

Background: Calcium-dependent activation of TRESK is mediated by calcineurin.
Results: The sensitivity of human TRESK to calcium is determined by the LQLP calcineurin-docking site.
Conclusion: The previously known PQIIIS and the novel LQLP sites cooperate for the regulation.
Significance: TRESK-induced hyperpolarization of the membrane potential is influenced by the LQLP-calcineurin interaction.
Keywords: Calcineurin, Calcium, Ion Channel, Potassium Channel, Protein Phosphatase 2B (PP2B), K2P, KCNK18, PXIXIT, Two-pore Domain
Abstract
Calcium-dependent activation of human TRESK (TWIK-related spinal cord K+ channel, K2P18.1) depends on direct targeting of calcineurin to the PQIIIS motif. In the present study we demonstrate that TRESK also contains another functionally relevant docking site for the phosphatase, the LQLP amino acid sequence. Combined mutations of the PQIIIS and LQLP motifs were required to eliminate the calcium-dependent regulation of the channel. In contrast to the alanine substitutions of PQIIIS, the mutation of LQLP to AQAP alone did not significantly change the amplitude of TRESK activation evoked by the substantial elevation of cytoplasmic calcium concentration. However, the AQAP mutation slowed down the response to high calcium. In addition, modest elevation of [Ca2+], which effectively regulated the wild type channel, failed to activate TRESK-AQAP. This indicates that the AQAP mutation diminished the sensitivity of TRESK to calcium. Even if PQIIIS was replaced by the PVIVIT sequence of high calcineurin binding affinity, the effect of the AQAP mutation was clearly detected in this TRESK-PVIVIT context. Substitution of the LQLP region with the corresponding fragment of NFAT transcription factor, perfectly matching the previously described LXVP calcineurin-binding consensus sequence, increased the calcium-sensitivity of TRESK-PVIVIT. Thus the enhancement of the affinity of TRESK for calcineurin by the incorporation of PVIVIT could not compensate for or prevent the effects of LQLP sequence modifications, suggesting that the two calcineurin-binding regions play distinct roles in the regulation. Our results indicate that the LQLP site is a fundamental determinant of the calcium-sensitivity of human TRESK.
Introduction
Background (leak) potassium channels are responsible for the hyperpolarizing outward potassium flux in a great variety of native cell types (1–5). Members of the K2P channel family, characterized by four transmembrane segments and two pore-loop forming domains (4TM/2P) in each subunit of the functional dimer, give rise to background K+ conductance. Among the fifteen members of this K+ channel family, TWIK-related spinal cord K+ channel, K2P18.1 (TRESK)2 is the only one that is regulated (i.e. substantially activated) by the elevation of cytoplasmic calcium concentration (6–9).
Activation of TRESK does not depend on the direct binding of calcium ion to the channel. We previously reported that the calcium/calmodulin-dependent protein phosphatase calcineurin mediates the effect (6). Three serine residues (Ser-264, Ser-274, and Ser-276) have been identified by alanine-scanning mutagenesis as the putative targets of the phosphatase in the mouse channel (6, 8, 10). These residues are also conserved in human TRESK as Ser-252, Ser-262, and Ser-264. The first serine is phosphorylated by protein kinase A (10), and this reaction permits the binding of 14-3-3 adapter protein (10, 11). Microtubule-affinity regulating (MARK) kinases are currently the only known enzymes that phosphorylate the other two serines of functional importance. Accordingly, the overexpression of MARK accelerates the return of the K+ current to the resting state after the calcium-dependent activation of TRESK in Xenopus oocytes (8).
In addition to the enzymatic interaction, calcineurin is directly anchored to a PXIXIT-like site in TRESK (where X may be any amino acid) (12). This mechanism for the targeting of the phosphatase is unparalleled within the ion channel superfamily. The PXIXIT consensus motif was originally described in Nuclear Factor of Activated T cells (NFAT), and later its functional significance was also reported in several other calcineurin-binding proteins (13, 14). The affinities of the PQIIIS and PQIVID sequences of human and mouse TRESK channels for calcineurin (KD of 5 or 10 μm, respectively) proved to be higher than that of NFAT (KD = 25 μm for PRIEIT of NFAT1, also called NFATc2) (14). However, they did not reach the value characteristic for the optimized PXIXIT sequence, the VIVIT peptide (KD = 0.5 μm) (14, 15). The VIVIT peptide binds to a surface of calcineurin A subunit, distinct from the catalytic site, as apparent in the crystal structure of the complex (PDB entry 2P6B, (16)). In good accordance with this arrangement, the PQIIIS motif (Pro at position 200) is located sufficiently far from the substrate serines in the cytoplasmic loop of TRESK. Thus the catalytic site of docked calcineurin may reach and dephosphorylate the substrate residues.
Mutation of PQIVID to PQAVAD in mouse TRESK completely prevented the calcium-dependent activation of the K+ current, indicating that the docking of calcineurin to the channel is a prerequisite of the regulation (12). Unexpectedly, human TRESK behaved differently from its rodent counterpart. The PQAAAS mutant, in which all three isoleucines of the PQIIIS motif were replaced by alanines, was still stimulated by the calcium ionophore ionomycin. Because the PQIIIS site was certainly destroyed by the triple A mutation, we hypothesized that there may be another calcineurin-targeting sequence in human TRESK.
In addition to PXIXIT, a second calcineurin-binding motif was also identified in NFAT transcription factors (17–19). This motif is described by the LXVP consensus sequence, and its instances have also been found in other proteins (13, 14, 20). The LXVP motif was predicted to bind to a hydrophobic cleft at the interface of calcineurin A and B subunits (20, 21). This region is different from the surface binding to PXIXIT and also from the catalytic site. Thus calcineurin can simultaneously associate to the PXIXIT and LXVP motifs in the same interacting protein (20, 21). In contrast to PXIXIT, the binding cleft for LXVP is accessible only in the active conformation of the phosphatase (13, 22).
TRESK does not contain a perfect match to the LXVP consensus sequence. Considering that the first leucine and the fourth proline may be the cornerstones of the motif, we decided to investigate whether the LQLP sequence (Pro at position 243) constitutes a calcineurin-docking site in human TRESK. Although no other protein has been known to contain leucine instead of valine in the third position of the motif, we demonstrate that LQLP interacts with calcineurin and critically determines the calcium sensitivity of the channel.
EXPERIMENTAL PROCEDURES
Plasmids and Reagents
The cloning of human and mouse TRESK cDNAs and their subcloning into the plasmid suitable for in vitro cRNA synthesis and expression in Xenopus oocytes was previously described (6). Different mutant versions of these constructs were produced with QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) according to the manufacturer's instructions. To obtain GST-hTRESK(174–280) fusion protein, the coding sequence of fragment 174–280 of human TRESK was amplified by PCR and subcloned into pGEX-4T1 (Amersham Biosciences, Little Chalfont, UK) between the EcoRI and XhoI sites. For the construction of fragment 232–280, this plasmid was cleaved with BamHI and BglII and the compatible ends were ligated. Both the 174–280 and the 232–280 fragments contained the additional AAVERPHRD amino acids at their C terminus in addition to TRESK coding sequence. Production of GST-mTRESK(164–292) was previously described (12).
Ionomycin (calcium salt, Enzo Life Sciences, Farmingdale, NY) was dissolved in DMSO as 5 mm stock solution, and diluted further before the measurement. Carbachol (100 mm) and VIVIT peptide (3.85 mm, NFAT inhibitor, Calbiochem, La Jolla, CA) were dissolved in water. Chemicals of analytical grade were purchased from Sigma, Fluka, or Merck. Enzymes and kits of molecular biology applications were purchased from Qiagen (Chatsworth, CA), Ambion (Austin, TX), Thermo Scientific (Waltham, MA), New England Biolabs (Beverly, MA), and Stratagene.
Animals, Tissue Preparation, Xenopus Oocyte Microinjection
Mouse brain tissue derived from NMRI mouse strain (Toxicop, Hungary). Xenopus oocytes were prepared, the cRNA was synthesized and microinjected as previously described (6). Oocytes were injected 1 day after defolliculation. Fifty nanoliters of the appropriate RNA solution was delivered with Nanoliter Injector (World Precision Instruments, Saratosa, Florida). All treatments of the animals were conducted in accordance with state laws and institutional regulations. The experiments were approved by the Animal Care and Ethics Committee of Semmelweis University (XIV-I-001/2154-4/2012).
Two-electrode Voltage Clamp Measurements
Two-electrode voltage clamp experiments were performed three or 4 days after the microinjection of cRNA, as described previously (6). Low [K+] solution contained (in mm): NaCl 95.4, KCl 2, CaCl2 1.8, HEPES 5 (pH 7.5 adjusted with NaOH). High [K+] solution contained 80 mm K+ (78 mm Na+ of the low [K+] solution was replaced with K+). TRESK background K+ current was measured at the ends of 250 or 300 ms voltage steps to −100 mV applied in every 4 s. Because TRESK activation varied substantially between the different oocyte preparations (e.g. 3.8- or 11-fold activation for wild type TRESK in response to 500 nm ionomycin), groups of oocytes were compared only from the same preparation in all graphs and calculations.
GST Pulldown Assay
Cerebrum, cerebellum, and brainstem from two mice were homogenized for each experiment in 3 ml of ice cold solution A containing (in mm): KH2PO4 50, NaCl 50, MgCl2 2, β-mercaptoethanol 5, PMSF 1, benzamidine 1 (pH 7.0 with NaOH), supplemented with 5% glycerol. The lysate was centrifuged at 12,100 × g for 20 min at 4 °C. The supernatant was supplemented with CHAPS (to a final concentration of 1%), and with EGTA (to a final concentration of 2 mm) or CaCl2 (1 mm), and centrifuged at 12,100 × g for 10 min.
Glutathione S-transferase (GST) fusion proteins of human TRESK fragments 174–280 and 232–280, immobilized on 20 μl of glutathione-agarose (Sigma), were incubated with the cytosol preparation by gently rotating the beads for 1 h at 4 °C. The resins were first washed with 1.3 ml of solution A. The second washing step was a high salt wash for 5 min with solution A containing 1 m NaCl. The final (third and fourth) washing steps were also performed with solution A to remove residual salt. The washing buffers for the first three washing steps were also supplemented with 1 mm CaCl2 for the assays also containing Ca2+ in the binding reactions. The proteins were eluted from the resins with SDS sample buffer and separated on 12% Tris-glycine SDS-PAGE gels, followed by Coomassie Brilliant Blue staining.
Immunoblot Experiments
Denatured samples were separated by SDS-PAGE on 12% gels, and transferred to nitrocellulose membranes (Schleicher and Schuell, Keene, NH). Nonspecific binding sites of the membranes were blocked by 5% nonfat milk in PBS-T solution (phosphate-buffered saline containing 1% Tween 20). The primary antibodies in the different experiments were monoclonal anti-HA (HA.11, Covance, Princeton, NJ) 3000× in PBS-T containing 0.5% nonfat milk, rabbit polyclonal anti-calcineurin A (PP2B-A, H-209, sc-9070, Santa Cruz Biotechnology, Santa Cruz, CA) 500× in PBS-T containing 1% bovine serum albumin (BSA), or monoclonal anti-β-tubulin isotype III IgG (Sigma T5076) diluted 5000× in PBS-T containing 1% BSA. The secondary antibodies were anti-mouse or anti-rabbit antibodies, respectively, (horseradish peroxidase-conjugated IgG from goat, R05071 or R05072, Advansta, Menlo Park, CA) diluted 5000× or 10,000× in PBS-T containing 0.5% nonfat milk. The membranes were washed once after blocking and four to six times after the antibodies for 5–20 min in PBS-T. The bands were visualized by the enhanced chemiluminescence detection method (WesternBright ECL HRP, Advansta) according to the manufacturer's instructions. Densitometry analysis was performed with ImageJ 1.47v software written by Wayne Rasband (Research Services Branch, NIH, Bethesda, MD).
In Vitro Dephosphorylation of Radioactively Labeled Phosphoprotein Substrates
GST-hTRESK(174–280), GST-hTRESK(174–280)-AQAP, GST-mTRESK(164–292) and GST-mTRESK(164–292)-AQAP fusion proteins immobilized on 10 μl of glutathione-agarose were phosphorylated with Trx-His6-MARK2-T208E for 1 h at 30 °C in 50 μl volume of solution B containing (in mm): Tris-HCl 50 (pH 7.5), MgCl2 5, EGTA 2, β-mercaptoethanol 1.4, PMSF 0.5, benzamidine 0.5, supplemented with 20 μm Na2ATP and 100 kBq [32P-γ]ATP. Preparation of the thioredoxin-hexahistidine-tagged constitutively active MARK2 kinase, containing the T208E phospho-mimicking activator loop mutation, has previously been described (8). (MARK2 phosphorylates the functionally relevant calcineurin-substrate residues Ser-274 and Ser-276 in mouse TRESK, which correspond to Ser-262 and Ser-264 in the human channel.)
Subsequently, the radioactively labeled proteins were dephosphorylated with mouse brain cytosol for different time periods from 0 to 60 min. The cytosol was prepared as detailed in the “GST Pulldown Assay” section, but one brain was homogenized in 4 ml of solution A. This substantial dilution of the cytoplasm sufficiently slowed down the rate of dephosphorylation, enabling the analysis of its kinetics. Cytosol from one brain was used for 18 dephosphorylation reactions (10 μl resin/reaction). The cytosol was supplemented with 1 mm CaCl2 or 2 mm EGTA in the different reactions. The reactions were stopped by rapidly washing the resin twice with 1 ml solution C containing (in mm): Tris-HCl 20 (pH 7.5), NaCl 50, β-mercaptoethanol 1, followed by elution of the proteins from the resin with sample buffer and separation on 12% SDS-PAGE gels. The gels were stained with Coomassie Brilliant Blue, and their radioactivity was detected with phosphorimager (GS-525, Bio-Rad).
Statistics and Calculations
Data are expressed as means ± S.E. Statistical significance was estimated by Student's t test for independent samples. The statistical difference was considered to be significant at p < 0.05.
RESULTS
Combined Mutations of the PQIIIS and LQLP Sites Are Required to Prevent TRESK Regulation
Functional relevance of the PQIIIS calcineurin docking motif of human TRESK (Fig. 1A) has not yet been investigated. To gradually disintegrate the binding site, we designed three mutants: PQIIIA, PQIIAS, and PQAAAS. The mutations (in the listed order) progressively deteriorated the activation of the channel evoked by high (0.5 μm) concentration of ionomycin (Fig. 1B). However, in sharp contrast to the previously reported similar PQAVAD modification of mouse TRESK (12), the PQAAAS mutation of the human channel failed to eliminate the activation. Human TRESK-PQAAAS was still activated more than 2-fold by the elevation of cytoplasmic [Ca2+]. (The activation of the PQAAAS mutant was significantly smaller than that of the wild type channel, 2.4 ± 0.3-fold versus 11.0 ± 1.4-fold, respectively, n = 10 in both groups, p < 10−4, Fig. 1B). The PQAAAS mutation interfered with the calcineurin-dependent activation process. The apparently reduced activation was not the consequence of enhanced basal channel activity as indicated by the equally low benzocaine-sensitivity of wild type and PQAAAS mutant TRESK (Fig. 1B). (Benzocaine specifically inhibits the activated TRESK current in Xenopus oocytes (12).)
FIGURE 1.

Combined mutations of the PQIIIS and LQLP sites are required for the complete elimination of the calcium-dependent activation of human TRESK. A, transmembrane topology of human TRESK subunit and the PQIIIS and LQLP calcineurin docking motifs are illustrated. The regulatory serine residues of the channel are shown in the vicinity of the LQLP site. (The drawing is not to scale.) B, Xenopus oocytes expressing wild type (PQIIIS, wt.) or mutant (PQIIIA, PQIIAS and PQAAAS) TRESK channels were stimulated with the calcium ionophore ionomycin (0.5 μm, Iono.) after verifying the insensitivity of the basal K+ currents to benzocaine (1 mm, Benzo.). The extracellular [K+] was changed from 2 to 80 mm and back as indicated above the graph. The currents were measured at −100 mV by two-electrode voltage clamp, and normalized to their resting value in 80 mm [K+]. Note that the mutations of the PQIIIS site gradually deteriorated but did not completely eliminate the activation; the PQAAAS mutant was activated more than 2-fold by ionomycin. The gray error bars represent S.E. C, normalized activation in response to ionomycin (0.5 μm) is shown for three different TRESK constructs, all containing the PQAAAS mutation. The LQLP motif was not modified in TRESK-PQAAAS (see TLQLPP, wt. curve); it was disintegrated by AQAP mutation in TRESK-PQAAAS-TAQAPP (TAQAPP curve); or it was substituted with the corresponding sequence of NFATc1 (NFAT2) in TRESK-PQAAAS-YLAVPQ (YLAVPQ curve). Maximum activation levels at the end of the stimulation with ionomycin are indicated by dashed lines for the different mutants, as labeled on the right side of the panel. Note that the combined mutations of the PQIIIS and LQLP sites eliminated the calcium-dependent regulation (TAQAPP curve), but YLAVPQ functionally substituted for the TLQLPP sequence of TRESK (YLAVPQ curve).
Because the PQAAAS mutation alone did not completely prevent the calcium-dependent activation of human TRESK, we searched for another calcineurin-binding site in the channel. We examined the LQLP sequence, which is similar to the LXVP calcineurin-binding consensus motif, and is located in the vicinity of the serines targeted by the phosphatase (Fig. 1A). The two leucines of the LQLP motif were replaced by alanines in TRESK-PQAAAS. This additional AQAP mutation abolished the residual activation. TRESK-PQAAAS-AQAP has not been affected by ionomycin (1.07 ± 0.03-fold change of K+ current), whereas TRESK-PQAAAS was activated 2.27 ± 0.15-fold (n = 12 in both groups, p < 10−7, Fig. 1C). This indicates that the LQLP motif mediates the residual calcineurin-dependent activation, if PQIIIS is disabled.
If the LQLP site of TRESK functions analogously to the LxVP calcineurin-docking motif then its replacement with a canonical LXVP sequence should also reproduce calcium-dependent regulation. To test this assumption, we replaced the TLQLPP sequence of TRESK-PQAAAS with YLAVPQ from NFATc1 (NFAT2). We modified a region of six amino acids, since it has been reported that the tyrosine located N-terminally to LxVP also contributes to the binding of calcineurin (19), whereas the C-terminal second proline may interfere with the association (22). TRESK-PQAAAS-YLAVPQ was activated identically to TRESK-PQAAAS in response to ionomycin (Fig. 1C), indicating that the canonical LXVP motif from NFAT functionally substituted for the wild type LQLP sequence.
The AQAP Mutation Slows Down the Activation of Human TRESK Evoked by High Calcium, and Also Interferes with the Activation of the Mouse Channel
The contribution of the LQLP site to the calcium-dependent regulation was also examined by the AQAP mutation in the context of the wild type channel. The amplitude of the activation of human TRESK-AQAP in response to ionomycin (0.5 μm) was not significantly different from that of wild type TRESK (4.8 ± 0.5-fold (n = 21) versus 6.2 ± 0.6-fold (n = 19), respectively, p = 0.08, Fig. 2A). However, the activation was decelerated by the AQAP mutation, as illustrated in Fig. 2B on a shorter time scale. At the time point indicated with an asterisk, the degrees of partial activation during the application of the ionophore were significantly different between the AQAP mutant (3.1 ± 0.3-fold) and the wild type channel (4.9 ± 0.6-fold, p < 0.01, Fig. 2B). The rates of activation in the two groups (calculated as % activation/second) were also statistically different (data not shown). Thus the AQAP mutation clearly slowed down the activation in response to the robust elevation of [Ca2+] induced by high concentration of ionomycin, although the final level of activation was only mildly (if at all) affected. The AQAP mutation did not influence the kinase activity responsible for the inhibitory re-phosphorylation of human TRESK, as indicated by the unaltered percent recovery values during the long washout period following the ionomycin-stimulation (Fig. 2C).
FIGURE 2.
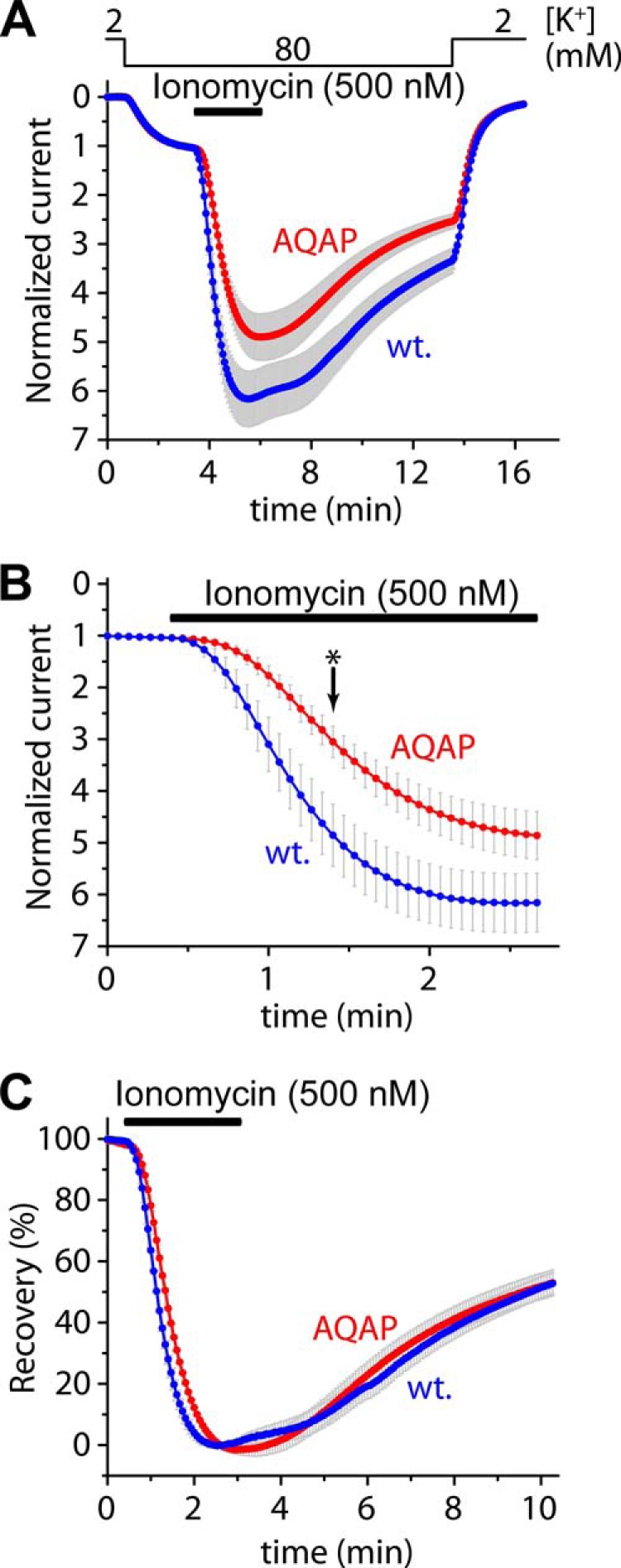
The AQAP mutation slows down the activation of human TRESK evoked by high concentration of ionomycin, but the inhibitory kinase reaction is not affected by this mutation. A, cells expressing wild type or AQAP mutant TRESK channels were stimulated with high (0.5 μm) concentration of ionomycin (as indicated by the horizontal black bar). Recovery from the activation was evaluated by a long washout period in 80 mm extracellular [K+], as indicated above the graph. The activations of the wild type and AQAP mutant channels were not significantly different at the end of the stimulation with ionomycin. B, activation kinetics of the currents (same as in panel A) during the stimulation with ionomycin are plotted on a shorter time scale. The AQAP mutation slowed down the activation process; the partial activation of the AQAP mutant is significantly smaller than that of the wild type channel at the time point indicated with an asterisk. C, percent recovery values were calculated from the recordings represented in panel A. The recovery from activation was identical in the cases of the wild type and AQAP mutant TRESK channels.
Mouse TRESK contains LQPP instead of the LQLP sequence characteristic for the human channel. We decided to examine whether LQPP destruction affects the activation of mouse TRESK, especially because the similar LAPP site of the Regulator of Calcineurin (RCAN1) was reported to interact with the phosphatase (23). The effects of the AQAP mutation on mouse TRESK were substantially different from those obtained in the case of the human channel (see the average currents in Fig. 3A, and the currents normalized to peak value in Fig. 3B). The basal current of mouse TRESK was increased more than 3-fold by the AQAP mutation (7.0 ± 1.6 μA), compared with the wild type channel (2.0 ± 0.6 μA, n = 4 in both groups, p < 0.05, Fig. 3C). The activation by ionomycin was smaller in the AQAP (1.8 ± 0.1-fold) than in the control group (5.1 ± 1.0-fold, p < 0.05, Fig. 3.D). The return of the K+ current to the resting state after the stimulation with ionomycin, calculated as the recovery in percent, was diminished by the AQAP mutation (8 ± 4% recovery for AQAP versus 48 ± 6% for the wild type channel at the end of the measurement, p < 0.01, Fig. 3E). The impaired activation of the AQAP mutant is compatible with the contribution of LQPP to the calcineurin-dependent activation, however, the increased basal current and reduced recovery from activation suggest that the AQAP mutation may have also interfered with the inhibitory kinase reaction targeting the mouse channel.
FIGURE 3.

In mouse TRESK, the AQAP mutation also interferes with the calcium-dependent activation. A, average currents of two groups of oocytes expressing wild type or AQAP mutant mouse TRESK are plotted. The cells were stimulated with ionomycin (Iono., 0.5 μm, as indicated by the horizontal black bar) in 80 mm extracellular [K+] (as shown above the graph). Basal K+ currents were estimated as I0, maximum activations in response to ionomycin as I1, and the recovery at the end of the measurement as I2. The small nonspecific leak currents measured in 2 mm [K+] were subtracted from the values of I0, I1 and I2 in further calculations but not in this graph. (Only plus or minus error bars (gray, S.E.) are shown.) B, same currents as in panel A were normalized to their peak values. The major part of the variation of data in panel A came from the different channel expression of the oocytes, and the response to ionomycin was rather uniform within the groups. C, average basal K+ currents of the wild type (wt.) and AQAP mutant (m.) channels (I0 in panel A) are illustrated in this column diagram. Basal currents of the AQAP mutant were significantly larger than those of the wild type channel. D, normalized activations (I1/I0) of the wild type, and AQAP mutant channels were calculated from the same data as represented in panel A. The apparent activation of the wild type channel exceeded that of the AQAP mutant. E, percent recovery from activation of the K+ currents at the end of the measurement (I2 in panel A) was calculated as indicated by the expression above the columns. The AQAP mutation diminished the return of the K+ current to the resting state after the stimulation with ionomycin.
The LQLP Site Is Required for the High Sensitivity of Human TRESK to Calcium
The slow activation of human TRESK-AQAP raised the suspicion that the LQLP site contributes to the calcium-sensitivity of the channel. We investigated this hypothesis by stimulating TRESK and its mutant versions with stepwise increasing concentrations (50, 100, 200, and 500 nm) of ionomycin (Fig. 4A). This stimulation protocol results in a slow elevation of [Ca2+] in the cytoplasm, suitable for the investigation of the response of TRESK to modest calcium levels. The final [Ca2+] at the end of this stepwise stimulation (in 500 nm ionomycin) could be lower than in the case of the prompt application of the same ionophore concentration, because the major source of calcium, the intracellular stores, are depleted during the prolonged treatment of the cell (24, 25).
FIGURE 4.
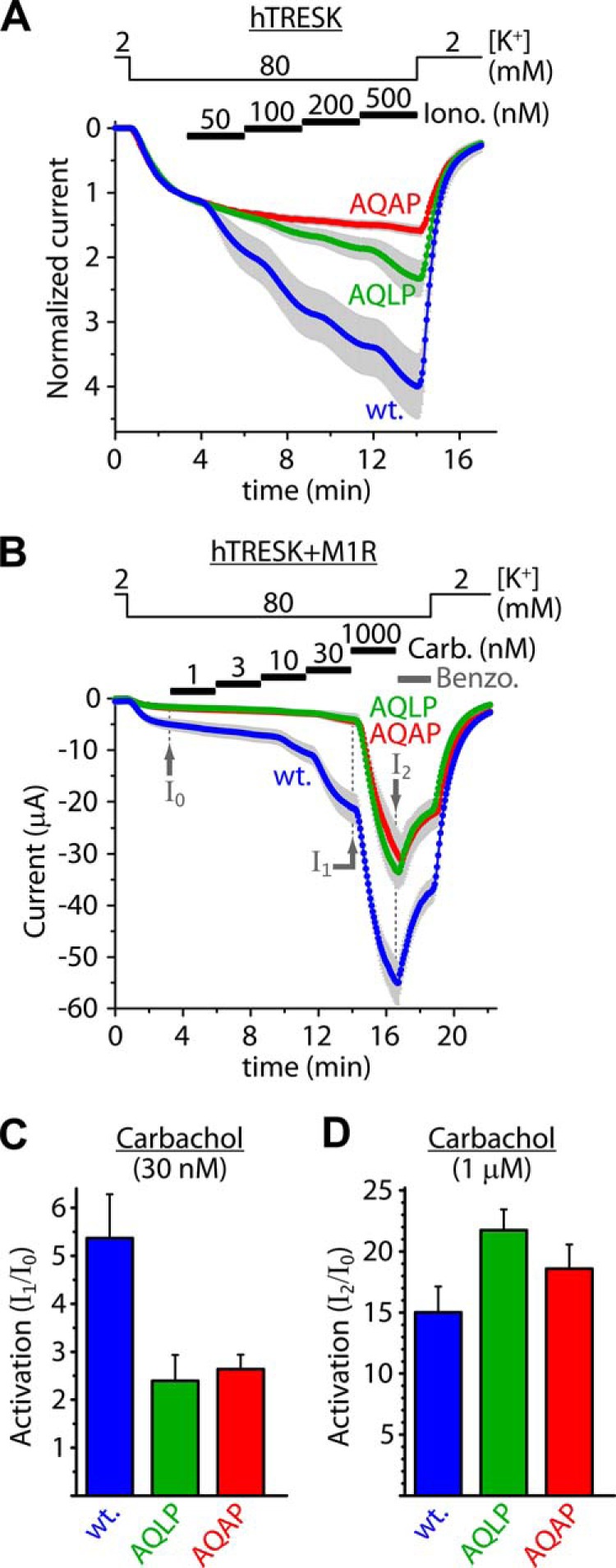
Alanine substitutions in the LQLP site reduce the sensitivity of human TRESK to calcium. A, cells expressing wild type (wt., LQLP) or mutant (AQLP and AQAP) TRESK channels were stimulated with stepwise increasing (50, 100, 200, and 500 nm) concentrations of ionomycin as indicated by the horizontal black bars. Note that the AQAP mutant was unresponsive to the modest elevation of cytoplasmic [Ca2+], in sharp contrast to the wild type channel. B, wild type (wt.), AQLP and AQAP mutant TRESK channels were coexpressed with M1 muscarinic receptor. Modest calcium signals were evoked by the application of progressively increasing (1, 3, 10, and 30 nm) concentrations of carbachol as indicated by the horizontal black bars. Subsequently, robust calcium signal was induced by 1 μm concentration of the agonist. Finally the sensitivity to benzocaine (1 mm) was tested as indicated by the horizontal gray bar. Note that the AQLP and AQAP mutants have not been activated by the low concentrations of carbachol, but they were responsive to high calcium. (The green AQLP and red AQAP curves overlap.) C, cumulative activations of the wild type (wt.), AQLP and AQAP mutant channels by low (1–30 nm) concentrations of carbachol were calculated as I1/I0. The wild type channel was more strongly activated than the AQLP and AQAP mutants. D, activations of the wild type, AQLP and AQAP mutant channels in response to the robust calcium signal (1 μm carbachol) were estimated as I2/I0. By high calcium, the AQLP and AQAP mutants were activated similarly to (or more than) the wild type channel.
Wild type TRESK was substantially activated by the stepwise ionomycin stimulation (4.0 ± 0.5-fold at the end of the 500 nm step, n = 10, Fig. 4A, wt. curve). However, TRESK-AQAP mutant was much less sensitive to the modest elevation of [Ca2+] under identical conditions (1.6 ± 0.1-fold activation, n = 12, p < 10−4, Fig. 4.A, AQAP curve). The activation of AQLP (L240A point) mutant was also attenuated (2.3 ± 0.3-fold, n = 12, Fig. 4.A, AQLP curve); intermediate between the wild type and AQAP. It is important to note that in the same oocyte preparation the wild type and the AQAP mutant channels were strongly and identically activated by the prompt application of 500 nm ionomycin, representing a high calcium stimulus (3.8 ± 0.4-fold and 3.9 ± 0.7-fold, respectively, n = 6 in both groups, curves not shown). These data clearly demonstrate that the mutation of the LQLP site abrogated the response of the channel to modest elevation of [Ca2+], whereas the sensitivity to high calcium was maintained.
To apply a more physiological stimulus than ionomycin, TRESK was coexpressed with M1 muscarinic acetylcholine (ACh) receptor. In this experiment, the effects of modest and high [Ca2+] levels could be tested in the same cell. Modest elevation of [Ca2+] was induced by stepwise increasing concentrations (1, 3, 10, and 30 nm) of carbachol. The final robust calcium signal was evoked by 1 μm concentration of the agonist (Fig. 4B). Modest receptor stimulation (steps up to 30 nm carbachol) activated wild type TRESK 5.4 ± 0.9-fold (n = 11, see wt. in Fig. 4, B and C). The same stimulation resulted in much weaker activation of TRESK-AQLP (2.4 ± 0.5-fold, n = 9, p < 0.02, AQLP) and TRESK-AQAP (2.6 ± 0.3-fold, n = 8, p < 0.03, AQAP in Fig. 4, B and C). These data reinforce the conclusion of the experiment with ionomycin that the sensitivity of TRESK to modest elevation of [Ca2+] is reduced by the AQLP and AQAP mutations.
In response to the robust stimulation of M1 receptor with 1 μm carbachol, the relative activations of the AQLP (21.7 ± 1.7-fold, p < 0.05) and AQAP mutants (18.6 ± 2.0-fold, ns) were equivalent to (or even higher than) that of the wild type channel (15.0 ± 2.1-fold, Fig. 4, B and D). This indicates that the calcium-dependent activation mechanism is also operational in the AQLP and AQAP mutants, but high calcium is required for its initiation. TRESK-AQLP and TRESK-AQAP were less activated by the modest elevation of [Ca2+] than wild type TRESK (I1/I0, Fig. 4, B and C), but the activation of the mutants in the same cells in response to high [Ca2+] was not less than that of the wild type channel (I2/I0, Fig. 4, B and D); these data together convincingly demonstrate that the sensitivity to calcium was shifted by the AQLP and AQAP mutations. The relative activation of the mutants by high calcium exceeded that of the wild type channel (Fig. 4D), because of their more prevalent basal inhibitory phosphorylation. (Dephosphorylation of TRESK by calcineurin could also be impeded by the mutation under resting (low) [Ca2+] conditions.) After the high calcium stimulation, the wild type and mutant channels attained a similarly activated state as indicated by their similar benzocaine-sensitivity (Fig. 4B).
The protein expression levels of wild type and AQAP mutant HA-tagged TRESK channels appeared to be equal in the oocyte plasma membrane (Fig. 5A). The different calcium-sensitivity of wild type and AQAP mutant TRESK was independent of the expression levels of the channel proteins (Fig. 5, B and C).
FIGURE 5.
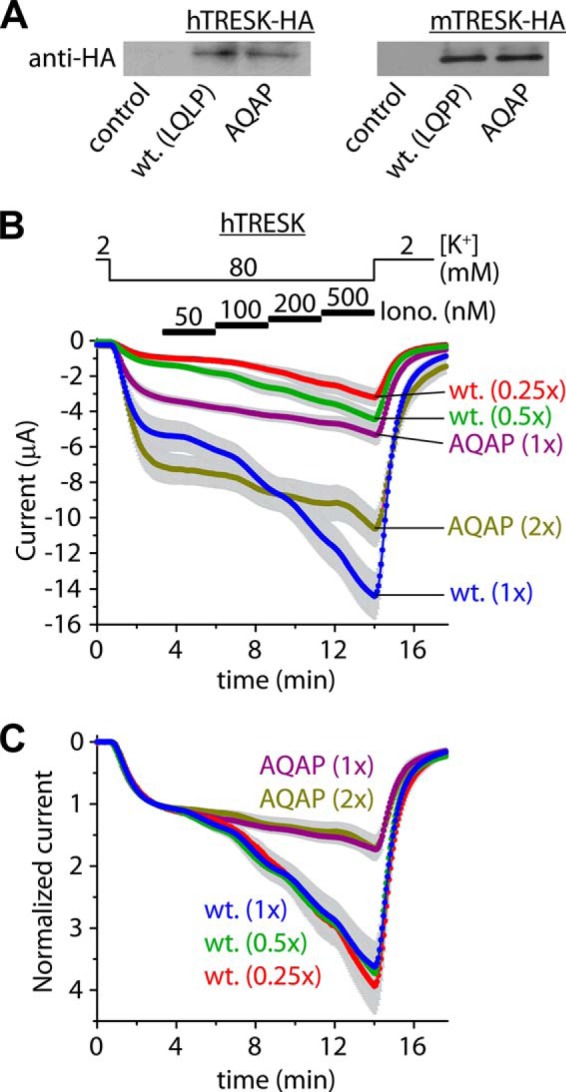
The effect of the AQAP mutation is independent of the expression levels. A, expression levels of TRESK channels containing the wild type (LQLP, wt.) sequence or the AQAP mutation in their intracellular loops, and the influenza hemagglutinin epitope (HA) at their C termini, were compared by anti-HA immunoblots. Expression levels of the LQLP (wt.) and AQAP constructs were similar both in the case of the human (hTRESK) and the mouse (mTRESK) channels. Control membrane preparations were prepared from an identical number of non-injected oocytes as used for the groups expressing hTRESK-HA and hTRESK-HA-AQAP (n = 17, 17) or mTRESK-HA and mTRESK-HA-AQAP (n = 40, 40). Identical amounts of injected cRNAs of the respective constructs were verified on denaturing agarose gels and identical protein content of the membrane preparations were checked by SDS-PAGE (not shown). B, expression levels of hTRESK (wt.) and hTRESK-AQAP (AQAP) were controlled by adjusting the amounts of cRNAs injected into the oocytes (as indicated on the right side). The five groups of cells (n = 22 for wt.(1x) and n = 11 or 12 for the other four groups), characterized by basal current amplitudes varying in line with the injected cRNA quantity, were stimulated by stepwise increasing concentrations of ionomycin (as in Fig. 4A). C, normalized currents from the same five groups of oocytes as in panel B. Note that the calcium sensitivity of the wild type and AQAP channels is independent of the expression levels. The wild type (blue, green, and red) and the AQAP (purple and olive) curves, respectively, overlap.
Mutations of the LQLP Site Are Also Effective in TRESK-PVIVIT Channel
We examined the calcium-dependent regulation, when PQIIIS was replaced by the PVIVIT sequence of high calcineurin binding affinity (14, 15). The average currents of TRESK-PVIVIT and the wild type channel appeared to be similar (Fig. 6A). In fact, TRESK-PVIVIT was less activated than TRESK in this experiment (7.0 ± 0.8-fold (n = 11) versus 11.5 ± 1.4-fold (n = 10), respectively, p < 0.01, normalized currents not shown). Thus the activation of the mutant did not exceed that of wild type TRESK, despite of the experimentally enhanced affinity of the binding site for calcineurin. In another oocyte preparation, normalized activation of TRESK-PVIVIT was not significantly different from that of the wild type channel (4.3 ± 0.3-fold (n = 11) versus 4.3 ± 0.5-fold (n = 10), respectively, curves not shown). These results suggest that the substitution of the native binding site with PVIVIT sequence decreased the basal inhibitory phosphorylation of TRESK in one oocyte preparation but not in the other. (Enhanced anchorage of calcineurin to TRESK-PVIVIT may result in the dephosphorylation of the channel under resting conditions, depending on the characteristics of the cell preparation, e.g. the basal cytoplasmic calcium concentration.)
FIGURE 6.

The loss- and gain-of-function mutations of the LQLP site are effective in the TRESK-PVIVIT context; the PVIVIT mutation is more effective in the wild type (LQLP) than in the AQAP background. A, average currents of oocytes expressing wild type TRESK (wt., gray curve) or TRESK-PVIVIT (PVIVIT, black curve) are compared. The PQIIIS site was replaced with the PVIVIT sequence of high calcineurin-affinity in the TRESK-PVIVIT mutant. The activation of the currents was evoked by ionomycin (0.5 μm, as indicated by the horizontal black bar). B, normalized responses of three different mutants of TRESK to stepwise increasing (50, 100, 200, and 500 nm) concentrations of ionomycin are illustrated. The PQIIIS motif was replaced with PVIVIT in all three mutants. The LQLP motif was not modified in TRESK-PVIVIT (see the LQLP curve); it was disabled by AQAP mutation in TRESK-PVIVIT-AQAP (AQAP curve); or the TLQLPP region was replaced by the corresponding fragment of NFAT in TRESK-PVIVIT-YLAVPQ (YLAVPQ curve). The partial activation of TRESK-PVIVIT-YLAVPQ (YLAVPQ) was significantly higher than that of TRESK-PVIVIT (LQLP) at the time point indicated with an asterisk, whereas the partial activation of TRESK-PVIVIT-AQAP (AQAP) was significantly reduced, compared with TRESK-PVIVIT (LQLP) at the time point indicated with a double asterisk. C, normalized responses of TRESK (wt.), TRESK-PVIVIT (PVIVIT), TRESK-AQAP (AQAP), and TRESK-PVIVIT-AQAP (PVIVIT+AQAP) to the same stimulation as in panel B are plotted. The partial activation of TRESK-PVIVIT (PVIVIT) was significantly higher than that of the wild type (wt.) channel at the time point indicated with an asterisk. In contrast, the sensitivity of TRESK-AQAP to low calcium was not enhanced by the PVIVIT mutation (compare PVIVIT+AQAP to AQAP curve). (Only plus or minus error bars are shown. The currents in panel C were measured from another oocyte preparation than those in panel B.)
Next, we asked whether LQLP was simply an auxiliary binding site for calcineurin and whether its major mechanism of action was to increase the overall affinity of TRESK for the phosphatase. If so, then the effects of its loss-of-function (AQAP) mutation could be compensated by the gain-of-function (PVIVIT) mutation of the main (PQIIIS) docking motif. Therefore we examined the effects of the AQAP mutation in TRESK-PVIVIT background (Fig. 6B). The AQAP mutation substantially reduced the sensitivity of TRESK-PVIVIT to modest elevation of [Ca2+] evoked by stepwise stimulation with 50–500 nm ionomycin (see AQAP (TRESK-PVIVIT-AQAP) and LQLP (TRESK-PVIVIT) curves in Fig. 6.B; 1.96 ± 0.17-fold activation for TRESK-PVIVIT-AQAP versus 2.98 ± 0.33-fold for TRESK-PVIVIT, at the time point indicated with double asterisk, n = 12 for both groups, p < 0.02). This indicates that LQLP is a major determinant of the overall affinity of TRESK for calcineurin (thus PVIVIT mutation cannot increase calcineurin binding as much as AQAP decreases it) or the two (PQIIIS and LQLP) sites are not functionally equivalent (and cross-compensation is impossible because of their independent functions).
TRESK activation was decreased by the reduction of the calcineurin binding affinity of the PQIIIS site by alanine substitutions (Fig. 1B). In contrast, the regulation of the wild type and TRESK-PVIVIT channels were identical (or highly similar, Fig. 6A), despite of the order of magnitude difference between the in vitro KD values (0.5 versus 5 μm) of their PXIXI(T/S) sites for calcineurin (14). Thus it is reasonable to assume that the binding of calcineurin to TRESK-PVIVIT is close to saturation under resting conditions. It was interesting to find that despite of this saturation, the calcium sensitivity of TRESK-PVIVIT could be increased by replacing its native TLQLPP site with YLAVPQ sequence of NFATc1 (compare YLAVPQ (TRESK-PVIVIT-YLAVPQ) and LQLP (TRESK-PVIVIT) curves in Fig. 6.B, 2.06 ± 0.25-fold activation for TRESK-PVIVIT-YLAVPQ versus 1.38 ± 0.06-fold for TRESK-PVIVIT, at the time point indicated with an asterisk, n = 12 for both groups, p < 0.02). It is evident from this result that calcineurin binds to YLAVPQ sequence with higher affinity than to the native TLQLPP motif of TRESK. It seems unlikely that YLAVPQ evoked its effect by further increasing the association of calcineurin to the nearly saturated TRESK-PVIVIT under resting conditions. It is more probable that the LQLP and PQIIIS sites of TRESK are involved in different phases of the activation process.
We have also examined the effect of the PVIVIT mutation on the sensitivity of the channel to modest elevation of [Ca2+]. In one oocyte preparation no difference was found between the activation of TRESK and TRESK-PVIVIT; the curves completely overlapped during the stepwise 50–500 nm ionomycin stimulation (curves not shown, 5.4 ± 0.8-fold (n = 9) versus 4.8 ± 0.8-fold (n = 13) activation at the end of the stimulation, respectively).
In another cell preparation, however, we could detect the difference: the PVIVIT mutant exhibited enhanced sensitivity to modest stimulation (compare wt. (TRESK) and PVIVIT (TRESK-PVIVIT) curves in Fig. 6C; 1.48 ± 0.05-fold activation for TRESK (n = 20) versus 1.82 ± 0.14-fold for TRESK-PVIVIT (n = 18), at the time point indicated with an asterisk, p < 0.02).
In the same cell preparation, we have also examined the effect of the PVIVIT mutation in TRESK-AQAP background. Interestingly, however, the PVIVIT mutation did not increase the sensitivity of TRESK-AQAP to modest [Ca2+] (compare AQAP and PVIVIT+AQAP curves in Fig. 6C). In high (200–500 nm) [ionomycin], the two curves deviated, although the difference did not reach significance (2.37 ± 0.28-fold activation for TRESK-AQAP (n = 11) versus 2.97 ± 0.38-fold for TRESK-PVIVIT-AQAP (n = 14), at the end of the measurement). Thus the effect of the PVIVIT mutation on the sensitivity of TRESK to modest [Ca2+] depended on the intact LQLP sequence.
Calcineurin Binds to the LQLP Site of TRESK in Vitro; the Interaction Is Calcium-dependent
Although the above functional data indicate the importance of the LQLP site in the regulation of TRESK, formal evidence is required to verify that LQLP interacts with calcineurin. To this end, we performed GST pulldown experiments from mouse brain cytosol with the cytoplasmic loop of human TRESK (amino acids 174–280) and also with the AQLP and AQAP mutant versions of this bait protein (Fig. 7A). The interaction of these constructs with calcineurin was tested in the presence or absence of calcium. We have previously reported (26) that the two intense bands of proteins interacting with the wild type bait were identified by mass spectrometry analysis as calcineurin A subunit and tubulin (see lane 4 in Fig. 7A; CnA and Tub). The AQLP and AQAP mutations drastically reduced the binding of calcineurin to TRESK loop in the presence of calcium (compare lanes 5 and 6 to lane 4 in Fig. 7A). This indicates that the LQLP site is a major determinant of the interaction between TRESK and calcineurin in the presence of calcium.
FIGURE 7.

The LQLP motif of TRESK is a calcineurin binding site. A, GST pulldown assays were performed from mouse brain cytosol with the cytoplasmic loop of human TRESK (wild type fragment 174–280, LQLP, lanes 1 and 4), and the AQLP (AQLP, lanes 2 and 5) and AQAP (AQAP, lanes 3 and 6) mutant versions of this bait protein. The binding of calcineurin was tested in the presence (1 mm, lanes 4–6) or in the absence of calcium (2 mm EGTA, lanes 1–3). The two known interacting proteins were marked on the right side of the Coomassie Blue-stained gel as tubulin (Tub.) and calcineurin A subunit (CnA). Note the abundant binding of calcineurin to the wild type bait protein in the presence of calcium (lane 4) and the drastic reduction of the interaction by the AQLP and AQAP mutations (lanes 5 and 6) or by the chelation of Ca2+ (lane 1). (The bait protein preparations contained several incompletely translated fragments in addition to the full-length product below 47 kDa, and a prominent contaminating bacterial protein band below 86 kDa.) B, GST pulldown reactions were performed with fragment 232–280 retaining only the LQLP motif (lanes 1–4), or with fragment 174–280 containing both intact (PQIIIS and LQLP) binding sites (lanes 5 and 6). The addition of mouse brain cytosol to the reaction (lane 1 is an only bait control), the presence of calcium (1 mm) or EGTA (2 mm) and the administration of VIVIT peptide (75 μm) were controlled as indicated in the table. The interacting partners, tubulin (Tub.) and calcineurin A (CnA), are labeled on the right side, and a faint contaminating band from the purification of fragment 232–280 is indicated with asterisks. The relevant region of the Coomassie Blue-stained gel was magnified and the contrast was adjusted for better visibility of the bands in the inset below the table. Note that the binding of calcineurin to fragment 232–280 was detected in the presence (lane 3), but not in the absence of calcium (lane 2). VIVIT peptide attenuated the interaction of calcineurin with fragment 174–280 (lane 5 versus 6), but did not enhance the binding of calcineurin to fragment 232–280 (lane 3 versus 4). C, four pairs of independent pulldown reactions were performed with GST-hTRESK(174–280) (wt.) and GST-hTRESK(174–280)-AQAP mutant (m.) as in lanes 4 and 6 of panel A. The samples were analyzed on SDS-PAGE gel followed by Coomassie Blue staining. D, immunoblot of the same samples as in panel C with anti-calcineurin A (anti-CnA) antibody. E, densitometry curve of the immunoblot in panel D, and column diagram of the densitometry counts. Significantly more calcineurin interacted with the wild type (LQLP) construct than with the AQAP mutant (p < 10−5). F, same membrane as in panel D was stripped and re-probed with anti-tubulin β3 (anti-TUBB3) antibody. The loaded amounts of the AQAP pulldown reactions were not less than those of the wild type.
The amount of calcineurin interacting with the wild type TRESK fragment was lower in the absence than in the presence of calcium (compare lane 1 to 4 in Fig. 7A). The relatively weak binding of calcineurin was not further reduced by the AQLP and AQAP mutations in the absence of calcium (compare lanes 2 and 3 to lane 1 in Fig. 7A). Identical results were obtained when the experiment in Fig. 7A was repeated with another bait protein, GST-hTRESK(174–247), which contained the PQIIIS and LQLP sites, but not the region of the Ser-252, Ser-262, Ser-264 substrate serines of calcineurin (results not shown). These data suggest that the binding of calcineurin to the LQLP site is calcium-dependent.
In the above pulldown assays, the cumulative effects of the PQIIIS and LQLP sites determined the interaction of the bait with calcineurin. In order to investigate the LQLP site separately, PQIIIS was removed from the bait protein by truncation. We could detect the calcium-dependent pulldown of calcineurin with this truncated TRESK fragment of amino acids 232–280, containing only the LQLP site but not PQIIIS (compare lane 2 to 3 in the enlarged inset of Fig. 7B). Nevertheless, the interaction of calcineurin with LQLP alone was much weaker than that with fragment 174–280 containing both binding sites (in the presence of calcium, compare lane 3 to 5 in Fig. 7B). Therefore we examined whether the occupation of the PXIXIT-binding site of calcineurin allosterically increases the affinity of the phosphatase for an LxVP ligand. Saturation of the PXIXIT-binding site of calcineurin with VIVIT peptide (15) failed to allosterically promote the association of the phosphatase to the bait containing only the LQLP motif (lanes 3 and 4 in Fig. 7B). Thus, in this respect, the mechanisms of binding to the two types of motifs are not coupled; however, the interaction of calcineurin with the LQLP site was verified to be calcium-dependent also in the case of the bait protein, which does not contain the PQIIIS motif.
Identity of the calcineurin A band was also verified by Western blot using specific antibody (Fig. 7, C and D). Densitometry and statistical analysis indicated that more calcineurin bound to the LQLP than to the AQAP construct (Fig. 7, D–F).
The Intracellular Loop of Wild Type TRESK Is More Rapidly Dephosphorylated in Vitro Than the AQAP Mutant
The different GST-TRESK constructs were radioactively labeled by phoshphorylation with recombinant MARK2 kinase. The kinetics of calcineurin-dependent dephosphorylation was estimated by the subsequent application of diluted brain cytosol to these phosphoproteins immobilized on glutathione agarose (Fig. 8). The human or mouse wild type TRESK sequences (including LQLP or LQPP, respectively) were more rapidly dephosphorylated in a calcium-dependent manner than the AQAP mutant versions of these substrate proteins. This in vitro result confirms that the altered dephosphorylation is responsible for the difference between the functional data of the wild type and AQAP mutant channels, obtained in the oocyte measurements.
FIGURE 8.

The in vitro calcium-dependent dephosphorylation of the wild type TRESK sequence is more rapid than that of the AQAP mutant. A, different substrate proteins, GST-hTRESK(174–280) (hTRESK), GST-hTRESK(174–280)-AQAP (hTRESK-AQAP), GST-mTRESK(164–292) (mTRESK), GST-mTRESK(164–292)-AQAP (mTRESK-AQAP) immobilized on glutathione resin, were in vitro phosphorylated in the presence of [32P-γ]ATP by constitutively active recombinant MARK2 kinase (Trx-His6-MARK2-T208E). Subsequently, they were dephosphorylated with diluted mouse brain cytosol in the presence or absence of calcium for different time periods (as indicated below the autoradiograms of SDS-PAGE gels). Note that the wild type TRESK sequences were more rapidly dephosphorylated than the AQAP mutants (see the 0, 15, 30, 60 min reactions in the left 4 columns of bands). For statistical analysis, three independent pairs of dephosphorylation reactions were performed in the presence or absence of calcium for 60 min with each substrate proteins (see the right 6 columns of bands). B, calcium-dependent dephosphorylation (illustrated in the column graph in percent) was calculated from the radioactive counts of these bands in panel A. The wild type versions of the substrate proteins were dephosphorylated more efficiently than the AQAP mutants (p < 0.002 for both hTRESK and mTRESK).
DISCUSSION
K2P background potassium channels are responsible for maintaining the highly negative resting membrane potential in different excitable cell types (1–5). Their regulation by voltage-independent mechanisms is of importance, since K2P channels generally determine the responsiveness of the cell to different excitatory stimuli and can also efficiently counter massive depolarization. The fifteen members of the K2P family are targeted by diverse regulatory mechanisms conferring temperature-, mechano-, or pH-sensitivity in the physiological range and modulated by a great variety of signaling pathways and protein interactions. The different excitable cell types typically express a specific subset of these channels to meet the regulatory requirements for their functions.
TRESK (K2P18) is abundantly expressed in the pseudounipolar neurons of dorsal root, trigeminal and other sensory ganglia (7, 27–31), together with a major fraction of TREK-2 (K2P10) and other minor K2P components (27, 32). TRESK is present in the plasma membrane of the cell body, as its single channel openings were detected in excised membrane patches of dorsal root ganglion (DRG) neurons (27), and perikaryons isolated form TRESK knock-out mice showed altered electrophysiological properties (28). TRESK function appeared to be partially compensated by other K+ channels in these cells (28), and a TRESK-related phenotype has not yet been described in the knock-out animals apart from a slightly increased mortality rate following anesthesia (33). Rat sciatic nerve axotomy induced hyperexcitability of nociceptive DRG neurons by decreasing TRESK mRNA expression (30), and the mutation of human TRESK was reported to be linked to a rare form of familial migraine (7), suggesting a role for the channel in pain disorders (31, 34–36).
Pharmacological separation of TRESK current from those of the other K2P channels in native cells has not yet been adequately resolved; TRESK current is usually approximated by the current component sensitive to the relatively nonspecific inhibitors lamotrigine (31, 36, 37), hydroxy-α-sanshool (38) or isobutylalkenyl amide (IBA) (30). Furthermore, it is a methodological challenge to reliably measure calcium-dependent TRESK activation under whole-cell patch clamp conditions (even in HEK cells overexpressing the channel (see Ref. (8)). Therefore we examined TRESK regulation by two-electrode voltage clamp of Xenopus oocytes, in a system characterized by minimally altered cytoplasmic composition during the measurement. This approach made it possible to investigate the effects of PXIXIT- and LXVP-like site modifications on a calcineurin-target protein with unprecedented time resolution.
Several lines of evidence indicate that the LQLP sequence functions as an LXVP-like calcineurin-docking site in TRESK. The AQAP mutation reduced the binding of calcineurin to the cytoplasmic loop of TRESK in vitro. A fragment of the loop containing only the LQLP but not the PQIIIS site also pulled down the phosphatase from brain cytosol. The interaction between LQLP and calcineurin was calcium-dependent, an established property of LXVP sites (20–22). These in vitro results are in good accordance with the functional data: alanine substitutions in the LQLP site reduced the sensitivity of TRESK to calcium, whereas the LQLP sequence was functionally substituted by the canonical LXVP motif from NFAT. Thus LQLP is the first reported LXVP-like calcineurin-docking site, which contains leucine instead of valine in the third position of the motif. Accordingly, the LXVP consensus sequence could be modified to LX(V/L)P.
Perhaps the first example of enhanced dephosphorylation by calcineurin of a short peptide fragment containing an LXVP site was documented in the case of DLDVPIPGRFDRRVSVCAE sequence of PKA regulatory subunit RII, although it was not evident in 1986 that the effect depended on the LxVP motif (39). This important example indicates that LXVP can be located relatively close to the dephosphorylated residue; the two sites are separated only by 9 amino acids. Therefore it is reasonable to assume that calcineurin, while it is docked to the LQLP site, can also dephosphorylate the regulatory serines of human TRESK in distances of 8, 18 and 20 residues.
The LXVP motif was first described in NFAT as a second binding site for calcineurin, in addition to PXIXIT (17–19). Interestingly, however, the 13 residues dephosphorylated by calcineurin in NFAT are not even C-terminal to the LXVP site, but they are distributed along a more than 200 amino acid long region positioned between the (N-terminal) PXIXIT and the (C-terminal) LXVP motifs (40). KSR2 (Kinase Supressor of Ras 2) also contains its 3 dephosphorylated residues N-terminally to its LXVP site, distributed in a region of about 200 amino acids (41). In contrast, the two substrate serines of human RCAN1 (Regulator of calcineurin 1) are located C-terminally to the LAPP motif in distances of 8 and 12 residues (23). In dynamin-related protein 1 (Drp1), LXVP and the phosphoserine are separated by 7 residues (42). This suggests that the mode of action of the LXVP-like binding sites may vary in the different calcineurin substrates. As indicated by the example of PKA RII peptide (20, 39), calcineurin, while it is docked to the LXVP-like site, can dephosphorylate substrate residues located nearby in the C-terminal direction in RCAN1, Drp1 and human TRESK (we may call them mode 1 LXVP substrates of calcineurin). However, in the other group of substrates (mode 2), as in NFAT and KSR2, three-dimensional movement of the substrate-phosphatase complex is required to bring the different substrate residues to the proximity of the active site, or calcineurin may dissociate from the LXVP motif during dephosphorylation.
Rodent TRESK contains LQPP instead of LQLP. This is highly similar to LAPP of RCAN1, which is a verified calcineurin-docking site (23). The significance of LQPP in the anchoring of calcineurin could not be unequivocally determined in the oocyte measurements, since the mutation of the motif also caused calcineurin-independent effects; presumably it interfered with the action of the TRESK-inhibitory kinase, or influenced the intrinsic channel activity of the protein. The contribution of the LQPP motif to the action of calcineurin was clearly demonstrated by the in vitro dephosphorylation experiment, where the AQAP mutation diminished dephosphorylation. Nevertheless, the PQAVAD mutation completely eliminated the activation of mouse TRESK (12), suggesting that LQPP is less effective in the mouse than LQLP in the human channel. These data indicate that the measurement of several kinetic parameters (e.g. basal activity, relative activation, rate of activation, recovery kinetics from activation) may be required for the interpretation of the effects of LXVP-like site mutations. It is apparent that several aspects of the kinase and phosphatase reactions are different between the human and mouse orthologs, and care must be taken if data are extrapolated from rodent experimental models to the physiology of human TRESK.
Mutation of LQLP to AQAP in human TRESK did not significantly influence the final level of activation in response to high calcium, but slowed down the regulatory process. AQAP mutation reduced the sensitivity of the channel to calcium, and this effect was also detected in TRESK-PVIVIT. Furthermore, the calcium sensitivity of TRESK-PVIVIT could be increased by the substitution of the native TLQLPP sequence with YLAVPQ, the canonical LXVP site of NFAT. The PVIVIT mutation itself could also increase the sensitivity of TRESK to modest elevation of [Ca2+] in an oocyte preparation probably characterized by relatively low occupancy of the PQIIIS calcineurin-binding site. However, this effect of the PVIVIT mutation required the presence of the intact LQLP site. Sensitivity of TRESK-AQAP to modest [Ca2+] has not been increased by the PVIVIT mutation in the same cell preparation. These results suggest that the function of the LQLP site is not equivalent to that of PQIIIS, and the mechanisms of action of these sites need further explanation. Regarding that the binding of calcineurin to the LQLP site is calcium-dependent, but it is generally accepted in the literature that the docking of the phosphatase to PXIXIT-like motifs is independent from [Ca2+] (13, 14, 43, 44), a plausible explanation for the results may be that the binding of calcineurin to TRESK is a process of two consecutive steps.
Calcineurin is anchored to PQIIIS under resting conditions. When the cytoplasmic [Ca2+] is elevated, calcineurin may dock to the LQLP motif and become adequately positioned for the dephosphorylation of the regulatory serines (Fig. 9). In other words, PQIIIS may be mainly responsible for the availability of calcineurin for the reaction, whereas LQLP for the efficient targeting of the activated phosphatase to the specific substrate serines. If LQLP is disabled then the reaction slows down, and this can be especially detrimental for the regulatory process, when the phosphatase spends only a limited fraction of time in the activated state in the case of modest stimulation. In accordance with the above model, LQLP may also attract a small amount of active calcineurin from solution or yet uncharacterized weak binding sites during the stimulation, and mediate the activation of TRESK-PQAAAS mutant.
FIGURE 9.

Interplay of the PQIIIS and LQLP calcineurin-docking motifs in the regulation of human TRESK. A, calcineurin binds to the PQIIIS motif under resting conditions, depending on the concentration of the phosphatase in the cell, but independently of the cytoplasmic [Ca2+]. B, LQLP-binding site of calcineurin becomes available in response to the calcium signal. The LQLP motif binds to calcineurin and brings the adjacent substrate residues in the proximity of the active site of the enzyme.
In summary, the LQLP site is a major determinant of the interaction of calcineurin with human TRESK, and significantly contributes to the calcium-sensitivity of the channel. Our results also provide general insight into the functions of the PXIXIT- and LXVP-like sites, and may impact on the research of other calcineurin-target proteins containing these motifs.
Acknowledgments
We thank Irén Veres and Alice Dobolyi for skillful technical assistance.
This work was supported by the Hungarian National Research Fund (OTKA K108496).
- TRESK
- TWIK-related spinal cord K+ channel, K2P18.1
- MARK
- microtubule-affinity regulating kinase
- NFAT
- nuclear factor of activated T cells.
REFERENCES
- 1. Enyedi P., Czirják G. (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev. 90, 559–605 [DOI] [PubMed] [Google Scholar]
- 2. Noël J., Sandoz G., Lesage F. (2011) Molecular regulations governing TREK and TRAAK channel functions. Channels 5, 402–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mathie A., Al-Moubarak E., Veale E. L. (2010) Gating of two pore domain potassium channels. J. Physiol. 588, 3149–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Plant L. D. (2012) A Role for K2P Channels in the Operation of Somatosensory Nociceptors. Front. Mol. Neurosci. 5, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lotshaw D. P. (2007) Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem. Biophys. 47, 209–256 [DOI] [PubMed] [Google Scholar]
- 6. Czirják G., Tóth Z. E., Enyedi P. (2004) The two-pore domain K+ channel, TRESK, is activated by the cytoplasmic calcium signal through calcineurin. J. Biol. Chem. 279, 18550–18558 [DOI] [PubMed] [Google Scholar]
- 7. Lafrenière R. G., Cader M. Z., Poulin J. F., Andres-Enguix I., Simoneau M., Gupta N., Boisvert K., Lafrenière F., McLaughlan S., Dubé M. P., Marcinkiewicz M. M., Ramagopalan S., Ansorge O., Brais B., Sequeiros J., Pereira-Monteiro J. M., Griffiths L. R., Tucker S. J., Ebers G., Rouleau G. A. (2010) A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat. Med. 16, 1157–1160 [DOI] [PubMed] [Google Scholar]
- 8. Braun G., Nemcsics B., Enyedi P., Czirják G. (2011) TRESK background K+ channel is inhibited by PAR-1/MARK microtubule affinity-regulating kinases in Xenopus oocytes. PLoS One 6, e28119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rahm A. K., Wiedmann F., Gierten J., Schmidt C., Schweizer P. A., Becker R., Katus H. A., Thomas D. (2014) Functional characterization of zebrafish K18.1 (TRESK) two-pore-domain K channels. Naunyn Schmiedebergs Arch. Pharmacol. 387, 291–300 [DOI] [PubMed] [Google Scholar]
- 10. Czirják G., Enyedi P. (2010) TRESK background K+ channel is inhibited by phosphorylation via two distinct pathways. J. Biol. Chem. 285, 14549–14557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Czirják G., Vuity D., Enyedi P. (2008) Phosphorylation-dependent binding of 14–3-3 proteins controls TRESK regulation. J. Biol. Chem. 283, 15672–15680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Czirják G., Enyedi P. (2006) Targeting of calcineurin to an NFAT-like docking site is required for the calcium-dependent activation of the background K+ channel, TRESK. J. Biol. Chem. 281, 14677–14682 [DOI] [PubMed] [Google Scholar]
- 13. Roy J., Cyert M. S. (2009) Cracking the phosphatase code: docking interactions determine substrate specificity. Sci. Signal. 2, re9. [DOI] [PubMed] [Google Scholar]
- 14. Li H., Rao A., Hogan P. G. (2011) Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 21, 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aramburu J., Yaffe M. B., López-Rodriguez C., Cantley L. C., Hogan P. G., Rao A. (1999) Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285, 2129–2133 [DOI] [PubMed] [Google Scholar]
- 16. Li H., Zhang L., Rao A., Harrison S. C., Hogan P. G. (2007) Structure of calcineurin in complex with PVIVIT peptide: portrait of a low-affinity signalling interaction. J. Mol. Biol. 369, 1296–1306 [DOI] [PubMed] [Google Scholar]
- 17. Liu J., Masuda E. S., Tsuruta L., Arai N., Arai K. (1999) Two independent calcineurin-binding regions in the N-terminal domain of murine NF-ATx1 recruit calcineurin to murine NF-ATx1. J. Immunol. 162, 4755–4761 [PubMed] [Google Scholar]
- 18. Liu J., Arai K., Arai N. (2001) Inhibition of NFATx activation by an oligopeptide: disrupting the interaction of NFATx with calcineurin. J. Immunol. 167, 2677–2687 [DOI] [PubMed] [Google Scholar]
- 19. Park S., Uesugi M., Verdine G. L. (2000) A second calcineurin binding site on the NFAT regulatory domain. Proc. Natl. Acad. Sci. U.S.A. 97, 7130–7135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grigoriu S., Bond R., Cossio P., Chen J. A., Ly N., Hummer G., Page R., Cyert M. S., Peti W. (2013) The molecular mechanism of substrate engagement and immunosuppressant inhibition of calcineurin. PLoS Biol. 11, e1001492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodríguez A., Roy J., Martínez-Martínez S., López-Maderuelo M. D., Niño-Moreno P., Ortí L., Pantoja-Uceda D., Pineda-Lucena A., Cyert M. S., Redondo J. M. (2009) A conserved docking surface on calcineurin mediates interaction with substrates and immunosuppressants. Mol. Cell 33, 616–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martínez-Martínez S., Rodríguez A., López-Maderuelo M. D., Ortega-Pérez I., Vázquez J., Redondo J. M. (2006) Blockade of NFAT activation by the second calcineurin binding site. J. Biol. Chem. 281, 6227–6235 [DOI] [PubMed] [Google Scholar]
- 23. Mehta S., Li H., Hogan P. G., Cunningham K. W. (2009) Domain architecture of the regulators of calcineurin (RCANs) and identification of a divergent RCAN in yeast. Mol. Cell. Biol. 29, 2777–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoshida S., Plant S. (1992) Mechanism of release of Ca2+ from intracellular stores in response to ionomycin in oocytes of the frog Xenopus laevis. J. Physiol. 458, 307–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thurman C. L., Burns J. S., O'Neil R. G. (2000) Identifying the Ca++ signalling sources activating chloride currents in Xenopus oocytes using ionomycin and thapsigargin. Cell Signal. 12, 629–635 [DOI] [PubMed] [Google Scholar]
- 26. Enyedi P., Veres I., Braun G., Czirják G. (2014) Tubulin binds to the cytoplasmic loop of TRESK background K+ channel in vitro. PLoS One, 9, e97854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kang D., Kim D. (2006) TREK-2 (K2P10.1) and TRESK (K2P18.1) are major background K+ channels in dorsal root ganglion neurons. Am. J. Physiol. Cell Physiol. 291, C138–C146 [DOI] [PubMed] [Google Scholar]
- 28. Dobler T., Springauf A., Tovornik S., Weber M., Schmitt A., Sedlmeier R., Wischmeyer E., Döring F. (2007) TRESK two-pore-domain K+ channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J. Physiol. 585, 867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoo S., Liu J., Sabbadini M., Au P., Xie G. X., Yost C. S. (2009) Regional expression of the anesthetic-activated potassium channel TRESK in the rat nervous system. Neurosci. Lett. 465, 79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tulleuda A., Cokic B., Callejo G., Saiani B., Serra J., Gasull X. (2011) TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol. Pain 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu P., Xiao Z., Ren F., Guo Z., Chen Z., Zhao H., Cao Y. Q. (2013) Functional analysis of a migraine-associated TRESK K+ channel mutation. J. Neurosci. 33, 12810–12824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marsh B., Acosta C., Djouhri L., Lawson S. N. (2012) Leak K+ channel mRNAs in dorsal root ganglia: relation to inflammation and spontaneous pain behaviour. Mol. Cell Neurosci. 49, 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chae Y. J., Zhang J., Au P., Sabbadini M., Xie G. X., Yost C. S. (2010) Discrete change in volatile anesthetic sensitivity in mice with inactivated tandem pore potassium ion channel TRESK. Anesthesiology 113, 1326–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mathie A. (2010) Ion channels as novel therapeutic targets in the treatment of pain. J. Pharm. Pharmacol. 62, 1089–1095 [DOI] [PubMed] [Google Scholar]
- 35. Zhou J., Yang C. X., Zhong J. Y., Wang H. B. (2013) Intrathecal TRESK gene recombinant adenovirus attenuates spared nerve injury-induced neuropathic pain in rats. Neuroreport 24, 131–136 [DOI] [PubMed] [Google Scholar]
- 36. Guo Z., Cao Y. Q. (2014) Over-Expression of TRESK K+ Channels Reduces the Excitability of Trigeminal Ganglion Nociceptors. PLoS One 9, e87029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang D., Kim G. T., Kim E. J., La J. H., Lee J. S., Lee E. S., Park J. Y., Hong S. G., Han J. (2008) Lamotrigine inhibits TRESK regulated by G-protein coupled receptor agonists. Biochem. Biophys. Res. Commun. 367, 609–615 [DOI] [PubMed] [Google Scholar]
- 38. Bautista D. M., Sigal Y. M., Milstein A. D., Garrison J. L., Zorn J. A., Tsuruda P. R., Nicoll R. A., Julius D. (2008) Pungent agents from Szechuan peppers excite sensory neurons by inhibiting two-pore potassium channels. Nat. Neurosci. 11, 772–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blumenthal D. K., Takio K., Hansen R. S., Krebs E. G. (1986) Dephosphorylation of cAMP-dependent protein kinase regulatory subunit (type II) by calmodulin-dependent protein phosphatase. Determinants of substrate specificity. J. Biol. Chem. 261, 8140–8145 [PubMed] [Google Scholar]
- 40. Macian F. (2005) NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5, 472–484 [DOI] [PubMed] [Google Scholar]
- 41. Dougherty M. K., Ritt D. A., Zhou M., Specht S. I., Monson D. M., Veenstra T. D., Morrison D. K. (2009) KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol. Cell 34, 652–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Slupe A. M., Merrill R. A., Flippo K. H., Lobas M. A., Houtman J. C., Strack S. (2013) A calcineurin docking motif (LXVP) in dynamin-related protein 1 contributes to mitochondrial fragmentation and ischemic neuronal injury. J. Biol. Chem. 288, 12353–12365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aramburu J., Garcia-Cózar F., Raghavan A., Okamura H., Rao A., Hogan P. G. (1998) Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol. Cell 1, 627–637 [DOI] [PubMed] [Google Scholar]
- 44. Garcia-Cozar F. J., Okamura H., Aramburu J. F., Shaw K. T., Pelletier L., Showalter R., Villafranca E., Rao A. (1998) Two-site interaction of nuclear factor of activated T cells with activated calcineurin. J. Biol. Chem. 273, 23877–23883 [DOI] [PubMed] [Google Scholar]