

Background: Viral RNA polymerase nucleotide incorporation fidelity contributes to pathogenesis.
Results: A poliovirus mutator strain and its polymerase have been characterized.
Conclusion: Pathogenesis requires optimal polymerase fidelity; not all mutations in a viral genome are linked to polymerase fidelity.
Significance: Mutator and antimutator phenotypes can be harnessed for the development of viral prophylaxis and antiviral therapies.
Keywords: Poliovirus, RNA Polymerase, RNA Virus, Viral Polymerase, Viral Replication
Abstract
The ability of an RNA virus to exist as a population of genetically distinct variants permits the virus to overcome events during infections that would otherwise limit virus multiplication or drive the population to extinction. Viral genetic diversity is created by the ribonucleotide misincorporation frequency of the viral RNA-dependent RNA polymerase (RdRp). We have identified a poliovirus (PV) RdRp derivative (H273R) possessing a mutator phenotype. GMP misincorporation efficiency for H273R RdRp in vitro was increased by 2–3-fold that manifested in a 2–3-fold increase in the diversity of the H273R PV population in cells. Circular sequencing analysis indicated that some mutations were RdRp-independent. Consistent with the population genetics theory, H273R PV was driven to extinction more easily than WT in cell culture. Furthermore, we observed a substantial reduction in H273R PV virulence, measured as the ability to cause paralysis in the cPVR mouse model. Reduced virulence correlated with the inability of H273R PV to sustain replication in tissues/organs in which WT persists. Despite the attenuated phenotype, H273R PV was capable of replicating in mice to levels sufficient to induce a protective immune response, even when the infecting dose used was insufficient to elicit any visual signs of infection. We conclude that optimal RdRp fidelity is a virulence determinant that can be targeted for viral attenuation or antiviral therapies, and we suggest that the RdRp may not be the only source of mutations in a RNA virus genome.
Introduction
Major challenges to virus survival occur during interactions with the host. Viruses struggle with host defense mechanisms, diverse cellular environments in different tissues, anatomic restrictions such as the blood-brain barrier, and host-to-host transmission. The outcomes of these multiple selective pressures determine tissue tropism and, ultimately, the pathogenic result of an infection. In this dynamic and changing environment, genetic diversity in RNA virus populations appears to be critical for fitness and survival and likely contributes to pathogenesis. For example, a poliovirus (PV)6 mutant expressing a high fidelity (antimutator) RNA-dependent RNA polymerase (RdRp) with a change of Gly-64 to Ser (G64S) displays an attenuated phenotype in susceptible mice. G64S efficiently replicates in a number of tissues but is restricted in its ability to access the central nervous system. Treatment of viral stocks with chemical mutagens to increase the number of mutations in the G64S genome to wild-type levels leads to a significant increase in neuropathogenesis. These observations provide compelling evidence that reduced population diversity restricts tissue tropism and determines an attenuated phenotype (1–4).
Both the number of genetic variants in a population and the speed with which they are created likely determine the efficiency with which a virus can navigate the dynamic environment in the infected host. Mathematical models on population genetics, however, identified a theoretical limit to the number of mutations (extent of genetic diversity) a haploid genome can withstand when the yield of offspring (fecundity) is fixed (5). It is therefore assumed that RNA viruses exhibit the highest mutation frequency tolerable, but the experimental evidence is limited to circumstances in which viruses are exposed to chemical mutagenesis (6–8).
Indeed, existence of an RNA virus on the brink of extinction has been the explanation for the sensitivity of viruses to nucleoside analogues with ambiguous base-pairing capacity (6, 8, 9). This class of nucleoside analogues has been termed lethal mutagens (6, 8, 9). The most extensively characterized lethal mutagen is ribavirin, a drug currently included in the standard of care for the treatment of hepatitis C virus infection (6, 8–10). A complication with the interpretation of these experiments arises from the fact that lethal mutagens can have pleiotropic effects on cells, not the least of which is utilization of the compounds by cellular polymerases.
One approach to explore the limits on viral population diversity with the least collateral damage is creation and characterization of a virus variant with a mutator phenotype. Poliovirus is an ideal model system to address this question because of the myriad tools available to study this virus (11, 12). However, the only PV RdRp derivative known to exhibit a mutator phenotype yielded a dead virus, for reasons that may be unrelated to extinction catastrophe (13). Viable mutator strains have been identified for coxsackievirus B3 (14) and murine and severe acute respiratory syndrome coronaviruses (15, 16). For coxsackievirus B3, mutations were in the active site of the RdRp. However, for coronaviruses, mutations were in a putative proofreading exonuclease (16). Because antimutator strains were not available for these systems, the question of an optimal fidelity could not be addressed directly but was inferred (15, 16).
Here, we describe a PV variant with an RdRp derivative exhibiting a mutator phenotype (H273R). GMP misincorporation and ribavirin triphosphate utilization efficiencies by H273R RdRp were significantly increased in vitro, leading to increased population diversity and ribavirin sensitivity of the virus in cells. H273R was driven to extinction more easily than WT in cell culture, indicating that WT virus is on the verge of the maximal mutation rate that can be tolerated without a loss in fitness. Interestingly, we observed a substantial reduction in H273R virulence, measured as the ability to cause paralysis in a susceptible mouse model (17). Reduced virulence was attributable to the inability of H273R to sustain replication in tissues/organs that are normally infected by PV. Despite the attenuated phenotype, H273R was capable of replicating in mice to levels sufficient to induce a protective immune response at doses in which inactivated virus would fail to elicit a protective response. These data demonstrate that population diversity is optimized for viral fitness. In combination with data for the high fidelity G64S variant, we conclude that the ability of RNA viruses to manipulate their genetic diversity confers upon a population a fast, effective mechanism to adapt to dynamic environments. However, substantial constraints exist on the sequence space that can be sampled without consequences for viral virulence.
MATERIALS AND METHODS
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco, and the Pennsylvania State University (assurance number A3400-01 UCSF and A3141-01 PSU). All efforts were made to minimize animal suffering.
Cells and Viruses
HeLa S3 cells were obtained from the American Type Culture Collection (ATCC) and grown in DMEM/F-12 plus 10% fetal bovine serum, 100 units/ml penicillin, and 100 units/ml streptomycin. PV type 1 Mahoney was used throughout this study.
Construction of H273R RdRp (3Dpol) Expression Plasmid
The H273R mutation was introduced into a modified 3Dpol-coding sequence by using overlap-extension PCR. In brief, the H273R clone was produced by overlap-extension PCR with oligonucleotides 3D-PV-AvrII-rev (5′-CCT GAG TGT TCC TAG GAT CTT TAG T-3′), 3D-PV-H273R-for (5′-CTA AAC CAC TCA CAC AGG CTG TAC AAG AAT AAA ACA-3′), and 3D-PV-PstI-for (5′-CCT GAG TGT TCC TAG GAT CTT TAG T-3′), 3D-PV-H273R-rev (5′-TGT TTT ATT CTT GTA CAG CCT GTG TGA GTG GTT TAG-3′) and with pET26Ub-3D-BPKN-I92T (18) as a template. The final product of the second PCR was purified and digested with PstI and NheI and ligated into pET26Ub-3D-BPKN-I92T that had been digested with the same enzymes. The mutation was confirmed by DNA sequencing (Nucleic Acid Facility, Pennsylvania State University).
Expression and Purification of PV RdRp Proteins
WT, G64S, and H273R RdRp derivatives were expressed in Escherichia coli by using a ubiquitin fusion system as described previously (18) and then purified as described previously (1).
Purification, 5′-32P Labeling of RNA Oligonucleotides, and Annealing of RNA Sym/Sub Substrates
RNA oligonucleotides were purified by denaturing PAGE as described previously (19). Concentrations were determined by measuring the absorbance at 260 nm using a Nanodrop spectrophotometer and using the appropriate calculated extinction coefficient. RNA oligonucleotides were end-labeled by using [γ-32P]ATP and T4 polynucleotide kinase. Reactions, typically 100 μl, contained 1 μm [γ-32P]ATP, 100 μm RNA oligonucleotide, 1× Kinase Buffer, and 0.4 units/μl T4 polynucleotide kinase. Reactions were incubated at 37 °C for 60 min and then placed at 65 °C for 10 min to heat-inactivate T4 polynucleotide kinase. RNA sym/sub primer-templates were produced by annealing 10–20 μm RNA oligonucleotides in T10E1 (10 mm Tris, pH 8.0, 1 mm EDTA) in a Progene Thermocycler (Techne). Annealing reactions were heated to 90 °C for 1 min and slowly cooled (5 °C/min) to 10 °C.
PV RdRp-catalyzed Nucleotide Incorporation
Nucleotide incorporation experiments were performed as described previously (20). Briefly, reactions were performed in 50 mm HEPES, pH 7.5, 10 mm 2-mercaptoethanol, 60 μm ZnCl2, and 5 mm MgCl2. All reactions were performed at 30 °C. Reactions were assembled by incubating 1 μm PV RdRp with 1 μm sym/sub RNA primer-template (0.5 μm duplex) for 3 min, allowing equilibration to 30 °C, and then rapidly mixing with the appropriate NTP substrate. Rapid mixing/quenching experiments were performed by using a model RQF-3 chemical quench-flow apparatus (KinTek Corp., Austin, TX). Reactions were quenched by addition of 0.5 m EDTA to a final concentration of 0.3 m. Products were analyzed by denaturing PAGE. Gels were visualized by using a PhosphorImager and quantified by using ImageQuant software (GE Healthcare).
Determination of Kinetic Parameters Kd, app and kpol
Data were fit by nonlinear regression using the program KaleidaGraph (Synergy Software, Reading, PA). Time courses at fixed nucleotide concentration were fit to Equation 1,
where A is the maximal concentration of product formed; kobs is the observed first-order rate constant describing product formation; t is the time, and C is a constant. The apparent dissociation constant (Kd, app) and maximal rate for nucleotide incorporation (kpol) were determined using Equation 2,
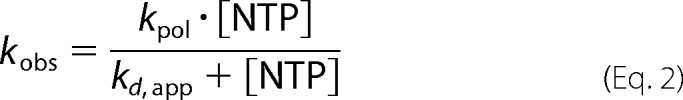 |
Construction of Mutated Viral cDNA Clones and Replicons
To introduce the H273R mutation into the 3Dpol-coding sequence of viral cDNA, pMovRA, PCR was performed with oligonucleotides PV-3D-AvrII-rev, PV-3D-AflII-for (5′-AAC GAT CCC AGG CTT AAG ACA GAT TTT GAG-3′) and the pET26Ub-3D-BPKN-I92T-Arg-273 plasmid as template. PCR products were purified and digested with AvrII and AflII. The digested PCR product was ligated into pUC18-BglII-EcoRI-3CD vector (21), a subclone of the viral cDNA. From this vector, the fragment between BglII and EcoRI was cloned into the viral cDNA plasmid (pMovRA). To introduce mutations into the subgenomic replicon, pRLucRA, the fragment between the BglII and AvrII sites from the mutated pUC18-BglII-EcoRI-3CD subclone vector was ligated into pRLucRA. DNA sequencing was used to verify the integrity of all clones. The H273K and H273Q mutations were introduced into the 3Dpol-coding sequence of viral cDNA, pMovRA, and subgenomic replicon, pRLucRA, by using overlap-extension PCR. In brief, PCR was performed with oligonucleotides 3D-PV-BglII-for (5′-TAG AGG ATC CAG ATC TTG GAT GCC A-3′), 3D-PV-H273K-rev (5′-TTG TTT ATT CTT GTA CAG TTT GTG TGA GTG GTT TAG-3′), 3D-PV-H273Q-rev (5′-TTG TTT ATT CTT GTA CAG CTG GTG TGA GTG GTT TAG-3′), and 3D-PV-EcoRI-ApaI-poly(A)-rev (5′-CGC TCA ATG AAT TCG GGC CCT TTT TTT TTT TTT TTT TTT TCT CC-3′), 3D-PV-H273K-for (5′-CTA AAC CAC TCA CAC AAA CTG TAC AAG AAT AAA ACA-3′), and 3D-PV-H273Q-for (5′-CTA AAC CAC TCA CAC CAG CTG TAC AAG AAT AAA ACA-3′) and with pMo-3D-BPKN-I92T as a template. The final product of the second PCR was purified and digested with BglII and ApaI and ligated into pMo-3D-BPKN-I92T and pRLuc-3D-BPKN-I92T that had been digested with the same enzymes. The mutation was confirmed by DNA sequencing (Nucleic Acid Facility, Pennsylvania State University).
RNA Transcription
The pMovRA and pRLucRA plasmids were linearized with EcoRI and ApaI, respectively, and purified with Qiaex II suspension (Qiagen) by following the manufacturer's protocol. RNA was then transcribed from the linearized plasmid DNAs in a 20-μl reaction mixture containing 350 mm HEPES, pH 7.5, 32 mm magnesium acetate, 40 mm dithiothreitol (DTT), 2 mm spermidine, 28 mm nucleoside triphosphates, 0.025 μg/μl linearized DNA, and 0.025 μg/μl T7 RNA polymerase. The reaction mixture was incubated for 3 h at 37 °C, and magnesium pyrophosphate was removed by centrifugation for 2 min. The supernatant was transferred to a new tube, and RQ1 DNase (Promega) was used to remove the template. The RNA concentration was determined by measuring absorbance at 260 nm, assuming that an A260 of 1 was equivalent to 40 μg/ml, and the RNA quality was verified by 0.8% agarose gel electrophoresis.
Infectious Center Assays
HeLa cells were transfected by electroporation with 5 μg of viral RNA transcript, and these cells were serially diluted and plated onto HeLa cell monolayers. Cells were allowed to adhere to the plate for 1 h at 37 °C, and then the medium/PBS was aspirated. Cells were covered with 1× DMEM/F-12 plus 10% fetal bovine serum, 100 units/ml penicillin, 100 units/ml streptomycin, and 1% agarose. After 2–4 days of incubation, the agarose overlay was removed, and the cells were stained with crystal violet.
Virus Isolation, Titer, and One-step Growth Curves
HeLa cells were transfected by electroporation with 5 μg of viral RNA transcript, added to HeLa cell monolayers and incubated at 37 °C. Upon cytopathic effect, viruses were harvested by three repeated freeze-thaw cycles, and cell debris was removed by centrifugation, and virus titers were performed. The resulting virus stocks were passaged four times on HeLa cell monolayers at a low multiplicity of infection (m.o.i. of 0.01). For plaque assays, virus was serially diluted in PBS and placed on cells in 6-well plates to allow the virus to adsorb to the cells, and PBS was removed and replaced with media containing 1% agarose. After 2–4 days of incubation, the agarose overlay was removed, and cells were stained with crystal violet. Plaques were counted to determine virus titer in pfu/ml. To analyze one-step virus growth, cells were infected with virus at a m.o.i. of 10. Virus was allowed to adsorb, and cells were washed with PBS and media added. The cells were incubated at 37 °C for various times post-infection. Virus was harvested by three repeated freeze-thaw cycles, and virus titers were performed.
RNA Isolation, cDNA Synthesis, and Sequencing to Confirm the Presence of the H273R Mutation
Viral RNA was isolated with QIAamp viral RNA purification kit (Qiagen), as recommended by the manufacturer. The 3Dpol cDNA was prepared from purified viral RNA by reverse transcription with Moloney murine leukemia virus-RT (New England Biolabs) with oligonucleotide 3D-BamHI-rev (5′-GCG GGA TCC TTA CTA AAA TGAGTC AAG CCA ACG GCG GTA-3′). The resulting DNA product was then PCR-amplified using SuperTaq DNA polymerase (Ambion) and oligonucleotides 3D-BamHI-rev and 3D-SacII-for (5′-GCG CCG CGG TGG AGG TGA AAT CCA GTG GAT GAG A-3′) as primers. The presence of Arg-273 was determined by sequencing of the nucleic acid obtained in second PCR step with oligonucleotide 3D-PV-PstI-for (5′-GGA GTG ATA ACA GGT TCT GCA GTG GGG TGC GAT-3′).
Northern Blot Analysis
Total RNA was isolated using TRI Reagent, separated on a 1% native agarose gel, and transferred to a nylon membrane by passive capillary blotting, and then hybridization was performed using PCR-based probes to the three-dimensional region of the PV genome essentially as described previously (22).
Subgenomic Replicon Assays
HeLa cells were transfected by electroporation with 5 μg of replicon RNA transcript, transferred to pre-warmed media in microcentrifuge tubes, and incubated at 37 °C. At each time point, cells were pelleted by centrifugation; media were removed, and cells were lysed using cell culture lysis reagent (Promega). Lysates were left on ice until luciferase and protein concentration assays were performed, at which point the lysates were centrifuged to remove cellular debris and nuclei. Assays were performed by mixing an equal volume of clarified lysate with luciferase assay substrate (Promega), and luciferase quantification was carried out as described by the supplier with a Junior LB 9509 luminometer (Berthold). Relative light units were adjusted to total protein concentration in the samples. Protein concentration was measured at 595 nm using the Bradford reagent (Bio-Rad). For experiments performed in the presence of ribavirin, HeLa cells were pretreated with ribavirin prior to transfection, and then transfected cells were transferred to media containing the same concentration of ribavirin.
Genomic Sequencing for Mutational Frequency
Twenty four viral isolates belonging to each of the WT and H273R populations were obtained by plaque isolation. Viral RNA was extracted and purified. PV cDNAs were generated by RT-PCR using viral RNA. The RT reaction was performed using the oligo(dT) primer. Two PCR products were obtained for each viral isolate, a product spanning the 5′-UTR and capsid coding region (nucleotides 300–3300) and a product spanning 3CD and 3′-UTR (relative light units 5800–7441). Primers for the 5′-product were 5′-CAGAGTGTAGCTTAGGC-3′ (forward) and 5′-GGTGGACGCGGGCACC-3′ (reverse). Primers for the 3′-product were 5′-CAGGGATATCTAAATCTC-3′ (forward) and 5′-CTCCGAATTAAAGAAAAATTTACCC-3′ (reverse). Direct sequencing was performed on PCR products.
Mutational Frequency Determination from Viral Stocks by Circular Sequencing
HeLa cell monolayers (8 × 106 cells) in 100 mm plates were infected with 8 × 104 pfu of either WT or H273R PV (m.o.i. of 0.01). Upon cytopathic effect, virus was harvested by three freeze-thaw cycles; cell debris was removed by centrifugation and Nonidet P-40 (Nonidet P-40) added to 0.5% final concentration. The viral supernatant was mixed at a 1:1 ratio with a solution of 20% PEG-8000 and 1 m NaCl, incubated on ice at 4 °C for 12–24 h, and then centrifuged at 8000 × g for 10 min at 4 °C. The pellet was carefully washed with 50 mm Tris-HCl, pH 8.0, 10 mm NaCl, and then suspended in 50 mm Tris-HCl, pH 8.0, 10 mm NaCl (1 ml per 10 ml of viral supernatant). Viral RNA was extracted using TRIzol reagent, suspended in RNase-free water, and then used for circular sequencing as described previously (23, 24).
Guanidine Resistance Assay
HeLa cells were infected with 106 pfu of either WT or H273R PV in the presence of 3 mm guanidine hydrochloride and then washed and overlaid with agarose media containing 3 mm guanidine hydrochloride. Cells were incubated for 3–4 days at 37 °C before being stained with crystal violet. Plaques were counted to determine the guanidine resistant frequency (guar/106 pfu).
Effect of Ribavirin on Virus Titer
HeLa cells were infected with either WT, G64S, or H273R PV at an m.o.i. of 0.01 or 5 in the presence of 0–1000 μm ribavirin. Upon cytopathic effect, virus was harvested by three repeated freeze-thaw cycles, and virus titers were performed.
Ribavirin Sensitivity Assay
HeLa cell monolayers in 6-well plates were pretreated for 1 h with various concentrations of ribavirin and then infected with 50 pfu of WT (ribavirin-sensitive), G64S (ribavirin-resistant), or H273R PV, incubated for 20 min to allow for virus adsorption, and then washed and overlaid with 0.5% agarose media containing the same concentration of ribavirin. Plates were incubated for 3–4 days at 37 °C before overlays were removed, and monolayers were stained with crystal violet. Plaques were counted and compared with the untreated (0 mm ribavirin) control dish.
Determination of IC50
To determine the IC50 value, the percentage of plaques was plotted as a function of ribavirin concentration and fit to a sigmoidal dose-response Equation 3,
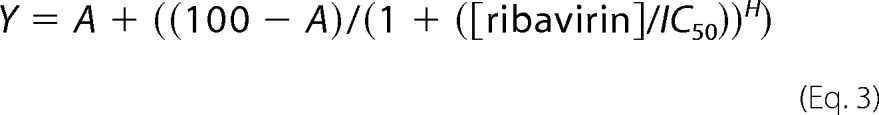 |
where Y is the % of plaques relative to untreated cells; A is the minimum % of plaques, and H is the Hill slope.
Virus Competition Experiment
HeLa cells were infected at an m.o.i. of 10 with a virus mixture composed of WT and H273R PV at a ratio of 1:9. After adsorption, input virus was removed, and infection was allowed to proceed to cell lysis. Virus from the supernatants of infected cells was harvested by three freeze-thaw cycles and used to re-infect fresh HeLa cell monolayers (passages 1–3). At each passage, viral RNA was extracted for quantitation using RT-PCR. RT-PCR experiments amplified polymerase-coding region, and direct sequencing of the PCR product was performed to determine the proportion of progeny virus belonging to the WT or H273R populations.
Virus Dilution
HeLa cells were infected with either WT, G64S, or H273R PV at a m.o.i. ranging from 1 × 10−8 to 10. Virus was allowed to adsorb, and cells were washed with PBS and media were added. The cells were incubated at 37 °C for 6 h post-infection. Virus was harvested by three repeated freeze-thaw cycles and virus titers were performed. Extinction titers were performed by standard plaque assay on the isolated viral stock. Undiluted viral stocks that gave no plaques were considered below the limit of detection (1 pfu).
Serial/Blind Passage
HeLa cells were infected with P0 virus at m.o.i. of 0.1, 0.01, and 0.001, were allowed to proceed for 8 h, and then viruses were harvested by three repeated freeze-thaw cycles and titered (P1). HeLa cells were infected with P1 virus at m.o.i. of 0.1, 0.01, and 0.001 and were allowed to proceed for 8 h, and the viruses were then harvested by three repeated freeze-thaw cycles (P2, virus titers were not performed at this point). The titer of P2 virus stock was assumed to be the same as the P1 virus stock for each virus-m.o.i. pair. This titer was then used to calculate the amount of virus required to achieve m.o.i. of 0.1, 0.01, or 0.001 for subsequent passage infections. After all eight passages were completed, viruses were collected after each passage was titered.
Extinction of Viral Populations at Fixed Number of Viral Genomes
Viral RNA was purified from virus stocks by using QIAamp viral RNA purification kit (Qiagen) and used for RT-qPCR to determine genome copies. HeLa cells were infected with either WT or H273R PV virus that corresponds to a total of 3.18 × 101, 3.18 × 102, 3.18 × 103, 3.18 × 104, and 3.18 × 105 viral RNA genomes, and replication was allowed to proceed for 8 h. After 8 h, total RNA from infected cells and supernatant was purified by RNeasy Plus mini kit (Qiagen), and viruses were harvested by three repeated freeze-thaw cycles. For real time qPCR analysis performed by the Genomics Core Facility of the Pennsylvania State University, DNase-treated RNA was reverse-transcribed using the High Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) and the protocol provided with the kit. Quantification by real time qPCR was done by adding 10 or 20 ng of cDNA in a reaction with 2× TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City CA) in a volume of 20 μl, with primers 5′-ACCCCTGGTAGCAATCAATATCTTAC-3′ (forward) and 5′-TTCTTTACTTCACCGGGTATGTCA-3′ (reverse) and probe 5′-[6-Fam]-TGTGCGCTGCCTGAATTTGATGTGA-3′ in a 7300 real time qPCR system (Foster City CA) machine. A standard curve was generated using in vitro transcribed RNA.
Mouse Infection, Protection, and Tissue Tropism
Mice were bred and housed in standard ventilated caging for all experiments. Protocols for animal studies were approved by Pennsylvania State University and University of California at San Francisco Institutional Animal Care and Use Committee (IACUC). All experiments were performed in accordance to guidelines and regulations overseen by the IACUC. Virus stocks were generated in serum-free media, harvested, and titered. Four- to 6-week-old outbred (ICR) mice transgenic for the PV receptor (cPVR) were infected with PV at the indicated titer (pfu) by either intraperitoneal (i.p.) injection in 3 ml of serum-free media (or mock-infected with 3 ml of serum-free media alone) or by intramuscular (i.m.) or intracranial injection. Mice were observed for 14 days for signs of disease and were euthanized upon showing dual limb paralysis or paralysis such that their ability to obtain food and water was compromised; this was in accordance with approval by the IACUC at Pennsylvania State University. PD50 values were determined by the Reed and Muench method. Surviving mice were challenged 1 month after initial infection using the same methods with 5PD50 of WT PV (1 × 108 pfu) by i.p. injection and observed for 14 days as above. For tissue tropism studies, whole organs (brain or spleen) were harvested from five mice that had been infected intravenously with 108 or 109 pfu of WT or H273R PV. Each day following infection, five mice from each group were sacrificed, and tissues were removed, washed, weighed, and homogenized in 2 ml of PBS with an Ultraturrax T8 homogenizer (IKA Works Inc., Wilmington, NC). Homogenates were clarified of cell debris and stored at −80 °C. Tissue homogenates were then titered for virus on HeLa cells by standard plaque assay.
Antibody Neutralization Assay
Serum from immunized mice was collected and pooled according to immunized dose at day 25 post-inoculation. Serum was diluted, mixed with 50 pfu WT PV, incubated at 37 °C for 2 h, and then added to HeLa cell monolayers. Cells were incubated for 2 days at 37 °C before being stained with crystal violet.
Virus Purification and Transmission Electron Microscopy
To obtain highly purified and concentrated viral stocks, three 10-mm plates of HeLa cells were infected with WT or H273R PV at an m.o.i. of 1. Upon cell death, media and cells were collected and subjected to three freeze-thaw cycles. Cellular debris was removed by centrifugation in Beckman JLA-16.250 rotor at 6000 rpm for 15 min at room temperature. The pH of the supernatant was adjusted to 7–8, and the virus was precipitated with 8% (w/v) polyethylene glycol (PEG 8000) (American Bioanalytical) and 0.3 m NaCl for 16 h at 4 °C while constantly rotating. Precipitated virus was centrifuged at 6000 rpm for 30 min at room temperature. The pellet was resuspended in 0.5 ml of TN buffer (50 mm Tris, pH 8.0, 10 mm NaCl). Two hundred fifty microliters of this suspension were then overlaid onto a 1.5-ml 30% sucrose cushion (30% sucrose, 30 mm Tris, pH 8.0, 0.1 mm NaCl) and centrifuged in a Sorvall S55S-582 swinging bucket rotor at 190,000 × g for 14 h at 4 °C. The pellet was resuspended in 100 μl of TN buffer with 2% deoxycholic acid. Virus solution was dialyzed overnight using a 50–100-kDa cutoff membrane against 1 liter of TN buffer at 4 °C. After dialysis, virus solution was subjected to the second purification step through a 30% sucrose cushion. The final pellet was resuspended in 100 μl of TN buffer and used to prepare samples for transmission electron microscopy (TEM). Titer of the purified virus was determined essentially as described above for one-step growth curve assay. The purified virus was negatively stained on either 200- or 400-mesh Formvar carbon-coated copper grids with 2% uranium acetate aqueous solution following standard protocols by the Electron Microscopy facility at Pennsylvania State University. Transmission electron microscopy images were collected at the Electron Microscopy facility on a JEM 1200 EXII microscope. To determine the fraction of empty viral particles, at least 10 images for each virus were analyzed.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 4.0 (La Jolla, CA). The S.E. and S.D. values are indicated where appropriate. Survival curves were performed using the product limit method of Kaplan and Meier and survival curve comparisons using the log rank test as provided by GraphPad Prism 4. p values are indicated.
RESULTS
H273R Substitution in PV RdRp Produces a Mutator Phenotype in Vitro
We previously reported a PV RdRp derivative (N297E) that exhibits a mutator phenotype (13). In performing that study, we identified an adventitious mutation in our N297E RdRp clone that changed His-273 to Arg. Like the high fidelity G64S RdRp derivative of PV (1–4), amino acid 273 is located in the fingers subdomain of the enzyme, remote from the active site (Fig. 1A). To address the impact of the H273R mutation on PV RdRp activity, we engineered the H273R mutation into the WT PV RdRp expression construct (18). We then evaluated H273R RdRp fidelity in biochemical assays using our self-complementary RNA-primed RNA template (Fig. 1B) (10, 19, 20), in which the first templating nucleotide is uridine (underlined in Fig. 1B). The H273R RdRp derivative was compared with WT and G64S RdRp derivatives. Under conditions in which all three enzymes exhibited the same efficiency of correct nucleotide incorporation (n + 1 product in lanes marked ATP in Fig. 1C), H273R RdRp was more efficient at GMP misincorporation and ribavirin incorporation than both WT and G64S RdRps (Fig. 1C). The reduced fidelity of nucleotide addition by H273R RdRp was further supported by the appearance of products greater than n + 1 when ATP and RTP were used as substrates (lanes 5 and 7 in Fig. 1C). These products of misincorporation were not as abundant for WT and G64S RdRps (Fig. 1C). These data suggest that H273R RdRp is a mutator polymerase.
FIGURE 1.

H273R substitution in PV RdRp produces a mutator phenotype in vitro. A, structure of PV RdRp showing the location of His-273 and Gly-64 (CPK, red and green, respectively). Palm, fingers, and thumb subdomains are indicated. Conserved structural motifs are colored as follows: A, red; B, green; C, yellow; D, blue; E, purple; F, orange; G, black. B, primed template used is referred to as sym/sub-U and contains uridine as the first templating base (underlined). C, reaction products from PV RdRp-catalyzed nucleotide incorporation using the indicated nucleotide and sym/sub-U. H273R RdRp incorporated GMP and ribavirin monophosphate more efficiently than both WT and the high fidelity variant G64S RdRp. Note the additional misincorporated nucleotides (>n + 1) that are observed with H273R RdRp. D, H273R RdRp is less faithful than WT. Kinetic parameters for correct and incorrect nucleotide incorporation catalyzed by WT, H273R, and G64S RdRp are shown. The relative fidelity or error frequency is shown, (kpol/Kd, app)corr/(kpol/Kd, app)incorr. Values are rounded to one significant figure. a, values for WT and G64S RdRp were taken from previous studies (1, 20).
To establish the magnitude of the fidelity difference between H273R and WT and to understand the kinetic basis for this difference, we performed a quantitative pre-steady-state kinetic characterization of H273R RdRp (Fig. 1D). These results were compared with those obtained previously for WT and G64S RdRps (1, 10, 20). The GMP misincorporation efficiency and ribavirin monophosphate incorporation efficiency of H273R RdRp were elevated by 2- and 5-fold, respectively, relative to WT (Fig. 1D). In both cases, the misincorporation was caused by an increase in the apparent affinity (Kd, app) for the incorrect nucleotide and an increase in the observed rate constant for nucleotide incorporation (kpol) (Fig. 1D). These results indicate that the recombinant, purified PV RdRp carrying the H273R substitution has a lower capacity to discriminate between the correct and the incorrect nucleotide during the polymerization reaction.
Replication Characteristics of H273R PV in Cell Culture
An infectious cDNA clone of Mahoney type 1 PV genome was engineered to encode an RdRp harboring the H273R substitution such that two transversion mutations would be required to restore the wild-type amino acid. H273R RNA produced by in vitro transcription of the mutated cDNA was infectious in HeLa cells, albeit with a 1.4-fold lower specific infectivity than observed for WT RNA (Fig. 2A). The H273R population produced an equal number of plaques at three different temperatures (34, 37, and 39 °C), but the plaque size of the mutant was slightly smaller than observed for WT (Fig. 2B), suggesting that the fitness of H273R may be somewhat reduced. Four consecutive passages of H273R at low multiplicities of infection showed no evidence of genetic changes in polymerase-coding sequence, even at the codon for position 273. Analyses of the growth kinetics of H273R under one-step growth conditions in HeLa cells revealed no significant differences relative to WT (Fig. 2C). However, using a more sensitive assay, a luciferase-expressing subgenomic replicon, we observed a subtle delay in H273R replication with respect to WT (see 2.5–5 h post-transfection, Fig. 2D), but by 6 h the mutant reached WT levels of replication. Evaluation of RNA accumulation by Northern blotting did not reveal this difference (Fig. 2E). We conclude that the growth properties and genetic stability of H273R are similar to those observed for WT PV, thus permitting us to evaluate the biological consequences of increasing mutation frequency both in cell culture and in a small animal model of infection.
FIGURE 2.
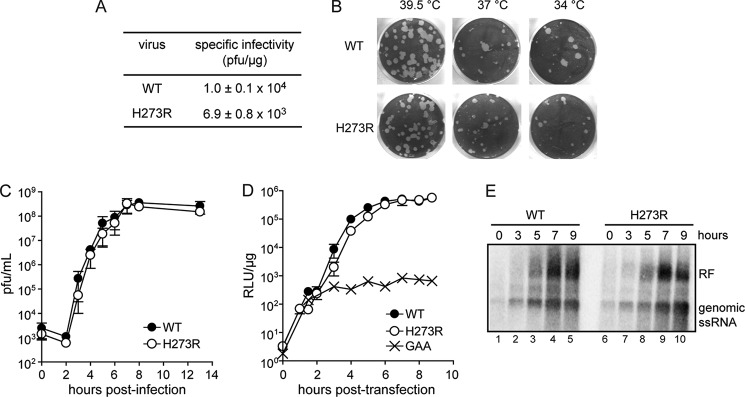
H273R PV is viable and replicates stably in cell culture. A, infectious center assay. HeLa cells were transfected by electroporation with 5 μg of viral RNA, and cells were serially diluted and plated onto HeLa cell monolayers. Cells were washed and overlaid with agarose media, and after 2–4 days of incubation, the agarose overlay was removed, and the cells were stained with crystal violet. Data are means from three independent experiments. Errors represent ± S.E. B, evaluation of the plaque phenotype and temperature sensitivity of H273R PV. HeLa cells were infected with 50 pfu of either WT or H273R PV, washed, and overlaid with agarose media. Cells were incubated for 3–4 days at the indicated temperature before being stained with crystal violet. C, kinetics of virus growth for H273R PV. Viral titer (pfu/ml) was plotted as a function of time post-infection. Data are means from three independent experiments. Error bars represent ± S.E. D, kinetics of RNA synthesis using a luciferase-expressing, subgenomic replicon. Luciferase specific activity is reported in relative light units (RLU) per μg of total protein in the extract. GAA represents a control for translation of input RNA without replication. The signature GDD motif of the RdRp was changed to GAA. Data are means from four independent experiments. Error bars represent ± S.E. E, Northern blot analysis. HeLa cells were infected with either WT or H273R PV, and at different times post-infection the total RNA was isolated, and Northern blots were performed. Genomic single-stranded (ss) RNA and replicative form (RF) are indicated.
H273R PV Expresses a Mutator Phenotype in Cell Culture
We determined the genetic diversity of H273R populations after four passages in cell culture by cloning and sequencing fragments of the genome encompassing capsid and polymerase coding regions derived from viral RNA isolated from plaques (Fig. 3A) (1, 4). A 50% increase in mutations was observed in H273R, averaging 3.0 mutations per genome (Fig. 3A). A phenotypic assay for the presence of viral mutants resistant to guanidine (guar), an inhibitor of PV genome replication, was consistent with the sequencing analysis. H273R exhibited a 3-fold increase in guar frequency relative to WT (Fig. 3A) (1, 4). The observed mutation frequencies from sequencing reflect averages at multiple locations within a specific genomic region (VP1), whereas the phenotypic assay (guar) provides the frequency of a specific transition mutation in the genome. Both experimental observations support the conclusion that the arginine substitution at position 273 of the PV RdRp significantly increases the virus mutation rate.
FIGURE 3.

H273R PV expresses a mutator phenotype in cell culture. A, sequencing analysis and guanidine resistance frequency confirms the increased mutation rate for H273R PV in tissue culture. Shown are the total number of mutations observed over the total number of nucleotides sequenced and the calculated average number of mutations per genome. A significant difference in the number of mutations was observed between WT and H273R PV (p < 0.002, Mann-Whitney U test). a, values reported for WT and G64S PV virus are taken from Ref. 4. The calculated guanidine resistance frequency for WT and H273R PV after infection of HeLa cells with 106 pfu in the presence of 3 mm guanidine hydrochloride is shown. Data are means from four independent experiments. B, ribavirin sensitivity. 50 pfu of WT, G64S, or H273R PV were used to infect HeLa cell monolayers pretreated with the various concentrations of ribavirin. Plaque numbers were plotted against ribavirin concentration normalized to untreated (0 mm) control. The solid line represents the fit of the data to a sigmoidal dose response equation (four-parameter logistic model), yielding IC50 values of 0.55 ± 0.03, 2.05 ± 0.10, and 0.062 ± 0.003 mm for WT, G64S, and H273R PV, respectively. Data are means from three independent experiments. Error bars represent ± S.E. C and D, susceptibility of H273R PV to ribavirin is reduced at high m.o.i. Shown is the percentage of surviving virus for WT, G64S, and H273R PV after infecting HeLa cells at either low (m.o.i. of 0.01, C) or high (m.o.i. of 5, D) m.o.i. in the presence of increasing concentrations of ribavirin. Titers were normalized to untreated (0 μm) control. The titer (pfu/ml) after infection at low m.o.i. for WT, G64S, and H273R PV in the absence of ribavirin was 3.5 × 108, 1.8 × 108, and 1.6 × 108, respectively. The titer (pfu/ml) after infection at high m.o.i. for WT, G64S, and H273R PV in the absence of ribavirin was 1.9 × 107, 4.7 × 107, and 1.8 × 107, respectively. Data are means from three independent experiments. Error bars represent ± S.E. p < 0.05 for all samples compared with WT by Student t test. E, percentage of relative light units (RLU) per μg at 5 h post-transfection for WT, G64S, and H273R subgenomic replicon in the absence or presence of ribavirin. Data are means from three independent experiments. Error bars represent ± S.E.
Ribavirin is a nucleoside analogue with known antiviral properties (25). Ribavirin is a mutagen to PV and other RNA viruses (9, 10). A high fidelity variant of the PV RdRp, G64S, exhibits reduced sensitivity to ribavirin in cell culture (1–4). We predicted that low fidelity variants of the PV RdRp, such as H273R, would exhibit enhanced sensitivity to ribavirin. Indeed, H273R demonstrated a substantial increase in sensitivity to the drug (Fig. 3, B–D). Quantification of this experiment revealed a 9-fold decrease in the concentration of ribavirin required to reduce the number of plaques by half relative to the untreated population (IC50, Fig. 3B). The effect of ribavirin was more pronounced at low multiplicities of infection (compare Fig. 3, C with D) either because certain mutations can be tolerated at a high m.o.i., because associated virus functions can be complemented, or because at a low m.o.i. the virus undergoes multiple rounds of replication. These experiments further support the conclusion that the H273R substitution results in a mutator phenotype.
The results presented above using luciferase activity produced from translation and replication of a subgenomic replicon suggested that replication of H273R may be slower than WT (Fig. 2D). However, this observation might result simply from the reduced specific activity of luciferase caused by the increased mutational load in the luciferase-coding sequence. To test this possibility, we used the mutagenic activity of ribavirin to determine whether or not changes in replication fidelity alone will produce effects on luciferase-specific infectivity that are unrelated to RNA copy number. The specific activity of luciferase varied as a function of polymerase fidelity (Fig. 3E). We conclude that information on replication kinetics and/or yield should not be inferred from reporter enzyme activity when the virus variant exhibits a fidelity phenotype.
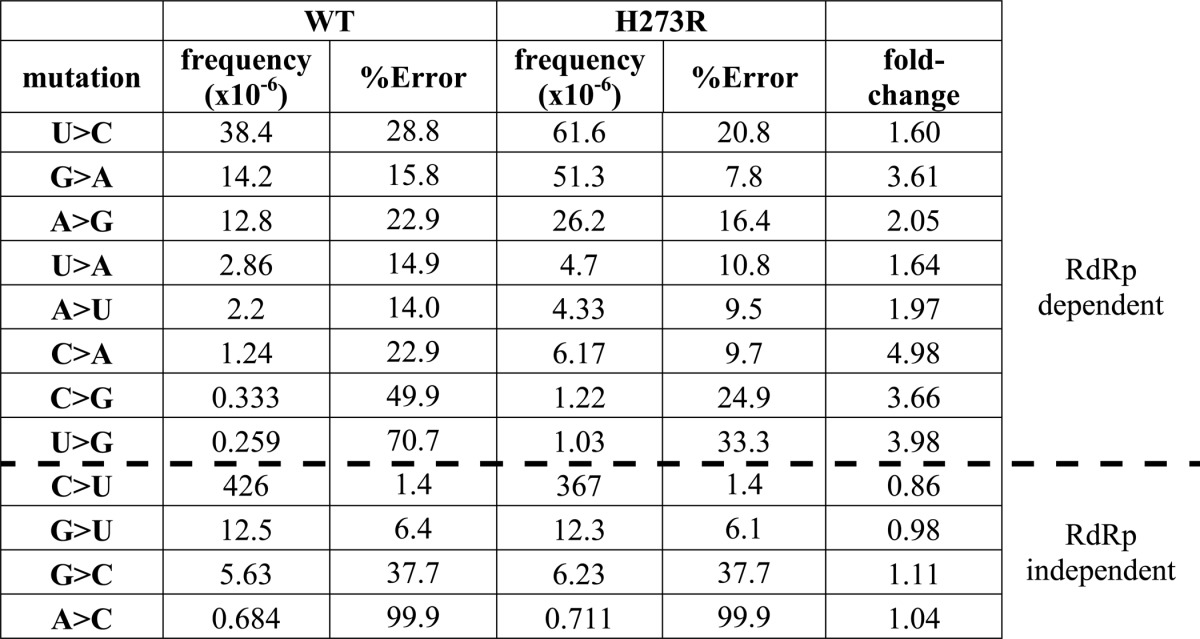
A new deep-sequencing approach has been developed that permits the entire mutational spectrum of an RNA virus to be elucidated (23, 24). This approach is referred to as circular sequencing, because fragmented RNA is circularized and the circularized template was copied reiteratively in a process of rolling-circle reverse transcription (23, 24). Multiple copies of a single sequence permits error correction sufficient to call mutations present at a frequency lower than 10−6 (23, 24). The most striking finding was that the fold-change in mutation frequency observed for H273R PV relative to WT PV could be separated into two categories (Table 1). The first category showed a clear increase in mutation frequency for H273R PV, consistent with a mutator polymerase. The second category showed no change at all in the mutation frequency for H273R PV (Table 1). The ranked order of mutation types was identical when H273R PV was compared with WT PV (Table 1). Collectively, these data suggest two mechanisms (RdRp-dependent versus RdRp-independent) driving the genetic diversity of the PV population in HeLa cells, an unexpected yet exciting finding.
TABLE 1.
Mutational spectrum determined by using CirSeq
H273R PV Exhibits Reduced Fitness in Cell Culture Relative to WT PV
Lethal mutagenesis is a prediction of population genetics theory, which suggests that a 1-log increase in the progeny per generation (fecundity) would be required to preclude a unit change in the mutation rate from causing extinction of the viral population (5). Thus, there should be a fitness penalty associated with the increased mutation frequency observed for H273R. To examine this prediction, we competed H273R with WT at an initial WT/H273R ratio of 1:9 (Fig. 4A). Serial passages revealed a fitness deficit in H273R relative to WT (Fig. 4A). By the third passage, WT represented the majority of the population and continued to increase in subsequent passages.
FIGURE 4.

H273R PV exhibits reduced fitness in cell culture relative to WT PV. A, WT out-competes H273R PV. Shown is the percentage of WT and H273R PV remaining after 1–4 serial passages. The initial virus mixture contained a ratio of WT/H273R, 1:9. B, dilution drives H273R PV to extinction before WT and G64S. Shown is the surviving progeny virus (pfu) for WT, G64S, and H273R PV at 6 h post-infection using the indicated inoculum (pfu). Data are means from five independent experiments. Error bars represent ± S.E. The lower limit of detection is one plaque and is indicated by the dashed line. C, polymerase fidelity controls adaptation. Shown is the surviving progeny virus (pfu) after serial/blind passage of WT, G64S, and H273R PV at an initial m.o.i. of 0.001. D, inoculum size and mutation frequency affect sustainability of virus population. Shown is the surviving progeny virus (pfu) after serial/blind passage of H273R PV at an initial m.o.i. of 0.001, 0.01, and 0.1. E, phenotypic changes of cell culture-adapted WT PV population. Shown is the surviving progeny virus (pfu) after serial/blind passage of WT PV at an initial m.o.i. of 0.001, 0.01, and 0.1. For experiments shown in C–E, HeLa cell monolayers were infected at the indicated m.o.i. of P1 virus, were allowed to proceed for 8 h, and then viruses were harvested. The titer of virus (pfu/ml) produced during P2 for each virus/m.o.i. pair was assumed to be constant (i.e. dose-response ratio), and this titer was then used to calculate the amount of the virus required to achieve m.o.i. of 0.1, 0.01, or 0.001 for subsequent passage infections. After all eight passages were completed, viruses collected after each passage were titered. F, fecundity for WT, H273R, and G64S PV. Fecundity was calculated as the ratio of progeny (pfu) to inoculum (pfu). Shown is the calculated range for each virus. G–I, progeny of virus relative to the starting inoculum for each infection from the data shown in B for WT, H273R, and G64S PV. Data are means from five independent experiments. Error bars represent ± S.E.
Fitness defects associated with the increased mutation frequency of H273R should also manifest as an increase in the concentration of virus required for establishment of an infection because population genetics theory predicts a requirement for a finite number of viable genomes to avoid extinction (5). Thus, this minimal concentration should be greater for those virus populations with a high mutation load (WT and H273R) than for a high replication fidelity variant (G64S) (5). We evaluated the yield of virus produced 6 and 8 h post-infection over a 9-log range of virus concentrations, expressed as m.o.i. (Fig. 4B). Six-hour time frame was chosen because the virus multiplication cycle should be essentially completed for all viruses, but all progeny should still reside within the infected cells. Extinction occurred at an m.o.i. (pfu/cell) of 10−6 for H273R, at 10−7 for WT, and at 10−8 for G64S PV (Fig. 4B). This result provides additional experimental evidence supporting RNA virus population dynamics predicted by population genetics theory.
Given that RNA viruses are thought to exist close to their tolerable mutation load, we anticipated that H273R would be susceptible to lethal mutagenesis. To test this hypothesis, we compared the fecundity of H273R with that of WT and G64S at a low m.o.i. over eight generations. The fecundity of WT increased during passage, whereas that of G64S PV was unchanged and that of H273R was substantially decreased (Fig. 4C). The fact that the only difference between these PV variants was their RdRp replication fidelity reveals a striking link between RdRp error rate, fitness, and evolution of RNA virus populations. Interestingly, the loss of fitness of H273R was not observed at higher multiplicities of infection (Fig. 4D, WT control is shown in Fig. 4E). This observation is consistent with the theory that extinction of virus populations through lethal mutagenesis should depend on burst size (5). These results establish a clear relationship between viral population diversity and fitness in cell culture. It appears that PV has evolved to an optimal mutation rate, and this mutation rate can be modulated by amino acid substitutions within the RdRp. The fitness defect resulting from a higher load of detrimental mutations is best revealed at low multiplicities of infection, where we observed population extinction (Fig. 4B) and maximal fecundity (Fig. 4, F and G). Of note, we observed a near 2-fold increase in the number of empty particles produced by H273R relative to both WT and G64S PVs (Fig. 5), suggesting that determinants of packaging are sensitive to mutational load and therefore may be useful to ensure that only the most fit genomes are disseminated.
FIGURE 5.
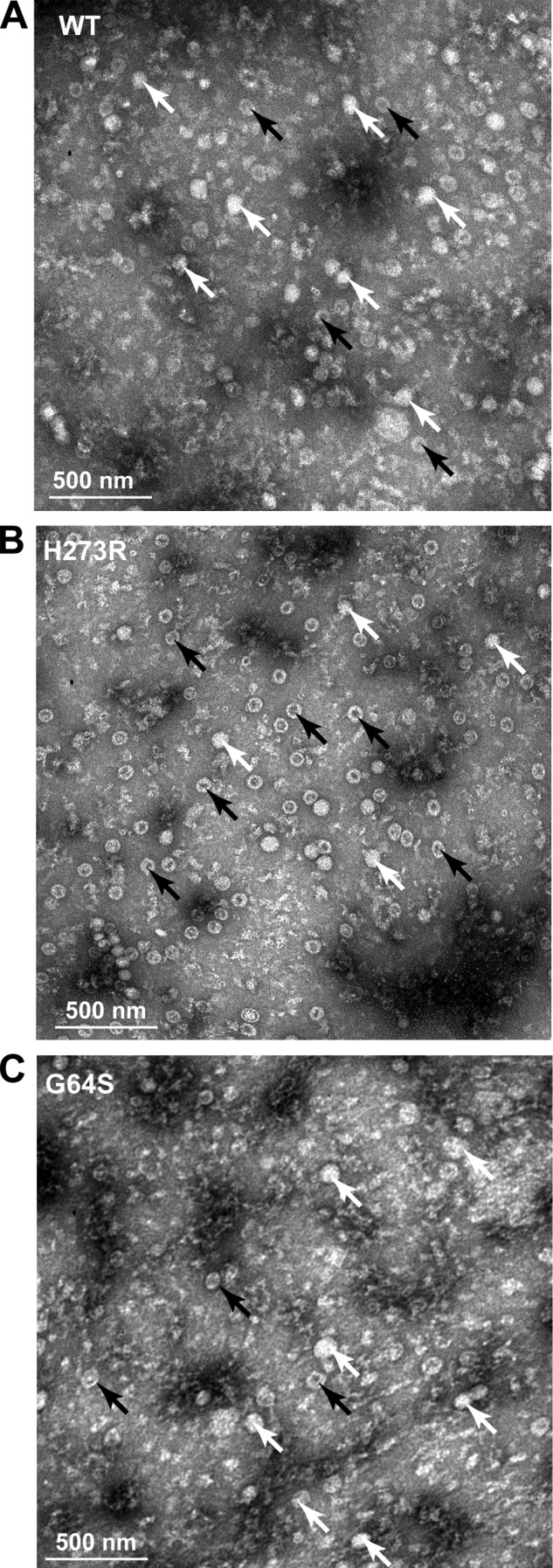
H273R PV identifies RNA packaging as a checkpoint for functional genomes. TEM analysis of the purified viruses. Representative TEM images at magnification of ×30 are shown. White bar in each image represents 500 nm. White arrows point to virus particles containing RNA, and black arrows point to empty viral particles. TEM images are shown of WT (A), H273R (B), and G64S PV (C). Empty particles constitute 35 ± 5, 55 ± 5, and 33 ± 5% of total viral particles for WT, H273R, and G64S PV, respectively.
All of the experiments presented above were performed using the pfu as the quantifiable measure of infectious virus. When we performed blind serial passages of H273R at low multiplicities of infection, we routinely observed extinction as shown in Fig. 4, C and D. However, if we quantified the viral titer, again using pfu, to determine the inoculum, then extinction was never observed. We eventually reconciled these differences by realizing that pfu is itself a phenotype, and the number of genomes per pfu could change from one virus to another. We used digital PCR to quantify standards used in conventional RT-qPCR experiments to measure virus titers as genomes or genomes/ml. For WT PV, 1 pfu equated to 50 ± 10 genomes; this value increased to 500 ± 100 genomes for H273R PV. The value for genomes/pfu measured was dependent on several factors, including m.o.i., m.o.i. used for serial passage, and number of passages. We were able to use this quantitative approach to corroborate the observation that H273R is more susceptible to extinction than WT (Fig. 6).
FIGURE 6.
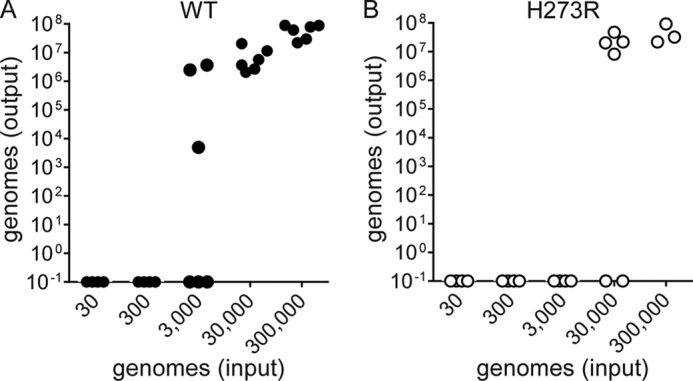
Extinction of viral populations at fixed number of viral genomes. Outcome of infection of WT and H273R PV at fixed number of viral genomes. Total genome copies (output) for WT (A) and H273R (B) at 8 h post-infection using the indicated inoculum that corresponds to 3.18 × 101, 3.18 × 102, 3.18 × 103, 3.18 × 104, and 3.18 × 105 viral genomes (input). Data are shown from six independent experiments for each virus. H273R proceeds to extinction prior to WT after infection at a lower number of viral genomes. HeLa cells were infected with an equivalent number of genomes for WT and H273R; replication was allowed to proceed for 8 h and then total RNA and viruses were harvested. RT-qPCR was performed using purified RNA to determine genome copies.
We conclude that the H273R substitution reduces replication fidelity of the PV RdRp, which results in a mutator phenotype. This mutation appears to drive the virus population over the maximal tolerated mutation frequency, which over time leads to extinction of finite virus populations. We thus propose that H273R should be incapable of sustaining an infection in vivo due to the inability to overcome the effect of bottlenecking on population size during infection.
H273R Is Attenuated in Vivo
Thus far, the data suggest that fitness defects associated with the low replication fidelity of H273R should manifest under conditions in which the size of the viral population is forced below the limit required for sustainability. Given the diverse and dynamic nature of the environment within an infected host, we predicted that H273R would be attenuated in vivo, where the virus is challenged by host defense mechanisms, diverse cellular environments in different tissues, and anatomical restrictions, which impose bottlenecks and restrain virus population size. Indeed, H273R virus presented a highly attenuated phenotype in mice where onset of paralysis was delayed and observed only at very high viral doses (Fig. 7, A–F). The 50% paralysis dose (PD50) for H273R was more than 200-fold higher than for WT (Fig. 7, A–F). Furthermore, intraperitoneal inoculation revealed that the low fidelity H273R virus exhibited restricted tissue tropism. Although both WT and H273R viruses were readily isolated over several days from the spleen and muscle, the H273R mutant was unable to establish infection and replicate effectively in the spinal cord and brain (Fig. 7G), despite these being principal sites of WT PV replication. Interestingly, intracranial inoculation showed that H273R PV replicated in brain equivalently to WT (Fig. 7H). However, H273R PV was still unable to make it to the spinal cord (Fig. 7I). These data are consistent with restrictions in neurons giving rise to the observed phenotype.
FIGURE 7.

H273R PV is attenuated in vivo. A–F, H273R PV is attenuated in the cPVR mouse model. cPVR mice (4–6 weeks old) were infected by either intraperitoneal (A–C) or intramuscular (D–F) injection with serum-free media containing WT (A and D) or H273R PV (B and E) at the indicated titer (pfu) and observed for 14 days. Mice that developed the following symptoms, two-limb paralysis or paralysis that compromised the ability to get food and water, were euthanized. The number of mice per group was n = 15 (i.p.) and n = 5 (i.m.). PD50 values by i.m. injection (C) for WT and H273R PV were 2 × 107 and 1 × 109 pfu, respectively. PD50 values by i.m. injection (F) for WT and H273R PV were 2 × 105 and 6 × 107 pfu, respectively. PD50 values were determined by the Reed and Muench method. G–I, viral tissue tropism. Shown are the virus titers (pfu/g) at the indicated days post-infection in tissue collected from mice infected intravenously (G) with 107 pfu of WT or H273R PV or infected intracranially (H and I) with 105 pfu of WT or H273R PV. The statistical significance of differences in virus load in brain (H) or in spinal cord (I) between WT and H273R after intracranial inoculation was determined by the Mann-Whitney test. Error bars represent ± S.E.
H273R Elicits a Protective Immune Response
We next determined the vaccine potential of H273R. First, we tested the ability of low fidelity H273R PV to elicit humoral immunity following a single intraperitoneal dose. Neutralization assays using sera collected from immunized animals 4 weeks post-infection revealed that H273R PV elicited PV-specific neutralizing antibodies and exhibited immunogenicity comparable with both WT PV and the Sabin type 1 vaccine strain (Fig. 8A).
FIGURE 8.
H273R PV elicits a protective immune response. A, antibody neutralization assay. Neutralizing antibody titers (reciprocal of the serum dilution able to reduce the number of plaques to 50% of 50 pfu WT PV) are shown for individual trials (solid circles) and as trial means (lines). Titers for PBS-immunized mice were below the detection level of the assay (indicated by dashed line). B, mouse protection studies. Mice immunized by i.p. injection with H273R PV or mock immunized with serum-free media alone (naive) were challenged 4 weeks after immunization with 5PD50 of WT PV (1 × 108 pfu) by i.p. injection and observed for 14 days. Mice that developed two-limb paralysis or paralysis that compromised the ability to get food and water were euthanized. The number of mice per group was n = 5. Survival curves for mice immunized with H273R PV at 107 pfu or greater were significantly higher than those for mice immunized with control serum-free media (p value of 0.014, Kaplan-Meier test).
To further evaluate the immune response elicited by H273R, we challenged the vaccinated mice with a lethal (5PD50) intraperitoneal inoculation of WT PV 1 month after immunization. Protection of mice from lethal infection correlated with neutralizing antibody levels. Thus, mice inoculated with doses of H273R at or above 107 pfu exhibited complete protection against lethal challenge by WT PV (Fig. 7B). Therefore, even though H273R is attenuated in its ability to replicate and cause disease in mice, this low fidelity variant elicits a strong immune response that confers protection.
H273R Phenotype Is Unique
Because H273R is the only viable PV mutant with a mutator phenotype, we asked whether other substitutions at this position would yield viable mutants with perturbed polymerase fidelity. We constructed H273K and H273Q PVs. Neither of these viruses were viable (Fig. 9A). Interestingly, inviability was not attributable to inactive polymerase as evaluation of these variants in the context of a subgenomic replicon revealed high levels of replication (Fig. 9B). Additional studies will be required to fully understand this observation.
FIGURE 9.
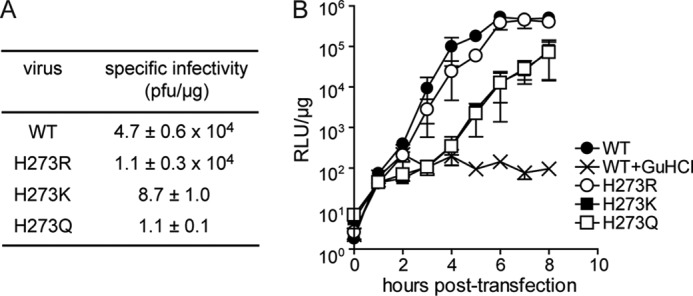
Mutations at position 273 other than Arg significantly impact PV replication. A, infectious center assay. HeLa cells were transfected by electroporation with 5 μg of viral RNA; cells were serially diluted and plated onto HeLa cell monolayers. Cells were washed and overlaid with agarose media and after 2–4 days of incubation; the agarose overlay was removed, and the cells were stained with crystal violet. Data are means from three independent experiments. Errors represent ± S.E. Plaques were not observed for H273A, H273N, H273G, H273P, and H273F mutants. B, kinetics of RNA synthesis using a luciferase-expressing subgenomic replicon. Luciferase-specific activity is reported in relative light units (RLU) per microgram of total protein in the extract. WT + guanidine HCl (Gu-HCl) represents a control for translation of input RNA without replication. Guanidine HCl is an inhibitor of PV replication. Data are means from three independent experiments. Error bars represent ± S.E. Replication was not observed for H273A, H273N, H273G, H273P, and H273F mutants.
DISCUSSION
An understanding of how genetic diversity of an RNA virus population impacts its interaction with the cell and, ultimately, the outcome of infection for the host have important implications for the development of antiviral therapies and vaccine strategies. Nucleoside analogues with ambiguous base-pairing capacity exhibit antiviral activity (6, 8–10). These agents, which include the clinically used ribavirin, have been termed lethal mutagens and are thought to act by increasing the genetic variation of the viral population beyond the extinction threshold (6, 8–10). Viral populations exhibiting decreased or increased genetic diversity are attenuated but replicate in the host and elicit a protective immune response (4, 14, 16, 26). Together, these observations suggest an optimal genetic diversity for a viral population to survive.
Mutation selection balance is a fundamental tenet of population genetics theory (5). Reflection of this theory on the evolutionary biology of RNA viruses reinforces the requirement for a minimal fecundity to sustain a viral population at a given mutation rate (5). These theoretical considerations relate to survival of viral populations and not the ability to cause disease. Only if survival of viral populations alone is sufficient for pathogenesis will these studies contribute to the prediction of viral virulence. Forcing a virus population to extinction is a laudable goal, but forcing a virus population to expression of an avirulent phenotype may be of great practical utility and more achievable.
This study reports the discovery of a viable PV mutant, H273R, that expresses a mutator phenotype. This low fidelity variant in combination with the high fidelity PV, G64S (1–4), provides a panel of viruses producing populations with genetic diversity both higher and lower than WT. Both mutants are attenuated in an animal model (Fig. 7) (3, 4), suggesting the existence of an optimal genetic diversity for a pathogenic outcome of infection. Replication clearly occurs in vivo (Figs. 7 and 8) (3, 4). This observation demonstrates that virus fitness in tissue culture is insufficient to predict disease in a more realistic, complex host. Attenuation appears to correlate with both a reduction of viral load and duration of infection in one or more specific organs/tissues. Viral load and persistence contribute to virus dissemination and consequential disease. Both viral load and persistence will be impacted by fecundity of the viral population. The viral population required for maximal fecundity and evasion of host immune defenses in one host tissue may be different from what is optimal in a different tissue.
Population genetics predicts a log-linear relationship between fecundity and mutation rate, thus providing the theoretical basis for lethal mutagenesis (5). Here, we provide empirical evidence to support the conclusion that population genetics theory applies to a positive-strand RNA virus in cell culture (Fig. 10A). A 2-fold decrease in mutation frequency leads to a 5-fold increase in fecundity; a 3-fold increase in mutation frequency leads to a 10-fold decrease in fecundity (Fig. 4). The reduced fecundity manifests as enhanced sensitivity to extinction when the population size is reduced (Fig. 4B) or when the time required for multiplication is fixed (Fig. 4D). Both of these experimental observations are likely akin to responses of the H273R population to bottlenecks in the host that contribute to the attenuated phenotype.
FIGURE 10.
PV obeys population genetics theory, but optimal fitness and virulence may be inextricably linked. A and B, shown is a plot of fecundity (log10) versus mutations per genome for WT, G64S, and H273R PV. Fecundity was calculated as progeny (pfu) per inoculum (pfu) for each virus. The average fecundity was calculated using the inocula from 10−3 to 10−1 for each virus. The solid red line depicts the threshold between viral survival and viral extinction by lethal mutagenesis, which exhibits a log-linear relationship between fecundity and the number of mutations. The limits of mutational load and fecundity that will support PV viability is shown as a green rectangle. The orange circle (B) represents viruses exhibiting a mutational load and fecundity near that of WT capable of exhibiting the most virulent phenotype. The solid line represents the fit of the data to a line yielding a y-intercept of 4.5 ± 0.7 and a slope of −1.5 ± 0.3. C, population speed represents a third dimension of viral population dynamics. In addition to mutation frequency and population size, the enhanced attenuation of K359R PV relative to G64S PV demands that our consideration of viral population genetics and dynamics be extended to include rate/efficiency of nucleotide addition as a third dimension.
Our studies suggest that PV fecundity may be determined directly by genome replication efficiency. This suggestion derives from the observation that there is either an inverse correlation or no correlation at all between the infecting viral dose and the amount of progeny produced (Fig. 4, G–I). The virus concentration independence is consistent with a cis-acting process, of which genome replication is a prime candidate. Because the 3-fold increase in mutation rate of H273R PV leads to as much as a 2-log reduction in fecundity relative to WT PV (compare Fig. 4, G and H), it is likely that genome replication represents a fitness peak that is sensitive to mutation. However, the ability of the fecundity to remain essentially constant over a 5-log range of infecting viral dose while undergoing mutagenesis is indeed a robust trait, which may explain the inability of a mutagen alone to eradicate a viral infection (27).
We and others have suggested that the sensitivity of RNA viruses to lethal mutagens/mutagenesis reflects the existence of the viral population on the “threshold of extinction.” This position is best illustrated by the line dividing extinction and survival in Fig. 10A. Placing the data on WT, G64S, and H273R PVs reported here in this context reveals that PV clearly does not reside on the threshold of extinction. A plot of mutations per genome versus fecundity defined a line that predicts a maximal fecundity of 103 and certain extinction at a value of 4.5 mutations per genome (Fig. 10B). This plot also makes it very evident that the number of solutions that will lead to WT virulence is likely small (e.g. the region circled in Fig. 10B) relative to the viable, attenuated possibilities defined by G64S and H273R PVs (boxed region Fig. 10B). Recent studies of the attenuated K359R PV mutant, which exhibits a 10-fold reduction in RNA synthesis relative to WT (28), suggest that a third dimension of speed should be considered when considering viral population dynamics (Fig. 10C). The magnitude of this axis is not currently known, hence the question marks in Fig. 10C.
The development of circular sequencing and its application to PV provided a complete, quantitative description of the genetic variation within the PV population (Table 1) (23, 24). Application of this technique to the H273R PV population suggested that not all observed mutation frequencies were dependent on RdRp fidelity (Table 1). Interestingly, the most abundant mutation in the PV population (C-to-U transition) may not be attributable to the viral polymerase. Conversion of cytidine to uridine occurs via deamination; whether a cellular deaminase is required remains to be determined. The other possible RdRp-independent mutations are transversions that cannot be explained as easily. Mutations like these in a DNA system would likely be attributed to a “damaged” templating base, for example an oxidized guanine (29). Oxidative damage of RNA occurs (30) but has not been studied for PV. It is also unknown how efficient and faithful the RdRp is during damage bypass. Perhaps this observation derives from the mutant showing increased promiscuity only in certain contexts. Additional studies will be necessary to clarify this observation.
The ability to design ab initio RNA virus mutants with altered fidelity could represent a substantial advance for vaccine development. Our current understanding of the structure, function, and mechanism of viral polymerases remains insufficient to guide such an effort. The viable fidelity mutants of PV both originate from substitutions at sites of the polymerase remote from the active site (Fig. 1A). It has recently been suggested that PV RdRp fidelity, and likely the fidelity of all RdRps, is influenced by dynamics of the enzyme (31, 32). Remote-site residues would therefore be predicted to alter networks controlling dynamics and could be considered to function by an allosteric mechanism (32, 33). Empirical evidence for this possibility was obtained for the antimutator G64S RdRp (32, 33). Whether or not the mutator H273R RdRp uses a similar mechanism remains to be determined. Whatever the mechanism, there is likely some complexity and much to learn because a G64S,H273R RdRp retains insufficient activity to observe virus replication in cells.7
This work was supported, in whole or in part, by National Institutes of Health Grant R01 AI045818 from NIAID (to C. E. C.).
J. J. Arnold and C. E. Cameron, unpublished observations.
- PV
- poliovirus
- RdRp
- RNA-dependent RNA polymerase
- m.o.i.
- multiplicity of infection
- TEM
- transmission electron microscopy
- qPCR
- quantitative PCR
- IUCAC
- Institutional Animal Care and Use Committee
- sym/sub
- symmetrical/substrate.
REFERENCES
- 1. Arnold J. J., Vignuzzi M., Stone J. K., Andino R., Cameron C. E. (2005) Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J. Biol. Chem. 280, 25706–25716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pfeiffer J. K., Kirkegaard K. (2003) A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. U.S.A. 100, 7289–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pfeiffer J. K., Kirkegaard K. (2005) Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 1, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vignuzzi M., Stone J. K., Arnold J. J., Cameron C. E., Andino R. (2006) Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bull J. J., Sanjuán R., Wilke C. O. (2007) Theory of lethal mutagenesis for viruses. J. Virol. 81, 2930–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson J. P., Daifuku R., Loeb L. A. (2004) Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 58, 183–205 [DOI] [PubMed] [Google Scholar]
- 7. Domingo E., Gonzalez-Lopez C., Pariente N., Airaksinen A., Escarmis C. (2005) Population dynamics of RNA viruses: the essential contribution of mutant spectra. Arch. Virol. Suppl. 2005, 59–71 [DOI] [PubMed] [Google Scholar]
- 8. Graci J. D., Cameron C. E. (2008) Therapeutically targeting RNA viruses via lethal mutagenesis. Future Virol. 3, 553–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hong Z., Cameron C. E. (2002) Pleiotropic mechanisms of ribavirin antiviral activities. Prog. Drug Res. 59, 41–69 [DOI] [PubMed] [Google Scholar]
- 10. Crotty S., Maag D., Arnold J. J., Zhong W., Lau J. Y., Hong Z., Andino R., Cameron C. E. (2000) The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 6, 1375–1379 [DOI] [PubMed] [Google Scholar]
- 11. Racaniello V. R. (2006) One hundred years of poliovirus pathogenesis. Virology 344, 9–16 [DOI] [PubMed] [Google Scholar]
- 12. Whitton J. L., Cornell C. T., Feuer R. (2005) Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3, 765–776 [DOI] [PubMed] [Google Scholar]
- 13. Korneeva V. S., Cameron C. E. (2007) Structure-function relationships of the viral RNA-dependent RNA polymerase: fidelity, replication speed, and initiation mechanism determined by a residue in the ribose-binding pocket. J. Biol. Chem. 282, 16135–16145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gnädig N. F., Beaucourt S., Campagnola G., Bordería A. V., Sanz-Ramos M., Gong P., Blanc H., Peersen O. B., Vignuzzi M. (2012) Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. U.S.A. 109, E2294–E2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rozen-Gagnon K., Stapleford K. A., Mongelli V., Blanc H., Failloux A. B., Saleh M. C., Vignuzzi M. (2014) Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog. 10, e1003877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith E. C., Blanc H., Vignuzzi M., Denison M. R. (2013) Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 9, e1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crotty S., Hix L., Sigal L. J., Andino R. (2002) Poliovirus pathogenesis in a new poliovirus receptor transgenic mouse model: age-dependent paralysis and a mucosal route of infection. J. Gen. Virol. 83, 1707–1720 [DOI] [PubMed] [Google Scholar]
- 18. Gohara D. W., Ha C. S., Kumar S., Ghosh B., Arnold J. J., Wisniewski T. J., Cameron C. E. (1999) Production of “authentic” poliovirus RNA-dependent RNA polymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expr. Purif. 17, 128–138 [DOI] [PubMed] [Google Scholar]
- 19. Arnold J. J., Cameron C. E. (2000) Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub). J. Biol. Chem. 275, 5329–5336 [DOI] [PubMed] [Google Scholar]
- 20. Arnold J. J., Cameron C. E. (2004) Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+. Biochemistry 43, 5126–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gohara D. W., Crotty S., Arnold J. J., Yoder J. D., Andino R., Cameron C. E. (2000) Poliovirus RNA-dependent RNA polymerase (3Dpol): structural, biochemical, and biological analysis of conserved structural motifs A and B. J. Biol. Chem. 275, 25523–25532 [DOI] [PubMed] [Google Scholar]
- 22. Oh H. S., Pathak H. B., Goodfellow I. G., Arnold J. J., Cameron C. E. (2009) Insight into poliovirus genome replication and encapsidation obtained from studies of 3B-3C cleavage site mutants. J. Virol. 83, 9370–9387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Acevedo A., Brodsky L., Andino R. (2014) Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505, 686–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lou D. I., Hussmann J. A., McBee R. M., Acevedo A., Andino R., Press W. H., Sawyer S. L. (2013) High throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc. Natl. Acad. Sci. U.S.A. 110, 19872–19877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graci J. D., Cameron C. E. (2006) Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 16, 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vignuzzi M., Wendt E., Andino R. (2008) Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 14, 154–161 [DOI] [PubMed] [Google Scholar]
- 27. Pariente N., Sierra S., Lowenstein P. R., Domingo E. (2001) Efficient virus extinction by combinations of a mutagen and antiviral inhibitors. J. Virol. 75, 9723–9730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weeks S. A., Lee C. A., Zhao Y., Smidansky E. D., August A., Arnold J. J., Cameron C. E. (2012) A Polymerase mechanism-based strategy for viral attenuation and vaccine development. J. Biol. Chem. 287, 31618–31622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Delaney S., Jarem D. A., Volle C. B., Yennie C. J. (2012) Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radic. Res. 46, 420–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Z., Malla S., Shin B., Li J. M. (2014) Battle against RNA oxidation: molecular mechanisms for reducing oxidized RNA to protect cells. Wiley Interdiscip. Rev. RNA 5, 335–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cameron C. E., Moustafa I. M., Arnold J. J. (2009) Dynamics: the missing link between structure and function of the viral RNA-dependent RNA polymerase? Curr. Opin. Struct. Biol. 19, 768–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moustafa I. M., Shen H., Morton B., Colina C. M., Cameron C. E. (2011) Molecular dynamics simulations of viral RNA polymerases link conserved and correlated motions of functional elements to fidelity. J. Mol. Biol. 410, 159–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang X., Welch J. L., Arnold J. J., Boehr D. D. (2010) Long-range interaction networks in the function and fidelity of poliovirus RNA-dependent RNA polymerase studied by nuclear magnetic resonance. Biochemistry 49, 9361–9371 [DOI] [PMC free article] [PubMed] [Google Scholar]