

Abstract
It is well established that immunization with attenuated malaria sporozoites induces CD8+ T cells that eliminate parasite-infected hepatocytes. Liver memory CD8+ T cells induced by immunization with parasites undergo a unique differentiation program and have enhanced expression of CXCR6. Following immunization with malaria parasites, CXCR6-deficient memory CD8+ T cells recovered from the liver display altered cell-surface expression markers as compared to their wild-type counterparts, but they exhibit normal cytokine secretion and expression of cytotoxic mediators on a per-cell basis. Most importantly, CXCR6-deficient CD8+ T cells migrate to the liver normally after immunization with Plasmodium sporozoites or vaccinia virus, but a few weeks later their numbers severely decrease in this organ, losing their capacity to inhibit malaria parasite development in the liver. These studies are the first to show that CXCR6 is critical for the development and maintenance of protective memory CD8+ T cells in the liver.
Keywords: malaria, memory CD8+ T cells, Plasmodium, chemokine receptor, liver
CD8+ T-cell responses represent a major mechanism of immune protection against intracellular infections caused by viruses, bacteria and parasites. These T-cell responses are induced by natural exposure to infectious pathogens or vaccination, and while their protective efficacy is well-recognized, the development of robust CD8+ T-cell responses is still not an integral part of most vaccine development programs. In part, this is due to significant knowledge gaps regarding the biology of these T-cell responses and the mechanisms involved in the maintenance of long-term protective memory responses.
CD8+ T cells play a significant protective role in a number of infections affecting the liver, such as viral hepatitis, listeriosis, and malaria. Numerous studies in experimental models and humans have clearly shown that CD8+ T cells specific for antigens expressed by Plasmodium parasites during the liver stages are an important component of the antiparasite immune response induced after immunization with radiation-attenuated malaria sporozoites (γ-spz) [1, 2]. Immunization with sporozoites through mosquito bites or intradermal injection induces CD8+ T-cell responses in the skin-draining lymph nodes [3]. After priming, activated CD8+ T cells migrate to the liver, where they develop a memory transcriptional signature that is unique, compared with that of splenic memory cells; in particular, these cells have increased expression of the chemokine receptors CXCR3 and CXCR6, as well as differences in expression of the transcription factor Eomes and cell cycle genes, such as Ki67 [4]. These liver-homing antigen-specific memory CD8+ T cells are capable of recognizing parasite antigen presented by hepatocytes [5] and eliminating Plasmodium-infected cells [6].
A critical feature of effector and memory CD8+ T cells is their capacity to migrate and establish residency in nonlymphoid tissues. After priming, CD8+ T cells upregulate distinct homing receptors, which direct their migration to different anatomical locations, where they confer protective immunity against infectious pathogens [7–9]. The chemokine receptors CCR4 and CCR10 are highly expressed on skin-homing memory T cells [10], while most T cells homing to intestinal sites express the integrin α4β7 [11, 12] and CCR9 [13, 14]. It is also known that the integrin CD103 (αE) is critical for establishing T-cell residence in the skin [9], gut [15], and brain [16]. The molecules required for homing to the liver are less well characterized. Our microarray data showing upregulation of CXCR3 and CXCR6 on antigen-specific CD8+ T cells in the liver suggest that these may be likely candidates, while CCR5 has also been implicated in T-cell trafficking to the liver [17]. In support of possible roles for these 3 chemokine receptors in homing to the liver, all 3 have been observed to be upregulated in liver-infiltrating lymphocytes in hepatitis C virus–infected patients [18]. Moreover, in functional studies with murine cytomegalovirus Cxcr3−/− effector CD8+ T cells were found to be defective in homing to the liver, while CCR5 is associated with hepatic graft-versus-host disease (GVHD). While CXCR6 has been implicated in the migration of CD8+ T cells to the liver in a GVHD model [19], it is also highly expressed on natural killer T cells (NKT cells), which are preferentially enriched in the liver. Indeed mice lacking CXCR6 have fewer NKT cells in their livers, indicating an important role for CXCR6 in the liver-homing of thymic NKT cells [20].
In view of the role that CCR5, CXCR3, and CXCR6 appear to play in these models and of our recent findings indicating that liver-resident CD8+ T cells against malaria parasites display increased expression of some of these chemokine receptors [4], we investigated the contribution of these receptors in the recruitment and retention of protective CD8+ T cells in the liver of mice immunized with γ-spz.
MATERIALS AND METHODS
Mice
C57Bl/6 mice aged 5–8 weeks were purchased from the National Cancer Institute (Frederick, MD). Ccr5−/− (N10) and Cxcr6−/− (N10) [21, 22] were purchased from Jackson Laboratory (Bar Harbor, ME). Cxcr3−/− mice were obtained from Dr Craig Morrell [23]. OT-1 (SIINFEKL-specific TCR) transgenic mice were obtained from Dr David Sacks (National Institute of Allergy and Infectious Diseases, Bethesda, MD). Cxcr6−/− mice were bred with OT-1 (CD45.1+) mice to generate Cxcr6−/− OT-1 transgenic mice. Age-and sex-matched Cxcr6+/− mice were used as recipient mice in adoptive transfer experiments. All mice were housed and bred in the animal facility at Johns Hopkins University. Experiments involving mice were approved by the Institutional Animal Care and Use Committee of Johns Hopkins University.
Parasites, Immunizations, Quantification of Parasite Burden, and Adoptive Transfer
The generation of Plasmodium berghei CS5M parasites and irradiation and immunization of mice was performed as described elsewhere [5]. Quantification of liver-stage parasites was performed as previously described [24]. For immunization with vaccinia-OVA, mice were given 2 × 106 plaque-forming units (PFU) of virus intravenously following adoptive transfer of OT-I, as described elsewhere [5]. Adoptive transfer of T cells was performed by intravenous injection of 5000 TCR-transgenic Cxcr6+/− (CXCR6+) or Cxcr6−/− (CXCR6−) OT-I cells T cells (CD45.1+) from spleen into naive CD45.2+ Cxcr6+/− heterozygous C57Bl6 recipients. For the in vivo homing assay, mice that received naive CXCR6+ or CXCR6− OT-I were subsequently immunized with 1 × 105 γ-spz or 2 × 106 PFU rVV-OVA. Between 6 and 8 days after immunization, spleens were harvested, and numbers of CD45.1+ OT-I were estimated by flow cytometry with anti-CD45.1 and anti-CD8 antibodies. Equal numbers of OT-I CD8+ T cells (approximately 0.5 × 106) were then adoptively transferred to naive CD45.2+ recipient mice. Twenty hours after transfer, spleen and liver were excised, and the numbers of donor CD8+CD45.1+ cells in these organs were determined by flow cytometry.
Lymphocyte Isolation and Ex Vivo Stimulation
Tissues were harvested on day 3–90 after immunization as indicated. Lymphocytes were isolated as described previously [25]. In brief, liver was perfused with Hank's balanced salt solution (HBSS), homogenized, and resuspended in 35% Percoll gradient (GE Healthcare). Lungs were treated with collagenase II (1 mg/mL) at 37°C for 30 minutes, followed by Percoll gradient as described above. Lymphocytes were cocultured with El4 cells pulsed with or without SIINFEKL peptide (10 µg/mL) for 4 hours at 37°C in the presence of brefeldin A and monensin. Intracellular staining was performed using Cytofix/perm kit (BD Biosciences).
Antibodies and Flow Cytometry
H2-Kb/SIINFEKL tetramer was generated in house. Antibodies were purchased from eBioscience or BD unless stated otherwise: PE-, PE-Cy7–, or APC-conjugated CD45.1 (A20); PE-Cy7–conjugated CD8 (53–6.7) and interferon γ (IFN-γ; XMG1.2), PE-conjugated CXCR6 (R and D), CCR5 (HM-CCR5 [7A4]), CD127 (A7R34), and CD25 (PC61.5); APC-conjugated CXCR3 (CXCR3-173), KLRG1 (2F1), and interleukin 2 (IL-2; ES6-5H4); and Pacific blue–conjugated tumor necrosis factor α (TNF-a; MP6-XT22). PE-conjugated BCL-2 (BD Biosciences) was performed using FoxP3 buffer set (eBioscience). All experiments were performed on a FACSCalibur or LSRII flow cytometer (BD).
BrdU Staining
Immunized mice were injected with 1.5 mg BrdU (Sigma-Aldrich) intraperitoneally daily for 10 days. BrdU+ cells were detected using a PE-conjugated BrdU flow kit (BD Biosciences).
Data Analysis
Fluorescence-activated cell sorting data were analyzed using FlowJo software (TreeStar). Unless otherwise stated, statistical analysis was performed using Prism 4 software (GraphPad Software) by 2-tailed Student t tests. Significant level was set to an α of 0.05.
RESULTS
CXCR6 Contributes to Memory CD8+ T-Cell Development and Maintenance in the Liver
To investigate the potential roles of CCR5, CXCR3, and CXCR6 in the establishment of liver-homing CD8+ T-cell populations, we examined the expression of these receptors in liver-stage-specific CD8+ T cells. In these experiments, we used P. berghei CS5M, a P. berghei strain, which we have described previously, in which the native H-2Kd–restricted epitope SYPSAEKI from the circumsporozoite protein was substituted with the H-2Kb–restricted epitope SIINFEKL. Importantly, these parasites can prime SIINFEKL-specific CD8+ T cells including OT-I TCR transgenic CD8+ T cells, and the insertion of the SIINFEKL epitope does not affect parasite growth or development [5].
To examine the expression of these chemokine receptors on antigen-specific CD8+ T cells, we transferred a low number of naive CD45.1+ OT-I cells into CD45.2+ recipient mice prior to immunization with γ-spz. One month after immunization, memory CD8+ T cells were obtained from spleen and liver to compare the expression of chemokine receptors in SIINFEKL-Tetramer+ (Tet+) CD8+ T cells. We found that spleen or liver memory cells did not differ regarding the expression of CCR5, but the expression of CXCR3 and CXCR6 on memory CD8+ T cells residing in the liver was consistently higher than that observed in spleen memory CD8+ T cells (Figure 1A). These results confirm previous findings that we obtained following immunization of H2-Kd mice with the related parasite Plasmodium yoelii [4].
Figure 1.
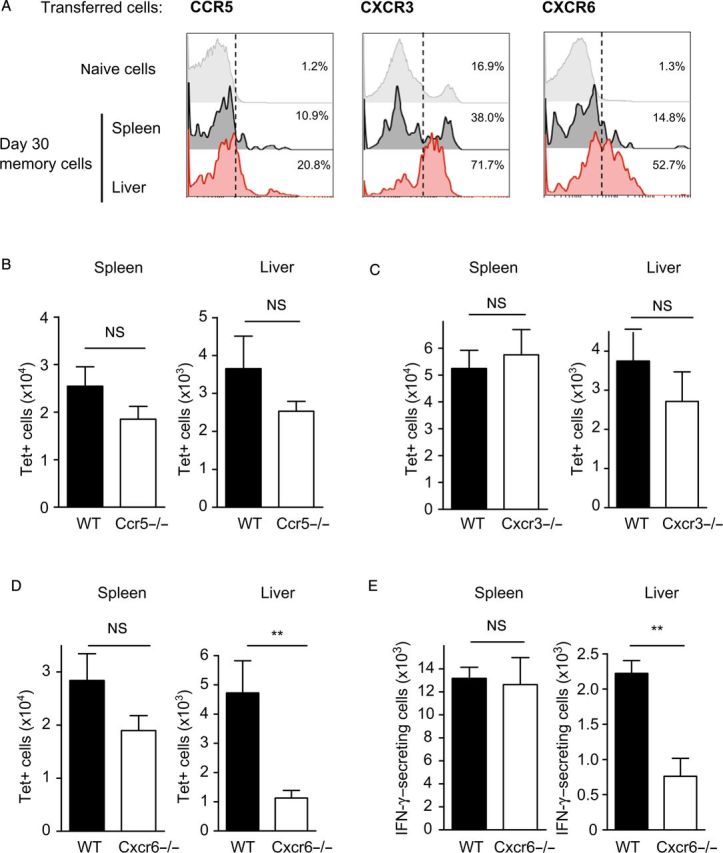
Expression of chemokine receptors on antigen-specific memory CD8+ T cells after irradiated sporozoite immunization. A, Naive CD45.1+ OT-I were adoptively transferred to C57BL/6 recipient mice 1 day prior to immunization with γ-spz. Representative histograms showing the expression of CXCR3, CCR5, and CXCR6 gated on memory OT-I cells 1 month after immunization. B–D, Ccr5−/− (B), Cxcr3−/− (C), and Cxcr6−/− (D) mice were immunized, and total numbers of tetramer+ cells were determined 1 month after immunization. E, Number of interferon γ–expressing cells determined by ex vivo stimulation with SIINFEKL peptide (10 μg/mL) in spleen and liver of immunized Cxcr6+/− and Cxcr6−/− mice. Data shown are results from 1 experiment, which are representative of 2 independent experiments (n = 3–4 per group). Error bars show mean ± standard error of the mean. **P < .01. Abbreviations: IFN-γ, interferon γ; NS, not significant; WT, wild type.
Taking advantage of the fact that a range of chemokine-receptor-knockout mice are available on the H2-Kb (C57Bl/6) background, we investigated the possible role of CCR5, CXCR3, or CXCR6 in the development of memory CD8+ T cells specific for P. berghei CS5M in the spleen and liver, using mice lacking these chemokine receptors. We found that the endogenous SIINFEKL-specific CD8+ T-cell response in the spleen and liver of immunized mice was not significantly different between wild-type and CCR5- and CXCR3-deficient mice (Figure 1B and 1C). In contrast, we observed a major reduction in both the total numbers of SIINFEKL-tetramer–positive cells, as well as the number of IFN-γ–producing SIINFEKL-specific CD8+ T cells in the livers of CXCR6-deficient mice, as compared to wild-type mice (Figure 1D and 1E) or heterozygous Cxcr6+/− mice (Supplementary Figure 1).
CXCR6 Is Critical to Establish Memory CD8+ T Cells in the Liver
The results described above suggested a role for CXCR6 in establishing memory CD8+ T-cell populations in the liver. To further characterize these findings and to determine whether the decreased number in memory cells in the liver depended on the expression of this chemokine receptor on T cells, we backcrossed our SIINFEKL-specific OT-I mice to the Cxcr6−/− mice. We then transferred low numbers of naive OT-I, Cxcr6−/− cells carrying the congenic marker CD45.1 to CD45.2 Cxcr6+/− recipients prior to immunization with γ-spz. Control mice were treated identically except they received Cxcr6+/− OT-I cells. We used Cxcr6+/− mice as recipients instead of wild-type C57Bl/6 mice because the gene targeting of Cxcr6 includes the insertion of enhanced green fluorescent protein (eGFP) in this locus, and we wished to avoid possible allogenic rejection of eGFP+ transferred cells in wild-type animals. Therefore, transferred Cxcr6−/− cells are hereafter referred to as CXCR6−, and Cxcr6+/− cells are referred to as CXCR6+. Consistent with the results obtained in Figure 1D, when compared to the response developed by CXCR6+ CD8+ cells, the number of CXCR6− CD8+ cells present in the liver 25 days after immunization is significantly reduced (Figure 2B and 2C). Similar results were observed 3 months after immunization (data not shown). This defect in memory formation appeared to be liver specific as the numbers of memory CXCR6− cells recovered from spleen and lungs were comparable to those found with CXCR6+ cells. These experiments recapitulate results obtained in Cxcr6−/− animals (Figure 1D and 1E) and are particularly stringent as we are comparing knockouts to heterozygous rather than wild-type controls.
Figure 2.
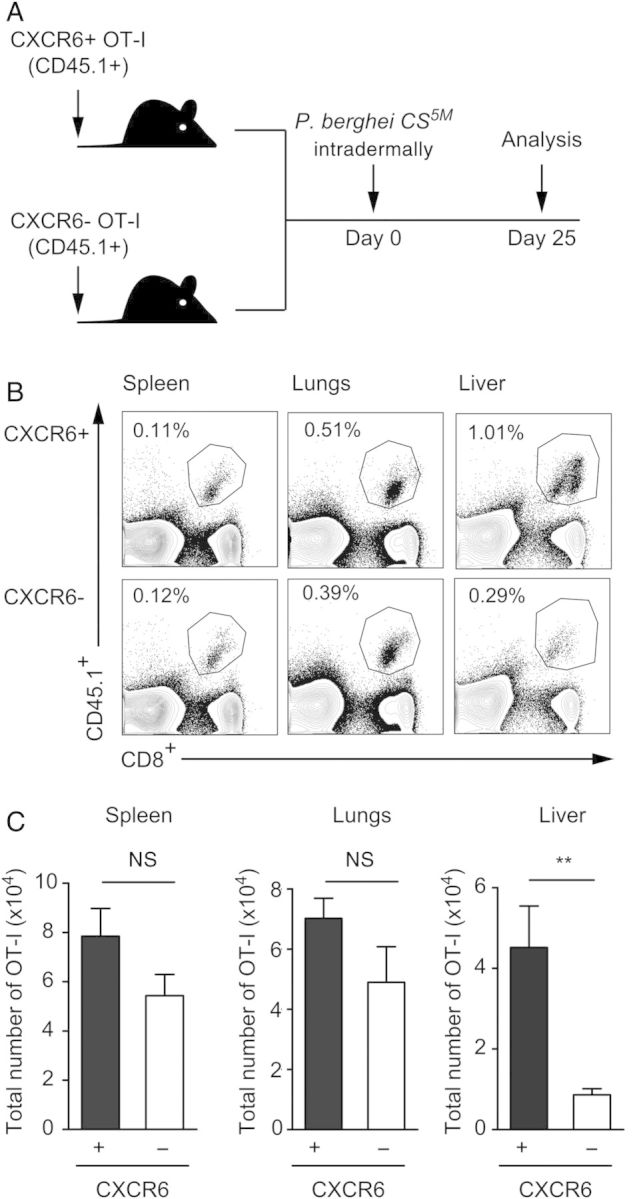
Expression of CXCR6 on CD8+ T cells is critical for the establishment of a memory population in the liver. A, Schematics of the experimental design, of which naive CXCR6+ or CXCR6− CD45.1+ OT-I cells were adoptively transferred to Cxcr6+/− C57BL/6 recipients prior to immunization with γ-spz. B and C, Representative fluorescence-activated cell sorting plots gated on total lymphocytes (B) and total numbers of OT-I cells in spleen, lungs, and liver of mice receiving CXCR6+ OT-I or CXCR6-OT-I cells on day 25 after immunization (C). Data shown are results from 1 experiment, which are representative of 6 independent experiments with 3–4 mice per group. Data are mean ± standard error of the mean. **P < .01. Abbreviation: NS, not significant.
Cxcr6−/− CD8+ T Cells Undergo Normal Priming and Initial Expansion in Tissues
To identify the causes underlying the reduction numbers of memory CXCR6− CD8+ T cells in the liver, we performed experiments to determine whether this was the result of an ineffective CD8+ T-cell priming, reduced homing to the liver, or a defect in homeostatic proliferation in the liver. For this purpose, we performed experiments similar to those described in Figure 2A, in which naive CD45.1+ CXCR6+ or CXCR6− OT-1 cells were transferred to CXCR6+ CD45.2+ recipients, which were subsequently immunized intradermally with γ-spz. The induction of CD8+ T cells was evaluated 3 days after immunization, and we found similar numbers of primed CXCR6+ or CXCR6− OT-1 cells in the skin-draining lymph node (SDLN) and spleen (Figure 3A). Similarly, on day 9 after immunization, we found no significant differences in the number of effector OT-I CXCR6+ or CXCR6− CD8+ T cells present in the spleen, lungs, or liver of immunized animals (Figure 3B). These results clearly indicate that priming of activated CD8+ T cells was not impaired in the absence of CXCR6, and, in addition, they suggest that the early liver migration of activated T cells is not affected. This apparently normal migration to the liver was confirmed in experiments using an in vivo homing assay, in which equal numbers of CXCR6+ or CXCR6− primed OT-I cells were transferred to naive mice and the number of cells homing to this organ evaluated 20 hours later. As shown in Figure 3C, we found similar numbers of CXCR6+ or CXCR6− cells in both the liver and spleen of recipient mice, indicating that the reduction in liver-resident memory is not likely caused by a defect in liver-homing of primed CD8+ T cells. Finally, we determined that CXCR6 deficient memory T cells in the spleen and liver display a normal homeostatic proliferation as indicated by their BrdU incorporation one month after immunization (Figure 3D). Taken together, these results indicate that CXCR6 deficiency does not affect T-cell priming, trafficking, or homeostatic proliferation of antigen-specific effector CD8+ T cells.
Figure 3.

CXCR6 is not required during priming, homing, or homeostatic proliferation of CD8+ T cells. A and B, Representative fluorescence-activated cell sorting plot gated on total lymphocytes and total numbers of OT-I cells recovered from spleen, lung, and liver of immunized mice on day 3 (A) and day 9 (B) after immunization. C, For the in vivo homing assay, equal number of effector CXCR6+ and CXCR6− OT-I cells were adoptively transferred to naive wild-type recipients. Twenty hours after adoptive transfer, total numbers of donor cells in spleen and liver in recipient mice were recovered. D, To measure homeostatic proliferation, mice receiving CXCR6+ or CXCR6− OT-I were immunized with γ-spz. One month later, mice were administered BrdU for 10 days. Percentages are of BrdU+ among memory OT-I in spleen and liver. The data are results from 1 experiment, which are representative of 2 (A, C, and D) and 4 (B) independent experiments with 3–4 mice per group. Data are mean ± standard error of the mean. Abbreviations: dLN, draining lymph node; NS, not significant; P. berghei, Plasmodium berghei.
Cxcr6−/− CD8+ T Cells Display Altered Differentiation Markers
Our finding that CXCR6 was not apparently important for homing to the liver was surprising, given the canonical role of chemokines and their receptors in migration. However, we next investigated the hypothesis that CXCR6 may be important for differentiation into memory in the liver. Functional analysis of memory cells revealed that CXCR6+ or CXCR6− memory OT-I cells from the spleen, lungs, or liver produced comparable amounts of IFN-γ, TNF-α ,and IL-2 (Figure 4A). Similarly, no differences were detected in the expression of cytolytic mediators such as perforin (not shown) and granzyme B (Supplementary Figure 2). However an analysis of surface markers associated with memory differentiation, including CD62L, CXCR3, CD127 (IL-7R), KLRG1, and CD27, revealed some striking differences between CXCR6− and CXCR6+ malaria-specific liver CD8+ T cells. Compared with malaria-specific CXCR6+ cells in the liver, CXCR6− cells had reduced expression of CXCR3 and were predominantly KLRG1hi—a phenotype that is characteristic of short-lived effector cells and terminally differentiated T-effector memory [26, 27]. On the contrary, no major differences were observed between CXCR6+ and CXCR6− memory OT-I cells in the spleen. We speculate that effector OT-I lacking CXCR6 may undergo faster contraction during the acute response and thus form smaller memory cell populations. In support of this, we observed lower expression of Bcl-2, an antiapoptotic molecule (Figure 4C) on day 9 in CXCR6− CD8+ cells present in both spleen and liver. We therefore speculate that effector cells lacking this chemokine receptor may have an intrinsic survival disadvantage. This deficiency may become particularly critical for memory cells in nonlymphoid organs such as the liver, where there may be a shortage of the cytokine or chemokine interactions required to sustain the formation of memory cells.
Figure 4.

Functional and phenotypical characterization of CD8+ T cells in the absence of CXCR6. A, Cytokine production in memory CXCR6+ and CXCR6− OT-I was evaluated after 4-hour ex vivo stimulation with cognate peptide. Data are pooled results of 2 independent experiments. B, Expression of CD62L, CD127, CD27, KLRG1, and CXCR3 among OT-I in spleen and liver on day 30 after immunization. Data shown are results from 1 experiment, which are representative of 2 independent experiments. C, Day 9 after immunization with γ-spz, the expression of BCL2 was evaluated among CXCR6+ or CXCR6− effector OT-I cells, shown as representative histograms (Ci; left) and percentage of BCL2+ (Cii; right) among gated OT-I . Data shown are results from 1 experiment, which are representative of 2 independent experiments with 3–4 mice per group. Data are mean ± standard error of the mean. *P < .05. Abbreviations: IFN-γ, interferon γ; IL-2, interleukin 2; TNF-α, tumor necrosis factor α.
Vaccine-Induced CXCR6-Deficient Memory CD8+ T Cells Fail to Inhibit Parasite Development in the Liver
To determine whether the failure of CXCR6− memory CD8+ T cells to establish a large memory population in the liver is a unique feature of parasite-induced CD8+ T cells, we immunized mice with a recombinant vaccinia virus (rVV) expressing SIINFEKL [28]. In this system, mice receiving CXCR6+ or CXCR6− naive CD8+ OT-1 cells were immunized with rVV, and the number of CD8+ T cells were evaluated 9 and 25 days after immunization. We found comparable CD8+ T cells in the spleen and liver 9 days after immunization (Supplementary Figure 3), but at day 25 we found a major reduction in the number of memory cells in the liver of mice receiving CXCR6− CD8+ T cells (Figure 5A), although they maintained their capacity to produce IFN-γ (Figure 5B), thus reproducing the phenotype we observed in mice that received CXCR6− cells and were immunized with sporozoites.
Figure 5.

Immunized mice harboring Cxcr6−/− memory CD8+ T cells do not inhibit the development of Plasmodium liver stages. Total numbers of OT-I cells in spleen and liver in mice on day 25 after immunization with recombinant vaccinia virus (rVV). A, Naive Cxcr6+/− or Cxcr6−/− OT-I were adoptively transferred to recipient mice, which were later immunized with rVV. B, Percentage of interferon γ (IFN-γ)–expressing cells among gated CD45.1+ OT-I. C, Seven days or 21 days after rVV immunization, mice were challenged with live Plasmodium berghei CS5M. Liver parasite burden was determined by reverse-transcription polymerase chain reaction 40 hours later. Data shown are results from 1 experiment, which are representative of 2 independent experiments with 3–5 mice per group. Data are mean ± standard error of the mean. *P < .05 and **P < .01. Abbreviations: NS, not significant; rRNA, ribosomal RNA.
We took advantage of the fact that CXCR6− OT-I cells were unable to form a large liver-memory CD8+ T-cell population in response to rVV, to determine whether the lack of this population impaired protection against live Plasmodium parasites. As rVV immunization generates CD8+ T cells that recognize P. berghei CS5M–parasitized hepatocytes but does not induce antiparasite antibodies or CD4+ T cells, this viral system provided a clean experimental set up to evaluate and compare the protective capacities of CXCR6+ and CXCR6− CD8+ memory T cells, in the absence of other protective mechanisms. When mice receiving naive CXCR6+ or CXCR6− OT-I cells were immunized with rVV and challenged 7 days later with live parasites, we observed a substantial inhibition of parasite development in mice harboring both memory cell types (Figure 5C). It should be noted that immunization with wild-type VV has no effect on parasite development, as shown in our previous studies [29]. In contrast, when mice were challenged with sporozoites 3 weeks after rVV immunization, the liver parasite load was reduced only in mice receiving CXCR6+ cells but not in those receiving CXCR6− cells. These results indicate that after immunization with VV, CXCR6 is not required during the early effector phase of the antimalaria response, but it is essential to establish a long-term protective memory cell population in the liver.
DISCUSSION
This present study demonstrates a critical role for CXCR6 in the maintenance of liver-resident memory CD8+ T cells induced after immunization with infectious pathogens. Our studies using CXCR6+ and CXCR6− CD8+ T cells show that the expression of CXCR6 does not appear to be critical for priming CD8+ T cells in lymphoid organs and is not required for homing and effector function in the liver during the first few days after immunization. However, we found that CXCR6 is critically required for the maintenance of long-term memory populations in the liver, as the number of memory cells and their protective capacity are drastically reduced as early as 3 weeks after immunization. The requirement for this chemokine receptor appears to be organ specific, because in the spleen and lungs the numbers of CXCR6− memory cells were comparable to those found for CXCR6+ memory T cells. Importantly, in mice harboring CXCR6− CD8+ T cells we did not observe increased numbers of memory cells in the spleen or lungs, indicating that the decreased numbers of memory cells in the liver may not result from a redistribution of liver memory cells into these organs.
Memory CD8+ T cells in the liver play an important role in controlling the earliest phase of the liver-stage infection by malaria parasites. A number of studies indicate that the antiparasite activity of CD8+ T cells depends on the direct recognition of parasite antigen in hepatocytes [3], and upon parasite challenge, these T cells are poised to provide an immediate antimicrobial response. Indeed, recent in vivo imaging studies showed that a few hours after infection, activated T cells form clusters around parasite-infected hepatocytes [6] and eliminate infected hepatocytes via direct recognition, without detectable bystander killing [30]. Considering that CXCR6+ or CXCR6− memory cells do not differ with regard to their functional properties, the inability of CXCR6− memory cells to inhibit parasite development likely results from the severely decreased numbers of antigen-specific memory cells in the liver. This is consistent with the observation that CD8+ T-cell–mediated protection closely depends on the frequency of antigen-specific memory T cells established before parasite challenge [31]. In fact, unlike other infectious models, CD8+ T-cell–mediated inhibition of parasite development in the liver can only occur during the liver stages (ie, during the 44-hour window after sporozoite challenge and before the establishment of the blood stages), precluding the possibility that recall memory responses can develop and confer protection.
While the secretion of cytokines and expression of cytotoxicity mediators such as granzyme B in CXCR6+ and CXCR6− cells appear to be similar on a per-cell basis, we found altered expression levels of surface/activation markers in memory CD8+ T cells lacking CXCR6, suggesting that this receptor is involved in modulating the tissue-specific phenotype on CD8+ T cells in the liver, as described in a previous study on gut-specific CD8+ T cells [15]. Although both CXCR6+ and CXCR6− effector CD8+ T cells express similar high levels of KLRG1 immediately after priming (data not shown), we observed that CXCR6+ memory CD8+ T cells in the liver are mostly KLRG1lo while CXCR6- memory cells are predominantly KLRG1hi, suggesting that CXCR6 may participate in the differentiation/survival of memory CD8+ T cells in the liver. In fact it has been described in other systems that KLRG1hi is a phenotype associated with short-lived effector CD8+ T cells and also a marker of terminal differentiation [26, 27], while KLRGlo cells have been identified as the precursors of skin-resident memory CD8+ T cells in herpes simplex virus infection [32].
The studies described in this report indicate that the expression of CXCR6 on CD8+ T cells is a critical requirement to establish long-lived memory T-cell populations in the liver. Since the size and protective capacity of memory populations in the liver are known to decrease with time, understanding the mechanisms by which memory CD8+ T cells are maintained in the liver should open new avenues of research to develop immunization strategies aimed at ensuring long-term survival of memory CD8+ T cells specific for infectious pathogens and resident in nonlymphoid tissues.
Supplementary Data
Notes
Acknowledgments. We thank Juan Yu, for technical support; Xiaodong Jiang, for his comments on the manuscript; and the Bloomberg Family Foundation, for the continued support.
Financial support. This work was supported by the National Institutes of Health (grant AI44375) and the Johns Hopkins Malaria Research Institute (predoctoral fellowship to S.-W. T.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Overstreet MG, Cockburn IA, Chen Y-C, Zavala F. Protective CD8 T cells against Plasmodium liver stages: immunobiology of an “unnatural” immune response. Immunol Rev. 2008;225:272–83. doi: 10.1111/j.1600-065X.2008.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seder RA, Chang L-J, Enama ME, et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science. 2013;341:1359–65. doi: 10.1126/science.1241800. [DOI] [PubMed] [Google Scholar]
- 3.Chakravarty S, Cockburn IA, Kuk S, Overstreet MG, Sacci JB, Zavala F. CD8+ T lymphocytes protective against malaria liver stages are primed in skin-draining lymph nodes. Nat Med. 2007;13:1035–41. doi: 10.1038/nm1628. [DOI] [PubMed] [Google Scholar]
- 4.Tse S-W, Cockburn IA, Zhang H, Scott AL, Zavala F. Unique transcriptional profile of liver-resident memory CD8(+) T cells induced by immunization with malaria sporozoites. Genes Immun. 2013;14:302–9. doi: 10.1038/gene.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cockburn IA, Tse S-W, Radtke AJ, et al. Dendritic cells and hepatocytes use distinct pathways to process protective antigen from plasmodium in vivo. PLoS Pathog. 2011;7:e1001318. doi: 10.1371/journal.ppat.1001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cockburn IA, Amino R, Kelemen RK, et al. In vivo imaging of CD8+ T cell-mediated elimination of malaria liver stages. Proc Natl Acad Sci U S A. 2013;110:9090–5. doi: 10.1073/pnas.1303858110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol. 2009;9:153–61. doi: 10.1038/nri2496. [DOI] [PubMed] [Google Scholar]
- 8.Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature. 2012;483:227–31. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackay LK, Stock AT, Ma JZ, et al. Long-Lived Epithelial Immunity by Tissue-Resident Memory T (TRM) Cells in the Absence of Persisting Local Antigen Presentation. Proc Natl Acad Sci U S A. 2012;109:7037–42. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reiss Y, Proudfoot AE, Power CA, Campbell JJ, Butcher EC. CC chemokine receptor (CCR)4 and the CCR10 ligand cutaneous T cell-attracting chemokine (CTACK) in lymphocyte trafficking to inflamed skin. J Exp Med. 2001;194:1541–7. doi: 10.1084/jem.194.10.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berlin C, Berg EL, Briskin MJ, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–95. doi: 10.1016/0092-8674(93)90305-a. [DOI] [PubMed] [Google Scholar]
- 12.Lefrançois L, Parker CM, Olson S, et al. The role of beta7 integrins in CD8 T cell trafficking during an antiviral immune response. J Exp Med. 1999;189:1631–8. doi: 10.1084/jem.189.10.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zabel BA, Agace WW, Campbell JJ, et al. Human G protein-coupled receptor GPR-9–6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J Exp Med. 1999;190:1241–56. doi: 10.1084/jem.190.9.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunkel EJ, Campbell JJ, Haraldsen G, et al. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: Epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J Exp Med. 2000;192:761–8. doi: 10.1084/jem.192.5.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting Edge: Gut Microenvironment Promotes Differentiation of a Unique Memory CD8 T Cell Population. J Immunol. 2006;176:2079–83. doi: 10.4049/jimmunol.176.4.2079. [DOI] [PubMed] [Google Scholar]
- 16.Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A. 2010;107:17872–79. doi: 10.1073/pnas.1010201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murai M, Yoneyama H, Harada A, et al. Active participation of CCR5+CD8+ T lymphocytes in the pathogenesis of liver injury in graft-versus-host disease. J Clin Invest. 1999;104:49–57. doi: 10.1172/JCI6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boisvert J, Kunkel EJ, Campbell JJ, Keeffe EB, Butcher EC, Greenberg HB. Liver-infiltrating lymphocytes in end-stage hepatitis C virus: subsets, activation status, and chemokine receptor phenotypes. J Hepatol. 2003;38:67–75. doi: 10.1016/s0168-8278(02)00328-8. [DOI] [PubMed] [Google Scholar]
- 19.Sato T, Thorlacius H, Johnston B, et al. Role for CXCR6 in recruitment of activated CD8+ lymphocytes to inflamed liver. J Immunol. 2005;174:277–83. doi: 10.4049/jimmunol.174.1.277. [DOI] [PubMed] [Google Scholar]
- 20.Geissmann F, Cameron TO, Sidobre S, et al. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3:e113. doi: 10.1371/journal.pbio.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuziel WA, Dawson TC, Quinones M, et al. CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis. 2003;167:25–32. doi: 10.1016/s0021-9150(02)00382-9. [DOI] [PubMed] [Google Scholar]
- 22.Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, Littman DR. The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J Immunol. 2000;165:3284–92. doi: 10.4049/jimmunol.165.6.3284. [DOI] [PubMed] [Google Scholar]
- 23.Srivastava K, Cockburn IA, Swaim A, et al. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe. 2008;4:179–87. doi: 10.1016/j.chom.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruña-Romero O, Hafalla JC, González-Aseguinolaza G, Sano G, Tsuji M, Zavala F. Detection of malaria liver-stages in mice infected through the bite of a single Anopheles mosquito using a highly sensitive real-time PCR. Int J Parasitol. 2001;31:1499–502. doi: 10.1016/s0020-7519(01)00265-x. [DOI] [PubMed] [Google Scholar]
- 25.Cockburn IA, Chakravarty S, Overstreet MG, Garcia-Sastre A, Zavala F. T cell responses expand when antigen presentation overcomes T cell self-regulation. J Immunol. 2008;180:64–71. doi: 10.4049/jimmunol.180.1.64. [DOI] [PubMed] [Google Scholar]
- 26.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–40. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joshi NS, Cui W, Chandele A, et al. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–95. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hickman HD, Takeda K, Skon CN, et al. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol. 2008;9:155–65. doi: 10.1038/ni1557. [DOI] [PubMed] [Google Scholar]
- 29.Rodrigues M, Li S, Murata K, et al. Influenza and vaccinia viruses expressing malaria CD8+ T and B cell epitopes. Comparison of their immunogenicity and capacity to induce protective immunity. J Immunol. 1994;153:4636–48. [PubMed] [Google Scholar]
- 30.Cockburn IA, Tse S-W, Zavala F. CD8+ T cells eliminate liver stage Plasmodium parasites without detectable bystander effect. Infect Immun. 2014;82:1460–64. doi: 10.1128/IAI.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt NW, Podyminogin RL, Butler NS, et al. Memory CD8 T cell responses exceeding a large but definable threshold provide long-term immunity to malaria. Proc Natl Acad Sci U S A. 2008;105:14017–22. doi: 10.1073/pnas.0805452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackay LK, Rahimpour A, Ma JZ, et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14:1294–301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.