

SUMMARY
Arabidopsis VIM/ORTH family proteins, which have SRA-domain methylcytosine-binding activity, are required for the maintenance of DNA methylation and epigenetic gene silencing in heterochromatic regions. In this study, endogenous targets of the VIM proteins in epigenetic silencing were identified through a genome-wide transcript profiling analysis in a vim1/2/3 triple mutant. We furthermore revealed the molecular mechanisms by which the VIM proteins regulate epigenetic silencing by modulating DNA methylation and histone modification in cooperation with MET1.
Key words: VIM/ORTH, SRA, MET1, epigenetic silencing, DNA methylation, histone modification.
Abstract
Methylcytosine-binding proteins containing SRA (SET- and RING-Associated) domain are required for the establishment and/or maintenance of DNA methylation in both plants and animals. We previously proposed that Arabidopsis VIM/ORTH proteins with an SRA domain maintain DNA methylation and epigenetic gene silencing in heterochromatic regions. However, their endogenous targets of epigenetic gene silencing have not been analyzed globally and the mechanisms by which VIM proteins coordinate DNA methylation and epigenetic silencing are largely unknown. In this study, a genome-wide transcript profiling analysis revealed 544 derepressed genes in a vim1/2/3 triple mutant, including 133 known genes. VIM1 bound to promoter and transcribed regions of the up-regulated genes in vim1/2/3 and VIM deficiency caused severe DNA hypomethylation in all sequence contexts at direct VIM1 targets. We found a drastic loss of H3K9me2 at heterochromatic chromocenters in vim1/2/3 nuclei. Furthermore, aberrant changes in transcriptionally active and repressive histone modifications were observed at VIM1 targets in vim1/2/3. VIM1-binding capacity to target genes was significantly reduced in the met1 background, indicating that VIM1 primarily recognizes CG methylation deposited by MET1. Overall, our data indicate that VIM proteins regulate genome-wide epigenetic gene silencing through coordinated modulation of DNA methylation and histone modification status in collaboration with MET1.
INTRODUCTION
DNA methylation is an essential epigenetic transcriptional repression mechanism that affects numerous biological processes such as development and oncogenesis in multi-cellular eukaryotes (Goll and Bestor, 2005; Klose and Bird, 2006; Henderson and Jacobsen, 2007). DNA methylation is found primarily in the CG sequence context in animals, while DNA methylation in plants exists in three sequence contexts: CG, CHG (where H is A, C, or T), and asymmetric CHH (Chan et al., 2005; Goll and Bestor, 2005). A genome-wide study of DNA methylation revealed that 24% of CG, 6.7% CHG, and 1.7% CHH sites in the Arabidopsis genome are methylated (Cokus et al., 2008). In Arabidopsis, CG methylation is maintained primarily by the DNMT1 DNA methyltransferase subfamily protein DNA METHYLTRANSFERASE 1 (MET1), whereas CHROMOMETHYLASE 3 (CMT3) maintains CHG methylation (Kankel et al., 2003; Saze et al., 2003). DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) catalyzes methylation at asymmetric CHH sites by de novo DNA methylation (Cao and Jacobsen, 2002). DRM3, a catalytically mutated paralog of DRM2, is responsible for the establishment of de novo DNA methylation in all sequence contexts in the RNA-directed DNA methylation process by stimulating the activity of DRM2 (Henderson et al., 2010).
Concerted changes in DNA methylation and histone modification modulate the composition, structure, and dynamics of chromatin, and thereby regulate gene expression by controlling the condensation and accessibility of genomic DNA (Bird, 2002; Kouzarides, 2007; Reik, 2007). Recent studies in Arabidopsis revealed an interaction web that tightly coordinates DNA methylation and histone modification. For example, CMT3 maintains CHG methylation in cooperation with several histone methyltransferases, SU(VAR)3–9 HOMOLOG (SUVH) proteins such as KRYPTONITE/SUVH4, SUVH5, and SUVH6 (Ebbs and Bender, 2006; Johnson et al., 2007; Law and Jacobsen, 2010). The Arabidopsis SUVH family proteins appear to be recruited to target loci by preferential binding to methylated cytosine via a SET- and RING-associated (SRA) domain (Arita et al., 2008; Rajakumara et al., 2011). A further example of molecular linker between DNA methylation and histone modification is a JmjC domain-containing histone demethylase, INCREASED IN BONSAI METHYLATION 1 (IBM1). An Arabidopsis mutation defective in IBM1 causes increased histone H3 Lys 9 dimethylation (H3K9me2) levels and concomitant CHG hypermethylation (Saze et al., 2008; Miura et al., 2009). Mutation of the gene encoding histone H3 acetyltransferase, INCREASED DNA METHYLATION 1 (IDM1), in Arabidopsis also results in elevated levels of cytosine methylation (Qian et al., 2012). MET1 has an important role in maintaining histone H3 Lys 27 trimethylation (H3K27me3) patterning at specific loci (Deleris et al., 2012), and in regulating locus-directed heterochromatin silencing in cooperation with HISTONE DEACETYLASE 6 (HDA6) (To et al., 2011). Moreover, a genome-wide analysis demonstrated a strong correlation between DNA methylation and H3K9 methylation (Bernatavichute et al., 2008).
Several lines of evidence support that molecular coupling of DNA methylation and histone modification might be partially mediated through methylcytosine-binding proteins. For example, a human methyl CG-binding protein 2 (MeCP2) is able to recruit histone deacetylases to the methylated region and also associates with histone methyltransferase activity, both of which result in transcriptional repression (Jones et al., 1998; Nan et al., 1998; Fuks et al., 2003). A mammalian SRA-domain-containing methylcytosine-binding protein, Ubiquitin-like with PHD and RING Finger 1 (UHRF1; also known as Np95 or ICBP90), preferentially binds to the methylated CG residues of hemi-methylated DNA and associates with DNMT1 during replication (Bostick et al., 2007; Sharif et al., 2007; Achour et al., 2008; Liu et al., 2013). Moreover, UHRF1 has been implicated in the maintenance of histone modification through association with histone methyltransferase and deacetylase (Unoki et al., 2004; Sharif et al., 2007; Karagianni et al., 2008). Arabidopsis homologs of UHRF1, the VARIANT IN METHYLATION/ORTHRUS (VIM/ORTH) family proteins, also function as methylcytosine-binding proteins (Johnson et al., 2007; Woo et al., 2007). The VIM proteins are involved in the regulation of DNA methylation and epigenetic gene silencing at heterochromatic regions (Woo et al., 2007, 2008). In addition, a recent genome-wide DNA methylome analysis revealed that CG and CHG methylation was strongly decreased in the vim1 vim2 vim3 triple mutant (hereafter designated vim1/2/3) (Stroud et al., 2013). However, the roles of the VIM proteins in histone modification have not been investigated.
Studies involving Arabidopsis VIM proteins enhanced our understanding of the mechanistic basis for VIM-mediated epigenetic gene silencing. The VIM proteins recognize methylcytosine in any sequence context, with preferential affinity for hemi-methylated CG sites (Bostick et al., 2007; Johnson et al., 2007; Woo et al., 2007; Yao et al., 2012). UHRF1 binds both 5-methylcytosine and 5-hydroxymethylcytosine (5hmC) sites with similar affinity, whereas VIM1 binds to 5hmC sites with significantly lower affinity than it binds to 5mC sites (Frauer et al., 2011; Yao et al., 2012). It was also reported that VIM1 possesses E3 ubiquitin protein ligase activity (Kraft et al., 2008). VIM1 is associated with NtSET1, a tobacco SU(VAR)3–9 protein, indicating that VIM1 might recruit H3K9 methyltransferases during heterochromatin formation (Liu et al., 2007). However, endogenous targets of the VIM proteins for epigenetic gene silencing have not been analyzed using a genome-wide screen. Moreover, the mechanisms by which the VIM proteins coordinate maintenance of DNA methylation and epigenetic gene silencing are largely unknown.
In this study, a genome-wide expression microarray analysis was performed in the vim1/2/3 triple mutant to identify the targets of the VIM proteins. We identified 544 derepressed loci in vim1/2/3, including 133 genes encoding proteins of known function or those similar to known proteins. VIM1 bound to both the promoter and transcribed regions of the derepressed genes in vim1/2/3. Furthermore, VIM deficiency resulted in strong DNA hypomethylation in all sequence contexts at the direct targets of VIM1, and a clear reduction in H3K9me2 was observed at condensed heterochromatic regions in the vim1/2/3 mutant. The vim1/2/3 mutation also led to significant changes in transcriptionally active and repressive histone modification at the VIM1 targets. VIM1-binding capacity to its target genes was substantially reduced by the met1 mutation, suggesting that VIM1 binds its targets primarily via recognition of CG methylation. Taken together, these data strongly suggest that the VIM proteins regulate genome-wide epigenetic gene silencing through modulation of DNA methylation and histone modification in collaboration with MET1.
RESULTS
Genome-Wide Identification of Genes Misregulated in the vim1/2/3 Mutant
To obtain a global view of target loci for the VIM proteins in the Arabidopsis genome, we conducted a genome-wide gene expression profiling in 14-day-old wild-type (WT) (Columbia (Col) ecotype) and vim1/2/3 mutant plants using an Arabidopsis gene expression microarray (4×44K from Agilent Technologies). Five hundred and forty-four loci were transcriptionally up-regulated in the vim1/2/3 mutant when compared with WT plants (fold change ≥ 5.0 and p-value < 0.05), with differential gene expression observed in the 5.0–65.6-fold range (Supplemental Table 1). Of the 544 loci, 216 loci (39.7%) were annotated as various types of transposons or related elements (TEs), including CACTA-like transposase, hAT-like transposase, Mutator-like transposase, Sadhu noncoding retrotransposon, gypsy-like retrotransposon, copia-like retrotransposon, and non-LTR retrotransposon family (Figure 1A and Supplemental Table 1). Genes encoding unknown proteins (154 loci), pseudogenes (28 loci), and noncoding RNAs (ncRNAs) (13 loci) were also up-regulated in the vim1/2/3 mutant (Figure 1A and Supplemental Tables 1 and 2). Notably, 133 genes (24.4%) of known function or similar to those of known function (hereafter designated ‘known genes’) were up-regulated in vim1/2/3 (Figure 1A and Supplemental Table 3). These data indicate that the VIM1, VIM2, and VIM3 proteins have functions in maintenance of transcriptional silencing at more than 500 discrete loci throughout the genome, in addition to the previously described repression of highly repetitive heterochromatic regions (Woo et al., 2007, 2008).
Figure 1.
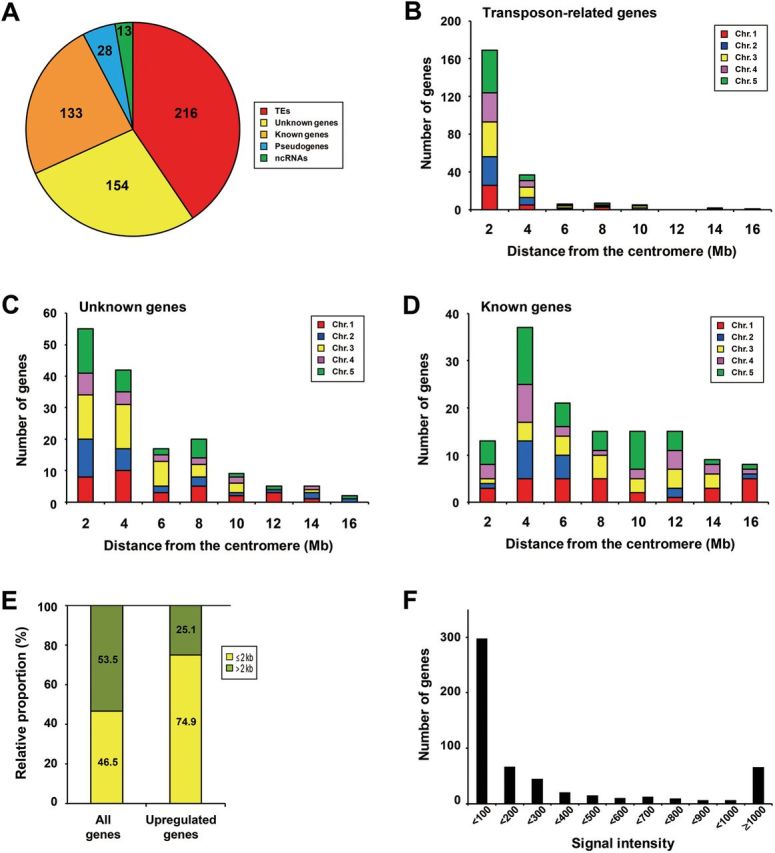
The VIM Proteins Are Required for Genome-Wide Transcriptional Gene Silencing.
(A) Categorization of loci up-regulated in the vim1/2/3 mutant in comparison with wild-type (WT): transposons or related elements (TEs) (red); genes for unknown proteins (yellow); genes for known proteins (orange); pseudogenes (blue); ncRNAs (green).
(B–D) Chromosomal positions of up-regulated TEs (B), unknown genes (C), and known genes (D) with respect to the centromere. Results for individual chromosomes are shown with the indicated colors.
(E) Relative portions of genes positioned close to TEs (within 2kb) in the up-regulated genes in vim1/2/3 and the all annotated Arabidopsis genes included in the microarray analyses. The p-value of enrichment for genes proximal to TEs was calculated using the hypergeometric distribution, based on the information about 31,189 TE annotations provided by the TAIR10 version of the Arabidopsis reference genome.
(F) Transcript levels of genes up-regulated in vim1/2/3 in comparison with WT plants. The number of genes within the indicated ranges of signal intensity from the microarray data in WT plants is shown.
Next, we examined whether the derepressed loci in vim1/2/3 were distributed randomly throughout the genome. We divided the 544 up-regulated loci into three classes, namely transposon-related genes, unknown genes, and known genes. Loci in the three classes were separately plotted with respect to their distance from the centromeres (Figure 1B–1D). Transposon-related genes displayed an extreme degree of clustering towards the pericentromeric regions, with 74.4% of transposons located within 2Mb of a centromere (Figure 1B). Unknown genes also exhibited a high degree of clustering towards the pericentromeric regions, with 35.5% within 2Mb and 62.6% within 4Mb of a centromere (Figure 1C). By contrast, known genes were more evenly distributed across the chromosomes, with only 9.6% of the genes located within 2Mb of a centromere (Figure 1D). Interestingly, we also found that among the up-regulated genes in vim1/2/3 a significantly higher proportion of genes were positioned close to TEs (within 2kb) in comparison to the all annotated Arabidopsis genes (Figure 1E). This observation implies that proximity to TE might be an important determinant of the derepression of gene expression in vim1/2/3.
Nearly half of the loci up-regulated in vim1/2/3 (298 of 544, 53.6%) were strongly silenced (signal intensity < 100) in WT plants (Figure 1F and Supplemental Table 1), indicating that massive reactivation of silenced genes occurred in vim1/2/3. In addition, 66 loci that were highly expressed in WT plants (11.9%; signal intensity ≥ 1000) were up-regulated in the vim1/2/3 mutant. We then asked whether the transcriptional activation observed in vim1/2/3 depends on DNA methylation. The data from a genome-wide DNA methylation analysis of Arabidopsis indicated that 20.2% and 56.0% of the expressed genes excluding known TEs and pseudogenes are methylated and unmethylated, respectively (Zilberman et al., 2007). Based on the data from Zilberman et al. (2007), genes with DNA methylation were substantially enriched among the unregulated genes in vim1/2/3 (Supplemental Figure 1).
It is noteworthy that 69 genes were significantly down-regulated in vim1/2/3 in comparison with WT plants (fold change ≥ 0.2 and p-value < 0.05) (Supplemental Table 4). Notably, 68.1% (47 of 69 loci) were known genes, while only two TEs were down-regulated in the vim1/2/3 mutant (Supplemental Figure 2A). Chromosomal positions of the down-regulated loci were evenly distributed across the chromosomes (Supplemental Figure 2B). In contrast to the up-regulated genes, about half of the loci down-regulated in vim1/2/3 (29 of 69, 42.0%) were highly expressed in WT plants (signal intensity ≥ 1000), whereas only three loci were strongly silenced (signal intensity < 100) in WT plants (Supplemental Figure 2C). Taken together, these results suggest that the VIM proteins regulate gene silencing on a genome-wide scale.
Properties of the Derepressed Loci in the vim1/2/3 mutant
Given that VIM1, VIM2, and VIM3 are essential components for maintenance of DNA methylation and epigenetic transcriptional silencing at heterochromatic regions (Woo et al., 2008), significant derepression of silenced transposons and pseudogenes in vim1/2/3 was easily predicted. Notably, we also found that 13 ncRNAs were up-regulated in the vim1/2/3 mutant with respect to WT. Although the up-regulated ncRNAs are randomly distributed throughout the genome, at least one TE was positioned either close to or inside the majority of the ncRNAs (10 out of 13 ncRNAs) (Supplemental Table 2). We selected two ncRNAs (At2g06562 and At4g15242) for validation of differential expression by reverse transcription polymerase chain reaction (RT–PCR) analysis and found that transcript levels of the two ncRNAs were markedly higher in vim1/2/3 than in the WT plants (Supplemental Figure 3A).
As mentioned above, 133 known genes were derepressed in the vim1/2/3 mutant (Supplemental Table 3). These included well-characterized epigenetically regulated genes such as MEDEA (MEA) (Kinoshita et al., 1999; Vielle-Calzada et al., 1999), FWA (Soppe et al., 2000; Kankel et al., 2003), and SUPPRESSOR OF drm1 drm2 cmt3 (SDC) (Henderson and Jacobsen, 2008). One of the predominant gene families derepressed in vim1/2/3 was β-galactosidase-related genes. Although expression of most of the 17 β-galactosidase genes (AtBGAL1 to 17) remained unchanged in vim1/2/3 (the most substantial increase among the BGAL genes was found in BGAL10 (3.36-fold increase, p = 0.004)), nearly 50% of β-galactosidase-related-genes represented on the array (10 of 21 putative β-galactosidase-related genes) were dramatically up-regulated in the vim1/2/3 mutant (Supplemental Table 5). Two putative β-galactosidase genes (At3g44070 and At5g35890) were selected to verify the microarray data by RT–PCR analysis. Transcripts of two putative β-galactosidase genes were either not detected or expressed at a low level in WT plants but increased in steady-state RNA levels in vim1/2/3 (Supplemental Figure 3B). The up-regulated putative β-galactosidase genes in vim1/2/3 shared several distinct characteristics. First, according to the publicly available Arabidopsis microarray data accessible through Genevestigator (Zimmermann et al., 2004), four β-galactosidase genes were generally expressed at low levels but were preferentially expressed in specific organ(s) (Supplemental Table 5). At3g44070 and At5g01080 exhibited extremely preferential expression in stamens. At4g29200 and At5g24480 were preferentially expressed in roots and the shoot apex, respectively. Second, similarly to the arrangement of ncRNAs, at least one TE was positioned close to, or inside, seven β-galactosidase genes. Third, nine β-galactosidase genes are highly methylated in the promoter and/or transcribed regions, according to publicly available DNA methylation data sets (Lister et al., 2008).
Data from Genevestigator indicated that 39 of the 133 known genes derepressed in the vim1/2/3 mutant were expressed at very low levels throughout development but that their expression was markedly up-regulated in specific organ(s) or developmental stage(s). These included preferential up-regulation in endosperm (12 genes including MEA and AGAMOUS-LIKE90 (AGL90)), stamens (nine genes including MICROSPORE-SPECIFIC PROMOTER 2 (MSP2)), and roots (five genes including MORPHOGENESIS OF ROOT HAIR 6 (MRH6)) (Supplemental Table 3). We chose 11 of the known genes, including three specifically expressed in endosperms (AGL87, AGL90, and CYP705A32), a stamen-specific gene (MSP2), and a gene preferentially expressed in roots (MRH6), for validation with RT–PCR. Nine of the 11 genes exhibited higher transcript levels in vim1/2/3 than in the WT (Supplemental Figure 3C); however, transcript levels of two genes (AGL87 and MRH6) were similar in WT and in vim1/2/3 plants (data not shown). Collectively, these data demonstrate that widespread transcriptional activation occurs in the vim1/2/3 mutant.
VIMs and MET1 Share Common Targets for Epigenetic Gene Silencing
To address whether gene derepression in vim1/2/3 was directed by DNA methylation, quantitative RT–PCR (qRT–PCR) analysis was used to investigate whether mutations in the DNA methyltransferase genes MET1, CMT3, and DRM2 affected the silencing of putative VIM targets. All 13 genes examined had higher transcript levels in vim1/2/3 than WT in the range of 2.7-fold (ENHANCED SILENCING PHENOTYPE 4 (ESP4)) to 1655.7-fold (At3g44070, a β-galactosidase gene) (Figure 2). As indicated in Figure 2, expression of the 13 genes was significantly misregulated in at least one of the three DNA methyltransferase mutants, supporting the hypothesis that up-regulation in the vim1/2/3 mutant might be due to DNA hypomethylation.
Figure 2.

Increased Expression of Putative VIM Targets in DNA Methyltransferase Mutants.
qRT–PCR analysis was performed with mRNA isolated from 14-day-old wild-type (WT), vim1/2/3, met1-1, cmt3, and drm2 plants. Relative expression levels of the genes whose expression was up-regulated in vim1/2/3 and in one of the three DNA methyltransferase mutants (A) and genes whose expression was significantly changed in vim1/2/3 and in at least two DNA methyltransferase mutants (B) are shown. Relative gene expression levels for qRT–PCR were normalized to the reference genes (ACT2 and UBQ10), and are displayed with respect to WT. The error bars represent standard error (SE) of three biological replicates. Numbers above bars indicate significantly different fold change in transcript levels of mutant in comparison to WT (≥ 2.0-fold change; p < 0.05).
We classified the up-regulated genes in vim1/2/3 into two groups: group I contained genes whose expression was up-regulated in one of the three DNA methyltransferase mutants (Figure 2A), and group II contained genes whose expression was significantly misregulated in at least two of the DNA methyltransferase mutants (Figure 2B). For eight genes in group I, six of which were significantly derepressed in the met1 mutant, although ESP4 and MSP2 were only up-regulated in cmt3 and drm2, respectively (Figure 2A). Overall, 11 of the 13 genes were strongly up-regulated in the met1 mutant, while only three and four genes were significantly derepressed in cmt3 and drm2, respectively (Figure 2). These data suggest that VIM and MET1 share common targets for epigenetic gene silencing.
Derepressed Genes in vim1/2/3 Are the Direct Targets of VIM1
To investigate whether the genes activated in vim1/2/3 are directly targeted by VIM proteins, we employed a chromatin immunoprecipitation-quantitative real-time PCR (ChIP–qPCR) assay on nuclei prepared from WT and transgenic Arabidopsis plants constitutively expressing Flag-VIM1. Genomic DNA was immunoprecipitated with anti-Flag antibody and used as template for qPCR. Four genes in group I (At1g47350, At2g06562, ESP4, and MSP2) and three genes in group II (At3g44070, At3g53910, and QQS) shown in Figure 2 were selected for ChIP–qPCR analysis, and two primer sets were designed for each gene for amplification of promoter and transcribed regions (Supplemental Figure 4 and Supplemental Table 6).
The VIM1 protein was significantly enriched in both the promoter and transcribed regions in all seven genes tested (Figure 3). No enrichment of VIM1 was observed in the negative control sequence UBIQUITIN 10 (UBQ10), whose expression did not differ between WT and vim1/2/3 (data not shown). These data suggest that VIM1 physically interacts with the genes derepressed in vim1/2/3. We also observed that VIM1 had three distinct chromatin-binding patterns: (1) similar binding levels within the promoter and transcribed regions of the target genes, as in At2g06562, At3g44070, At3g53910, and QQS (Figure 3A); (2) preferential binding to the promoter region rather than the transcribed region, as in At1g47350 (Figure 3B); and (3) preferential binding to the transcribed regions of the targets, as in ESP4 and MSP2 (Figure 3C). These results suggest that VIM1 binds to the regulatory or transcribed regions of genes whose expression was up-regulated in vim1/2/3, implying that VIM1 likely has a direct function in epigenetic gene silencing.
Figure 3.
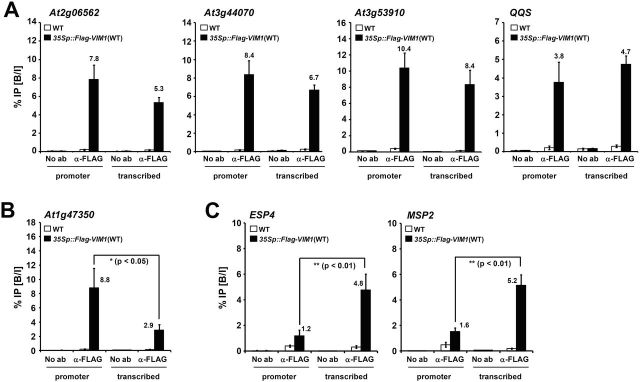
VIM1 Associates Directly with the Chromatins of the Derepressed Genes in the vim1/2/3 Mutant.
(A) ChIP analysis of Flag-VIM1 with promoter and transcribed regions of At2g06562, At3g44070, At3g53910, and QQS.
(B) VIM1 binding to the At1g47350 promoter region.
(C) VIM1 binding to the transcribed regions of ESP4 and MSP2. Chromatin fragments isolated from wild-type (WT) and transgenic plants constitutively expressing Flag-VIM1 (35Sp::Flag-VIM1(WT)) nuclei were immunoprecipitated by antibodies against Flag. Input and precipitated chromatin were analyzed by qPCR. The bound-to-input ratio (% IP (B/I)) plotted against input chromatin from both WT and transgenic plants is shown (y-axis). Numbers above bars indicate the bound-to-input ratio of the VIM1 association with each gene in 35Sp::Flag-VIM1 transgenic plants that are significantly different from that in WT (p < 0.05). Error bars represent SE from at least four biological replicates. No ab, control samples without antibodies in the immunoprecipitations steps; α-Flag, samples precipitated with anti-Flag antibody.
Derepression of VIM1 Targets Is Associated with DNA Hypomethylation of Promoter and/or Transcribed Regions
We previously proposed that the VIM proteins are essential for the maintenance of DNA methylation at heterochromatic regions (Woo et al., 2007, 2008). The DNA methylation status of the putative VIM1 targets was therefore examined to determine whether transcriptional activation in the vim1/2/3 mutant is due to changes in DNA methylation. The promoter and transcribed regions of seven up-regulated genes in vim1/2/3 were bisulfite-sequenced (Supplemental Figure 4). For all seven genes, DNA methylation levels were significantly reduced in vim1/2/3 when compared to WT (Figure 4). For example, almost complete DNA demethylation was observed in vim1/2/3 for all sequence contexts in three genes (At3g44070, ESP4, and MSP2) (Figure 4C, 4E, and 4F). By contrast, partial DNA hypomethylation was observed in vim1/2/3 in the other four genes tested (At1g47350, At2g06562, At3g53910, and QQS) (Figure 4A, 4B, 4D, and 4G). These data indicate that release of transcriptional silencing in the vim1/2/3 mutant is associated with DNA hypomethylation of the promoter and/or transcribed regions.
Figure 4.

DNA Hypomethylation of Promoter and Transcribed Regions in VIM1 Targets.
(A–G) The DNA methylation status of VIM1 targets was analyzed by bisulfite sequencing in both wild-type (WT) and vim1/2/3 plants. Genomic DNA was treated with sodium bisulfite and amplified with primers specific to the promoter and transcribed regions of each gene. The percentage cytosine methylation is indicated for each genotype, as determined at CG, CHG, and CHH sites for at least 24 clones. H represents A, T, or C.
The DNA methylation patterns of the tested genes had characteristics in common with WT plants. All seven genes had high levels of CG methylation but relatively low levels of CHG and CHH methylation, and were highly methylated within the promoter and transcribed regions, or in parts of the genes at least (Figure 4). Four genes (At2g06562, At3g44070, At3g53910, and QQS) in the WT plant contained significant levels of DNA methylation within the promoter as well as in the transcribed regions (Figure 4B–4D and 4G). Preferential DNA methylation within the promoter of At1g47350 was observed in WT plants (Figure 4A), and extremely preferential DNA methylation was noted in the transcribed regions of ESP4 and MSP2 (Figure 4E and 4F). Differential DNA methylation patterns in promoters and transcribed regions of the VIM1 targets correlated with preferential VIM1-binding activity to those regions (Figures 3 and 4), suggesting that VIM1 binds to target sequences via its methylcytosine-binding activity.
The vim1/2/3 Mutation Leads to Aberrant Changes in Transcriptionally Active and Repressive Histone Modifications at the VIM1 Targets
To investigate further whether the VIM proteins regulate the expression of target genes by altering histone modifications, we assessed the levels of histone H3 lysine 4 trimethylation (H3K4me3), H3K9me2, histone H3 lysine 9/14 acetylation (H3K9/K14ac), and H3K27me3 in WT and vim1/2/3 plants using ChIP–qPCR at the genes analyzed for DNA methylation (Figure 5). Immunoprecipitates were amplified using primers that located within the regions examined by bisulfite sequencing to determine whether DNA methylation and histone modification were correlated (Supplemental Figure 4).
Figure 5.
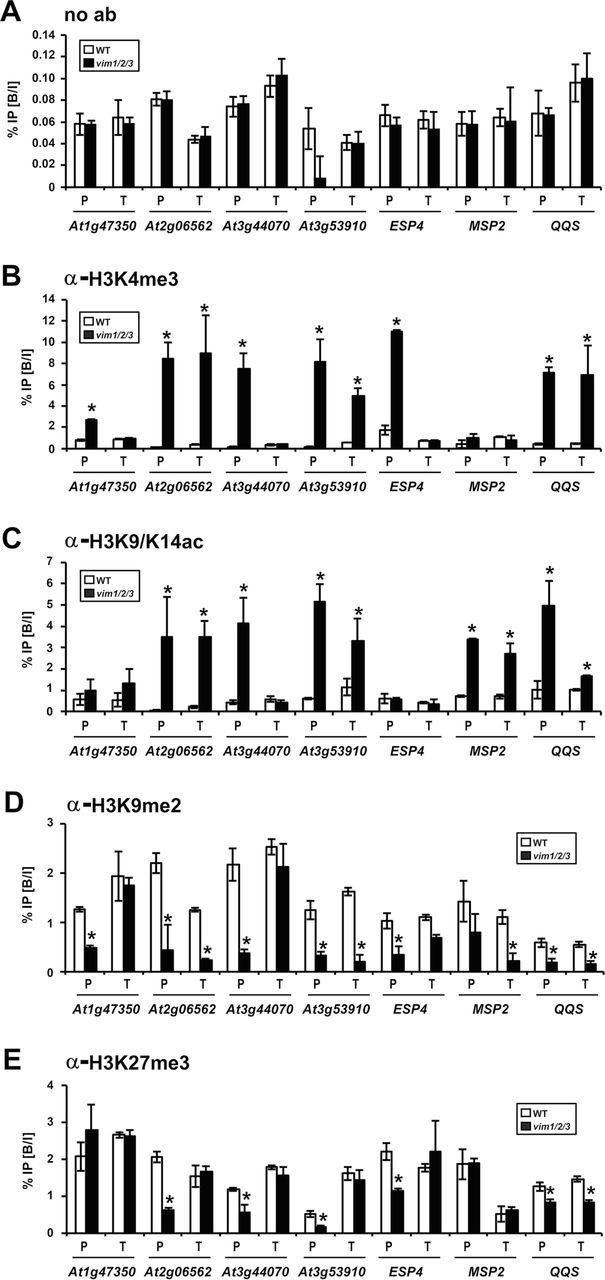
Changes in Active and Repressive Histone Marks at VIM1 Targets.
ChIP–qPCR analysis of VIM1 targets with no antibodies (A) and with antibodies against H3K4me3 (B), H3K9/K14ac (C), H3K9me2 (D), and H3K27me3 (E). Chromatin fragments isolated from nuclei of 14-day-old wild-type (WT) and vim1/2/3 plants were immunoprecipitated using the indicated antibodies. Input and precipitated chromatin were analyzed by qPCR. The bound-to-input ratio (% IP (B/I)) plotted against input chromatin from both WT and vim1/2/3 mutant plant is shown (y-axis). The error bars represent SE from at least three biological replicates. Asterisks above bars indicate a significant change of histone mark in vim1/2/3 compared to WT (p < 0.05). P, promoter region; T, transcribed region.
All of the genes tested demonstrated a significant increase in at least one active histone mark in the vim1/2/3 mutant. Among the seven genes, At2g06562, At3g53910, and QQS harbored substantial enrichment of two active histone marks (H3K4me3 and H3K9/K14ac) within the promoter and transcribed regions in the vim1/2/3 mutant (Figure 5B and 5C). In case of MSP2, the accumulation of H3K9/K14ac, but not H3K4me3 was enhanced by the vim1/2/3 mutation (Figure 5B and 5C). These results suggest that the vim1/2/3 triple mutation prompted an increase in active histone marks at the target genes.
We next characterized inactive histone modification status across the same regions of the selected VIM1 target genes. We observed that significant reductions in H3K9me2 and H3K27me3 marks at the promoter and/or transcribed regions of the loci including At2g06562, At3g44070, At3g53910, ESP4, and QQS (Figure 5D and 5E). Substantial reductions in the H3K9me2 mark, but not H3K27me3, were observed in At1g47350 and MSP2 (Figure 5D and 5E). As observed for active histone marks, the H4K9me2 and H3K27me3 reduction in the vim1/2/3 mutation was more prevalent in promoter regions than in transcribed regions (Figure 5D and 5E). The changes in H3K9me2 at the VIM1 target genes in the vim1/2/3 mutant were more pronounced than changes in H3K27me3 (Figure 5D and 5E). Overall, these data suggest that the VIM1 target genes are transcriptionally activated by DNA hypomethylation and active histone mark enrichment as well as loss of inactive histone modifications in the vim1/2/3 mutant. These data further indicate that VIM proteins maintain the silenced status of the target genes through modulating DNA methylation and histone modification.
The vim1/2/3 Mutation Results in a Drastic Reduction in H3K9me2 at Heterochromatic Chromocenters
Using antibodies that recognize H3K4me3 (associated with transcriptionally active chromatin) and H3K9me2 (typically associated with repressive heterochromatin), we next performed immunolocalization experiments to investigate whether VIM deficiency also affects global histone modification patterns. In WT nuclei, immunolocalization of H3K4me3 yielded a diffuse nuclear distribution that was visually punctuated with dark holes representing condensed heterochromatin (Figure 6A). Although VIM deficiency led to a drastic increase in H3K4me3 when VIM1 target chromatin was examined (Figure 5B), significant difference was not observed between vim1/2/3 and WT nuclei with H3K4me3 immunolocalization (Figure 6A). H3K9me2 in WT nuclei was localized at conspicuous heterochromatic chromocenters distinguished through DAPI staining (Figure 6B). By contrast, the H3K9me2 signal was significantly reduced and redistributed away from DAPI-stained chromocenters in vim1/2/3 nuclei (Figure 6B). We then used protein gel blot analysis to compare the proportions of H3K4me3 and H3K9me2 in enriched histone fractions. Similar levels of H3K4me3 were observed in WT and vim1/2/3, but H3K9me2 abundance was significantly lower in the vim1/2/3 mutant (0.43-fold compared to WT) (Figure 6C and 6D). Thus, these data suggest that the VIM proteins are required for the overall presence of heterochromatic histone marks, but might act in a rather locus-specific manner for the deposition of transcriptionally active histone marks.
Figure 6.

Immunolocalization of H3K4me3 and H3K9me2 in Wild-Type and vim1/2/3 Nuclei.
Detection of H3K4me3 (A) and H3K9me2 (B) in nuclei isolated from wild-type (WT) and the vim1/2/3 mutant. DAPI-stained (blue signals), FITC immunostained (green signals), and merged images of leaf nuclei from WT and vim1/2/3 are indicated. Bar = 5 μm.
(C) Analysis of H3 lysine methylation from WT and vim1/2/3 plants. H3 lysine methylation levels were assessed by a protein gel blot analysis with antibodies against H3K4me3 (α-H3K4me3) or H3K9me2 (α-H3K9me2). α-H3 was used as loading control.
(D) Quantitation of H3K4me3, H3K9me2, and H3 band intensities from (C) and two additional independent experiments. The H3 lysine methylation levels in WT and vim1/2/3 were normalized to the total H3 level, which was set at 1 (y-axis). The error bars indicate SE of the mean from three independent experiments. Numbers above bars indicate a significant change of histone mark in vim1/2/3 compared to WT (p < 0.05).
Deposition of VIM1 on Target Genes Is Primarily Dependent on MET1
Given that vim1/2/3 displays similar patterns of genome-wide DNA methylation with met1 (Stroud et al., 2013) and the majority of the examined VIM target genes were up-regulated in the met1 mutant (Figure 2), we hypothesized that MET1 activity is required for proper functions of the VIM proteins to maintain the silent status of the target genes. To test this possibility, we assessed VIM1-binding activity at the promoters of the target genes by ChIP–qPCR analysis in plants constitutively expressing Flag-VIM1 in WT and met1-1 backgrounds. Significantly higher levels of VIM1-precipitated DNA were recovered from WT than from the met1-1 mutant for the promoter regions of four genes (At1g47350, At2g06562, At3g44070, and At3g53910) (Figure 7). The met1-1 mutation also reduced VIM1 binding at the promoter regions of ESP4, MSP2, and QQS, with a weaker degree than at the promoter regions of At1g47350, At2g06562, At3g44070, and At3g53910 (Figure 7). This finding indicates that significantly lower amounts of VIM1 were bound at the target sites in the met1-1 mutant than in WT. Our result suggests that VIM1 primarily recognizes CG methylation deposited by MET1 for target binding but that CHG and/or CHH methylation also have roles in VIM1 binding to target sequences. Taken together, we propose that MET1 is important for the deposition of VIM1 at its target sequences, and that VIM1 acts as an essential component of the MET1-mediated DNA methylation pathway.
Figure 7.
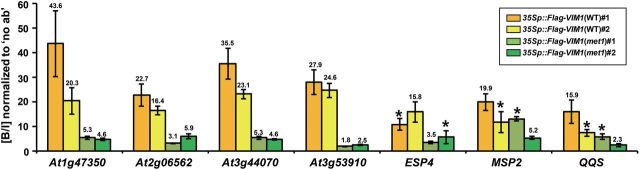
VIM1 Binds the Promoters of Its Target Genes in a MET1-Dependent Manner.
ChIP analysis of VIM1 associated with the promoter regions of At1g47350, At2g06562, At3g44070, At3g53910, ESP4, MSP2, and QQS in Arabidopsis plants constitutively expressing Flag-VIM1 in wild-type (WT) and met1-1 backgrounds. Chromatin fragments were immunoprecipitated from two independent transgenic lines overexpressing Flag-VIM1 in WT (35Sp::Flag-VIM1(WT)) and met1-1 (35Sp::Flag-VIM1(met1-1)) plants using an anti-Flag antibody. Both the input chromatin and the precipitated products were analyzed by qPCR, and the bound-to-input ratio (% IP (B/I)) in samples precipitated with anti-Flag antibody (α-Flag) was normalized to the ratio in no antibody samples (set at 1). The error bars represent SE from at least three biological replicates. Numbers above bars indicate the normalized (B/I) of VIM1 association with the target genes in the indicated genotype that are significantly different from one another (p < 0.05). Asterisks indicate normalized (B/I) in WT and met1-1 backgrounds that do not significantly differ.
DISCUSSION
VIM family proteins, which have SRA-domain methylcytosine-binding activity, are required for the maintenance of DNA methylation and epigenetic gene silencing at heterochromatic regions (Woo et al., 2007, 2008). In addition, a recent genome-wide methylome analysis revealed that vim1/2/3 strongly causes global CG and CHG hypomethylation (Stroud et al., 2013). However, the molecular mechanisms underlying VIM protein activity in epigenetic gene regulation remain to be fully elucidated, and their endogenous targets of epigenetic gene silencing had not been analyzed on a genome-wide scale.
In this study, we compared the genome-wide transcription profiles of WT and vim1/2/3 triple mutant plants and identified more than 500 loci that require the VIM proteins for epigenetic gene silencing. Our study revealed several interesting features of the genes that were derepressed in the vim1/2/3 mutant. First, the majority of the activated genes in vim1/2/3 were transposon-related and genes of unknown function (Figure 1 and Supplemental Table 1), which supports the hypothesis that VIM proteins are important for silencing in heterochromatic regions. Genomic location analysis of the approximately 400 transposon-related genes and unknown genes reactivated in vim1/2/3 indicated that VIM proteins regulate epigenetic gene silencing throughout the genome, but with a preference for loci near the centromeres (Figure 1 and Supplemental Table 1). Second, our genome-wide analysis also revealed that more than 100 genes of known function or with similarity to known genes were derepressed in vim1/2/3 (Figure 1 and Supplemental Table 3). This indicates that the role of VIM proteins is not restricted solely to the highly repetitive heterochromatic regions and transposons. Third, a significant portion of the derepressed genes in vim1/2/3 was located close to TEs (Figure 1E), suggesting that, at least in some cases, aberrant expression may have been due to defective epigenetic regulation of nearby TEs; these findings are similar to previously reported cases in which transposons affect gene expression of proximal protein-coding genes (Slotkin and Martienssen, 2007; Popova et al., 2013). Lastly, of the 133 known genes derepressed in vim1/2/3, 39 were expressed at a low level throughout development but their expression was markedly up-regulated in specific organ(s) or developmental stage(s) in WT plants (Supplemental Table 3). This observation suggests that epigenetic regulation mediated by the VIM proteins is important for gene regulation and activation under specific temporal and spatial circumstances.
We have addressed whether the VIM proteins are involved in maintaining the silenced status of target genes through modulation of DNA methylation and histone modification in this study. An important role for VIM proteins in DNA methylation is indicated by the observation that all of the direct targets of VIM1 examined in this study lost DNA methylation in all sequence contexts in the vim1/2/3 triple mutant (Figure 4). It was further indicated that release of transcriptional silencing in vim1/2/3 was associated with DNA hypomethylation of the promoter and/or transcribed regions at the direct targets of VIM1 (Figure 4). In addition, active chromatin marks, such as H3K4me3 and H3K9/K14ac, significantly increased at the VIM1 targets in vim1/2/3, whereas marks of repressive chromatin, such as H3K9me2 and H3K27me3, decreased (Figure 5). Furthermore, the VIM deficiency resulted in a significant loss of H3K9me2 at heterochromatic chromocenters (Figure 6). These findings strongly suggest that the VIM proteins silence their targets by regulating both active and repressive histone modifications. Taken together, we concluded that the VIM proteins play important roles in the coordinated modulation of histone modification and DNA methylation status in epigenetic transcriptional regulation. This conclusion is consistent with previous findings that changes in DNA methylation are tightly associated with changes in covalent modifications of histones, forming a complex regulatory network contributing to the transcriptional state of chromatin (Esteve et al., 2006; Cedar and Bergman, 2009).
It was previously reported that the levels of centromeric small RNA in vim1 were not different from WT, although the vim1 mutation induced centromere DNA hypomethylation (Woo et al., 2007). However, considering the studies proposing that small-interfering RNAs (siRNAs) function in the re-establishment of DNA methylation and gene silencing when DNA methylation is lost in DNA hypomethylation mutants like met1 and ddm1 (Mathieu et al., 2007; Mirouze et al., 2009; Teixeira et al., 2009), we could not rule out the possibility that VIM deficiency in vim1/2/3 caused changes in siRNA levels at the direct targets of VIM1. Furthermore, some genes that are known to be silenced via the RNA-dependent DNA methylation process (e.g. SDC) (Supplemental Table 1) were derepressed in vim1/2/3. This finding suggests that epigenetic gene silencing established by VIM proteins might also involve changes of siRNAs in addition to DNA methylation and histone modification. Investigating the effects of VIM deficiency on siRNAs at the direct targets will help us to elucidate the detailed mechanisms by which VIM proteins regulate genome-wide epigenetic gene silencing.
It is noteworthy that a genome-wide DNA methylome analysis demonstrated the strong resemblance between vim1/2/3 and met1 in global CG and CHG hypomethylation patterns (Stroud et al., 2013). In addition, a recent genome-wide transcriptome analysis reported a remarkable overlap between the sets of genes differentially expressed in vim1/2/3 and met1 (Shook and Richards, 2014). Consistently with these data, our result that the majority of the genes derepressed in vim1/2/3 were up-regulated in met1 (11 out of 13 genes) (Figure 2) further supports an important functional connection between the VIM proteins and MET1. We also observed that VIM1-binding capacity to its target genes correlated with DNA methylation (Figures 3 and 4) and was significantly decreased in the met1 mutant (Figure 7). Furthermore, the VIM deficiency caused a significant decrease in H3K9me2 marks at the heterochromatic chromocenters (Figure 6B), which is consistent with previous observations in the met1 mutant (Tariq et al., 2003). We therefore propose that the VIM proteins are deposited at target sequences primarily via recognition of CG methylation established by MET1 and thus act as essential components of the MET1-mediated DNA methylation pathway.
As described for UHRF1, a mammalian homolog of VIM1 (Bostick et al., 2007; Sharif et al., 2007; Achour et al., 2008), the VIM proteins may mediate the loading of MET1 onto their hemi-methylated targets through direct interactions with MET1, stimulating MET1 activity to ensure appropriate propagation of DNA methylation patterns during DNA duplication. Equally, it is possible that the VIM proteins may indirectly interact with MET1 by constituting a repressive machinery complex. It can therefore be postulated that either the VIM proteins or MET1 serves as a guide for histone-modifying enzyme(s). VIM1 physically interacts with a tobacco histone methyltransferase NtSET1 (Liu et al., 2007), which supports the notion that VIM1 might play a role in ensuring the link between DNA methylation and histone H3K9 methylation. Conversely, MET1 physically interacts with HDA6 and MEA, which are involved in maintaining the inactive state of their target genes by establishing repressive histone modifications (Liu et al., 2012; Schmidt et al., 2013). Given that VIM1 binds to histones, including H3 (Woo et al., 2007), and is capable of ubiquitylation (Kraft et al., 2008), we hypothesize that the VIM proteins directly modify histones. Although no incidences of histone ubiquitylation by the VIM proteins have been reported to date, it is noteworthy that UHRF1 is able to ubiquitylate H3 in vivo and in vitro (Citterio et al., 2004; Jenkins et al., 2005; Karagianni et al., 2008; Nishiyama et al., 2013). Moreover, UHRF1-dependent H3 ubiquitylation is a prerequisite for the recruitment of DNMT1 to DNA replication sites (Nishiyama et al., 2013). These findings support the hypothesis that the VIM proteins act as a mechanistic bridge between DNA methylation and histone modification via histone ubiquitylation. Future challenges will include identification of the direct targets of each VIM protein through genome-wide screening. Further experiments combining genome-wide analyses on DNA methylation and histone modification in vim1/2/3 will contribute to our understanding of their molecular functions within the context of epigenetic gene silencing, and will help us to elucidate how these epigenetic marks are interconnected through the VIM proteins. Collectively, our study provides a new perspective on the interplay between the two major epigenetic pathways of DNA methylation and histone modification in gene silencing.
METHODS
Plant Materials and Growth Conditions
Arabidopsis thaliana ecotype Columbia (Col) was used as the parent strain for all mutants in this study. The met1-1 (Kankel et al., 2003), vim1/2/3 (Woo et al., 2008), and 35Sp::Flag-VIM1 transgenic lines (Woo et al., 2007) were identical to those previously described. The T-DNA insertion lines for cmt3 (SALK_148381) and drm2 (SALK_150863) were obtained from the Salk T-DNA insertion collection (Alonso et al., 2003). To generate met1-1 mutant plants constitutively expressing Flag-VIM1, a construct containing a full-length VIM1 cDNA recombined into pEarleyGate202 (Earley et al., 2006) was introduced into the met1-1 plants by standard infiltration protocols. Plants were grown in a controlled environmental chamber at 22°C under long-day conditions (16h light per day).
Microarray Analysis
Microarray analyses were performed using an Arabidopsis (v4) gene expression microarray (4×44K from Agilent Technologies Inc., USA) through a custom service offered by GenomicTree, Inc. (Seoul, Republic of Korea). Total RNA from four biological replicates from 14-day-old WT and vim1/2/3 mutant plants was extracted using the RNeasy plant kit (Qiagen, USA), Cy3 or Cy5 labeled, and hybridized to the array slides. Slides were washed and then scanned using a microarray scanner, and digitized data were normalized using GeneSpring GX 10 (Agilent Technologies Inc., USA). Genes with large fold change values (fold change ≥ 5.0 or ≤ 0.2) and high statistical significance (p < 0.05), were considered to be up-regulated or down-regulated in vim1/2/3 in comparison with WT. The microarray data were deposited to GEO (Accession No. GSE55956).
RNA Isolation, RT–PCR, and qRT–PCR
Total RNA for RT–PCR and qRT–PCR was extracted from 14-day-old soil-grown plants using WelPrep total RNA isolation reagents (Welgene, Republic of Korea), according to the manufacturer’s instructions. First-strand cDNA synthesis was performed using the ImProm II Reverse Transcriptase system kit (Promega, USA), and was followed by PCR or qPCR. PCR products were visualized on a 1% agarose gel stained with ethidium bromide and imaged digitally using a UV video capture system. After performing qPCR (CFX96 Touch Real-Time PCR Detection System, Bio-Rad, USA), transcript levels were calculated using the comparative threshold (CT) method, with ACT2 (At3g18780) and UBQ10 (At4g05320) used as internal controls. Gene-specific primers used for PCR are listed in Supplemental Table 6.
ChIP–qPCR
For each experiment, 2g of 14-day-old plants were cross-linked in 1% formaldehyde solution under vacuum until the tissue became translucent. After washing twice with cold de-ionized water, tissue was ground in liquid N2 and extraction of chromatin was performed as described in Gendrel et al. (2002). To evaluate binding activity of VIM1 to its target genes, nuclei were prepared from WT plants overexpressing Flag-VIM1 and met1-1 mutant plants constitutively expressing Flag-VIM1, and sonicated chromatin samples were precipitated using an anti-Flag antibody (Sigma-Aldrich, USA). To assess the status of histone modification at the VIM1 targets, nuclei were prepared from WT and vim1/2/3 plants, and the chromatin samples were immunoprecipitated with anti-H3K4me3 (Millipore, USA), anti-H3K9me2 (Millipore, USA), anti-H3K9/K14ac (Abcam, USA), and anti-H3K27me3 (Abcam, USA) antibodies. Immunoprecipitated DNA was purified using the Qiaquick PCR purification kit (Qiagen, USA), and used for qPCR to examine the enrichment of target genes. Primers used are listed in Supplemental Table 6.
Bisulfite Sequencing
Genomic DNA (2 μg) prepared from 14-day-old WT and vim1/2/3 plants was bisulfite treated using the EpiTech Bisulfite Kit (Qiagen, USA) according to the manufacturer’s protocols. Bisulfite-modified DNA was used as template in a PCR with specific primers (listed in Supplemental Table 6). PCR products were TA-cloned into pGEM-T Easy (Promega, USA) and individual clones were sequenced using the T7 primer. At least 24 individual clones were sequenced for each locus from two independent bisulfite sequencing experiments.
Histone Immunostaining
Immunostaining analyses were performed with rosette leaves, as described, with minor modifications (Ay et al., 2009). Briefly, after post-fixation in 4% formaldehyde/1 phosphate-buffered saline (PBS), leaves were washed in 1 PBS then blocked in 3% BSA/1 PBS. Nuclei were incubated overnight at 4°C with anti-H3K9me2 (1:100 dilution; Abcam, USA) or anti-H3K4me3 (1:100 dilution; Abcam, USA) in 3% BSA/1 PBS. After washing in 1 PBS three times, nuclei were incubated with Alexa Fluor® 488 fluorochrome-conjugated secondary antibody (Invitrogen, USA) in PBS, and were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, USA) in PBS. Nuclei were examined using a Zeiss Duo LSM700 confocal microscope (Carl Zeiss, Inc., Germany). The images were pseudo-colored, merged, and processed using Adobe Photoshop (Adobe Systems, USA).
Protein Gel Blot Analysis
Protein gel blot analysis was performed according to Probst et al. (2004) with minor modifications. Briefly, 500mg of 14-day-old plant tissue was ground in liquid N2 and transferred to 1ml of histone extraction buffer (10mM Tris–HCl (pH 7.5), 2mM EDTA, 0.25M HCl, 5mM 2-mercaptoethanol, and protease inhibitors), followed by sonication for 10min and centrifugation for 10min. Total soluble proteins were aggregated with 5% trichloroacetic acid and repelleted by centrifugation at 12000rpm for 30min. Pellets were washed three times with acetone containing 0.1% 2-mercaptoethanol, and re-suspended in SDS-UREA buffer (8M urea, 1% SDS, 12.5mM Tris–HCl (pH 6.8), 1mM EDTA, and protease inhibitors). Proteins were separated electrophoretically on a 15% SDS–PAGE gel and transferred to Immobilon PVDF membranes (Millipore, USA). Histone proteins were probed for methylation using appropriate antibodies (α-H3K4Me3, Upstate, USA; α-H3K9Me2, α-H3, Abcam, USA) and were detected using SuperSignal West Pico (Thermo Fisher Scientific Inc., USA).
SUPPLEMENTARY DATA
Supplementary Data are available at Molecular Plant Online.
FUNDING
This work was supported by grants from the National Research Foundation of Korea (NRF) funded by the Government of the Republic of Korea: the Research Center Program of the Institute for Basic Science (IBS) (grant number CA1208) and the Basic Science Research Program (grant numbers 2012R1A1A3004599 and 2010-0021862).
Supplementary Material
ACKNOWLEDGMENTS
We thank M.J. Lee and H.S. Kim for excellent technical assistance. No conflict of interest declared.
REFERENCES
- Achour M., Jacq X., Ronde P., Alhosin M., Charlot C., Chataigneau T., Jeanblanc M., Macaluso M., Giordano A., Hughes A.D., et al. (2008). The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene. 27, 2187–2197 [DOI] [PubMed] [Google Scholar]
- Alonso J.M., Stepanova A.N., Leisse T.J., Kim C.J., Chen H., Shinn P., Stevenson D.K., Zimmerman J., Barajas P., Cheuk R., et al. (2003). Genome-wide insertional mutagenesis of Arabidopsis thaliana . Science. 301, 653–657 [DOI] [PubMed] [Google Scholar]
- Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M. (2008). Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 455, 818–821 [DOI] [PubMed] [Google Scholar]
- Ay N., Irmler K., Fischer A., Uhlemann R., Reuter G., Humbeck K. (2009). Epigenetic programming via histone methylation at WRKY53 controls leaf senescence in Arabidopsis thaliana . Plant J. 58, 333–346 [DOI] [PubMed] [Google Scholar]
- Bernatavichute Y.V., Zhang X., Cokus S., Pellegrini M., Jacobsen S.E. (2008). Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana . PLoS One. 3, e3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 [DOI] [PubMed] [Google Scholar]
- Bostick M., Kim J.K., Esteve P.O., Clark A., Pradhan S., Jacobsen S.E. (2007). UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 317, 1760–1764 [DOI] [PubMed] [Google Scholar]
- Cao X., Jacobsen S.E. (2002). Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 12, 1138–1144 [DOI] [PubMed] [Google Scholar]
- Cedar H., Bergman Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304 [DOI] [PubMed] [Google Scholar]
- Chan S.W., Henderson I.R., Jacobsen S.E. (2005). Gardening the genome: DNA methylation in Arabidopsis thaliana . Nat. Rev. Genet. 6, 351–360 [DOI] [PubMed] [Google Scholar]
- Citterio E., Papait R., Nicassio F., Vecchi M., Gomiero P., Mantovani R., Di Fiore P.P., Bonapace I.M. (2004). Np95 is a histone-binding protein endowed with ubiquitin ligase activity. Mol. Cell Biol. 24, 2526–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus S.J., Feng S., Zhang X., Chen Z., Merriman B., Haudenschild C.D., Pradhan S., Nelson S.F., Pellegrini M., Jacobsen S.E. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 452, 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleris A., Stroud H., Bernatavichute Y., Johnson E., Klein G., Schubert D., Jacobsen S.E. (2012). Loss of the DNA methyltransferase MET1 Induces H3K9 hypermethylation at PcG target genes and redistribution of H3K27 trimethylation to transposons in Arabidopsis thaliana . PLoS Genet. 8, e1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley K.W., Haag J.R., Pontes O., Opper K., Juehne T., Song K., Pikaard C.S. (2006). Gateway-compatible vectors for plant functional genomics and proteomics. Plant J. 45, 616–629 [DOI] [PubMed] [Google Scholar]
- Ebbs M.L., Bender J. (2006). Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell. 18, 1166–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve P.O., Chin H.G., Smallwood A., Feehery G.R., Gangisetty O., Karpf A.R., Carey M.F., Pradhan S. (2006). Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 20, 3089–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauer C., Hoffmann T., Bultmann S., Casa V., Cardoso M.C., Antes I., Leonhardt H. (2011). Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One. 6, e21306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F., Hurd P.J., Wolf D., Nan X., Bird A.P., Kouzarides T. (2003). The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 278, 4035–4040 [DOI] [PubMed] [Google Scholar]
- Gendrel A.V., Lippman Z., Yordan C., Colot V., Martienssen R.A. (2002). Dependence of heterochromatic histone H3 methylation patterns on the Arabidopsis gene DDM1. Science. 297, 1871–1873 [DOI] [PubMed] [Google Scholar]
- Goll M.G., Bestor T.H. (2005). Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514 [DOI] [PubMed] [Google Scholar]
- Henderson I.R., Jacobsen S.E. (2007). Epigenetic inheritance in plants. Nature. 447, 418–424 [DOI] [PubMed] [Google Scholar]
- Henderson I.R., Jacobsen S.E. (2008). Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev. 22, 1597–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson I.R., Deleris A., Wong W., Zhong X., Chin H.G., Horwitz G.A., Kelly K.A., Pradhan S., Jacobsen S.E. (2010). The de novo cytosine methyltransferase DRM2 requires intact UBA domains and a catalytically mutated paralog DRM3 during RNA-directed DNA methylation in Arabidopsis thaliana . PLoS Genet. 6, e1001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins Y., Markovtsov V., Lang W., Sharma P., Pearsall D., Warner J., Franci C., Huang B., Huang J., Yam G.C., et al. (2005). Critical role of the ubiquitin ligase activity of UHRF1, a nuclear RING finger protein, in tumor cell growth. Mol. Biol. Cell. 16, 5621–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L.M., Bostick M., Zhang X., Kraft E., Henderson I., Callis J., Jacobsen S.E. (2007). The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr. Biol. 17, 379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.L., Veenstra G.J., Wade P.A., Vermaak D., Kass S.U., Landsberger N., Strouboulis J., Wolffe A.P. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191 [DOI] [PubMed] [Google Scholar]
- Kankel M.W., Ramsey D.E., Stokes T.L., Flowers S.K., Haag J.R., Jeddeloh J.A., Riddle N.C., Verbsky M.L., Richards E.J. (2003). Arabidopsis MET1 cytosine methyltransferase mutants. Genetics. 163, 1109–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagianni P., Amazit L., Qin J., Wong J. (2008). ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol. Cell Biol. 28, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita T., Yadegari R., Harada J.J., Goldberg R.B., Fischer R.L. (1999). Imprinting of the MEDEA polycomb gene in the Arabidopsis endosperm. Plant Cell. 11, 1945–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose R.J., Bird A.P. (2006). Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97 [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (2007). Chromatin modifications and their function. Cell. 128, 693–705 [DOI] [PubMed] [Google Scholar]
- Kraft E., Bostick M., Jacobsen S.E., Callis J. (2008). ORTH/VIM proteins that regulate DNA methylation are functional ubiquitin E3 ligases. Plant J. 56, 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law J.A., Jacobsen S.E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., O’Malley R.C., Tonti-Filippini J., Gregory B.D., Berry C.C., Millar A.H., Ecker J.R. (2008). Highly integrated single-base resolution maps of the epigenome in Arabidopsis . Cell. 133, 523–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Yu Y., Ruan Y., Meyer D., Wolff M., Xu L., Wang N., Steinmetz A., Shen W.H. (2007). Plant SET- and RING-associated domain proteins in heterochromatinization. Plant J. 52, 914–926 [DOI] [PubMed] [Google Scholar]
- Liu X., Gao Q., Li P., Zhao Q., Zhang J., Li J., Koseki H., Wong J. (2013). UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 4, 1563. [DOI] [PubMed] [Google Scholar]
- Liu X., Yu C.W., Duan J., Luo M., Wang K., Tian G., Cui Y., Wu K. (2012). HDA6 directly interacts with DNA methyltransferase MET1 and maintains transposable element silencing in Arabidopsis . Plant Physiol. 158, 119–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu O., Reinders J., Caikovski M., Smathajitt C., Paszkowski J. (2007). Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell. 130, 851–862 [DOI] [PubMed] [Google Scholar]
- Mirouze M., Reinders J., Bucher E., Nishimura T., Schneeberger K., Ossowski S., Cao J., Weigel D., Paszkowski J., Mathieu O. (2009). Selective epigenetic control of retrotransposition in Arabidopsis . Nature. 461, 427–430 [DOI] [PubMed] [Google Scholar]
- Miura A., Nakamura M., Inagaki S., Kobayashi A., Saze H., Kakutani T. (2009). An Arabidopsis jmjC domain protein protects transcribed genes from DNA methylation at CHG sites. EMBO J. 28, 1078–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X., Ng H.H., Johnson C.A., Laherty C.D., Turner B.M., Eisenman R.N., Bird A. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 393, 386–389 [DOI] [PubMed] [Google Scholar]
- Nishiyama A., Yamaguchi L., Sharif J., Johmura Y., Kawamura T., Nakanishi K., Shimamura S., Arita K., Kodama T., Ishikawa F., et al. (2013). Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 502, 249–253 [DOI] [PubMed] [Google Scholar]
- Popova O.V., Dinh H.Q., Aufsatz W., Jonak C. (2013). The RdDM pathway is required for basal heat tolerance in Arabidopsis . Mol. Plant. 6, 396–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst A.V., Fagard M., Proux F., Mourrain P., Boutet S., Earley K., Lawrence R.J., Pikaard C.S., Murfett J., Furner I., et al. (2004). Arabidopsis histone deacetylase HDA6 is required for maintenance of transcriptional gene silencing and determines nuclear organization of rDNA repeats. Plant Cell. 16, 1021–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W., Miki D., Zhang H., Liu Y., Zhang X., Tang K., Kan Y., La H., Li X., Li S., et al. (2012). A histone acetyltransferase regulates active DNA demethylation in Arabidopsis . Science. 336, 1445–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakumara E., Law J.A., Simanshu D.K., Voigt P., Johnson L.M., Reinberg D., Patel D.J., Jacobsen S.E. (2011). A dual flip-out mechanism for 5mC recognition by the Arabidopsis SUVH5 SRA domain and its impact on DNA methylation and H3K9 dimethylation in vivo . Genes Dev. 25, 137–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W. (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 447, 425–432 [DOI] [PubMed] [Google Scholar]
- Saze H., Mittelsten Scheid O., Paszkowski J. (2003). Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 34, 65–69 [DOI] [PubMed] [Google Scholar]
- Saze H., Shiraishi A., Miura A., Kakutani T. (2008). Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana . Science. 319, 462–465 [DOI] [PubMed] [Google Scholar]
- Schmidt A., Wohrmann H.J., Raissig M.T., Arand J., Gheyselinck J., Gagliardini V., Heichinger C., Walter J., Grossniklaus U. (2013). The Polycomb group protein MEDEA and the DNA methyltransferase MET1 interact to repress autonomous endosperm development in Arabidopsis . Plant J. 73, 776–787 [DOI] [PubMed] [Google Scholar]
- Sharif J., Muto M., Takebayashi S., Suetake I., Iwamatsu A., Endo T.A., Shinga J., Mizutani-Koseki Y., Toyoda T., Okamura K., et al. (2007). The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 450, 908–912 [DOI] [PubMed] [Google Scholar]
- Shook M.S., Richards E.J. (2014). VIM proteins regulate transcription exclusively through the MET1 cytosine methylation pathway. Epigenetics. 9, 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin R.K., Martienssen R. (2007). Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285 [DOI] [PubMed] [Google Scholar]
- Soppe W.J., Jacobsen S.E., Alonso-Blanco C., Jackson J.P., Kakutani T., Koornneef M., Peeters A.J. (2000). The late flowering phenotype of FWA mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol. Cell. 6, 791–802 [DOI] [PubMed] [Google Scholar]
- Stroud H., Greenberg M.V., Feng S., Bernatavichute Y.V., Jacobsen S.E. (2013). Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell. 152, 352–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq M., Saze H., Probst A.V., Lichota J., Habu Y., Paszkowski J. (2003). Erasure of CpG methylation in Arabidopsis alters patterns of histone H3 methylation in heterochromatin. Proc. Natl Acad. Sci. U S A. 100, 8823–8827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira F.K., Heredia F., Sarazin A., Roudier F., Boccara M., Ciaudo C., Cruaud C., Poulain J., Berdasco M., Fraga M.F., et al. (2009). A role for RNAi in the selective correction of DNA methylation defects. Science. 323, 1600–1604 [DOI] [PubMed] [Google Scholar]
- To T.K., Kim J.M., Matsui A., Kurihara Y., Morosawa T., Ishida J., Tanaka M., Endo T., Kakutani T., Toyoda T., et al. (2011). Arabidopsis HDA6 regulates locus-directed heterochromatin silencing in cooperation with MET1. PLoS Genet. 7, e1002055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki M., Nishidate T., Nakamura Y. (2004). ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene. 23, 7601–7610 [DOI] [PubMed] [Google Scholar]
- Vielle-Calzada J.P., Thomas J., Spillane C., Coluccio A., Hoeppner M.A., Grossniklaus U. (1999). Maintenance of genomic imprinting at the Arabidopsis medea locus requires zygotic DDM1 activity. Genes Dev. 13, 2971–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo H.R., Dittmer T.A., Richards E.J. (2008). Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis . PLoS Genet. 4, e1000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo H.R., Pontes O., Pikaard C.S., Richards E.J. (2007). VIM1, a methylcytosine-binding protein required for centromeric heterochromatinization. Genes Dev. 21, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q., Song C.X., He C., Kumaran D., Dunn J.J. (2012). Heterologous expression and purification of Arabidopsis thaliana VIM1 protein: in vitro evidence for its inability to recognize hydroxymethylcytosine, a rare base in Arabidopsis DNA. Protein Expr. Purif. 83, 104–111 [DOI] [PubMed] [Google Scholar]
- Zilberman D., Gehring M., Tran R.K., Ballinger T., Henikoff S. (2007). Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 39, 61–69 [DOI] [PubMed] [Google Scholar]
- Zimmermann P., Hirsch-Hoffmann M., Hennig L., Gruissem W. (2004). GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol. 136, 2621–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.