

Background: MTHFD1 is the primary source of one-carbon units for thymidylate synthesis.
Results: MTHFD1 localizes to the nucleus in folate deficiency and S- and G2/M phases in mammalian cells to support de novo thymidylate biosynthesis.
Conclusion: MTHFD1 nuclear localization explains the incorporation of formate into thymidylate during de novo thymidylate biosynthesis.
Significance: Nuclear localization of MTHFD1 protects DNA by limiting uracil misincorporation into DNA.
Keywords: Cell Compartmentalization, DNA Synthesis, Folate, Homocysteine, Nucleoside/Nucleotide Metabolism, MTHFD1, One-carbon Metabolism, Uracil
Abstract
Folate-mediated one-carbon metabolism is a metabolic network of interconnected pathways that is required for the de novo synthesis of three of the four DNA bases and the remethylation of homocysteine to methionine. Previous studies have indicated that the thymidylate synthesis and homocysteine remethylation pathways compete for a limiting pool of methylenetetrahydrofolate cofactors and that thymidylate biosynthesis is preserved in folate deficiency at the expense of homocysteine remethylation, but the mechanisms are unknown. Recently, it was shown that thymidylate synthesis occurs in the nucleus, whereas homocysteine remethylation occurs in the cytosol. In this study we demonstrate that methylenetetrahydrofolate dehydrogenase 1 (MTHFD1), an enzyme that generates methylenetetrahydrofolate from formate, ATP, and NADPH, functions in the nucleus to support de novo thymidylate biosynthesis. MTHFD1 translocates to the nucleus in S-phase MCF-7 and HeLa cells. During folate deficiency mouse liver MTHFD1 levels are enriched in the nucleus >2-fold at the expense of levels in the cytosol. Furthermore, nuclear folate levels are resistant to folate depletion when total cellular folate levels are reduced by >50% in mouse liver. The enrichment of folate cofactors and MTHFD1 protein in the nucleus during folate deficiency in mouse liver and human cell lines accounts for previous metabolic studies that indicated 5,10-methylenetetrahydrofolate is preferentially directed toward de novo thymidylate biosynthesis at the expense of homocysteine remethylation during folate deficiency.
Introduction
Folate-dependent one-carbon (1C)3 metabolism is a network of interconnected metabolic pathways involved in the de novo synthesis of adenosine, guanosine, and thymidylate nucleotides and the remethylation of homocysteine to methionine (1). Disruption of this metabolic network can result from insufficient dietary intake of B vitamins (folate, vitamin B12, or riboflavin) and/or polymorphisms in genes that encode folate-utilizing enzymes or the interactions among these factors. Such disruptions increase the risk for pathologies that include certain cancers, cardiovascular and neurodegenerative disease, and developmental anomalies (2). These pathologies are associated with changes in blood biomarkers including elevated serum homocysteine, increased uracil in lymphocyte DNA, and DNA hypomethylation (3). The mechanisms and associated pathways underlying folate-associated pathologies have yet to be established in part because the interconnectedness of the pathways within the 1C metabolic network limits the ability to assign a single pathway to a pathological outcome (4).
The hydroxymethyl group of serine and mitochondrially derived formate are the two major sources of folate-activated 1C units (Fig. 1). Serine hydroxymethyltransferase (SHMT) converts serine and tetrahydrofolate (THF) to glycine and 5,10-methylene-THF and exists as three isoforms: SHMT1, which is localized to the cytosol and nucleus, and SHMT2 and SHMT2α, which are encoded by SHMT2 through alternative promoter usage. SHMT2 localizes to mitochondria, whereas SHMT2α localizes to the cytosol and nucleus and is functionally redundant with SHMT1 (5). Expression of SHMT1 is limited to tissues including liver, kidney, colon, and the neural epithelium at specific developmental stages, whereas SHMT2 is ubiquitously expressed (3, 6, 7). Formate, generated from serine and glycine catabolism in the mitochondria, is the primary source of 1C units for cytoplasmic 1C metabolism and is converted to 10-formyl-THF, 5,10-methenyl-THF, and ultimately 5,10-methylene-THF in the cytosol by the three enzyme activities encoded by the MTHFD1 gene (1).
FIGURE 1.

One-carbon metabolism is required for the synthesis of purines, thymidylate (dTMP), and methionine. Mitochondrial-derived formate is a major source of 1C units and can enter the cytoplasm to function as a one-carbon unit for folate metabolism. The synthesis of dTMP occurs in the nucleus and mitochondria. At S phase, the enzymes of the thymidylate (dTMP) synthesis pathway undergo SUMO-dependent translocation to the nucleus. The remethylation of homocysteine to methionine by MTR requires vitamin B12. The one carbon is labeled in bold. The inset shows the thymidylate synthesis cycle, which involves the enzymes, SHMT1, SHMT2a, TYMS, and dihydrofolate reductase (DHFR). DHF, dihydrofolate; MTR, methionine synthase; GNMT, glycine N-methyltransferase; MTHFR, 5-methenylenetetrahydrofolate reductase.
Pathways within the 1C network compete for a limited pool of reduced intracellular THF cofactors (8). This competition is most acute for the partitioning of 5,10-methylene-THF at a critical branch point where it can be directed toward de novo thymidylate synthesis or irreversibly reduced to 5-methyl-THF by the enzyme methylenetetrahydrofolate reductase, which commits folate cofactors for homocysteine remethylation to methionine and subsequently S-adenosylmethionine synthesis (9). Results from mathematical models and isotope tracer studies indicated that 1C moieties are preferentially partitioned to de novo thymidylate biosynthesis via kinetic competition for 5,10-methylene-THF between thymidylate synthase (TYMS) and methylenetetrahydrofolate reductase, especially in folate deficiency (10). This competition is affected by SHMT1 expression, as changes in SHMT1 expression result in nearly proportional changes in rates of de novo thymidylate biosynthesis, as SHMT1-derived 5,10-methylene-THF is preferentially partitioned to thymidylate synthesis (9). The preferential enrichment of SHMT-derived 1Cs into thymidylate was recently explained by the observation that the enzymes of the thymidylate cycle, SHMT1, SHMT2α, TYMS, and dihydrofolate reductase, translocate to the nucleus during the S-phase of the cell cycle (11) and after ultraviolet radiation exposure (12, 13), accounting for the partitioning of SHMT-derived 5,10-methylene-THF to thymidylate synthesis (11). However, SHMT does not play a primary catalytic role in nuclear de novo thymidylate biosynthesis, as isotope tracer studies demonstrate that >70% of the 1Cs consumed in de novo thymidylate biosynthesis are derived from formate through MTHFD1 as well as the observation that catalytically inactive SHMT mutants also increase the flux of SHMT1-derived 1C units to thymidylate synthesis (14). Subsequently, SHMT1 and SHMT2α were shown to function as essential scaffold proteins that anchor the thymidylate synthesis pathway enzymes to the nuclear lamina and interact with DNA at the replication fork (14). This model, however, does not account for the observation that formate is the primary source of 1C units for thymidylate synthesis (9) or fully explain the mechanism whereby folate cofactors are preferentially partitioned to de novo thymidylate synthesis during folate deficiency.
MTHFD1 provides 5,10-methylene-THF from formate and THF for both homocysteine remethylation and thymidylate biosynthesis, but the mechanisms whereby it directs methylene-THF to thymidylate biosynthesis during folate depletion are not known (Fig. 1). In this study we show that MTHFD1 protein localizes to the nucleus in S-phase HeLa and MCF-7 cells and that MTHFD1 localization to the nucleus in mouse liver is responsive to folate depletion. Furthermore, we provide evidence that nuclear folate levels are resistant to depletion when cellular folate levels decline in mouse liver. These studies indicate that MTHFD1 senses intracellular folate levels in the cytosol and thereby provides a mechanism by which de novo thymidylate biosynthesis is preserved at the expense of homocysteine remethylation during folate deficiency.
EXPERIMENTAL PROCEDURES
Animal Models and Diet
All study protocols were approved by the Institutional Animal Care and Use Committee of Cornell University and conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The generation and genotyping of Mthfd1gt (15) and Shmt1−/− (16) mice have been described previously; mice were maintained under specific-pathogen free conditions. Mthfd1gt/+ and Mthfd1+/+ study animals were generated by crossing C57Bl/6 female mice to C57Bl/6 Mthfdgt/+ male mice. Shmt1+/+, Shmt1+/−, and Shmt1−/− were generated by intercrossing C57Bl/6 Shmt1+/− female and C57Bl/6 Shmt1+/− male mice. At weaning, male offspring (littermates) were randomly assigned to either AIN-93G (control (C) diet, Dyets, Inc., Bethlehem, PA) that contained 2 mg/kg folic acid or to a modified AIN-93G diet lacking folic acid (FD diet, Dyets, Inc., Bethlehem, PA). For liver nuclear isolation mice were maintained on diet for 22 ± 2 weeks, whereas for plasma formate analyses mice were maintained on diets for 6 weeks.
Tissue Harvesting
Mice were killed by carbon dioxide asphyxiation. Blood was collected in heparin-coated tubes by cardiac puncture. After washing in ice-cold PBS, liver pieces for whole cell lysate analysis or Lactobacillus casei microbiological folate assays were flash-frozen in liquid nitrogen then stored at −80 °C. Liver nuclei were subfractionated using an iodixanol gradient as described previously (14). Nuclear pellets were apportioned for both immunoblot assays and microbiological assays and flash-frozen at −80 °C.
Immunoblotting
Liver nuclei pellets were lysed at 4 °C in 50 mm Tris-HCl at pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 5 mm EDTA, 5 mm DTT, and 1:100 protease inhibitor cocktail (Sigma) and sonicated to shear DNA. Supernatant protein concentration was determined using Lowry-Bensadoun protein assay (17). Proteins (25 μg/lane) were separated on 12% SDS-PAGE gels and transferred to PVDF membranes (Millipore). MTHFD1 levels were determined using a 1:10,000 dilution of sheep anti-mouse MTHFD1 antibody, which has been previously described (15). The purity and equal loading of the nuclear fraction was verified by detection of cytoplasmic marker glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using α-GAPDH (Novus Biologicals, 1:120,000 dilution) and the nuclear marker Lamin A/C (Santa Cruz Biotechnology, 1:2,000 dilution). Horseradish peroxidase-conjugated anti-mouse IgG (Pierce) or anti-rabbit IgG (Pierce) secondary antibodies were diluted to 1:10,000 to detect GAPDH or Lamin A/C, respectively.
For quantifying proteins in cultured HeLa and MCF-7 nuclear and cytoplasmic extracts, cells were maintained at 37 °C in 5% CO2 in minimal essential media (αMEM) (Hyclone) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen). Nuclear and cytoplasmic extracts were prepared using a nuclear extract kit following the manufacturer's protocol (Active Motif) with a minor modification; the supernatant was strained through a 70-μm mesh cup. Immunoblots were performed as described above for mouse liver extracts.
Cell Culture and Cell Cycle Arrest
MCF-7 or HeLa cells were passaged in minimal essential media, α-modification (α-MEM, Hyclone) supplemented with 10% fetal bovine serum and penicillin/streptomycin. For cell cycle arrest, cells were either cultured in α-MEM or “defined α-MEM,” which lacks ribonucleotides, deoxyribonucleotides, folate, pyridoxine, methionine, serine, and glycine and was supplemented with 10% dialyzed, charcoal-treated fetal bovine serum, 10 μm methionine, 200 μm glycine, 250 μm serine, and 5 nm 5-(6R,S)-formyl-THF. Before arrest, cells were cultured for three passages in either folate-replete α-MEM or folate-deficient defined α-MEM. Cells were exposed for 24 h before harvest to 35 μm lovastatin, 1 mm hydroxyurea (HU), or 100 ng/ml nocodazole to arrest in G1 phase, S phase, and G2/M, respectively. Cell cycle arrest was verified by fluorescence-activated cell sorting (FACS). The Nuclei EZ Prep kit (Sigma) was used for nuclear isolation and was modified slightly by adding three additional washes of the nuclear pellet with 0.5% sodium ascorbate to preserve nuclear folates.
Nuclear Localization as a Function of S-phase Progression
HeLa cells were passaged in α-MEM supplemented with 10% fetal bovine serum and penicillin/streptomycin. For cell cycle arrest, cells were treated with 1 mm HU for 24 h, then washed with PBS and released into fresh α-MEM for 3, 6, 9, or 12 h. For cells collected at 9 and 12 h after HU release, 100 ng/ml nocodazole was added to prevent cell cycle progression through mitosis. The Active Motive nuclear extraction kit was used to prepare nuclear and cytosolic extracts.
Nuclear Total Folate Analysis
Folate concentrations from liver tissue, liver nuclear pellets, and MCF-7 cells were determined using the L. casei microbiological assay as described previously (18).
Quantitative Analysis of Uracil Concentrations in Liver
Uracil in liver nuclear DNA was determined using gas chromatography-mass spectrometry (GC/MS with negative chemical ionization) as previously described (16).
Quantitative Analysis of Plasma Formate
Plasma formate were quantified as described previously (19, 20).
Generation of MTHFD1-GFP Fusion Protein
The MTHFD1 cDNA open reading frame was amplified from the MGC IMAGE clone 3508998 (GenBankTM accession number BC009806.2) using PCR with forward primer 5′-ATGGCGCCAGCAGAAATCCTGAAC and reverse primer 5′-GGACTTGTCTTGTCCACTTACCTAATAAG under the following conditions: 95 °C for 45 s, 55 °C for 45 s, and 72 °C for 10 min. The PCR product was cloned into the pcDNA3.1/CT-GFP-TOPO vector (Invitrogen) to generate a fusion protein of MTHFD1-GFP per the manufacturer's instructions. Because this cDNA contained the G1958A polymorphic variant, the 1958A allele was mutated back to 1958G using the QuikChange II site-directed mutagenesis kit (Agilent Technologies) according to manufacturer's protocol.
Confocal Microscopy
The plasmid encoding the MTHFD1-GFP fusion protein was transfected into HeLa cells when 50% confluent using FuGENE 6. Cells were plated in duplicate in 6 well plates containing cover glass #1 (Fisher) on the bottom of each well and were allowed to grow for 36–48 h in α-MEM supplemented with 10% FBS. 5 μm DRAQ5 (Thermo Scientific) was used for nuclear staining according to the manufacturer's protocol. Cell fixation was performed as described (21) with minor modifications. Briefly, cells were washed 2 times with PBS and fixed with 4% formaldehyde in PBS for 10 min, washed 4 times with PBS, and mounted on microscopy slides with Fluoromount G (SouthernBiotech). Cells were visualized with the Leica SP2 confocal microscope at the Cornell microscopy facility. Nuclear and cytosolic signal intensity were quantified using Leica Lite software, and the ratio of fluorescence intensity in the nucleus to the cytosol was calculated for at least 20 individual cells per condition and graphed as the mean ± S.E. The statistical significance of the differences between the means was estimated using bilateral Student's t test for unpaired data with Bonferroni correction for multiple comparisons.
Nuclear Thymidylate Biosynthesis Assay
Nuclei (2 × 107 per condition) were isolated from HeLa cells arrested in S phase with 1 mm HU for 24 h and released into fresh medium for 3 h using a nuclear extraction kit (Active Motif) with the modification that all extraction buffers were supplemented with 50 mm β-mercaptoethanol. Purified nuclei were suspended in 100 μl of nuclear assay buffer per condition. The assay was conducted under three different experimental conditions: experiment 1, intact nuclei in the assay buffer; experiment 2, intact nuclei with ATP omitted from the assay buffer; experiment 3, intact nuclei with dUMP omitted from the assay buffer. The assay buffer contained 6 mm NADPH (Sigma), 50 mm β-mercaptoethanol, 25 mm HEPES, pH 7.5, 50 mm sucrose, 5 mm MgCl2, 25 mm KCl, 2 mm ATP, 1 mm dUMP (Sigma), and 370 μm [3H]formate (Moravek). Reactions were incubated for 18 h at 37 °C with shaking at 300 rpm. Nuclei were pelleted by centrifugation at 10,000 rpm for 5 min, and the supernatant was collected and treated with alkaline phosphatase for 1 h at 37 ºC. 25 μl was injected into a Phenomenex Synergi 4-μm Fusion-RP 80A 150 × 4.6 mm column using a binary buffer system with a flow rate of 1 ml/min. Buffer A consisted of 20 mm ammonium acetate, pH 4.5, and buffer B was acetonitrile. Buffer B concentration increased from 0 to 5% over a 0–4-min run time, then 5 to 40% over 4–20 min. Nucleosides were detected using a Shimadzu SPD-M20A diode array detector, monitoring wavelengths from 220 to 300 nm. 250-μl fractions were collected using a Shimadzu FRC-10A fraction collector; retention times of thymidine (∼8.6 min) were verified using an unlabeled internal thymidine standard for each sample. Fractions were mixed with 4 ml of Ecoscint (National Diagnostics) scintillation fluid and counted on a Beckman LS-6500 liquid scintillation counter in dual disintegrations/min mode. Experimental conditions were performed in duplicate, and the experiment was repeated twice.
RESULTS
MTHFD1 Localizes to S-phase Nuclei and Its Localization to Mouse Liver Nuclei Is Folate-responsive
MTHFD1 protein from both Mthfd1gt/+ and Mthfd1+/+ mice was present in liver nuclear extracts, and its nuclear localization was increased in animals placed on the FD diet for 22 ± 2 weeks (Fig. 2A) without an increase in whole cell MTHFD1 protein levels (Fig. 2B). In contrast, expression of thymidine kinase 1, a salvage pathway enzyme for thymidylate synthesis, was not significantly different between Mthfd1gt/+ and Mthfd1+/+ or affected by dietary folate. A human MTHFD1-GFP fusion protein expressed in HeLa (Fig. 3A) cells also localized to the nucleus. Confocal microscopy revealed co-localization of MTHFD1-GFP fluorescence and the fluorescent nuclear DNA stain DRAQ5 in S-phase-arrested HeLa cells; overlap was reduced during G1 and G2/M (Fig. 3A). The MTHFD1-GFP signal in the nucleus was nearly 2-fold higher during S-phase of the cell cycle (p < 0.001) than during G1 or at the G2/M transition (Fig. 3B). To determine if a common polymorphism in the human MTHFD1 gene, MTHFD1 G1958A (R653Q), which has been associated with risk for neural tube defects (22), affected MTHFD1 nuclear translocation, a fusion GFP-MTHFD1 (G1958A) was expressed in HeLa cells. The GFP-MTHFD1 R653Q variant accumulated in the nucleus, indicating that the G1958A variant does not prevent nuclear accumulation of MTHFD1 protein (Fig. 3B).
FIGURE 2.
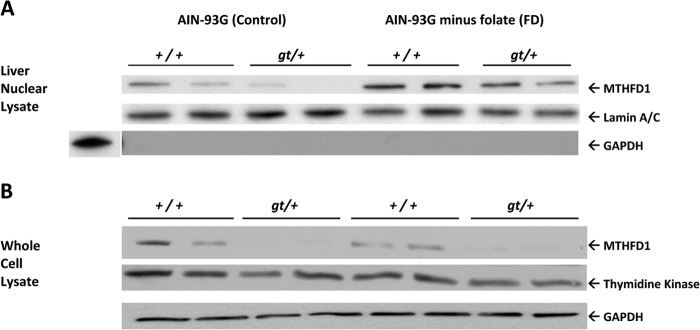
MTHFD1 protein levels in liver nuclei from wild type and Mthfd1gt/+ knockdown mice after 22 ± 2 weeks on diet. A, nuclear tissue lysate was probed with rabbit anti-human Lamin A/C antibody as the loading control. The same membrane was probed with mouse anti-human glyceraldehyde-3-phosphate as a control for nuclei purity. B, immunoblot analysis of total liver extract from wild type and Mthfd1gt mice. Total cell lysate was probed with mouse anti-human glyceraldehyde 3-phosphate as the loading control. Both membranes were treated with polyclonal sheep anti-mouse MTHFD1 antibody to detect MTHFD1 protein.
FIGURE 3.

MTHFD1 traffics to the nucleus in cell cycle-dependent manner. A, MTHFD1-GFP fusion protein localization within cell cycle-synchronized HeLa cells. HeLa cells were transfected with a plasmid encoding an MTHFD1-GFP fusion protein and arrested in G1, S, and G2/M phase. Two representative images for cells for the indicated phases of the cell cycle are shown. MTHFD1-GFP is colored green in the left panels with the nuclear stain Draq5 colored red on the middle panels; the merged images are in the right panels. B, quantitation of nuclear and cytoplasmic localization of MTHFD-GFP wt (black bars) and G1958A polymorphic variant (white bars) at each stage of the cell cycle, as described under “Experimental Procedures.” The ratios of fluorescence intensity in the nucleus to the cytosol were calculated for at least 20 individual cells per condition and are graphed as the mean ± S.E. The statistical significance p was assessed as a t test with Bonferroni correction for multiple comparisons and is represented as follows: NS (non-significant), ***, p < 0.001. Noc, nocodazole; Lpmb, leptomycin B. C, immunoblot analysis of MTHFD1 partitioning between the cytosolic and nuclear fractions in asynchronous HeLa cells, and in HeLa cells arrested in S phase and released into fresh medium for the indicated time points. S phase cell cycle arrest and release were confirmed by FACS analysis.
When HeLa cells were blocked in S-phase using hydroxyurea and then released from the cell-cycle block so that cells could progress through S-phase, nuclear localization of MTHFD1, SHMT1, and PCNA (proliferating cell nuclear antigen) increased with the percentage of cells in S-phase (Fig. 3C).
Effect of Cell Cycle and Dietary Folate Restriction on Nuclear Folate Accumulation
Whole cell liver folate levels in mice decreased by ∼40–55% relative to the control diet (p = <0.001, Table 1) after 22 ± 2 weeks on the FD diet. Mthfd1 genotype did not affect cellular or nuclear folate levels. Interestingly, liver nuclear folate levels were resistant to depletion in mice fed the FD diet (Table 1).
TABLE 1.
Nuclear liver folate levels are resistant to folate depletion
Male mice were weaned onto and maintained on folate-sufficient and folate-deficient diets for 22 ± 2 weeks. Total cellular and nuclear folates n = 3–8 per group were measured. Values represent the mean ± S.E. NS, no significant difference.
| Genotype | AIN-93G (control) |
AIN-93G − folic acid (FD) |
||
|---|---|---|---|---|
| Whole cell folate | Nuclear folate | Whole cell folate | Nuclear folate | |
| fmol/μg protein | fmol/μg protein | fmol/μg protein | fmol/μg protein | |
| Mthfd1+/+ | 99.0 ± 14.4 | 29.2 ± 6.2 | 63.4 ± 6.2 | 28.9 ± 5.2 |
| Mthfd1gt/+ | 104.3 ± 2.7 | 32.3 ± 5.2 | 44.7 ± 9.5 | 28.8 ± 8.0 |
| p Value, diet effect | <0.001 | NS | ||
| p Value, genotype effect | NS | NS | ||
| p Value, diet x genotype effect | NS | NS | ||
In MCF-7 cells, cell cycle progression did not affect nuclear folate levels. MCF-7 cells were cultured for three passages in either folate-sufficient α-MEM or folate-deficient defined α-MEM and then arrested in G1, S, or G2/M phase of the cell cycle, as described under “Experimental Procedures.” Cell cycle arrest was verified using FACS analysis. Nuclear folate levels did not vary with cell cycle in either folate-replete or folate-deficient culture medium (p > 0.1). During each phase of the cell cycle, whole cell folate levels were significantly lower when cultured in depleted media (Table 2, p < 0.001). Interestingly, nuclear folate levels were lower in folate-deplete media during G1 and at G2/M of the cell cycle (p < 0.05, Table 2), but not during S-phase, the phase of the cell cycle when the enzymes required for de novo thymidylate synthesis are present in the nucleus.
TABLE 2.
Nuclear folate levels are not affected by cell cycle arrest or folate depletion in MCF-7 cells
Values are represented as the mean ± S.D. with n = 3. Intracompartmental differences with cell cycle and folate content of the media were analyzed using Student's t test and a Bonferroni correction for multiple (9) comparisons, and data were considered significantly different if p < 0.05. p values are shown for significant comparisons (6R,S)-5-CHOTHF, (6R,S)5-formyltetrahydrofolate.
| Culture media | Cell cycle arrest | Whole cell folate | Nuclear folate |
|---|---|---|---|
| fmol/μg protein | fmol/μg protein | ||
| α-MEM | G1 | 24.6 ± 0.9 | 1.40 ± 0.19 |
| S | 23.8 ± 1.2 | 1.67 ± 0.29 | |
| Folate-replete | G2/M | 23.3 ± 0.9 | 1.65 ± 0.25 |
| Defined MEM with 5 nm (6R,S)-5-CHOTHF | G1 | 13.0 ± 0.7 (p < 0.001 compared to replete) | 0.76 ± 0.06 (p < 0.05 compared to replete) |
| S | 12.9 ± 0.4 (p < 0.001 compared to replete) | 1.12 ± 0.17 | |
| Folate-depleted | G2/M | 12.5 ± 0.8 (p < 0.001 compared to replete) | 0.84 ± 0.04 (p < 0.05 compared to replete) |
Mthfd1gt/+ Mice Are Protected from Uracil Misincorporation in Liver Nuclear DNA
It has been shown that Mthfd1gt/+ mice accumulate less uracil in colon genomic DNA as compared with Mthfd1+/+ littermates (23). Here it is shown that Mthfd1gt/+ liver DNA is also resistant to uracil accumulation (Table 3, p = 0.013) at 25 ± 2 weeks of age. Uracil content in liver genomic DNA was not affected by the FD diet (Table 3).
TABLE 3.
Liver nuclear uracil in DNA in Mthfd1gt/+ mice
DNA uracil of Mthfd1+/+ and Mthfd1gt/+ independent of diet are significantly different as analyzed by 2-way analysis of variance (n = 5 and the values are stated as the means ± S.E. of the mean). NS, no significant difference.
| Diet | Mthfd1 Genotype | Uracil DNA content |
|---|---|---|
| pg/μg of DNA | ||
| AIN-93G (control) | Mthfd1+/+ | 0.23 ± 0.01 |
| Mthfd1gt/+ | 0.17 ± 0.01 | |
| AIN-93G minus folate (FD) | Mthfd1+/+ | 0.21 ± 0.02 |
| Mthfd1gt/+ | 0.19 ± 0.02 | |
| p Value, diet effect | NS | |
| p Value, genotype effect | 0.013 | |
| p Value, diet x genotype effect | NS |
Formate Is Incorporated into Thymidylate in Isolated HeLa Cell Nuclei
The ability of isolated HeLa cell nuclei to synthesize dTMP from dUMP and formate was investigated. Intact nuclei were incubated with [3H]formate, ATP, and NADPH to support the MTHFD1-catalyzed conversion of [3H]formate to 5,10-[3H]methylene-THF and with dUMP. After incubation, the presence of [3H]thymidylate was examined by HPLC (Fig. 4). [3H]Thymidylate was present in the reaction mixtures, and its levels were increased upon the addition of excess dUMP and ATP.
FIGURE 4.
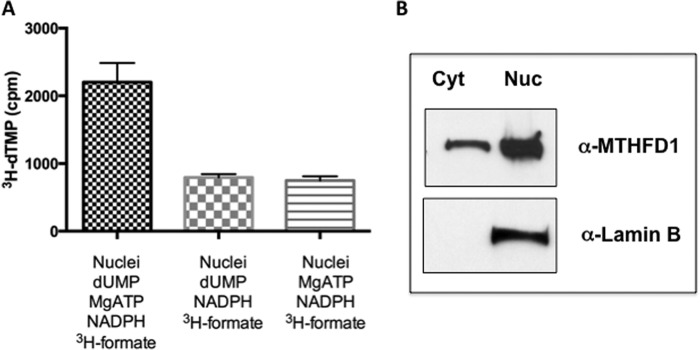
Formate is utilized for thymidylate biosynthesis in purified nuclei. A, nuclei (2 × 107 per condition) were isolated from HeLa cells arrested in S phase with 1 mm HU and released into fresh media for 3 h. The capacity to convert dUMP and [3H]formate to [3H]dTMP was determined in reactions that contained 1) intact nuclei with all the components of the reaction, 2) intact nuclei with the components of the reaction lacking ATP, and 3) intact nuclei with the components of the reaction lacking dUMP as described under “Experimental Procedures.” All reactions were performed in duplicate, and the experiment was repeated two times. Variation is expressed as the S.D. B, nuclei preparation in A was verified by a Western blot with anti-lamin B antibody. Cyt, cytosol; Nuc, nucleus.
Mthfd1gt/+ Mice Exhibit Increased Plasma Formate When Placed on a Folate-deficient Diet
Formate derived from mitochondria is a primary source of cytoplasmic 1C units (24). The 10-formyl-THF synthetase activity of the MTHFD1 protein generates 10-formyl-THF from formate and THF. Mthfd1gt/+ mice placed on a folate-deficient (FD) diet exhibited 25% higher plasma formate than Mthfd1+/+ littermates on the FD diet (Fig. 5A, p = 0.02). There was no difference between plasma formate levels when Mthfd1gt/+ mice consumed a folate-replete control diet for 5 weeks (Fig. 5A, p > 0.05). Taken together, these data indicate that Mthfd1gt/+ mice do not effectively utilize or trap intracellular formate when MTHFD1 protein translocates to the nucleus under folate-deficient conditions, leading to its accumulation in plasma. The SHMT isozymes provide a second entry point for one-carbons by converting serine and THF to glycine and 5,10-methylene-THF. The Shmt1 knock-out mouse displays perturbations in 1C metabolism including increased uracil in nuclear DNA and increased liver AdoMet (25), but plasma formate from Shmt1+/− and Shmt1−/− mice did not differ from Shmt1+/+ mice after 6 weeks on either control or FD diet (Fig. 5B, p > 0.05).
FIGURE 5.
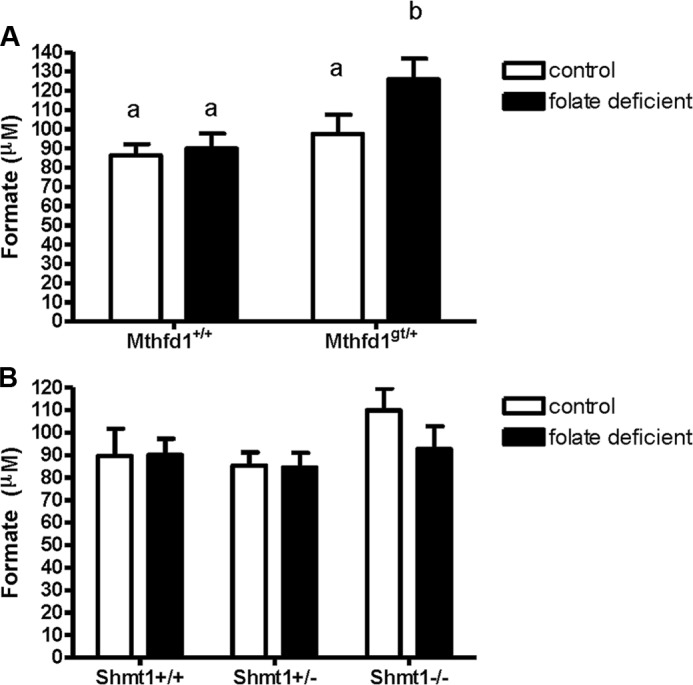
Plasma formate levels as a result of decreased MTHFD1 and SHMT1 expression in mice. A, formate levels from wild-type and Mthfd1gt/+ knockdown mice after 6 weeks consuming control (white bars) or folate-deficient diet (black bars). B, formate levels from wild-type, Shmt1+/−, and Shmt1−/− mice after 6 weeks on control (white bars) or folate-deficient diet (black bars). Data were analyzed using a two-way analysis of variance with a Scheffe post-hoc test for multiple comparisons; groups not connected by the same letter are significantly different (p < 0.05, n = 10 per group; values are shown as the means ± S.E.).
DISCUSSION
This study demonstrates that MTHFD1 translocates to the nucleus in both human and mouse cells in a cell cycle-dependent manner (Figs. 2 and 3) and that nuclear MTHFD1 levels are increased in folate deficiency. This finding accounts for previous observations that 5,10-methylene-THF is preferentially directed toward de novo thymidylate synthesis at the expense of homocysteine remethylation under folate-deficient conditions (8, 10). Whereas this observation was previously explained by kinetic competition for 5,10-methylene-THF cofactors between the enzymes methylenetetrahydrofolate reductase and TYMS, the recent discovery that TYMS and de novo thymidylate synthesis occur in different cellular compartments called into question the conclusion that simple kinetic competition explains the preferential partitioning of 5,10-methylene-THF to de novo thymidylate biosynthesis. Rather, MTHFD1 generates the 5,10-methylene-THF cofactor in the nucleus for thymidylate biosynthesis and in the cytosol for homocysteine remethylation. The enrichment of MTHFD1 in the nucleus at the expense of cytosolic MTHFD1 levels during folate deficiency accounts for the observed protection of de novo thymidylate biosynthesis at the expense of homocysteine remethylation.
This observation that MTHFD1 translocates to the nucleus also provides a mechanism to explain previous isotope tracer studies that demonstrate that formate is the major source of one-carbon units for de novo nuclear thymidylate synthesis (9). In this study MTHFD1 was shown to localize to the nucleus during S phase of the cell cycle with the enzymes required for de novo thymidylate biosynthesis. In the nucleus, SHMT functions as a scaffold that tethers the enzymes of the thymidylate cycle (SHMT, TYMS, dihydrofolate reductase) and the DNA replication machinery to the nuclear lamina such that thymidylate is synthesized at the replication fork (14). The presence of MTHFD1 in the nucleus accounts for observations that formate is the primary source of single carbons for de novo thymidylate biosynthesis. The experiments presented herein also suggest that the two enzymes that synthesize 5,10-methylene-THF, SHMT and MTHFD1, respond independently and uniquely to a limited one-carbon supply. Whereas SHMT1 expression is increased during folate deficiency to enhance nuclear de novo thymidylate biosynthesis (25), MTHFD1 nuclear import is enhanced in the absence of an increase in protein expression to support de novo thymidylate biosynthesis at the expense of homocysteine remethylation. Under folate-depleted conditions, nuclear MTHFD1 is enriched without increased whole cell MTHFD1 protein levels, indicating that enhanced nuclear localization likely results from increased nuclear accumulation as opposed transcriptional and/or translational up-regulation. Taken together, these patterns suggest that SHMT1 and MTHFD1 respond to limited 1C supply and protect the integrity of thymidylate synthesis using distinct mechanisms. The mechanism by which MTHFD1 translocates into the nucleus remains to be established. The primary structure of MTHFD1 does not contain a nuclear localization signal (NLS), although non-canonical three-dimensional NLSs are known to exist on other proteins. Similarly, the primarily cytosolic enzyme glycine N-methyltransferase, which transfers the methyl group from S-adenosylmethionine to glycine, was also shown to translocate to the nucleus in response to folate deficiency, where it also served to increase de novo purine and thymidylate synthesis and limit uracil in DNA (26). Glycine N-methyltransferase also lacks a canonical nuclear localization signal, and the mechanism by which it enters the nucleus remains unknown (27).
The preferential enrichment of folate cofactors in the nucleus during folate deficiency provides a second mechanism whereby de novo thymidylate synthesis is favored over homocysteine remethylation in states of cellular folate depletion. After 22 weeks of dietary folate deficiency, mouse liver nuclei are resistant to folate depletion even when whole cell levels fall by 50% (Table 1). Nuclear folate concentrations do not vary by cell cycle, which is surprising given that the enzymes involved in de novo thymidylate synthesis are enriched in the nucleus in S-phase and mostly absent in G1. However, nuclear folate concentrations are protected from cellular folate depletion during S-phase of the cell cycle in cultured MCF-7 cells (Table 2). Reduced MTHFD1 expression does not affect nuclear folate levels (Table 1). It is not known how folates accumulate in the nucleus nor is the identity of the binding proteins that sequester folate cofactors during G1 known.
Mice do not effectively trap or utilize mitochondrially derived formate in the cytosol for homocysteine remethylation when both folate and MTHFD1 are limiting, as evidenced by the increase in plasma formate in Mthfd1gt/+ mice fed a folate-deficient diet (Fig. 4). This observation supports our previous findings that Mthfd1gt/+ mice fed a folate-deficient diet exhibit impaired homocysteine remethylation without the accumulation of uracil in nuclear DNA, which occurs when de novo thymidylate biosynthesis is impaired.
Minor impairments in de novo thymidylate biosynthesis capacity causes elevated uracil in nuclear DNA and susceptibility to folate-responsive neural tube defects (28, 29). Surprisingly, Mthfd1gt/+ animals do not develop neural tube defects even when fed a folate-deficient diet (28) and exhibit decreased levels of uracil in nuclear DNA in both liver (Table 3) and colon (23). The decrease in uracil in DNA is not explained by increased thymidylate salvage pathway activity as thymidine kinase 1 is not up-regulated in Mthfd1gt/+ liver (Fig. 2B). The observation of increased folate and MTHFD1 enrichment in the nucleus during folate deficiency provides a mechanism that accounts for the lack of a neural tube defect phenotype in Mthfd1gt/+ mice and why these mice are protected from uracil accumulation in DNA and exhibit marked impairments in homocysteine remethylation (15, 30, 31).
Acknowledgments
We thank Cheryll Perry Navestad and Danielle Blemur for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant R37DK58144 (to P. J. S.).
- 1C
- one carbon
- MTHFD1
- methylenetetrahydrofolate dehydrogenase 1
- SHMT
- serine hydroxymethyltransferase
- TYMS
- thymidylate synthase
- gt
- gene-trap
- αMEM
- minimal essential media
- HU
- hydroxyurea
- FD
- folate-deficient.
REFERENCES
- 1. Fox J. T., Stover P. J. (2008) Folate-mediated one-carbon metabolism. Vitam. Horm. 79, 1–44 [DOI] [PubMed] [Google Scholar]
- 2. Stover P. J. (2004) Physiology of folate and vitamin B12 in health and disease. Nutr. Rev. 62, S3–S12 [DOI] [PubMed] [Google Scholar]
- 3. Beaudin A. E., Stover P. J. (2009) Insights into metabolic mechanisms underlying folate-responsive neural tube defects: a minireview. Birth Defects Res. A Clin. Mol. Teratol. 85, 274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scotti M., Stella L., Shearer E. J., Stover P. J. (2013) Modeling cellular compartmentation in one-carbon metabolism. Wiley Interdiscip. Rev. Syst. Biol. Med. 5, 343–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson D. D., Stover P. J. (2009) SHMT1 and SHMT2 are functionally redundant in nuclear de novo thymidylate biosynthesis. PLoS ONE 4, e5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Girgis S., Nasrallah I. M., Suh J. R., Oppenheim E., Zanetti K. A., Mastri M. G., Stover P. J. (1998) Molecular cloning, characterization and alternative splicing of the human cytoplasmic serine hydroxymethyltransferase gene. Gene 210, 315–324 [DOI] [PubMed] [Google Scholar]
- 7. Stover P. J., Chen L. H., Suh J. R., Stover D. M., Keyomarsi K., Shane B. (1997) Molecular cloning, characterization, and regulation of the human mitochondrial serine hydroxymethyltransferase gene. J. Biol. Chem. 272, 1842–1848 [DOI] [PubMed] [Google Scholar]
- 8. Suh J. R., Herbig A. K., Stover P. J. (2001) New perspectives on folate catabolism. Annu. Rev. Nutr. 21, 255–282 [DOI] [PubMed] [Google Scholar]
- 9. Herbig K., Chiang E. P., Lee L. R., Hills J., Shane B., Stover P. J. (2002) Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J. Biol. Chem. 277, 38381–38389 [DOI] [PubMed] [Google Scholar]
- 10. Green J. M., MacKenzie R. E., Matthews R. G. (1988) Substrate flux through methylenetetrahydrofolate dehydrogenase: predicted effects of the concentration of methylenetetrahydrofolate on its partitioning into pathways leading to nucleotide biosynthesis or methionine regeneration. Biochemistry 27, 8014–8022 [DOI] [PubMed] [Google Scholar]
- 11. Woeller C. F., Anderson D. D., Szebenyi D. M., Stover P. J. (2007) Evidence for small ubiquitin-like modifier-dependent nuclear import of the thymidylate biosynthesis pathway. J. Biol. Chem. 282, 17623–17631 [DOI] [PubMed] [Google Scholar]
- 12. Fox J. T., Stover P. J. (2009) Mechanism of the internal ribosome entry site-mediated translation of serine hydroxymethyltransferase 1. J. Biol. Chem. 284, 31085–31096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fox J. T., Shin W. K., Caudill M. A., Stover P. J. (2009) A UV-responsive internal ribosome entry site enhances serine hydroxymethyltransferase 1 expression for DNA damage repair. J. Biol. Chem. 284, 31097–31108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anderson D. D., Woeller C. F., Chiang E. P., Shane B., Stover P. J. (2012) Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J. Biol. Chem. 287, 7051–7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. MacFarlane A. J., Perry C. A., Girnary H. H., Gao D., Allen R. H., Stabler S. P., Shane B., Stover P. J. (2009) Mthfd1 is an essential gene in mice and alters biomarkers of impaired one-carbon metabolism. J. Biol. Chem. 284, 1533–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. MacFarlane A. J., Liu X., Perry C. A., Flodby P., Allen R. H., Stabler S. P., Stover P. J. (2008) Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J. Biol. Chem. 283, 25846–25853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bensadoun A., Weinstein D. (1976) Assay of proteins in the presence of interfering materials. Anal. Biochem. 70, 241–250 [DOI] [PubMed] [Google Scholar]
- 18. Suh J. R., Oppenheim E. W., Girgis S., Stover P. J. (2000) Purification and properties of a folate-catabolizing enzyme. J. Biol. Chem. 275, 35646–35655 [DOI] [PubMed] [Google Scholar]
- 19. Lamarre S. G., Molloy A. M., Reinke S. N., Sykes B. D., Brosnan M. E., Brosnan J. T. (2012) Formate can differentiate between hyperhomocysteinemia due to impaired remethylation and impaired transsulfuration. Am. J. Physiol. Endocrinol. Metab. 302, E61–E67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lamarre S. G., Morrow G., Macmillan L., Brosnan M. E., Brosnan J. T. (2013) Formate: an essential metabolite, a biomarker, or more? Clin. Chem. Lab. Med. 51, 571–578 [DOI] [PubMed] [Google Scholar]
- 21. Anderson D. D., Eom J. Y., Stover P. J. (2012) Competition between sumoylation and ubiquitination of serine hydroxymethyltransferase 1 determines its nuclear localization and its accumulation in the nucleus. J. Biol. Chem. 287, 4790–4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pangilinan F., Molloy A. M., Mills J. L., Troendle J. F., Parle-McDermott A., Signore C., O'Leary V. B., Chines P., Seay J. M., Geiler-Samerotte K., Mitchell A., VanderMeer J. E., Krebs K. M., Sanchez A., Cornman-Homonoff J., Stone N., Conley M., Kirke P. N., Shane B., Scott J. M., Brody L. C. (2012) Evaluation of common genetic variants in 82 candidate genes as risk factors for neural tube defects. BMC Med. Genet. 13, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. MacFarlane A. J., Perry C. A., McEntee M. F., Lin D. M., Stover P. J. (2011) Mthfd1 is a modifier of chemically induced intestinal carcinogenesis. Carcinogenesis 32, 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tibbetts A. S., Appling D. R. (2010) Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81 [DOI] [PubMed] [Google Scholar]
- 25. Macfarlane A. J., Perry C. A., McEntee M. F., Lin D. M., Stover P. J. (2011) Shmt1 heterozygosity impairs folate-dependent thymidylate synthesis capacity and modifies risk of Apc(min)-mediated intestinal cancer risk. Cancer Res. 71, 2098–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y. C., Lin W. L., Lin Y. J., Tang F. Y., Chen Y. M., Chiang E. P. (2014) A novel role of the tumor suppressor GNMT in cellular defense against DNA damage. Int. J. Cancer 134, 799–810 [DOI] [PubMed] [Google Scholar]
- 27. DebRoy S., Kramarenko I. I., Ghose S., Oleinik N. V., Krupenko S. A., Krupenko N. I. (2013) A novel tumor suppressor function of glycine N-methyltransferase is independent of its catalytic activity but requires nuclear localization. PLoS ONE 8, e70062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beaudin A. E., Abarinov E. V., Noden D. M., Perry C. A., Chu S., Stabler S. P., Allen R. H., Stover P. J. (2011) Shmt1 and de novo thymidylate biosynthesis underlie folate-responsive neural tube defects in mice. Am. J. Clin. Nutr. 93, 789–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beaudin A. E., Abarinov E. V., Malysheva O., Perry C. A., Caudill M., Stover P. J. (2012) Dietary folate, but not choline, modifies neural tube defect risk in Shmt1 knockout mice. Am. J. Clin. Nutr. 95, 109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Field M. S., Shields K. S., Abarinov E. V., Malysheva O. V., Allen R. H., Stabler S. P., Ash J. A., Strupp B. J., Stover P. J., Caudill M. A. (2013) Reduced MTHFD1 activity in male mice perturbs folate- and choline-dependent one-carbon metabolism as well as transsulfuration. J. Nutr. 143, 41–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ash J. A., Jiang X., Malysheva O. V., Fiorenza C. G., Bisogni A. J., Levitsky D. A., Strawderman M. S., Caudill M. A., Stover P. J., Strupp B. J. (2013) Dietary and genetic manipulations of folate metabolism differentially affect neocortical functions in mice. Neurotoxicol. Teratol. 38, 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]