

Background: PHF2 is a histone demethylase that possesses an H3K4me2/3 binding activity.
Results: PHF2 represses rDNA transcription depending on its H3K4me2/3 binding but not demethylase activity and does so by antagonizing PHF8 and recruiting SUV39H1. PHF2 also seems to generally repress Pol II transcription.
Conclusion: PHF2 represses rather than activates rDNA transcription.
Significance: This work reveals a potential general transcriptional repression function for PHF2.
Keywords: Histone Methylation, Nucleolus, Ribosomal Ribonucleic Acid (rRNA) (Ribosomal RNA), RNA Polymerase I, Transcription Regulation
Abstract
Regulation of rDNA transcription is central to cell growth and proliferation. PHF2 and PHF8 belong to a subfamily of histone demethylases that also possess a PHD domain-dependent di-/trimethylated histone 3 lysine 4 (H3K4me2/3) binding activity and are known to be enriched in the nucleolus. In this study, we show that, unlike PHF8 that activates rDNA transcription, PHF2 inhibits rDNA transcription. Depletion of PHF2 by RNA interference increases and overexpression of PHF2 decreases rDNA transcription, respectively, whereas simultaneous depletion of PHF8 and PHF2 restores the level of rDNA transcription. The inhibition of rDNA transcription by PHF2 depends on its H3K4me2/3 binding activity that is also required for PHF2 association with the promoter of rDNA genes but not its demethylase activity. We provide evidence that PHF2 is likely to repress rDNA transcription by competing with PHF8 for binding of rDNA promoter and by recruiting H3K9me2/3 methyltransferase SUV39H1. We also provide evidence that, whereas PHF8 promotes, PHF2 represses the transcriptional activity of RARα, Oct4, and KLF4 and a few PHF8 target genes tested. Taken together, our study demonstrates a repressive role for PHF2 in transcription by RNA polymerase I and II.
Introduction
In eukaryotic cells, the rDNA genes that encode rRNAs, the major components of ribosome, are arrayed in many copies at nucleolar organizer regions. Each mammalian rDNA gene is about 43 kb, includes a promoter and transcribed and non-transcribed regions, and is specifically transcribed by RNA polymerase I (Pol I)4 in nucleoli to produce pre-rRNA transcripts that will be processed into 18, 5.8, and 28 S rRNAs (Fig. 4A) (1, 2). rDNA transcription by Pol I requires the coordinated function of a transcription activator termed upstream binding factor (UBF) and the promoter selectivity factor (SL1 in humans and TIF-IB in mice), a complex that contains TBP and a few TAF proteins (3, 4). UBF activates rDNA transcription by recruiting Pol I and acts synergistically with SL1 to promote the assembly of Pol I preinitiation complex (5, 6).
FIGURE 4.

PHF2 inhibits the binding of PHF8 and UBF and recruits SUV39H1 to the rDNA promoter. A, schematic diagram of a rDNA gene showing its promoter and non-transcribed and transcribed parts. Arrows point to different areas of rDNA paired by the four primer sets used in ChIP (see Table 1). B, ChIP analysis showing PHF2 is highly enriched in the rDNA gene promoter. ChIP analysis for PHF2 association at four different regions of the rDNA gene was performed with HeLa cells stably expressing either ShPHF2–1 or control shRNA. The results were presented as percentage of input. Error bars, S.D.; n = 3. The knockdown of PHF2 by shPHF2–1 was verified by Western blot analysis. C, PHF2 requires PHD domain to bind rDNA promoter. HeLa cells transfected with FLAG-PHF2 or FLAG-PHF2-M20A were subjected to a ChIP assay with anti-FLAG antibody, and the promoter association was analyzed. Error bars, S.D.; n = 3. A fraction of the cells were subjected to Western blot with PHF2, FLAG, and β-actin antibodies. D, knockdown of PHF2 led to increased association of PHF8 and UBF with the rDNA promoter. HeLa cells stably expressing either shPHF2–1 or control shRNA were subjected to ChIP analysis for PHF8 (left) or UBF (right). Note that the -fold changes in rDNA occupancy between ShPHF2-1-treated and control cells are shown. Error bars, S.D.; n = 3. E, knockdown of PHF2 led to decreased association of SUV39H1 and reduced H3K9me3 methylation at the rDNA promoter. A ChIP assay was performed as in C using antibodies as indicated. Error bars, S.D.; n = 3. F, co-immunoprecipitation of ectopically expressed SUV39H1 and PHF2. HeLa cells were transfected with V5-SUV39H1 alone or together with FLAG-PHF2, and immunoprecipitation was performed with FLAG antibody, followed by Western blot analysis with rabbit V5 and mouse FLAG and GAPDH antibodies. G, co-immunoprecipitation of ectopically expressed SUV39H1 with endogenous PHF2. HeLa cells were transfected with or without Myc-SUV39H1, and immunoprecipitations were performed with anti-Myc antibody, followed by Western blot analysis using anti-PHF2 or PHF8 antibody, as indicated.
rDNA transcription is a fundamental determinant of cellular growth and proliferation. Cumulative studies indicate that rDNA transcription is tightly regulated via epigenetic mechanisms (7, 8). First, a subset of rDNA genes are silenced via formation of a repressive heterochromatin structure mediated by a chromatin remodeling complex, NoRC, that also recruits histone modification enzymes and DNA methyltransferase, DNMT1 (9–11). Second, NuRD, a histone deacetylase and chromatin remodeling complex, has been shown to render a subset of rDNA genes in a poised state (12). Third, the remaining actively transcribed rDNA genes are regulated at the steps of transcriptional initiation and elongation via reversible histone modifications (2, 8, 13). Consistently, various chromatin modification enzymes, including PCAF, SIRT1, SUV39H1, PHF2, and PHF8, have been shown to play a role in regulation of rDNA transcription (13–18).
PHF2 (also known as JHDM1E and KDM7B), PHF8 (also known as JHDM1F and KDM7C), and KIAA1718 (also known as JHDM1D and KDM7A) are more closely related members of the α-ketoglutarate/Fe2+-dependent dioxygenases characterized by the presence of a Jumonji domain (19, 20). Many but not all proteins of this family have been shown to possess histone/non-histone demethylase activity. The enzymatic activity for PHF8 has been extensively studied, and the general conclusion is that PHF8 possesses demethylase activities toward H3K9me1/2 (13, 18, 21–23) and H4K20me1 (23, 24). However, the exact demethylase activity for PHF2 is less clear. In one study, PHF2 was not detected for any histone demethylase activity, and the lack of demethylase activity was explained by one major amino acid alteration in the JmjC domain of PHF2 (25). In other studies, PHF2 has been respectively reported to exhibit an H3K9me1-specific activity (17), a protein kinase A (PKA)-dependent H3K9me2 demethylase activity (26), and H4K20me3 demethylase activity (27). In addition to the JmjC domain, PHF2, PHF8, and KIAA1718 are unique among the JmjC family proteins with possession of an N-terminal PHD (plant homeodomain) domain. These PHD domains have been shown to specifically bind H3K4me2/3 (13, 17, 21, 22, 25, 28, 29), an epigenetic mark commonly enriched at the promoter region and associated with transcriptional activation (19). Given that both PHF2 and PHF8 are enriched in nucleoli, PHF8 and PHF2 have been studied for their function in rDNA transcription. PHF8 was shown to interact with UBF and facilitate rDNA transcription, depending on its H3K4me3 binding and H3K9me1/2 demethylase activity (13, 17, 18), whereas PHF2 was also shown to activate rDNA transcription, depending on its H3K4me3 binding activity (17). In addition, PHF8 has been shown to interact with specific transcription factors, such as RARα (21) and NOTCH1 intracellular domain (30), as well as RNA polymerase II (Pol II) (28) and functions as a coactivator, whereas PHF2 has been shown to interact with CEBPA to regulate adipogenesis (31), with ARID5B to regulate PKA-dependent gene induction of Pepck and G6Pase (26), and with NF-κB to regulate proinflammatory gene programs (27).
In this study, we have characterized in more detail how PHF2 localizes to the nucleolus, and we found, surprisingly, that PHF2 inhibits rather than activates rDNA transcription. The inhibition of rDNA transcription is dependent on its H3K4me3 binding activity but not its demethylase activity. We present evidence that PHF2 may inhibit rDNA transcription by antagonizing PHF8 and by recruitment of corepressor SUV39H1. In addition, we present evidence that PHF2 also has a repression function for transcription by Pol II.
EXPERIMENTAL PROCEDURES
Plasmids, Antibodies, Enzymes, siRNAs, shRNAs, Primers, and Cell Lines
The plasmids for FLAG-PHF2, FLAG-PHF2-M20A, FLAG-PHF2-HD/AA(H249A/D251A), FLAG-PHF2ΔPHD, GFP-PHF2, GFP-PHF2ΔPHD, GFP-PHF2ΔJmjC, GFP-PHF2(1–414), GFP-PHF2(1–755), GFP-PHF2(749–1096), GFP-PHF2-M20A, GST-PHF2-PHD, GST-PHF2-PHD-M20A, GST-PHF8-PHD, GST-PHF8-PHD-M37A, FLAG-OCT4, FLAG-KLF4, and V5-SUV39H1 were constructed in our laboratory. FLAG-PHF8, GAL4-RARα, 4×UAS-TK-luc, IAP-luc, and Rex1-luc were described previously (21, 32–34). The two PHF2 shRNAs, ShPHF2-1 (against coding region) and ShPHF2-2 (against non-coding region) and PHF8 shRNA (shPHF8) were from Open Biosystems. Mouse monoclonal antibodies used in this study were as follows: FLAG, BrU, and β-actin from Sigma; UBF and Pol I from Santa Cruz Biotechnology, Inc.; GAPDH from Abmart. Commercial rabbit polyclonal antibodies used were as follows: H3K9me1 from Abcam, H3K9me2 from Upstate, nucleolin from Dr. Philippe Bouvet, and V5 from Invitrogen. Rabbit anti-PHF2 antibody was raised against purified recombinant GST-PHF2(830–1098), corresponding to the PHF2 C-terminal region amino acids 830–1098, and rabbit anti-PHF8 antibody was raised against GST-PHF8(2–251), corresponding to the PHF8 N-terminal region amino acids 2–251. Fluochore-conjugated secondary antibodies are from Jackson ImmunoResearch. DNase I and RNase A were from New England Biolabs. The siRNAs against PHF2 and PHF8 and quantitative PCR primers are listed in Table 1. Cells were routinely cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and antibiotics. Transient transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
TABLE 1.
Sequences for RT-qPCR and ChIP-qPCR primers and siRNAs
| Primer/siRNA | Sequence |
|---|---|
| RT-qPCR primers | |
| Pre-rRNA | 5′-GCCTTCTCTAGCGATCTGAGAG |
| Pre-rRNA | 3′-CCATAACGGAGGCAGAGACA |
| PHF2 mRNA | 5′-GCAAACGACTGCTGAAGAGG |
| PHF2 mRNA | 3′-ACTCCAGTGAGGGGTAAACG |
| PHF8 mRNA | 5′-ACTCCAGTGAGGGGTAAACG |
| PHF8 mRNA | 3′-CAAGTGCTACAAGTGTTCCGTG |
| JARIDIC mRNA | 5′-TGCATAAGCTGAAGGTTCGGG |
| JARIDIC mRNA | 3′-GCCACTCGCACTTTGTTGG |
| CDC40 mRNA | 5′-AGCAAAATCTCTTTGTGGCTGG |
| CDC40 mRNA | 3′-ACAATGGTGTTGACAGCTCCC |
| OCRL mRNA | 5′-TGATCCCCGGATCTGCCGAC |
| OCRL mRNA | 3′-GGTTGGGTGGAGGCCTCAGGA |
| ChIP-qPCR primers | |
| H4 | 5′-CGACGACCCATTCGAACGTCT |
| H4 | 3′-CTCTCCGGAATCGAACCCTGA |
| H13 | 5′-ACCTGGCGCTAAACCATTCGT |
| H13 | 3′-GGACAAACCCTTGTGTCGAGG |
| H27 | 5′-CCTTCCACGAGAGTGAGAAGCG |
| H27 | 3′-CTCGACCTCCCGAAATCGTACA |
| H42.9 | 5′-CCCGGGGGAGGTATATCTTT |
| H42.9 | 3′-CCAACCTCTCCGACGACA |
| siRNA | |
| PHF2 | 5′-GCUGGAAAUUCGAGAGCAATT |
| PHF2 | 3′-UUGCUCUCGAAUUUCCAGCCT |
| PHF8 | 5′-CUAUGUUGGUUCUGACAAATT |
| PHF8 | 3′-UUUGUCAGAACCAACAUAGTG |
Pull-down Assay with Biotinylated Histone Peptides
A pull-down assay with biotinylated histone H3 tail peptides (aa 1–21) containing or not containing H3K4me3 was performed essentially as described (35). For analyzing the binding of H3K4me3 in the context of full-length PHF2 proteins, the whole cell extracts derived from HeLa cells expressing the wild-type PHF2 or M20A mutant were used. For analyzing the binding of H3K4me3 in the isolated PHD domain, purified recombinant GST-PHF2-PHD or GST-PHF8-PHD or their mutants were used.
Co-immunoprecipitation
To detect the interaction between ectopic expressed SUV39H1 and PHF2, HEK293T cells were transfected with V5-SUV39H1 alone or together with FLAG-PHF2, and immunoprecipitation was performed with mouse anti-FLAG antibody followed by Western blot with rabbit V5, mouse FLAG, and GAPDH antibodies. For detection of co-immunoprecipitation with endogenous PHF2 and PHF8, as shown in Fig. 4G, HEK293T cells were transfected with or without Myc-SUV39H1, and immunoprecipitation was performed with mouse anti-Myc antibody followed by Western blot with anti-PHF2 or anti-PHF8 antibody, as indicated.
Fluorescence Microscopy
Indirect immunofluorescence was performed essentially as described (36). In brief, HeLa cells were transfected with FLAG-PHF2, FLAG-PHF8, or mutants as indicated, fixed, treated with DNase I or RNase A, and stained with antibody, as indicated. Nuclei were stained with Hoechst33342. For direct fluorescence, cells were transfected with the indicated GFP constructs and viewed for fluorescence.
Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed essentially as described (37). In brief, control HeLa cells and HeLa cells stably expressing shPHF2-1 were cross-linked with 1% formaldehyde for 10 min, sonicated, and immunoprecipitated with antibodies, as indicated, followed by quantitative PCR with primer pairs specific to different regions of rDNA genes, as summarized in Fig. 4A and Table 1.
Quantitative RT-PCR
RNA extraction and RT-PCR for pre-rRNA were performed as described (37) The RT-PCR analyses for PHF2, PHF8, JARIDIC, and CDC40 mRNA were performed as described (35) using primers listed in Table 1.
Luciferase Reporter Assay
HeLa cells were transfected with 4xUAS-TK-luc, Gal4-RARα, and various amounts of FLAG-PHF2 or control vector, as indicated, and cells were treated with or without 1 nm retinoic acid and subjected to a luciferase reporter assay according to the Promega Dual-Luciferase reporter assay kit as shown previously (21). For analyzing the effect of PHF2 and PHF8 on transcriptional activation by OCT4 and KLF4, HeLa cells were transfected with Rex1-Luc or IAP-Luc reporter and various amounts of FLAG-PHF2 or FLAG-PHF8, as indicated, and the luciferase reporter assay was carried out essentially as described (32).
BrUTP Incorporation Assay
BrUTP incorporation was performed essentially as described (38). Briefly, HeLa cells were transfected with GFP-tagged PHF2 or mutants. Two days after transfection, BrUTP was transfected into cells with Lipofectamine 2000 accordingly (38). Cells were then fixed and stained with BrU antibody and rhodamine-conjugated secondary antibody. Nuclei were stained with Hoechst 33342.
RESULTS
The PHF2 Nucleolus Localization Is Independent of Its H3K4me3 Binding and Putative Demethylase Activities
PHF2 was reported to be enriched in nucleoli (17), but the molecular determinant(s) of the nucleolus localization was not studied. Using an affinity-purified rabbit polyclonal antibody raised against PHF2 C-terminal region amino acids 830–1098, we confirmed the endogenous PHF2 in HeLa and 293T cells co-localized with the transfected FLAG tagged nucleolin (FLAG-NCL), a marker of nucleoli (Fig. 1A). Using GFP-tagged PHF2, we further confirmed the PHF2 nucleolus localization in HeLa cells (Fig. 1B) and other cell lines (data not shown). We thus generated various GFP-tagged PHF2 mutants and examined their subcellular localization in HeLa cells. The representative results in Fig. 1B show that the nucleolus localization of PHF2 is dependent neither on its H3K4me3 binding activity nor its demethylase activity, because the PHF2 mutants with deletion of either the PHD domain (ΔPHD) or JmjC domain (ΔJmjC) maintained a pattern of nucleolus-enriched localization. We found that the N-terminal region 1–755 exhibited a nucleolus location, whereas the more N-terminal fragment 1–414 was primarily cytoplasmic. On the other hand, the C-terminal region 749–1096 exhibited a nucleolus localization. Thus, our data suggest the existence of two independent nucleolus localization determinants in PHF2, although the exact sequences involved in the nucleolus localization remain to be determined.
FIGURE 1.

Characterization of PHF2 nucleolus localization. A, nucleolus localization for endogenous PHF2 in HeLa and 293T cells. HeLa or 293T cells were transfected with FLAG-tagged nucleolin (Flag-NCL), and double immunostaining was performed for FLAG tag and endogenous PHF2. B, the mapping of sequence determinant(s) for PHF2 nucleolus localization. HeLa cells were transiently transfected with GFP-tagged PHF2 and various deletion mutants, and their subcellular localization was determined by using a fluorescent microscope. Nuclei were stained with Hoechst 33342. C, diagram illustrating the various PHF2 deletion constructs used in B and a summary of their nucleolus localization status. D, the nucleolus localization of both PHF2 and PHF8 is sensitive to RNase A treatment but insensitive to DNase I treatment. The HeLa cells were transfected with either FLAG-PHF2 or FLAG-PHF8 and then treated with RNase A or DNase I at a final concentration of 100 μg/ml or 10 units/ml at 37 °C for 30 min before they were processed for immunofluorescent staining for FLAG-PHF2 or FLAG-PHF8 using anti-FLAG antibody. Immunofluorescent staining was also performed for endogenous nucleolin, which served as a control for nucleolus integrity. E, in vitro peptide pull-down assay examining the binding of FLAG-tagged PHF2, PHF2 with deletion of PHD domain (PHF2ΔPHD), and PHF2 with M20A mutation to control H3 and H3 peptide with H3K4me3. Both peptides contain the H3 N-terminal tail aa 1–21 with a C-terminal biotin moiety. F, in vitro peptide pull-down as in E but with purified recombinant GST-PHF2-PHD domain or GST-PHF8-PHD domain and their mutant as indicated.
We next tested whether the nucleolus localization of PHF2 is dependent on rDNA binding and/or RNA association. The results in Fig. 1D show that, much like the case of nucleolin, the nucleolus localization of PHF2 is dependent on RNA association because treatment of permeabilized HeLa cells with RNase A but not DNase I abolished its nucleolus localization.
By using mammalian expressed FLAG-PHF2 and an in vitro peptide pull-down assay, we confirmed the previous finding (17) that PHF2 binds preferentially the immobilized H3K4me3-containing histone H3 tail peptide (aa 1–21) in comparison with the control H3 peptide (Fig. 1E). Also in agreement with the previous report (17), deletion of PHD domain or mutation of a single key residue, Met20, to alanine within the PHD domain abolished the preferential H3K4me3 binding activity of PHF2 (Fig. 1E). We also confirmed by using recombinant GST-tagged PHD domain and an in vitro pull-down assay that the PHF2 PHD domain is sufficient for binding of H3K4me3 and that M20A mutation abolishes the H3K4me3 binding activity (Fig. 1F). As a comparison, the PHD domain from PHF8 is also sufficient for binding H3K4me3 peptide, and an equivalent M37A mutation abolishes the binding. Note that GST-PHF2-PHD exhibited a better binding of H3K4me3 peptide than GST-PHF8-PHD, a result consistent with a previous study (25).
PHF2 Suppresses rDNA Transcription
Although PHF2 and PHF8 appear to exhibit distinct histone demethylase activities, both proteins have been shown to activate rDNA transcription (13, 17, 18). We therefore were interested in the underlying mechanisms by which these two closely related but distinct demethylases regulate rDNA transcription. Toward this end, we established conditions to efficiently deplete PHF2 or PHF8 in HeLa and HEK293T cells by transfecting siRNA against either PHF2 (siPHF2) or PHF8 (siPHF8). The specific knockdown upon 2 days of siRNA transfection was confirmed by Western blot analysis of the corresponding cellular extracts using PHF2- or PHF8-specific antibody (Fig. 2A). We then measured the effect of knockdown of PHF2 and PHF8 on pre-rRNA synthesis by qRT-PCR. Surprisingly, we observed that, whereas PHF8 knockdown resulted in reduced levels of pre-rRNA synthesis, PHF2 knockdown led to increased levels of pre-rRNA synthesis in both HeLa and HEK293T cells (Fig. 2B). This effect of siPHF2 on rDNA transcription is unlikely to be cell type-specific because it was also observed in HCT116 and A549 cells (Fig. 2C). To minimize the possibility that the observed effect on rDNA transcription is due to an off-target effect of siPHF2, we made use of two different shPHF2 constructs to knock down PHF2 in HeLa and HEK293T cells (Fig. 2D). Endogenous PHF2 was significantly depleted by transfection of these two shRNAs, as demonstrated by Western blot analysis (Fig. 2D). Note that neither shRNA affected the level of endogenous PHF8 proteins, suggesting that both shRNAs are highly specific to PHF2 (Fig. 2D). Subsequent qRT-PCR analysis confirmed that knockdown of PHF2 by either of these two shRNAs led to increased levels of pre-rRNA transcripts, and the degree of this increase correlated with the extent of knockdown by these two shRNAs (Fig. 2E). Thus, our RNA interference experiments revealed an opposite role for PHF2 and PHF8 in control of rDNA transcription. In support of this idea, we found that, although knockdown of PHF2 or PHF8 led to increased or decreased pre-rRNA synthesis, simultaneous knockdown of both PHF2 and PHF8 nearly restored the pre-rRNA synthesis to the level of the control cells (Fig. 2F).
FIGURE 2.

PHF2 inhibits rather than activates rDNA transcription. A, depletion of PHF2 and PHF8 by specific siRNAs. HeLa or HEK293T cells were transfected with siRNA against either PHF2 (siPHF2) or PHF8 (siPHF8) for 2 days, and the effect on the levels of endogenous PHF2 and PHF8 was determined by Western blot. Actin served as a loading control. siCtrl, nonspecific control siRNA. Concentration for each siRNA is 100 nm. B, Knockdown of PHF2 and PHF8 has opposite effects on rRNA transcription. HeLa or HEK293T cells were treated with control, siPHF2, or PHF8 as in A, and the effects on the levels of pre-rRNA transcripts were determined by quantitative RT-PCR analysis. Error bars, S.D.; n = 3. C, knockdown of PHF2 in HCT116 and A549 cells also led to increased rDNA transcription. HCT116 and A549 cells were transfected with siCtrl or siPHF2 for 2 days and then collected for Western blot analysis for PHF2 (left) or the levels of pre-rRNA transcripts by RT-PCR (right). The data are shown as levels of pre-rRNA transcription in siPHF2-treated cells relative to that of siCtrl-treated cells. Error bars, S.D.; n = 3. D, specific knockdown of PHF2 in HeLa or HEK293T cells by two distinct PHF2 shRNA constructs. HeLa or HEK293T cells were stably transfected with two different PHF2 shRNAs, ShPHF2-1 and ShPHF2-2, followed by Western blot analyses with antibodies against PHF2, PHF8, and β-actin. shCtrl, the control shRNA vector. E, knockdown of PHF2 with either of two distinct shRNAs led to increased expression of pre-rRNA. The relative levels of pre-rRNA in the HeLa and HEK293T cells stably transfected with shPHF2-1, shPHF2-2, or control shRNA were measured by qRT-PCR. Error bars, S.D.; n = 3. F, simultaneous knockdown of PHF2 and PHF8 nearly restored the normal level of rDNA transcription. HeLa cells were transfected with siCtrl, siPHF2, siPHF8, and siPHF2 + siPHF8, respectively, and the effect on the levels of PHF2 and PHF8 proteins was revealed by Western blot analysis (left), and the effect on rDNA transcription was determined by qRT-PCR analysis (right). Error bars, S.D.; n = 3.
PHD but Not JmjC Domain Is Crucial for PHF2 to Inhibit rDNA Transcription
To investigate how PHF2 represses rDNA transcription, we next tested whether overexpression of PHF2 is able to repress endogenous rDNA transcription in HEK293T cells and, if it does so, the role of its H3K4me3 binding activity and putative demethylase activity in repression. We found that ectopic expression of an increasing amount of FLAG-tagged PHF2 led to progressive repression of endogenous rDNA transcription (Fig. 3A). This repressive activity is probably dependent on binding of H3K4me3 because the repression was not observed for the PHF2 mutant with deletion of the PHD domain (Fig. 3A). The expression of both wild-type and PHD deletion mutant PHF2 was verified by Western blot analysis (Fig. 3A, bottom).
FIGURE 3.

PHF2 inhibits rDNA transcription in an H3K4me2/3 binding activity-dependent and demethylase activity-independent manner. A, HEK293T cells transfected with increasing amounts of FLAG-PHF2, FLAG-ΔPHF2, or control vector, and the effects on rDNA transcription were determined by qRT-PCR analysis (top). The expression of FLAG-PHF2 and FLAG was revealed by Western blot analysis using anti-FLAG antibody (bottom). Error bars, S.D.; n = 3. B, HEK293T cells were transfected with increasing amounts of FLAG-PHF2, FLAG-PHF2-M20A, and FLAG-PHF2-HD/AA, and the effects on rDNA transcription were determined by qRT-PCR analysis (top). The expression of FLAG-PHF2, FLAG-PHF2-M20A, and FLAG-PHF2-HD/AA mutants was verified by Western blot analysis with anti-FLAG antibody (bottom). Error bars, S.D.; n = 3. C, restoration of rDNA repression by ectopic expression of wild-type and HD/AA mutant PHF2 but not M20A mutant in shPHF2-2-expressing HEK293T cells. HEK293T cells were infected with lentiviral shPHF2-2, which is against the non-coding region of PHF2, to establish shPHF2-2 stable expressing cells. These cells were then transiently transfected with vector alone, FLAG-PHF2, FLAG-PHF2-M20A, or FLAG-PHF2-HD/AA, as indicated, and the effects on rDNA transcription were determined by qRT-PCR analysis. Error bars, S.D.; n = 3. The expression of transfected PHF2 and its mutants was verified by Western blot with anti-FLAG antibody. D, a BrUTP incorporation assay demonstrated that PHF2 inhibits rRNA transcription, and this inhibition depends on PHD rather than the JmjC domain. HeLa cells were transfected with GFP-PHF2, GFP-PHF2-M20A, or GFP-PHF2ΔJmjC, and 2 days after transfection, 2 mm BrUTP was added to the medium for 30 min. Cells were then immunostained with anti-BrU antibody and rhodamine-conjugated goat anti-mouse secondary antibody. The bright BrU dots observed in control cells correspond to highly actively transcribed nucleolus regions. The transfected GFP-PHF2 and its mutants were revealed by direct fluorescence.
Previous structural analysis indicated that PHF2 binds H3K4me3 through a hydrophobic cage formed with residues Tyr7, Tyr14, Met20, and Trp29 in its PHD domain (17). Because our in vitro pull-down assay demonstrated that M20A substitution inactivated the H3K4me3 binding activity of PHF2 (Fig. 1, E and F), we tested further the role of H3K4me3 binding activity of PHF2 in repression of rDNA transcription by using the M20A mutant PHF2. For testing the potential role of PHF2 demethylase activity in rDNA repression, we used the HD/AA mutant that has both His249 and Asp251 residues substituted with alanine. His249 and Asp251 are believed to be required for binding of Fe2+, and their mutation would inactivate PHF2 demethylase activity (25, 27). Note that in the peptide pull-down assay, this mutant maintained an intact H3K4me3 binding activity (Fig. 1E).
When increasing amounts of FLAG-PHF2, FLAG-PHF2-M20A, or FLAG-PHF2-HD/AA plasmids were transfected into HEK293T cells, we confirmed by Western blot analysis the increased expression of the corresponding proteins (Fig. 3B, bottom). Again, whereas increasing amounts of FLAG-PHF2 led to progressively decreased expression of rDNA (Fig. 3B), the PHF2-M20A mutant was much less active in repression of rDNA transcription. In fact, at a low level of M20A expression that is closer to the endogenous level of PHF2, essentially no repression of rDNA transcription was observed (Fig. 3B). On the other hand, the PHF2-HD/AA mutant exhibited a repression of rDNA transcription comparable with that of wild-type PHF2, indicating that the putative demethylase activity is not required for repression.
To test further the distinct role of H3K4me3 binding and demethylase activity in repression of rDNA transcription, we made use of a shRNA (shPHF2–2) that targets the 3′-untranslated region of PHF2 mRNA to knock down the endogenous PHF2 and then performed rescue experiments by expressing FLAG-tagged PHF2, PHF2-M20A, or PHF2-HD/AA mutant. Subsequent qRT-PCR analysis revealed that, whereas knockdown of PHF2 resulted in increased pre-rRNA expression, this elevated pre-RNA expression could be partially suppressed by ectopic expression of either wild-type PHF2 or PHF2-HD/AA mutant but not the PHF2-M20A mutant (Fig. 3C). Taken together, these data indicate that the repression of rDNA transcription by PHF2 is largely dependent on its H3K4me3 binding activity and surprisingly is independent of its demethylase activity.
To ascertain that PHF2 represses rather than activates rDNA transcription, we next employed an independent assay for rDNA transcription. Because rDNA transcription accounts for more than half of the total RNA synthesis in proliferating cells, incorporation of BrUTP by a short pulse labeling followed by immunofluorescent staining using anti-BrU antibody can be used to measure the relative synthesis rate of rRNA synthesis in nucleoli (38, 39). We thus transfected HeLa cells with plasmids encoding GFP-tagged PHF2, PHF2-M20A, or PHF2-ΔJmjC mutant and carried out BrUTP pulse labeling experiments. As shown in the top panel of Fig. 3D, we observed bright dotlike BrU signals that represented rRNA synthesis in the nucleolus in the untransfected control cells. However, the BrU incorporation, as measured by BrU dot sizes and intensity, was much reduced in the GFP-PHF2-expressing cells. Importantly, no significant difference in BrU incorporation was observed between the untransfected control and GFP-PHF2-M20A mutant-expressing cells (Fig. 3D, middle). Note that the BrU dots co-localized exactly with GFP-PHF2-M20A proteins, indicating that these dots indeed represented rRNA synthesis in nucleoli. On the other hand, much like the cells expressing the wild-type GFP-PHF2, the cells expressing GFP-PHF2-ΔJmjC also showed significantly reduced BrU incorporation. The same results were observed when 293T cells were used (data not shown). Together, our RT-PCR analysis and BrUTP incorporation experiments provide compelling evidence that PHF2 represses rDNA transcription in an H3K4me3 binding-dependent and demethylase activity-independent manner.
PHF2 Inhibits the Binding of PHF8 to rDNA Genes
We next wished to investigate the underlying mechanism for repression of rDNA transcription by PHF2. Because PHF2 plays a role opposite to that of PHF8 in regulating rDNA transcription (Fig. 2) and PHF8 regulates rDNA transcription in an H3K4me3 binding-dependent manner (13), we tested the hypothesis that PHF2 may repress rDNA transcription by competing with PHF8 for binding of rDNA promoter. In support of this hypothesis, it was reported previously that the PHD domain of PHF2 has an ∼4-fold higher affinity for H3K4me3 than that of PHF8 (25), which is also in agreement with our in vitro peptide pull-down data in Fig. 1F. To test this hypothesis, we first analyzed the association of PHF2 with various regions of rDNA genes (Fig. 4A) in HeLa cells by a ChIP assay. As a control for PHF2 antibody specificity, we performed the ChIP assay using PHF2 knockdown HeLa cells (Fig. 4B). This analysis revealed a substantial enrichment of PHF2 in the promoter region of the rDNA gene, although the association with other regions of rDNA genes was also detected (Fig. 4B). To test whether the observed PHF2 promoter enrichment is dependent on its H3K4me3 binding activity, we expressed FLAG-tagged PHF2 and PHF2-M20A mutant in HeLa cells and compared their association with rDNA promoter by ChIP assay using anti-FLAG antibody. As shown in Fig. 4C, the wild-type PHF2, but not the PHF2-M20A mutant, was found to associate with the rDNA promoter. This finding provides evidence that the observed PHF2 rDNA promoter enrichment is probably dependent on its H3K4me3 binding activity, a result that is also consistent with the observed H3K4me3 peak in the rDNA promoter (37).
In support of our hypothesis that PHF2 inhibits PHF8 for binding of rDNA promoter, we found that knockdown of PHF2 led to increased association of PHF8 with the rDNA genes, especially at the promoter region (Fig. 4D). Furthermore, knockdown of PHF2 also led to an increased association of Pol I transcription factor UBF at the promoter. Thus, our ChIP analyses provide evidence that PHF2 inhibits PHF8 for binding of the rDNA promoter, which in turn may suppress the association of UBF with rDNA genes and consequently the repression of rDNA transcription.
PHF2 Recruits SUV39H1
We also examined by ChIP assay how knockdown of PHF2 affected histone modifications in the rDNA promoter. We found that knockdown of PHF2 led to a moderate increase in H3K4me3 and acetylated histone H3 Lys9 and a decrease in H3K9me3, whereas the level of histone H3 did not show any significant change (Fig. 4E). The observed increase in H3K4me3 and decrease in H3K9me3 were consistent with elevated rDNA transcription upon knockdown of PHF2 but were unlikely to be the direct effect of PHF2 knockdown because PHF2 is not known for H3K4me3 demethylase activity, not to mention for promoting H3K9me3. We found that knockdown of PHF2 also led to decreased association of H3K9me2/3 methyltransferase SUV39H1 with the rDNA promoter (Fig. 4E). On the other hand, knockdown of PHF2 had no effect on the association of TIP5 (10), the large subunit of NoRC complex, with the rDNA promoter (Fig. 4E), suggesting that PHF2 is unlikely to inhibit rDNA transcription by influencing the repressive function of NoRC.
Because our ChIP assay revealed a potential role for PHF2 in recruiting SUV39H1 to the rDNA promoter (Fig. 4E), we next examined whether PHF2 interacts with SUV39H1. By a co-immunoprecipitation assay, we found that V5-SUV39H1 interacted with FLAG-PHF2 when both were ectopically expressed in 293T cells (Fig. 4F). Furthermore, the co-immunoprecipitation assay demonstrated that ectopically expressed Myc-SUV39H1 interacted with endogenous PHF2 but not PHF8 in 293T cells (Fig. 4G). Taken together, our data provide evidence that PHF2 is likely to repress rDNA transcription by at least the following two mechanisms: inhibiting the binding of coactivator PHF8 and recruiting the corepressor SUV39H1.
PHF2 May Act as a General Corepressor for RNA Pol II-transcribed Genes
PHF8 has not only been shown to activate rDNA transcription but also RARα and other transcription factors and is likely to serve as a general coactivator through its association with RNA polymerase II (21, 28, 29). Given our finding that PHF2 antagonizes PHF8 in regulating rDNA transcription, we were interested in testing whether PHF2 may also repress Pol II-transcribed genes. Using a luciferase reporter assay, we first tested the effect of PHF2 on RARα activated transcription. We found that expression of an increasing amount of FLAG-PHF2 progressively diminished the transcriptional activation from a 4xUAS-TK-Luc reporter driven by GAL4-RARα (Fig. 5A). This result is contrary to our previous observation that expression of an increasing amount of FLAG-PHF8 led to a gradually increased activation by GAL4-RARα from the same reporter (21). We next compared the effect of ectopic expression of PHF2 and PHF8 on transcriptional activation by transcription factors OCT4 and KLF4. As shown in Fig. 5, B and C, PHF8 enhanced transcriptional activation by OCT4 and KLF4 in a dose-dependent manner, whereas PHF2 repressed their transcriptional activation also in a dose-dependent manner. Moreover, to test whether the endogenous PHF2 also plays an opposite role to PHF8 in transcription, we transfected HEK293T cells with shRNA against PHF2 or PHF8 and quantitatively measured the levels of several known PHF8 target genes by qRT-PCR. We first confirmed by qRT-PCR that the levels of PHF2 and PHF8 mRNA were specifically depleted by their corresponding shRNAs (Fig. 5D). RT-PCR analysis for PHF8 target genes (29) revealed that, whereas, as expected, knockdown of PHF8 led to reduced expression of JARID1C, CDC40, and OCR5, knockdown of PHF2 resulted in increased expression of these genes (Fig. 5D). As an internal control, qRT-PCR analysis showed that knockdown of PHF2 and PHF8 led to opposite effects on rDNA transcription. Together, these data indicate that, contrary to PHF8, PHF2 is likely to serve as a general transcriptional corepressor.
FIGURE 5.

PHF2 and PHF8 play opposite roles in transcriptional regulation by RARα, OCT4, and KLF4. A, PHF2 inhibits transactivation by Gal4-RARα. HeLa cells were transfected with 125 ng of 4xUAS-TK-luc reporter and GAL4-RARα, together with either vector alone or 100, 200, or 300 ng of FLAG-PHF2. Two days after transfection, the cells were treated with or without 1 nm retinoic acid for 6 h and then subjected to a luciferase reporter assay and Western blot with FLAG and β-actin antibodies. Error bars, S.D.; n = 3. B, PHF2 inhibits and PHF8 enhances transactivation by OCT4. HeLa cells were transfected with 100 ng of IAP-luc alone or together with an increasing amount (25, 50, 100, and 200 ng) of FLAG-PHF2 (PHF2) or FLAG-PHF8 (PHF8). The pSG5-FLAG vector was used to maintain the total amount of DNA in each transfection as 300 ng. A luciferase assay was performed 24 h after transfection. Error bars, S.D.; n = 3. The expression of FLAG-PHF2, FLAG-PHF8, and FLAG-OCT4 was detected by Western blot using FLAG antibody. C, PHF2 inhibits and PHF8 enhances transactivation by KLF4. The experiments were essentially as in B, except the Rex1-luc reporter and FLAG-KLF4 plasmids were used. Error bars, S.D.; n = 3. D, PHF2 inhibits the transcription of PHF8 target genes. HEK293T cells were stably transfected with ShPHF2-1, ShPHF2-2, shPHF8, or control shRNA, and the expression of pre-rRNA and PHF2, PHF8, JARIDIC, CDC40, and OCRL mRNAs was measured by qRT-PCR. Error bars, S.D.; n = 3.
DISCUSSION
In an attempt to better understand how histone modifications regulate rDNA transcription, we investigated how PHF2 and PHF8, two related histone demethylases that are known to be enriched in the nucleolus, regulate rDNA transcription. We show that the nucleolus localization is independent of the H3K4me2/3 binding activity and demethylase activity of PHF2 (Fig. 1). It is of note that PHF2 seems to contain two separate sequence determinants for nucleolus localization (Fig. 1C). The presence of more than one such sequence determinant may contribute to the widespread nucleolus localization observed for PHF2 in various cells that we have tested (data not shown). Much like nucleolin proteins, digestion with RNase A rather than DNase I abrogated the PHF2 nucleolus localization (Fig. 1D), suggesting that the PHF2 nucleolus localization relies on direct or indirect association with rRNA rather than binding of DNA.
Through a proteomics approach, we first identified PHF2 as a H3K4me2/3-specific binding protein (40). Wen et al. (17) later elegantly demonstrated via both biochemical and crystallographic studies that the PHD finger of PHF2 is responsible for the recognition of H3K4me2/3 mark. They also showed that the H3K4me2/3-PHD interaction is essential for PHF2 occupancy of rDNA genes (17). Our data are consistent with their data in these aspects. However, our data differ from theirs in that PHF2 represses rather than activates rDNA transcription. To ascertain this unexpected result, we employed, in addition to qRT-PCR analysis of pre-rRNA transcripts, an independent BrUTP labeling assay. We observed in this assay that overexpression of PHF2 repressed BrUTP incorporation into the nucleolus (Fig. 3D). Furthermore, in agreement with our qRT-PCR analyses, the BrUTP labeling assay demonstrated that the H3K4me2/3 binding activity is essential for repression of rDNA genes by PHF2, whereas the demethylase activity is dispensable (Fig. 3C). Collectively, our data suggest that PHF2 represses rather than activates rDNA transcription. The reason for the discrepancy between previous data and our present data is not clear, but it may lie in differences in the analysis of pre-rRNA transcripts.
Our study demonstrates that PHF2 represses rDNA transcription independent of its putative demethylase activity because neither deletion of JmjC domain nor HD/AA mutation affects the repression activity (Fig. 3). In this regard, it is noteworthy that, in our hands, we could not detect H3K9me2 demethylase activity for PHF2, although a weak H3K9me1 demethylase activity could be inconsistently detected in comparison with PHF8. Thus, our data are consistent with the results of Horton et al. (25) and suggest that PHF2 may not possess or may possess only a low constitutive H3K9me1/2 demethylase activity. In this regard, it has been reported that the H3K9me2 demethylase activity of PHF2 requires a PKA-dependent phosphorylation (26).
Our study provides evidence that PHF2 may repress rDNA transcription at least in part by inhibiting PHF8 association with rDNA. In support of this notion, we showed that knockdown of PHF2 led to increased association of PHF8 with rDNA promoters (Fig. 4D). We also observed increased association of UBF with the rDNA promoters upon PHF2 knockdown (Fig. 4D), in agreement with an early report that PHF8 interacts with UBF and is likely to enhance rDNA transcription by promoting UBF binding (13). The fact that double knockdown of PHF2 and PHF8 nearly restores the level of rDNA transcription to that of control cells is also consistent with the idea that PHF2 represses rDNA transcription at least in part by antagonizing PHF8. It should be pointed out that we could not exclude the possibility that PHF2 may repress transcription by competing with other H3K4me2/3-binding proteins, including KIAA1718. Furthermore, PHF2 is unlikely to inhibit rDNA transcription by promoting the silencing chromatin structure through NoRC because we found that knockdown of PHF2 did not affect the association of TIP5, the large subunit of NoRC, with the rDNA promoter.
Our study also presents evidence that PHF2 may repress rDNA transcription in part by recruiting SUV39H1 (Fig. 4, E–G). We observed that knockdown of PHF2 led to reduced association of SUV39H1 and a diminished level of H3K9me3 at the rDNA promoters and that PHF2 and SUV39H1 interact with each other. Taken together, we proposed a working model in Fig. 6 that PHF2 is likely to repress rDNA transcription by competing with PHF8 for binding of the H3K4me3 mark in the rDNA promoter and thus antagonizing transcriptional activation by PHF8 and by recruiting SUV39H1, which presumably contributes to repression via catalyzing H3K9me2/3.
FIGURE 6.
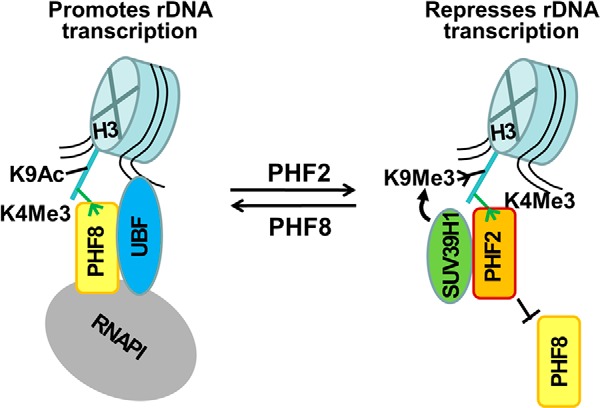
A working model on transcriptional repression by PHF2. PHF2 and PHF8 compete for binding of H3K4me3 mark. The binding of PHF8 enhances rDNA transcription in part by promoting the recruitment of UBF. The binding of PHF2 to the H3K4me3 in the rDNA promoter inhibits the binding of PHF8 to the same H3K4me3 mark, which results in reduced binding of UBF. PHF2 also recruits SUV39H1 to rDNA promoter. These two mechanisms are likely to work together to repress rDNA transcription. The same mechanisms may also apply to repression of Pol II target genes by PHF2. H3K4me3 and H3K9me3 are illustrated in short green and black lines, respectively, and the H3 tail is shown by a long blue line. Ac, acetylation. Me, methylation.
In addition to repressing rDNA transcription, our study also presents evidence that PHF2 may function as a general corepressor for transcription by RNA polymerase II. In contrast to PHF8, which enhances transcription by RARα, OCT4, and KLF4, PHF2 represses transcriptional activation by these transcription factors in reporter assays (Fig. 5). Furthermore, whereas knockdown of PHF8 led to reduced expression of a few known PHF8 target genes, including JARID1C, CDC40, and OCR5 (28, 29), knockdown of PHF2 resulted in elevation of their expression (Fig. 5D). Thus, whereas PHF2 has been reported to activate transcription in various contexts through its associated H3K9me2 or H4K20me3 demethylase activities (26, 27), our data raise the possibility that PHF2 may serve as a general transcription corepressor that is independent of its demethylase activity, presumably through antagonizing PHF8/KIAA1718 or other H3K4me2/3-binding proteins and recruiting SUV39H1. Future work is necessary to further investigate the exact function of PHF2 in transcriptional regulation and the underlying mechanisms.
Acknowledgments
We thank Dr. Shuo Dong for kindly providing FLAG-PHF8 and Gal4-RARα and Dr. Philippe Bouvet for the nucleolin antibody.
This study was supported by Ministry of Science and Technology of China Grant 2010CB944903; National Natural Science Foundation of China Grants 90919025, 30871381, 81272287, and 31371504; and Science Technology Commission of Shanghai Municipality Grants 09DJ1400400 and 11DZ2260300.
- Pol I
- RNA polymerase I
- Pol II
- RNA polymerase II
- JmjC
- Jumonji C
- PHD
- plant homeodomain
- RARα
- retinoic acid receptor α
- UBF
- upstream binding factor
- BrU
- bromouridine
- BrUTP
- 5-bromouridine 5′-triphosphate
- HD/AA
- H249A/D251A mutant
- H3K4me2/3
- di-/trimethylated histone 3 lysine 4
- H3K9me1/2/3
- mono-/di-/trimethylated histone 3 lysine 9.
REFERENCES
- 1. Prieto J. L., McStay B. (2005) Nucleolar biogenesis: the first small steps. Biochem. Soc. Trans. 33, 1441–1443 [DOI] [PubMed] [Google Scholar]
- 2. McStay B., Grummt I. (2008) The epigenetics of rRNA genes: from molecular to chromosome biology. Annu. Rev. Cell Dev. Biol. 24, 131–157 [DOI] [PubMed] [Google Scholar]
- 3. Russell J., Zomerdijk J. C. (2006) The RNA polymerase I transcription machinery. Biochem. Soc. Symp. 203–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beckmann H., Chen J. L., O'Brien T., Tjian R. (1995) Coactivator and promoter-selective properties of RNA polymerase I TAFs. Science 270, 1506–1509 [DOI] [PubMed] [Google Scholar]
- 5. Schnapp A., Grummt I. (1991) Transcription complex formation at the mouse rDNA promoter involves the stepwise association of four transcription factors and RNA polymerase I. J. Biol. Chem. 266, 24588–24595 [PubMed] [Google Scholar]
- 6. Tuan J. C., Zhai W., Comai L. (1999) Recruitment of TATA-binding protein-TAFI complex SL1 to the human ribosomal DNA promoter is mediated by the carboxy-terminal activation domain of upstream binding factor (UBF) and is regulated by UBF phosphorylation. Mol. Cell Biol. 19, 2872–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McKeown P. C., Shaw P. J. (2009) Chromatin: linking structure and function in the nucleolus. Chromosoma 118, 11–23 [DOI] [PubMed] [Google Scholar]
- 8. Grummt I., Längst G. (2013) Epigenetic control of RNA polymerase I transcription in mammalian cells. Biochim. Biophys. Acta 1829, 393–404 [DOI] [PubMed] [Google Scholar]
- 9. Santoro R., Li J., Grummt I. (2002) The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat. Genet. 32, 393–396 [DOI] [PubMed] [Google Scholar]
- 10. Zhou Y., Santoro R., Grummt I. (2002) The chromatin remodeling complex NoRC targets HDAC1 to the ribosomal gene promoter and represses RNA polymerase I transcription. EMBO J. 21, 4632–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li J., Längst G., Grummt I. (2006) NoRC-dependent nucleosome positioning silences rRNA genes. EMBO J. 25, 5735–5741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xie W., Ling T., Zhou Y., Feng W., Zhu Q., Stunnenberg H. G., Grummt I., Tao W. (2012) The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl. Acad. Sci. U.S.A. 109, 8161–8166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feng W., Yonezawa M., Ye J., Jenuwein T., Grummt I. (2010) PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nat. Struct. Mol. Biol. 17, 445–450 [DOI] [PubMed] [Google Scholar]
- 14. Muth V., Nadaud S., Grummt I., Voit R. (2001) Acetylation of TAF(I)68, a subunit of TIF-IB/SL1, activates RNA polymerase I transcription. EMBO J. 20, 1353–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murayama A., Ohmori K., Fujimura A., Minami H., Yasuzawa-Tanaka K., Kuroda T., Oie S., Daitoku H., Okuwaki M., Nagata K., Fukamizu A., Kimura K., Shimizu T., Yanagisawa J. (2008) Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133, 627–639 [DOI] [PubMed] [Google Scholar]
- 16. Yang L., Song T., Chen L., Kabra N., Zheng H., Koomen J., Seto E., Chen J. (2013) Regulation of SirT1-nucleomethylin binding by rRNA coordinates ribosome biogenesis with nutrient availability. Mol. Cell Biol. 33, 3835–3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wen H., Li J., Song T., Lu M., Kan P. Y., Lee M. G., Sha B., Shi X. (2010) Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J. Biol. Chem. 285, 9322–9326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu Z., Wang Y., Li X., Wang Y., Xu L., Wang X., Sun T., Dong X., Chen L., Mao H., Yu Y., Li J., Chen P. A., Chen C. D. (2010) PHF8 is a histone H3K9me2 demethylase regulating rRNA synthesis. Cell Res. 20, 794–801 [DOI] [PubMed] [Google Scholar]
- 19. Fortschegger K., Shiekhattar R. (2011) Plant homeodomain fingers form a helping hand for transcription. Epigenetics 6, 4–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suganuma T., Workman J. L. (2010) Features of the PHF8/KIAA1718 histone demethylase. Cell Res. 20, 861–862 [DOI] [PubMed] [Google Scholar]
- 21. Qiu J., Shi G., Jia Y., Li J., Wu M., Li J., Dong S., Wong J. (2010) The X-linked mental retardation gene PHF8 is a histone demethylase involved in neuronal differentiation. Cell Res. 20, 908–918 [DOI] [PubMed] [Google Scholar]
- 22. Horton J. R., Upadhyay A. K., Qi H. H., Zhang X., Shi Y., Cheng X. (2010) Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 17, 38–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qi H. H., Sarkissian M., Hu G. Q., Wang Z., Bhattacharjee A., Gordon D. B., Gonzales M., Lan F., Ongusaha P. P., Huarte M., Yaghi N. K., Lim H., Garcia B. A., Brizuela L., Zhao K., Roberts T. M., Shi Y. (2010) Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 466, 503–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu W., Tanasa B., Tyurina O. V., Zhou T. Y., Gassmann R., Liu W. T., Ohgi K. A., Benner C., Garcia-Bassets I., Aggarwal A. K., Desai A., Dorrestein P. C., Glass C. K., Rosenfeld M. G. (2010) PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 466, 508–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Horton J. R., Upadhyay A. K., Hashimoto H., Zhang X., Cheng X. (2011) Structural basis for human PHF2 Jumonji domain interaction with metal ions. J. Mol. Biol. 406, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baba A., Ohtake F., Okuno Y., Yokota K., Okada M., Imai Y., Ni M., Meyer C. A., Igarashi K., Kanno J., Brown M., Kato S. (2011) PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat. Cell Biol. 13, 668–675 [DOI] [PubMed] [Google Scholar]
- 27. Stender J. D., Pascual G., Liu W., Kaikkonen M. U., Do K., Spann N. J., Boutros M., Perrimon N., Rosenfeld M. G., Glass C. K. (2012) Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol. Cell 48, 28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fortschegger K., de Graaf P., Outchkourov N. S., van Schaik F. M., Timmers H. T., Shiekhattar R. (2010) PHF8 targets histone methylation and RNA polymerase II to activate transcription. Mol. Cell Biol. 30, 3286–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kleine-Kohlbrecher D., Christensen J., Vandamme J., Abarrategui I., Bak M., Tommerup N., Shi X., Gozani O., Rappsilber J., Salcini A. E., Helin K. (2010) A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol. Cell 38, 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yatim A., Benne C., Sobhian B., Laurent-Chabalier S., Deas O., Judde J. G., Lelievre J. D., Levy Y., Benkirane M. (2012) NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Mol. Cell 48, 445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Okuno Y., Ohtake F., Igarashi K., Kanno J., Matsumoto T., Takada I., Kato S., Imai Y. (2013) Epigenetic regulation of adipogenesis by PHF2 histone demethylase. Diabetes 62, 1426–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang H., Ma Y., Gu J., Liao B., Li J., Wong J., Jin Y. (2012) Reprogramming of somatic cells via TAT-mediated protein transduction of recombinant factors. Biomaterials 33, 5047–5055 [DOI] [PubMed] [Google Scholar]
- 33. Ben-Shushan E., Thompson J. R., Gudas L. J., Bergman Y. (1998) Rex-1, a gene encoding a transcription factor expressed in the early embryo, is regulated via Oct-3/4 and Oct-6 binding to an octamer site and a novel protein, Rox-1, binding to an adjacent site. Mol. Cell Biol. 18, 1866–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Evans P. M., Zhang W., Chen X., Yang J., Bhakat K. K., Liu C. (2007) Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J. Biol. Chem. 282, 33994–34002 [DOI] [PubMed] [Google Scholar]
- 35. Li J., Chu M., Wang S., Chan D., Qi S., Wu M., Zhou Z., Li J., Nishi E., Qin J., Wong J. (2012) Identification and characterization of nardilysin as a novel dimethyl H3K4-binding protein involved in transcriptional regulation. J. Biol. Chem. 287, 10089–10098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karagianni P., Amazit L., Qin J., Wong J. (2008) ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol. Cell Biol. 28, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cong R., Das S., Ugrinova I., Kumar S., Mongelard F., Wong J., Bouvet P. (2012) Interaction of nucleolin with ribosomal RNA genes and its role in RNA polymerase I transcription. Nucleic Acids Res. 40, 9441–9454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haukenes G., Kalland K. H. (1998) Visualization of ribosomal RNA (rRNA) synthesis in eukaryotic cells in culture. Methods Cell Sci. 19, 295–302 [Google Scholar]
- 39. Wei X., Somanathan S., Samarabandu J., Berezney R. (1999) Three-dimensional visualization of transcription sites and their association with splicing factor-rich nuclear speckles. J. Cell Biol. 146, 543–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chan D. W., Wang Y., Wu M., Wong J., Qin J., Zhao Y. (2009) Unbiased proteomic screen for binding proteins to modified lysines on histone H3. Proteomics 9, 2343–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]