomas. Ann Surg. 2002;235(3):424–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–283 [DOI] [PubMed] [Google Scholar]
- 76. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331 [DOI] [PubMed] [Google Scholar]
- 77. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303 [DOI] [PubMed] [Google Scholar]
- 78. Yang Y, Li CC, Weissman AM. Regulating the p53 system through ubiquitination. Oncogene. 2004;23(11):2096–2106 [DOI] [PubMed] [Google Scholar]
- 79. Oliner JD, Kinzler KW, Meltzer PS, et al. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358(6381):80–83 [DOI] [PubMed] [Google Scholar]
- 80. Momand J, Zambetti GP, Olson DC, et al. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69(7):1237–1245 [DOI] [PubMed] [Google Scholar]
- 81. Wang S, Zhao Y, Bernard D, et al. Targeting the MDM2-p53 protein-protein interaction for new cancer therapeutics. Top Med Chem. 2012;8:57–80 [Google Scholar]
- 82. Momand J, Jung D, Wilczynski S, et al. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26(15):3453–3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rosai J, Akerman M, Dal Cin P, et al. Combined morphologic and karyotypic study of 59 atypical lipomatous tumors. Evaluation of their relationship and differential diagnosis with other adipose tissue tumors (a report of the CHAMP Study Group). Am J Pathol. 1996;20(10):1182–1189 [DOI] [PubMed] [Google Scholar]
- 84. Pedeutour F, Forus A, Coindre JM, et al. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer. 1999;24(1):30–41 [PubMed] [Google Scholar]
- 85. Fletcher CD, Akerman M, Dal Cin P, et al. Correlation between clinicopathological features and karyotype in lipomatous tumors. A report of 178 cases from the Chromosomes and Morphology (CHAMP) Collaborative Study Group. Am J Pathol. 1996;148(2):623–630 [PMC free article] [PubMed] [Google Scholar]
- 86. Aleixo PB, Hartmann AA, Menezes IC, et al. Can MDM2 and CDK4 make the diagnosis of well differentiated/dedifferentiated liposarcoma? An immunohistochemical study on 129 soft tissue tumours. J Clin Pathol. 2009;62(12):1127–1135 [DOI] [PubMed] [Google Scholar]
- 87. Nakayama T, Toguchida J, Wadayama B, et al. MDM2 gene amplification in bone and soft-tissue tumors: association with tumor progression in differentiated adipose-tissue tumors. Int J Cancer. 1995;64(5):342–346 [DOI] [PubMed] [Google Scholar]
- 88. Singer S, Socci ND, Ambrosini G, et al. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res. 2007;67(14):6626–6636 [DOI] [PubMed] [Google Scholar]
- 89. Shen H, Maki CG. Pharmacologic activation of p53 by small-molecule MDM2 antagonists. Curr Pharm Des. 2011;17(6):560–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105(10):3933–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ray-Coquard I, Blay JY, Italiano A, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13(11):1133–1140 [DOI] [PubMed] [Google Scholar]
- 92. Yu S, Qin D, Shangary S, et al. Potent and orally active small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2009;52(24):7970–7973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vu BT, Vassilev L. Small-molecule inhibitors of the p53-MDM2 interaction. Curr Top Microbiol Immunol. 2011;348:151–172 [DOI] [PubMed] [Google Scholar]
- 94. Tovar C, Rosinski J, Filipovic Z, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A. 2006;103(6):1888–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Crago AM, Singer S. Clinical and molecular approaches to well differentiated and dedifferentiated liposarcoma. Curr Opin Oncol. 2011;23(4):373–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Carol H, Reynolds CP, Kang MH, et al. Initial testing of the MDM2 inhibitor RG7112 by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2013;60(4):633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. UniProtKB/Swiss-Prot. P11802 (CDK4_HUMAN). Available at: http://www.uniprot.org/uniprot/P11802. Accessed September 18, 2013 [Google Scholar]
- 98. Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–1438 [PubMed] [Google Scholar]
- 99. Hagen KR, Zeng X, Lee MY, et al. Silencing CDK4 radiosensitizes breast cancer cells by promoting apoptosis. Cell Div. 2013;8(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shimura T, Kakuda S, Ochiai Y, et al. Targeting the AKT/GSK3beta/cyclin D1/Cdk4 survival signaling pathway for eradication of tumor radioresistance acquired by fractionated radiotherapy. Int J Radiat Oncol Biol Phys. 2011;80(2):540–548 [DOI] [PubMed] [Google Scholar]
- 101. Shimura T, Noma N, Oikawa T, et al. Activation of the AKT/cyclin D1/Cdk4 survival signaling pathway in radioresistant cancer stem cells. Oncogenesis. 2012;1:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Flaherty KT, Lorusso PM, Demichele A, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;18(2):568–576 [DOI] [PubMed] [Google Scholar]
- 104. Dickson MA, Tap WD, Keohan ML, et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol. 2013;31(16):2024–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Toulany M, Rodemann HP. Potential of Akt mediated DNA repair in radioresistance of solid tumors overexpressing erbB-PI3K-Akt pathway. Transl Cancer Res. 2013;2(3):190–202 [Google Scholar]
- 106. Fedrigo CA, Grivicich I, Schunemann DP, et al. Radioresistance of human glioma spheroids and expression of HSP70, p53 and EGFr. Radiat Oncol. 2011;6:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Liu Y, Cui B, Qiao Y, et al. Phosphoinositide-3-kinase inhibition enhances radiosensitization of cervical cancer in vivo. Int J Gynecol Cancer. 2011;21(1):100–105 [DOI] [PubMed] [Google Scholar]
- 108. Price BD, Youmell MB. The phosphatidylinositol 3-kinase inhibitor wortmannin sensitizes murine fibroblasts and human tumor cells to radiation and blocks induction of p53 following DNA damage. Cancer Res. 1996;56(2):246–250 [PubMed] [Google Scholar]
- 109. Nagata Y, Takahashi A, Ohnishi K, et al. Effect of rapamycin, an mTOR inhibitor, on radiation sensitivity of lung cancer cells having different p53 gene status. Int J Oncol. 2010;37(4):1001–1010 [DOI] [PubMed] [Google Scholar]
- 110. Toulany M, Lee KJ, Fattah KR, et al. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Res. 2012;10(7):945–957 [DOI] [PubMed] [Google Scholar]
- 111. Tanno S, Yanagawa N, Habiro A, et al. Serine/threonine kinase AKT is frequently activated in human bile duct cancer and is associated with increased radioresistance. Cancer Res. 2004;64(10):3486–3490 [DOI] [PubMed] [Google Scholar]
- 112. Li HF, Kim JS, Waldman T. Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells. Radiat Oncol. 2009;4:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lee CM, Fuhrman CB, Planelles V, et al. Phosphatidylinositol 3-kinase inhibition by LY294002 radiosensitizes human cervical cancer cell lines. Clin Cancer Res. 2006;12(1):250–256 [DOI] [PubMed] [Google Scholar]
- 114. Kao GD, Jiang Z, Fernandes AM, et al. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem. 2007;282(29):21206–21212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mukherjee B, Tomimatsu N, Amancherla K, et al. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012;14(1):34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Moding EJ, Kastan MB, Kirsch DG. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat Rev Drug Discov. 2013;12(7):526–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gutierrez A, Snyder EL, Marino-Enriquez A, et al. Aberrant AKT activation drives well-differentiated liposarcoma. Proc Natl Acad Sci U S A. 2011;108(39):16386–16391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kim S, Dodd RD, Mito JK, et al. Efficacy of phosphatidylinositol-3 kinase inhibitors in a primary mouse model of undifferentiated pleomorphic sarcoma. Sarcoma. 2012;2012:680708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Marklein D, Graab U, Naumann I, et al. PI3K inhibition enhances doxorubicin-induced apoptosis in sarcoma cells. PloS One. 2012;7(12):e52898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hosaka S, Horiuchi K, Yoda M, et al. A novel multi-kinase inhibitor pazopanib suppresses growth of synovial sarcoma cells through inhibition of the PI3K-AKT pathway. J Orthop Res. 2012;30(9):1493–1498 [DOI] [PubMed] [Google Scholar]
- 121. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kim JR, Moon YJ, Kwon KS, et al. Tumor Infiltrating PD1-Positive Lymphocytes and the Expression of PD-L1 Predict Poor Prognosis of Soft Tissue Sarcomas. PloS One. 2013;8(12):e82870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8(6):467–477 [DOI] [PubMed] [Google Scholar]
- 124. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zeng J, Harris TJ, Lim M, et al. Immune Modulation and Stereotactic Radiation: Improving Local and Abscopal Responses. BioMed Res Int. 2013;2013:658126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sharma A, Bode B, Studer G, et al. Radiotherapy of human sarcoma promotes an intratumoral immune effector signature. Clin Cancer Res. 2013;19(17):4843–4853 [DOI] [PubMed] [Google Scholar]
- 127. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Candido KA, Shimizu K, McLaughlin JC, et al. Local administration of dendritic cells inhibits established breast tumor growth: implications for apoptosis-inducing agents. Cancer Res. 2001;61(1):228–236 [PubMed] [Google Scholar]
- 129. Nikitina EY, Gabrilovich DI. Combination of gamma-irradiation and dendritic cell administration induces a potent antitumor response in tumor-bearing mice: approach to treatment of advanced stage cancer. Int J Cancer. 2001;94(6):825–833 [DOI] [PubMed] [Google Scholar]
- 130. Teitz-Tennenbaum S, Li Q, Rynkiewicz S, et al. Radiotherapy potentiates the therapeutic efficacy of intratumoral dendritic cell administration. Cancer Res. 2003;63(23):8466–8475 [PubMed] [Google Scholar]
- 131. Finkelstein SE, Iclozan C, Bui MM, et al. Combination of external beam radiotherapy (EBRT) with intratumoral injection of dendritic cells as neo-adjuvant treatment of high-risk soft tissue sarcoma patients. Int J Radiat Oncol Biol Phys. 2012;82(2):924–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang D, Zhang Q, Eisenberg B, et al. Significant reduction of radiation related morbidities in the extremity sarcoma patients treated with image-guided radiotherapy to reduced target volume: results of RTOG 0630. In: ASTRO Annual Meeting. Atlanta, GA, 2013: Abstract 87 (suppl 2), p. S63. Int J Radiat Oncol Biol Phys. [Google Scholar]
- 133. Davis AM, O’Sullivan B, Turcotte R, et al. Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol. 2005;75(1):48–53 [DOI] [PubMed] [Google Scholar]
- 134. O’Sullivan B, Griffin AM, Dickie CI, et al. Phase 2 study of preoperative image-guided intensity-modulated radiation therapy to reduce wound and combined modality morbidities in lower extremity soft tissue sarcoma. Cancer. 2013;119(10):1878–1884 [DOI] [PubMed] [Google Scholar]
- 135. Wang D, Zhang Q, Kirsch D, et al. RTOG Phase II Trial of Preoperative Image Guided Radiotherapy (IG-RT) For Primary Soft Tissue Sarcoma of the Extremity: Acute Toxicity Report. In: ASTRO annual meeting. San Diego, CA, 2011: Abstract 81 (suppl. 2), p. S117. Int J Radiat Oncol Biol Phys [Google Scholar]
- 136. Chung PW, Deheshi BM, Ferguson PC, et al. Radiosensitivity translates into excellent local control in extremity myxoid liposarcoma: a comparison with other soft tissue sarcomas. Cancer. 2009;115(14):3254–3261 [DOI] [PubMed] [Google Scholar]
- 137. Canter RJ, Qin LX, Ferrone CR, et al. Why do patients with low-grade soft tissue sarcoma die? Ann Surg Oncol. 2008;15(12):3550–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jones JJ, Catton CN, O’Sullivan B, et al. Initial results of a trial of preoperative external-beam radiation therapy and postoperative brachytherapy for retroperitoneal sarcoma. Ann Surg Oncol. 2002;9(4):346–354 [DOI] [PubMed] [Google Scholar]
- 139. Smith MJ, Ridgway PF, Catton CN, et al. Combined management of retroperitoneal sarcoma with dose intensification radiotherapy and resection: Long-term results of a prospective trial. Radiother Oncol. 2014;110(1):165–171 [DOI] [PubMed] [Google Scholar]
- 140. Dalal KM, Kattan MW, Antonescu CR, et al. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Treasure T, Fiorentino F, Scarci M, et al. Pulmonary metastasectomy for sarcoma: a systematic review of reported outcomes in the context of Thames Cancer Registry data. BMJ Open. 2012;2(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Timmerman R, Paulus R, Galvin J, et al. Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA. 2010;303(11):1070–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Videtic GM, Stephans K, Reddy C, et al. Intensity-modulated radiotherapy-based stereotactic body radiotherapy for medically inoperable early-stage lung cancer: excellent local control. Int J Radiat Oncol Biol Phys. 2010;77(2):344–349 [DOI] [PubMed] [Google Scholar]
- 144. Fakiris AJ, McGarry RC, Yiannoutsos CT, et al. Stereotactic body radiation therapy for early-stage non-small-cell lung carcinoma: four-year results of a prospective phase II study. Int J Radiat Oncol Biol Phys. 2009;75(3):677–682 [DOI] [PubMed] [Google Scholar]
- 145. Onishi H, Araki T, Shirato H, et al. Stereotactic hypofractionated high-dose irradiation for stage I nonsmall cell lung carcinoma: clinical outcomes in 245 subjects in a Japanese multiinstitutional study. Cancer. 2004;101(7):1623–1631 [DOI] [PubMed] [Google Scholar]
- 146. Mehta N, Selch M, Wang PC, et al. Safety and efficacy of stereotactic body radiation therapy in the treatment of pulmonary metastases from high grade sarcoma. Sarcoma. 2013;2013:360214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Stragliotto CL, Karlsson K, Lax I, et al. A retrospective study of SBRT of metastases in patients with primary sarcoma. Med Oncol. 2012;29(5):3431–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Dhakal S, Corbin KS, Milano MT, et al. Stereotactic body radiotherapy for pulmonary metastases from soft-tissue sarcomas: excellent local lesion control and improved patient survival. Int J Radiat Oncol Biol Phys. 2012;82(2):940–945 [DOI] [PubMed] [Google Scholar]
- 149. Lovly CM, Gupta A, Lipson D, et al. Inflammatory Myofibroblastic Tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4(8):889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Butrynski JE, D’Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363(18):1727–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wong P, Roberge D, Berthelet F, et al. Treatment of recurrent central nervous system inflammatory myofibroblastic tumor with crizotinib. In: Canadian Neuro-Oncology Meeting. Halifax, NS, 2014 [Google Scholar]
- 152. Sun Y, Nowak KA, Zaorsky NG, et al. ALK inhibitor PF02341066 (crizotinib) increases sensitivity to radiation in non-small cell lung cancer expressing EML4-ALK. Mol Cancer Ther. 2013;12(5):696–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Su L, Sampaio AV, Jones KB, et al. Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell. 2012;21(3):333–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Blattmann C, Oertel S, Ehemann V, et al. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys. 2010;78(1):237–245 [DOI] [PubMed] [Google Scholar]
- 155. Brenner JC, Feng FY, Han S, et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012;72(7):1608–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Lee HJ, Yoon C, Schmidt B, et al. Combining PARP-1 inhibition and radiation in ewing sarcoma results in lethal DNA damage. Mol Cancer Ther. 2013;12(11):2591–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Shimizu A, O’Brien KP, Sjoblom T, et al. The dermatofibrosarcoma protuberans-associated collagen type Ialpha1/platelet-derived growth factor (PDGF) B-chain fusion gene generates a transforming protein that is processed to functional PDGF-BB. Cancer Res. 1999;59(15):3719–3723 [PubMed] [Google Scholar]
- 159. McArthur G. Dermatofibrosarcoma protuberans: recent clinical progress. Ann Surg Oncol. 2007;14(10):2876–2886 [DOI] [PubMed] [Google Scholar]
- 160. Novel Therapies in Glioblastoma. Neurol Res Int. 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Scappaticci FA, Fehrenbacher L, Cartwright T, et al. Surgical wound healing complications in metastatic colorectal cancer patients treated with bevacizumab. J Surg Oncol. 2005;91(3):173–180 [DOI] [PubMed] [Google Scholar]
- 162. Kesmodel SB, Ellis LM, Lin E, et al. Preoperative bevacizumab does not significantly increase postoperative complication rates in patients undergoing hepatic surgery for colorectal cancer liver metastases. J Clin Oncol. 2008;26(32):5254–5260 [DOI] [PubMed] [Google Scholar]
- 163. Yoshioka Y, Uehara K, Ebata T, et al. Postoperative complications following neoadjuvant bevacizumab treatment for advanced colorectal cancer. Surg Today. 2014;44(7):1300–1306 [DOI] [PubMed] [Google Scholar]
- 164. Dipetrillo T, Pricolo V, Lagares-Garcia J, et al. Neoadjuvant bevacizumab, oxaliplatin, 5-fluorouracil, and radiation for rectal cancer. Int J Radiat Oncol Biol Phys. 2012;82(1):124–129 [DOI] [PubMed] [Google Scholar]
- 165. Golshan M, Garber JE, Gelman R, et al. Does neoadjuvant bevacizumab increase surgical complications in breast surgery? Ann Surg Oncol. 2011;18(3):733–737 [DOI] [PubMed] [Google Scholar]
- 166. Cowey CL, Amin C, Pruthi RS, et al. Neoadjuvant clinical trial with sorafenib for patients with stage II or higher renal cell carcinoma. J Clin Oncol. 2010;28(9):1502–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Hellenthal NJ, Underwood W, Penetrante R, et al. Prospective clinical trial of preoperative sunitinib in patients with renal cell carcinoma. J Urol. 2010;184(3):859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Powles T, Kayani I, Blank C, et al. The safety and efficacy of sunitinib before planned nephrectomy in metastatic clear cell renal cancer. Ann Oncol. 2011;22(5):1041–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. O’Leary M, Krailo M, Anderson JR, et al. Progress in childhood cancer: 50 years of research collaboration, a report from the Children’s Oncology Group. Semin Oncol. 2008;35(5):484–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Houghton PJ, Morton CL, Tucker C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49(7):928–940 [DOI] [PubMed] [Google Scholar]
- 171. Fletcher CDM, World Health Organization, International Agency for Research on Cancer. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon, France: IARC Press; 2013 [Google Scholar]
- 172. Francis P, Namlos HM, Muller C, et al. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics. 2007;8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Hoffmann AC, Danenberg KD, Taubert H, et al. A three-gene signature for outcome in soft tissue sarcoma. Clin Cancer Res. 2009;15(16):5191–5198 [DOI] [PubMed] [Google Scholar]
- 174. Chibon F, Lagarde P, Salas S, et al. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat Med. 2010;16(7):781–787 [DOI] [PubMed] [Google Scholar]
- 175. Wong P, Hui A, Su J, et al. A Potential Prognostic micro-RNA Signature for Distant Metastasis in Soft-Tissue Sarcomas In: ASTRO 2012. Boston, MA, 2012: Abstract 84 (suppl 3), p. S23-24. Int J Radiat Oncol Biol Phys. [Google Scholar]
- 176. Linch M, Miah AB, Thway K, et al. Systemic treatment of soft-tissue sarcoma-gold standard and novel therapies. Nat Rev Clin Oncol. 2014;11(4):187–202 [DOI] [PubMed] [Google Scholar]
- 177. Sleijfer S, Bogaerts J, Siu LL. Designing transformative clinical trials in the cancer genome era. J Clin Oncol. 2013;31(15):1834–1841 [DOI] [PubMed] [Google Scholar]