

Background: The human 2-oxoglutarate dehydrogenase complex, comprising E1o, E2o, and E3 components, catalyzes conversion of 2-oxoglutarate to succinyl-CoA.
Results: Human E1o generates both a thiamin-enamine-derived radical and the reactive oxygen species, superoxide, and hydrogen peroxide.
Conclusion: Human E1o produces reactive oxygen species at a rate of <1% of succinyl-CoA under physiological conditions.
Significance: This work presents the novel discovery that the 5-carboxyl group affects enzymatic reactivity of 2-oxoglutarate.
Keywords: Dehydrogenase, Electron Paramagnetic Resonance (EPR), Mass Spectrometry (MS), Reactive Oxygen Species (ROS), Thiamin
Abstract
Herein are reported unique properties of the human 2-oxoglutarate dehydrogenase multienzyme complex (OGDHc), a rate-limiting enzyme in the Krebs (citric acid) cycle. (a) Functionally competent 2-oxoglutarate dehydrogenase (E1o-h) and dihydrolipoyl succinyltransferase components have been expressed according to kinetic and spectroscopic evidence. (b) A stable free radical, consistent with the C2-(C2α-hydroxy)-γ-carboxypropylidene thiamin diphosphate (ThDP) cation radical was detected by electron spin resonance upon reaction of the E1o-h with 2-oxoglutarate (OG) by itself or when assembled from individual components into OGDHc. (c) An unusual stability of the E1o-h-bound C2-(2α-hydroxy)-γ-carboxypropylidene thiamin diphosphate (the “ThDP-enamine”/C2α-carbanion, the first postdecarboxylation intermediate) was observed, probably stabilized by the 5-carboxyl group of OG, not reported before. (d) The reaction of OG with the E1o-h gave rise to superoxide anion and hydrogen peroxide (reactive oxygen species (ROS)). (e) The relatively stable enzyme-bound enamine is the likely substrate for oxidation by O2, leading to the superoxide anion radical (in d) and the radical (in b). (f) The specific activity assessed for ROS formation compared with the NADH (overall complex) activity, as well as the fraction of radical intermediate occupying active centers of E1o-h are consistent with each other and indicate that radical/ROS formation is an “off-pathway” side reaction comprising less than 1% of the “on-pathway” reactivity. However, the nearly ubiquitous presence of OGDHc in human tissues, including the brain, makes these findings of considerable importance in human metabolism and perhaps disease.
Introduction
The 2-oxo acid dehydrogenase multienzyme complexes have roles at key junctions in human metabolism (1). Whereas the pyruvate dehydrogenase complex (PDHc)3 produces acetyl-CoA at the entry to the citric acid cycle, the 2-oxoglutarate dehydrogenase complex (OGDHc; also known as α-ketoglutarate dehydrogenase complex) is a rate-limiting enzyme in the citric acid cycle and produces succinyl-CoA by oxidative decarboxylation of 2-oxoglutarate (OG; also known as α-ketoglutarate). The OGDHc is a multienzyme complex that contains multiple copies of three component enzymes (Scheme 1): a thiamin diphosphate (ThDP)-dependent 2-oxoglutarate dehydrogenase (E1o; EC 1.2.4.2), dihydrolipoylsuccinyl transferase (E2o; EC 2.3.1.61), and dihydrolipoyl dehydrogenase (E3; EC 1.8.1.4). The E1o and E2o carry out the principal reactions for succinyl-CoA formation, whereas the E3 reoxidizes dihydrolipoamide-E2o to lipoamide-E2o, the redox cofactor covalently amidated onto a single lysine residue of the E2o components. The OGDHc catalyzes one of the rate-limiting steps in the citric acid cycle (1–3), which is the common pathway for oxidation of fuel molecules, including carbohydrates, fatty acids, and amino acids. Reduced activities of key citric acid cycle enzymes, including OGDHc activity, has been linked to neurodegenerative disorders, such as Alzheimer and Parkinson disease and oxidative damage (4–6).
SCHEME 1.

Mechanism of OGDHc with alternative fates of the enamine.
Functionally competent E1o-h and E2o-h components were created for the first time. There are no structures for these components or, for that matter, for any intact E2 component from any source. Structures are available for the E3 component (7–12) and for N-terminally truncated (some 77 amino acids) Escherichia coli 2-oxoglutarate dehydrogenase (E1o-ec) (13). The availability of the human E1o and E2o components enabled us to characterize longer-lived ThDP-bound intermediates on E1o-h.
Although typical ThDP-assisted decarboxylation mechanisms are ionic in nature, there are important known exceptions; (a) pyruvate-ferredoxin oxidoreductase (14, 15) utilizes three tandem Fe4S4 clusters to oxidize the enamine intermediate to acetyl-CoA, and (b) pyruvate oxidase produces acetate in E. coli and acetyl phosphate in Lactobacillus plantarum, and both pyruvate-ferredoxin oxidoreductase and pyruvate oxidase (16) have been shown to support a postdecarboxylation-related radical that has spin density not only at the C2α atom but also around the thiazolium ring (henceforth referred to as the ThDP-enamine radical) (14–17). The observation of a similar radical on E1o-ec (18) raised the possibility that such a radical also exists on the E1o-h. There have been several reports that mammalian OGDHc could be a major source of the reactive oxygen species (ROS), superoxide radical anion and hydrogen peroxide, in isolated brain and skeletal muscle mitochondria (5, 19) and in the isolated synaptosomes (6), as was also confirmed with the isolated mammalian OGDHc (5, 6). Based on experiments with spin-trapping techniques and using the isolated mammalian OGDHc with a modified flavin cofactor of E3, it was concluded that E3-bound FAD is responsible for the production of radical species by OGDHc (20). The above conclusions were supported further by experiments with brain mitochondria isolated from E3-deficient mice that produce less H2O2 as compared with wild-type mice (5). Based on these studies, ROS generation by OGDHc was attributed to the E3 component (6, 21–24). It has been documented that OGDHc could produce H2O2 in the forward physiological reaction by oxidation of OG in the presence CoA and in the reverse reaction at high NADH/NAD+ ratio (no OG is needed). With the E3 component being shared by OGDHc, PDHc, and branched-chain 2-oxo acid dehydrogenase complex, OGDHc was reported to produce H2O2 at about twice the rate produced by PDHc and 4 times the rate produced by branched-chain 2-oxo acid dehydrogenase complex in isolated skeletal muscle mitochondria, and the rates were dependent on concentration of their specific substrates rather than on NADH/NAD ratio (19).
At the same time, it has recently been suggested that isolated E1 components of the OGDHc and PDHc could provide intermediates leading to ROS production (19). With individually expressed human E1o, E2o, and E3, we have an opportunity to investigate the redox chemistry on human OGDHc. We here demonstrate that E1o-h gives rise to a stable ThDP-derived radical corresponding to the C2(2α-hydroxy)-γ-carboxypropylidenethiamin diphosphate cation radical (ThDP-enamine radical), also detected on pyruvate oxidase and pyruvate-ferredoxin oxidoreductase and on E1o-ec earlier by electron paramagnetic resonance (EPR) spectroscopy (18). Circular dichroism (CD) and UV-visible spectroscopic evidence was found for a stable E1o-h-bound “enamine”/C2α-carbanion (the first ThDP-bound postdecarboxylation intermediate in Scheme 1), C2-(2α-hydroxy)-γ-carboxypropylidenethiamin diphosphate (ThDP-enamine), thereby completing the description of the chemistry in Reaction 1.
REACTION 1.

The relatively stable enzyme-bound enamine is the likely substrate for oxidation by O2, leading to the generation of superoxide anion and ThDP-derived radical.
The results indicate that E1o-h does produce both the ThDP-enamine radical and superoxide/H2O2. Generation of superoxide/H2O2 by E1o-h may have pathological relevance, especially under conditions of genetic deficiencies of the E2o-h due to mutations (25), polymorphism (26), or down-regulation (27, 28). In cultured human embryonic kidney cells, the reduction in E2o-h level augmented ROS generation and cell death (27). E2o-h deficiency also elevated the amyloid plaque burden, the levels of Aβ oligomers and nitrotyrosine, and the occurrence of spatial learning and memory deficits in mice (29), whereas decreased OGDHc activity in general and resultant prolonged oxidative stress are known to be part of the pathogenesis in Alzheimer disease (30–32).
Although the E3 component is common to all 2-oxo acid dehydrogenase complexes in a particular cell type, the chemistry on E1o is apparently significantly different from that on the PDHc. The findings with both human and bacterial E1os place on firm ground the dramatic differences between the pyruvate and OG-derived intermediates on ThDP, and all point to and are consistent with a role of the side chain carboxyl group (carbon 5) in the series of reactions taking place on ThDP.
EXPERIMENTAL PROCEDURES
Reagents
ThDP, NAD+, CoA, DTT, dl-α-lipoic acid, isopropyl 1-thio-β-d-galactopyranoside, thiamin·HCl, and benzamidine·HCl were from U.S. Biochemical Corp. 2,6-Dichlorophenolindophenol (DCPIP) was from Sigma-Aldrich, and Amplex Red fluorescent dye was from Molecular Probes, Inc. (Eugene, OR).
Protein Expression and Purification
Expression and purification of the E. coli E1o-ec, E2o-ec, and E3-ec was as reported earlier (33).
Construction of Plasmid and Expression and Purification of E1o-h and E2o-h
The genes encoding His6-tagged E1o-h and His6-tagged E2o-h were synthesized by DNA2.0, Inc. (Menlo Park, CA). The E1o-h gene optimized for expression in E. coli cells and the E2o-h gene were inserted into pET-22b(+) and pET-15b vectors, respectively, and the resulting plasmids were expressed in BL21 (DE3) cells. Cells were grown in LB medium supplemented with 50 μg/ml ampicillin containing 1 mm MgCl2 and 0.5 mm thiamin for E1o-h expression or 0.3 mm dl-α-lipoic acid for E2o-h expression. Protein expression was induced by 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 5 h at 37 °C for E2o-h and for 15 h at 25 °C for E1o-h. Cells were washed with 0.20 m KH2PO4 (pH 7.0) containing 0.15 m NaCl and stored at −20 °C. The harvested cells were dissolved in 50–70 ml of 50 mm KH2PO4 (pH 7.5) containing 0.3 m KCl, 2.0 mm ThDP, 5.0 mm MgCl2, and one protease inhibitor mixture tablet (Roche Applied Science). Cells were treated with lysozyme (0.60 mg/ml) at 4 °C for 20 min and were disrupted using a sonic dismembrator. The clarified lysate was loaded onto a nickel-Sepharose column (GE Healthcare) equilibrated with 50 mm KH2PO4 containing 0.30 m KCl, 0.50 mm ThDP, 1.0 mm MgCl2, and 30 mm imidazole (pH 7.5). The E1o-h was eluted with 300 mm imidazole in the same buffer. The E1o-h was dialyzed against 50 mm KH2PO4 (pH 7.5) containing 0.30 m NaCl, 0.50 mm ThDP, 2.0 mm MgCl2, and 1.0 mm benzamidine·HCl at 4 °C for 15 h. The protein was concentrated, and buffer was exchanged to 50 mm HEPES (pH 7.5) containing 0.15 m NaCl, 0.50 mm ThDP, 1.0 mm MgCl2, and 1 mm benzamidine·HCl and was stored at −20 °C. The His6-tagged E2o-h was purified in a manner similar to that reported for E2p-ec (34).
Expression and Purification of E3-h
For expression and purification of E3h, E. coli M15 cells were used as well as pQE-9 vector with the gene encoding E3-h as reported earlier (35) with some modifications. Cells were grown in LB medium supplemented with 100 μg/ml ampicillin, and E3-h expression was induced by 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 25 °C for 15 h. E3-h was purified using a nickel-Sepharose column (GE Healthcare). The protein was bound to the nickel-Sepharose column in the presence of 30 mm imidazole in 50 mm KH2PO4 (pH 7.5) containing 0.3 m KCl and was eluted with 300 mm imidazole in 50 mm KH2PO4 (pH 7.5) containing 0.3 m KCl. Protein was concentrated, and the buffer was exchanged to 50 mm KH2PO4 (pH 7.5) containing 0.20 m KCl, 1.0 mm benzamidine·HCl, and 25 μm FAD. The protein was stored at −80 °C.
Enzyme Activity Measurements; Overall Activity upon Reconstitution of OGDHc-h from Individual Components
For reconstitution of OGDHc-h from its individual components, E1o-h (0.13 mg; 4.6 μm subunits) was incubated for 30 min with E2o-h (0.65 mg; 60 μm subunits) and E3-h (0.65 mg; 52 μm subunits) with a mass ratio (μg/μg/μg) of 1:5:5 in 0.25 ml of 50 mm KH2PO4 (pH 7.5) containing 0.50 mm ThDP, 1.0 mm MgCl2, 0.15 m NaCl, and 1.0 mm benzamidine·HCl at 25 °C. A 0.010-ml aliquot of the reaction mixture was withdrawn after a 1-h incubation to start the reaction. The stoichiometry of components used for activity measurement was chosen from kinetic experiments where, at constant concentrations of E1o-h subunits (0.046 μm) and E3-h subunits (0.52 μm), the concentration of E2o-h subunits was varied (0.059–0.80 μm), leading to S0.5,E2o-h = 0.18 ± 0.08 μm. In a similar experiment with varied concentrations of E3-h subunits (0.026–0.53 μm) and constant concentrations of E1o-h (0.046 μm) and E2o-h (0.24 μm) subunits, an S0.5,E3-h of 0.17 ± 0.02 μm was calculated. The reaction medium contained the following in 1.0 ml: 0.10 m Tris-HCl (pH 7.5), 0.50 mm ThDP, 2.0 mm MgCl2, 2.0 mm OG, 2.0 mm DTT, 5 mm NAD+, 0.1–0.20 mm CoA at 37 °C. The reaction was initiated by the addition of CoA and OGDHc. Steady-state velocities were taken from the linear portion of the progress curve. One unit of activity is defined as the amount of NADH produced (μmol·min−1·mg E1o−1).
E1o-h Specific Activity
The E1o-h specific activity was measured in the reaction medium with the external oxidizing agent DCPIP (36) at 600 nm in the following reaction medium contained in 1 ml: 50 mm KH2PO4 (pH 7.0), 0.50 mm ThDP, 1.0 mm MgCl2, 2 mm OG, and DCPIP (0.08 mm) at 37 °C. The reaction was initiated by the addition of 0.01–0.015 mg of E1o-h. Steady-state velocities were taken from the linear portion of the progress curve recorded at 600 nm. One unit of activity is defined as the amount of reduced DCPIP produced (μmol·min−1·mg E1o-h−1).
Superoxide Production by E1o-h
Superoxide production by E1o-h was measured via reduction of acetylated cytochrome c (cyt c), as described earlier, with minor modifications, at pH 6.3, where the sensitivity of the method was optimal (22, 37–40). The initial reaction velocity was measured and converted to generated superoxide amount (nmol)·min−1·mg E1o-h−1, taking into account that the stoichiometry of the reaction between cyt c and superoxide is 1:1 (37). Assay conditions were the following in a 200-μl final volume in a microtiter plate-based experiment: 50 mm KH2PO4 (pH 6.3), 0.2 mm ThDP, 1 mm Mg2+, 0.1 mm CoA, 2 mm OG, 50 μm cyt c (all in final concentrations), and 64.44 μg of E1o-h. The concentration of cyt c was chosen to determine a linear dynamic range for the measurements (22, 37). Protein concentration was determined by the Bradford method (41). The reaction was initiated by the addition of OG (10 μl, 5%) after a 15-min incubation at 37 °C and after the baseline was stabilized. Measurements were carried out at a λmax of 550 nm of the reduced form of acetylated cyt c in enzyme immunoassay/radioimmune assay 96-well, flat-bottom, polystyrene plates (Corning Inc.) using a Spectramax M2 (Molecular Devices, Sunnyvale, CA) or a Victor 3 (PerkinElmer Life Sciences) multilabel counter spectrophotometer/fluorimeter. An extinction coefficient of 10,170 m−1 cm−1 (λmax of 550 nm) was determined for the fully reduced cyt c by sodium dithionite under the assay conditions. The superoxide dismutase from bovine erythrocytes was used to verify that the cyt c reduction was indeed caused by superoxide generated by the E1o-h; 100 units of superoxide dismutase was added to 200 μl of reaction mixture, where applied.
H2O2 Production by E1o-h
To detect H2O2 generated by E1o-h, the Amplex Red fluorescent dye (Molecular Probes) was used as reported earlier (6, 21, 42). In the presence of horseradish peroxidase (HRP), the Amplex Red reacts with H2O2 with a 1:1 stoichiometry, producing highly fluorescent resorufin. Fluorescence was detected in a Photon Technology International (Lawrenceville, NJ) Deltascan fluorescence spectrophotometer; the excitation wavelength was 550 nm, and the fluorescence emission was detected at 585 nm. A calibration curve was generated with known amounts of H2O2 at the end of each experiment. The reaction was initiated by the addition of the E1o-h or OG after a 15-min incubation at 37 °C of Amplex Red and HRP and after the stabilization of the fluorescence signal, generally 100–200 s after the addition of all required components. For calculation of the reaction rate, slopes of the curves detected in the absence of E1o-h were subtracted from the initial slopes measured in the presence of both E1o-h and OG. The reaction assay in 2 ml of 50 mm KH2PO4 (pH 6.3 or 7.3) contained the following components at the final concentrations: 1 unit of HRP, 1 μm Amplex Red, 0.1 mm EGTA, 0.2 mm ThDP, 0.4 mm ADP, 1 mm Mg2+, 0.12 mm CoA, 1 mm OG, and 64.4 μg of E1o-h, at 37 °C under continuous stirring. Statistical differences were evaluated using the Excel program with two-tailed Student's t tests assuming unequal variances and were accepted to be significant when p < 0.05.
Stopped-flow CD Spectroscopy
Kinetic traces were recorded on a Pi*-180 stopped-flow CD spectrometer (Applied Photophysics) using a 10-mm path length. The intermediates seen on steady-state CD at ∼298 and ∼352 nm were detected on stopped-flow CD at 297 and 365 nm, respectively, as dictated by the high sensitivity of the lamp at those wavelengths. Data from six or seven repetitive shots were averaged and fit to the appropriate equations (see the figure legends). For pre-steady-state rate determination of ThDP-bound intermediates, E1o-ec (38 μm concentration of active centers) in one syringe was mixed in the reaction cell with OG (4 mm) from the second syringe, and the reaction was monitored at 297 or 365 nm for 50 s at 20 °C. For CD experiments at 365 nm in the absence and presence of enamine acceptors, E1o-ec (38 μm concentration of active centers) from one syringe was mixed with OG (200 μm) placed in the second syringe, and the reaction was monitored for 50 s at 20 °C. The alternative enamine acceptors, E2o-ec(1–176) didomain (60 μm), DCPIP (50 μm), and glyoxylate (GA, 2 mm) were placed in the second syringe together with OG.
Rapid Chemical Quench and NMR Methods
The rapid reaction mixing experiments were carried out using a rapid chemical quench apparatus (Kintek RQF-3 model, Kintek Corp.), and NMR spectra were acquired on a Varian INOVA 600-MHz instrument using gradient carbon homonuclear single quantum coherence (gCHSQC; to enable observation of only those protons attached to 13C nuclei (43)). The water signal was suppressed by presaturation. For reconstitution with [C2,C6′-13C2]ThDP, the E1o-ec (0.12 mm, free of ThDP) was incubated with 1 eq of [C2,C6′-13C2]ThDP (0.12 mm) and 5 mm MgCl2 for 30 min on ice. [C2,C6′-13C2]ThDP-labeled E1o-ec was loaded into syringe A, and OG was loaded into syringe B in a rapid chemical quench apparatus. A quench solution (12.5% TCA in 1 m DCl/D2O) was loaded into syringe C. Enzyme and OG solutions were mixed rapidly in a 1:1 volume ratio by a computer-controlled drive-plate and incubated for predetermined times in a reaction loop. After this incubation period, the reaction mixture was mixed rapidly with quench solution from syringe C, and the resulting mixture was collected. For longer time scales (>20 s), the samples were assembled similarly into three different solutions. Next, equal volumes of [C2,C6′-13C2]ThDP-labeled E1o-ec (200 μl) and OG (200 μl) were mixed in an Eppendorf tube, and after predetermined times, the reaction was stopped by the addition of quench solution (200 μl). The contents of the syringes in 20 mm KH2PO4 (pH 7.4) were varied to monitor the following different reactions. (a) For E1o-ec with OG single turnover, syringe A contained [C2,C6′-13C2]ThDP-labeled E1o-ec (115 μm concentration of active centers), and syringe B contained OG (20 mm). The reaction was performed for a time period of 10–2000 ms. (b) For the E1o-ec and DCPIP reductase reaction, syringe A contained [C2,C6′-13C2]ThDP labeled E1o-ec (115 μm concentration of active centers), and syringe B contained OG (20 mm) and DCPIP (8 mm). The reaction was performed for 10–200 ms. (c) For the E1o-ec and succinic semialdehyde (SSA) reverse reaction, syringe A contained [C2,C6′-13C2]ThDP-labeled E1o-ec (115 μm concentration of active centers), and syringe B contained SSA (20 mm).
The quenched reaction mixture was collected and centrifuged at 15,700 × g for 20 min, the supernatant was separated and filtered through Gelman 0.45 μm discs, and then the filtrate was analyzed by gCHSQC NMR at 25 °C. Using [C2,C6′-13C2]ThDP in conjunction with the gCHSQC method enables observation of only those protons bound to 13C atoms, very useful for intermediate detection in the aromatic spectral region. The interpretation of NMR spectra was carried out as with pyruvate earlier (43).
Electron Paramagnetic Resonance Spectroscopy
For sample preparation, the following protocol was used: (a) E1o-h (50 mg/ml concentration of active centers = 0.449 mm) in 0.4 ml of 50 mm HEPES (pH 7.5) containing 0.15 m NaCl, 0.50 mm ThDP, and 1.0 mm MgCl2 was mixed with 10 mm OG at room temperature. The mixture was immediately transferred into an EPR tube and was flash-frozen in liquid nitrogen within 40 s of mixing all components. (b) E1o-ec (0.150 mm concentration of subunits) in 0.20 ml of 50 mm Tris-HCl (pH 7.4) containing 2 mm ThDP, 2 mm MgCl2, and 10% glycerol was mixed with 10 mm OG or 2-oxoadipate at room temperature and was transferred into an EPR tube. (c) For OGDHc-h assembly, E1o-h, E2o-h, and E3-h were mixed in 50 mm HEPES (pH 7.5) containing 0.50 mm ThDP, 1.0 mm MgCl2, and 0.15 m NaCl with a 0.222 mm concentration of subunits for each protein (ratio of E1o-h/E2o-h/E3-h subunits = 1:1:1). The reaction was started by the addition of 10 mm OG. After ∼30 s, the samples were flash-frozen in liquid nitrogen, and the sample was inserted into the EPR resonator. The following conditions were used for EPR experiments. X-band (9 GHz) EPR measurements were made using a Varian E-112 spectrometer interfaced to a PC using custom written software. A finger Dewar filled with liquid nitrogen and inserted into a TE102 rectangular EPR cavity maintained the sample at 77 K throughout the measurements. Spectrometer parameters used to acquire the spectrum in Fig. 4 are as follows: modulation amplitude, 5 G; microwave power, 0.05 milliwatt; receiver gain, 3.2 × 104; microwave frequency, 9.119 GHz, scan time, 0.5 min; time constant, 0.128 s; number of scans, 49. The field was calibrated using a standard sample of manganese doped in MgO (44). The concentration of the radical species was estimated by comparing the double integrated intensity of the radical signal with that of a 400-μmol Cu(II)-EDTA standard sample. Spectral simulations were carried out using previously described software (45) in which the Zeeman interaction is treated exactly and nuclear hyperfine coupling interactions are treated to first order. Parameters used for the simulation displayed under “Results and Discussion” are as follows: principal g-values g1 = 2.008, g2 = 2.005, g3 = 2.002; 14N (nuclear spin I = 1) hyperfine coupling principal values (in Gauss) = 1, 1, 13; unique principal value of 14N hyperfine coupling interaction oriented along g3; four isotropic 1H hyperfine coupling interactions (in gauss) = 5, 4, 3, and 3; Gaussian broadening function = 2.5 G half-width at half-height.
FIGURE 4.
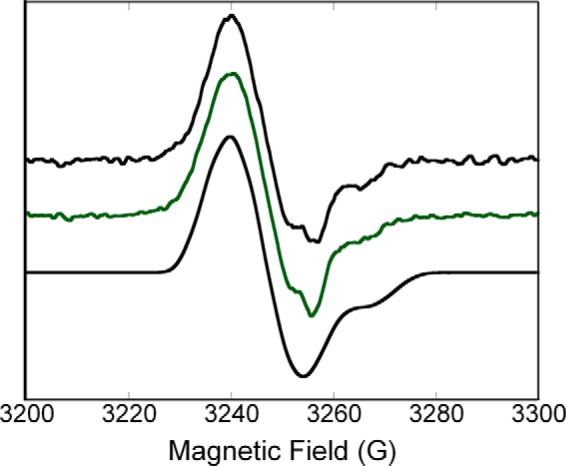
X-band (9 GHz) EPR spectra of the radical species generated in the E1o-h (top) and in the human oxoglutarate dehydrogenase complex (middle) along with a spectral simulation (bottom). Top, E1o-h (concentration of active centers = 0.449 mm) in 0.4 ml of 50 mm HEPES (pH 7.5) containing 0.15 m NaCl, 0.50 mm ThDP, and 1.0 mm MgCl2 was mixed with 10 mm OG at room temperature. The mixture was immediately transferred into an EPR tube and was flash-frozen in liquid nitrogen within 40 s of mixing all components. Middle, E1o-h, E2o-h, and E3 components were mixed at a concentration of subunits of 0.222 mm for each of the three components in 50 mm HEPES (pH 7.5) containing 0.50 mm ThDP, 2.0 mm MgCl2, and 0.15 m NaCl. The reaction was started by the addition of 10 mm OG, and sample was treated the same way as E1o-h above. Parameters used for the acquisition of the experimental spectra are presented under “Experimental Procedures.” Bottom, the parameters used for the simulation are consistent with the ThDP-enamine radical species (for details, see “Experimental Procedures”).
RESULTS AND DISCUSSION
Functional Competence of the E1o-h
Our interest in the catalytic mechanism of ThDP-dependent enzymes and the involvement of OGDHc in human pathology known in the literature prompted us to synthesize genes expressing E1o-h and E2o-h. Because data on OGDHc reported in the literature are mostly limited by mammalian sources, this is the first report on OGDHc-h assembled from individual recombinant components that produce active OGDHc-h. The identity of E1o-h and E2o-h was confirmed by Fourier transform MS analysis of peptic peptides resulting from pepsin treatment of E1o-h and E2o-h components (see supplemental Tables S1 and S2). The functional competence of E1o-h and E2o-h was confirmed by the following experiments.
Kinetic Studies
An E1o-h specific activity of 1.78 ± 0.40 μmol·min−1·mg E1o−1 (kcat = 6.7 s−1) was measured by monitoring the reduction of the external oxidizing agent 2,6-DCPIP at 600 nm (36) in the assay containing E1o-h and OG as compared with 0.34 μmol·min−1·mg E1o−1 reported by us earlier for E1o-ec (33) (Table 1) (for the composition of assay medium see “Experimental Procedures”). In this reaction, the enamine intermediate produced on E1o-h from OG is oxidized by DCPIP with the formation of succinate and reduced DCPIP (36). Although there is a recent report of an E1o-h specific activity of 0.30 μmol·min−1·mg protein−1, measured as the rate of carboligase reactivity with glyoxylate as an acceptor instead DCPIP, we cannot directly compare the results of the two assays (46).
TABLE 1.
The kinetic parameters of human OGDHc
| Overall OGDHc activitya | kcat/Km | E1o-h-specific assay | ROS activity (pH 6.3) |
|---|---|---|---|
| μmol·min−1·mg E1o−1 | s−1·mm−1 | μmol·min−1·mg E1o-h−1 | nmol·min−1·mg E1o-h−1 |
| 7.86 ± 0.74 | 200 | 1.78 ± 0.40 | H2O2 4.05 ± 0.10b |
| (kcat = 30 s−1) | (kcat = 6.7 s−1) | O2⨪ 2.06 ± 0.07 |
a Steady-state kinetic results obtained at pH 7.5; see details under “Experimental Procedures.”
b H2O2 generation at pH 7.3 was found to be quantitatively very similar to the value at pH 6.3 (4.45 ± 0.41 nmol·min−1·mg E1o-h−1).
An overall OGDHc activity of 7.86 ± 0.74 μmol·min−1·mg E1o-h−1 (kcat = 30 s−1) was measured for E1o-h reconstituted with E2o-h and E3h components once the stoichiometry of the components was optimized (for the composition of assay medium and the stoichiometry of the components see “Experimental Procedures” and the legend to Fig. 1). To our knowledge, this is the first report on the successful expression of recombinant human E1o and E2o components that could be assembled into active OGDHc-h. As a comparison, an E1o activity of 20 μmol·min−1·mg E1o−1 within the isolated pig heart OGDHc was calculated (46), in good accord with our data above (7.86 ± 0.74 μmol·min−1·mg E1o-h−1) and confirming the functional competence of the recombinant E1o-h and E2o-h components.
FIGURE 1.
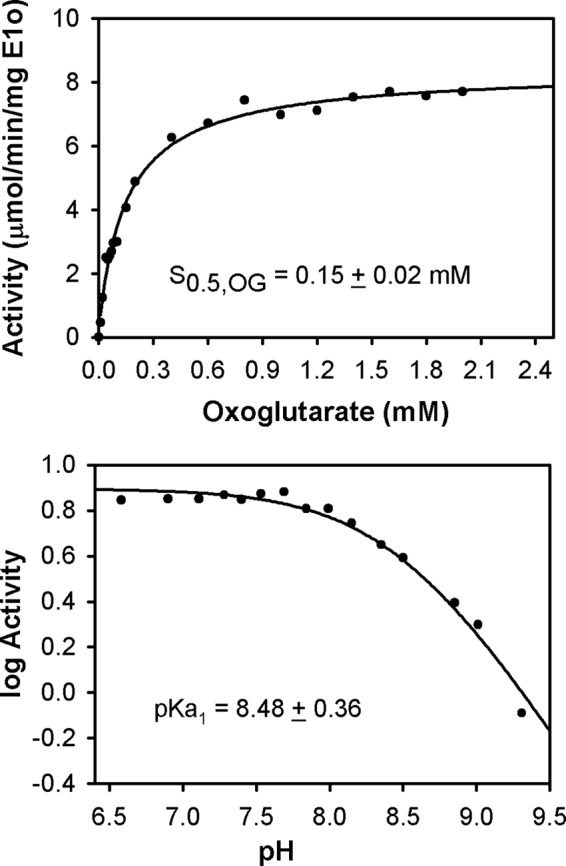
Kinetic parameters for human OGDHc. Top, dependence of the OGDHc activity on concentration of OG. The OGDHc was assembled from E1o-h, E2o-h, and E3 components at a mass ratio (μg/μg/μg) of 1:5:5, corresponding to the following concentrations of subunits: E1o-h (0.046 μm), E2o-h (0.60 μm), and E3 (0.52 μm). For experimental details, see “Experimental Procedures.” Data were fitted to a Hill equation (vo = vomax[OG]n/(S0.5n + [OG]n), and the trace is the regression fit line. nH = 0.92 ± 0.08. Bottom, pH dependence of OGDHc activity. The reaction was carried out in 0.1 m Tris·HCl in the pH range of 6.5–9.3. For details on OGDHc assembly and activity measurement, see “Experimental Procedures.” The values of activity were plotted to a curve defined by one ionizing group according to log(activity) = log(activitymax) − log(1 + 10(x − pKa1)), where x is the value of pH.
We estimate a S0.5,OG of 0.15 mm for E1o-h in the overall OGDHc assay (Fig. 1, top), similar to that reported for mammalian oxoglutarate dehydrogenase complexes (47–52). Also, values of S0.5,OG of 30 μm and S0.5,OG of 120 μm were reported for OG binding in two active centers of human heart OGDHc with an overall OGDHc activity of ∼6 μmol·min−1·mg protein−1 (53).
The pH dependence of the overall OGDHc-h activity displayed a pH-independent plateau near physiological pH (6.5–7.5) but reduced activity with increasing pH, with an apparent pKa of 8.48 (Fig. 1, bottom), which correlates well with the pH optimum of 6.65 over a relatively broad range (∼2 pH units) (51) and a pH optimum of 6.5–7.0 reported for mammalian OGDHcs (54). Our subsequent experiments were carried out in this pH-independent plateau region.
Circular Dichroism Studies with OG
CD titration of E1o-h by OG in the absence of E2o-h and E3 revealed the appearance of two CD bands (Fig. 2, top) similar to those observed with E1o-ec (Fig. 2, bottom), leading to the following conclusions: (a) a positive CD band at 302 nm, earlier assigned to the 1′,4′-iminopyrimidine tautomer (IP form) of either the first predecarboxylation or the second postdecarboxylation ThDP-bound intermediate (C2-(α-hydroxy)-γ-carboxypropyl-ThDP in Scheme 1) because it had been concluded that all ThDP intermediates with C2α tetrahedral substitution exist in their IP form at a pH near and above the pKa of the 4′-aminopyrimidinium conjugate acid form (55) and (b) a positive CD band at 347 nm (Fig. 2) that could tentatively be assigned to the enamine intermediate (the first postdecarboxylation intermediate in Scheme 1). Both E1o-h CD bands displayed dependence on the concentration of OG, resulting in an S0.5,OG of 0.14 mm (302 nm) and S0.5,OG of 0.09 mm (347 nm). Unlike on E1o-ec, according to the behavior of this CD band, this intermediate is not as stabilized on E1o-h as on E1o-ec, making its detection more difficult.
FIGURE 2.
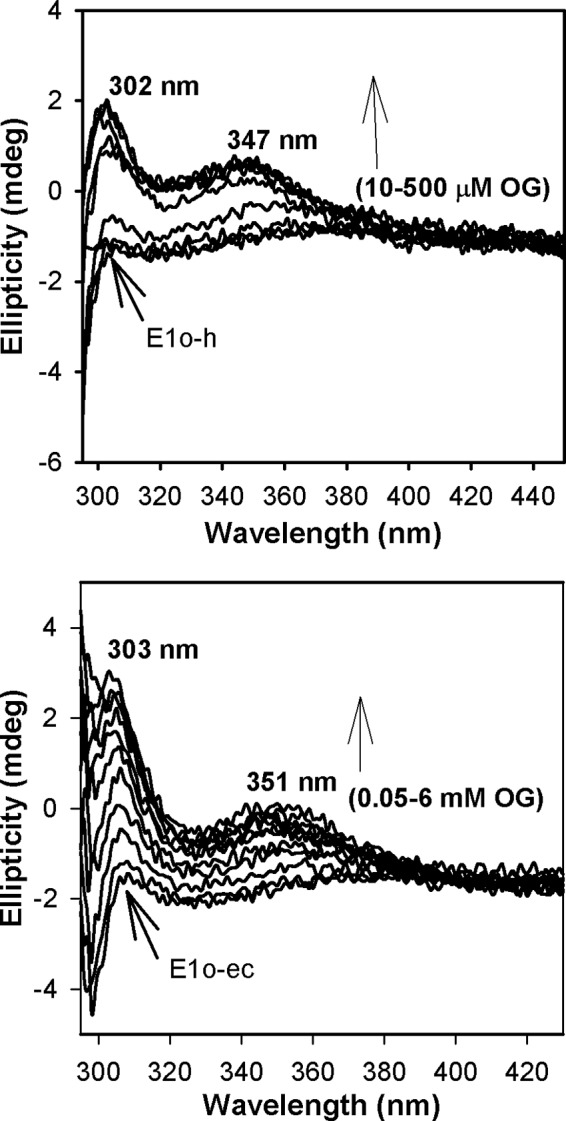
Circular dichroism titration of E1o-h and E1o-ec by OG. Top, E1o-h (18 μm active center concentration) in 50 mm KH2PO4 (pH 7.5) containing 0.20 mm ThDP and 1.0 mm MgCl2 was titrated by OG (10–500 μm) at 4 °C. Bottom, E1o-ec (20 μm active center concentration) was titrated by OG (0.05–6 mm) in 20 mm KH2PO4 (pH 7.0) containing 150 mm NaCl, 0.2 mm ThDP, and 1 mm MgCl2 at 20 °C.
Circular Dichroism Studies on E1o-h with Phosphonate and Phosphinate Analogues
Formation of the IP tautomer of ThDP-bound predecarboxylation intermediate in the active centers of E1o-h was evident from CD spectra of E1o-h titrated by phosphonate analogues of OG succinylphosphonate methyl ester (CD maximum at 296 nm, S0.5,succinylphosphonate methyl ester = 48 ± 19 μm) and succinylphosphonate (CD maximum at 304 nm) and also from acetylphosphinate, an analogue of pyruvate (CD maximum at 300 nm, S0.5,acetylphosphinate = 28 ± 2.6 μm), known to be a potent inhibitor of ThDP-dependent decarboxylases (33, 47, 56–63). In addition, the CD spectra of all tested phosphonate analogues displayed a the negative CD band around 330 nm, earlier assigned at Rutgers to the Michaelis complex (64), suggesting half-of-the site reactivity with one active center filled with ThDP-bound predecarboxylation intermediate and the second active center filled with Michaelis complex. Based on the data presented, we can conclude that the tested phosphonate and phosphinate analogues bind in the active centers of E1o-h according to the CD bands formed at 300, 302, and 304 nm, respectively (see Fig. 3 for an example), all assigned to stable C2α-tetrahedral predecarboxylation intermediate analogues in their 1′,4′-iminopyrimidinyl tautomeric forms, as demonstrated earlier for E1o-ec (33) and for E1p-ec (61). The ability of E1o-h to form this intermediate confirms that E1o-h is folded correctly with ThDP in the active V conformation. Earlier, the potency of succinylphosphonate inhibition of the OGDHc from bovine brain (47) and of E1o-ec (58) was demonstrated in kinetic experiments.
FIGURE 3.
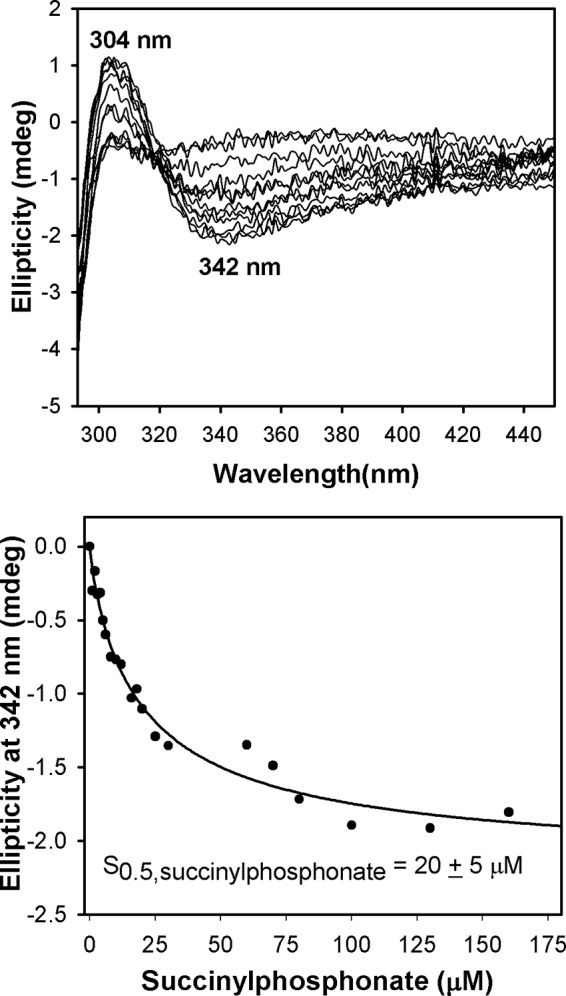
Circular dichroism titration of E1o-h by succinylphosphonate. Top, the E1o-h (1.68 mg/ml; concentration of active centers, 14.9 μm) in 20 mm KH2PO4 (pH 7.5) containing 0.2 mm ThDP and 2.0 mm MgCl2 was titrated by succinylphosphonate (1–130 μm). Two CD bands observed could be assigned to 1′,4′-iminotautomer of the first predecarboxylation intermediate (positive CD band at 304 nm) and to the Michaelis complex (negative CD band at 342 nm). Bottom, the dependence of the CD at 342 nm on concentration of succinylphosphonate, leading to S0.5,succinylphosphonate of 20 ± 5 μm. Data were fitted to a Hill equation (CDo = CDomax[SP]n/(S0.5n + [SP]n), and the trace is the regression fit line. nH = 0.86 ± 0.11.
EPR Detection of the ThDP-Enamine Radical on E1o-h
The X-band continuous wave EPR spectrum of the radical generated by reaction of E1o-h (445-μmol concentration of active centers) aerobically with 0.5 mm ThDP and 10 mm OG is shown in Fig. 4 (top). The spectrum has an overall line shape reminiscent of that previously assigned to thiamin-derived radical species, specifically C2α-hydroxyethylidene-thiamin diphosphate (15) and ThDP-enamine radicals (15). In order to determine the validity of the assignment to a thiamin-enamine radical for the experimental spectrum shown in Fig. 4, we generated spectral simulations using parameters (see “Experimental Procedures”) previously assigned for the ThDP-enamine radical (15). Although the simulation depicted in Fig. 4 (bottom) cannot be considered a unique fit at this stage, several points can be made regarding the parameters that provide support for the structural assignment of the radical. (a) The principal g-values used in the simulation are those measured for the ThDP-enamine radical using high frequency EPR (130 GHz) (15). Although the X-band (9 GHz) spectrum in Fig. 4 does not resolve the peaks associated with those g-values, the spectral position and shape require a g-matrix with values and anisotropy similar to those determined for the ThDP-enamine radical. Further confirmation of the g-values for the radical observed on E1o-h requires a high frequency EPR study. (b) A large, highly anisotropic nuclear spin = 1 hyperfine interaction is required to simultaneously fit both the low field and high field spectral features. The value used in the simulation in Fig. 4 is consistent with that used for the 14N in the ThDP-enamine radical and is diagnostic for hyperfine interaction expected for the 14N in the thiazolium ring. (c) 3–5 isotropic hyperfine couplings originating from spin = ½ nuclei (most likely 1H) are required to adequately reproduce the spectral shape. This fit cannot be achieved by simply convolving a line shape function with the spectrum generated using the g-values and 14N hyperfine interaction mentioned previously. The requirement of including 3–5 protons with isotropic couplings in the range of 3–5 G is consistent with what would be expected for a π-type radical mainly localized to a thiazolium cation ring that has β-type protons at the various ring positions. The resolution observed in the X-band spectrum (Fig. 4) does not allow for a more detailed assignment of the protons contributing to the spectrum (e.g. determination of whether a CH2-CH2 moiety has replaced a methyl group at the C2α position following OG decarboxylation). Indeed, a more rigorous assignment of g-values, 14N hyperfine values, and 1H hyperfine values will require experiments using high frequency EPR, isotopic substitution, electron nuclear double resonance, and/or electron spin echo envelope modulation. However, the X-band CW spectrum and simulation displayed in Fig. 4, together with the arguments presented above, provide strong support for the assignment of the radical species to a ThDP-derived species, probably a ThDP-enamine radical.
Upon assembly of OGDHc from equal numbers of subunits of E1o-h, E2o-h, and E3 components, the thiamin-enamine radical was once more readily detected from OG with no CoA present (Fig. 4, middle), indicating that assembly to OGDHc per se does not affect formation of the thiamin-enamine radical.
Radical species were also detected in samples of E1o-ec reacted aerobically with both OG and 2-oxoadipate in the presence of ThDP. The spectra (not shown), although lower in intensity, displayed a line shape similar to that reported previously for E1o-ec (18) and for the E1o-h/OG/ThDP system studied here (Fig. 4). The E1o-ec EPR spectra are consistent with a ThDP-enamine radical species, in agreement with the assignment made in the previous EPR/electron nuclear double resonance study of E1o-ec (18) and for E1o-h (Fig. 4).
An important further result drawn from the EPR studies is the assessment of the concentration of ThDP-enamine radicals at the E1o-h and E1o-ec active centers. Spin quantification measurements determined the amount of radical to be approximately (a) 0.9 μm for the E1o-h/OG/ThDP system (0.449 mm concentration of E1o-h active centers; 0.2% occupancy), (b) ∼0.3 μm for both the E1o-ec/OG/ThDP and E1o-ec/2-oxoadipate/ThDP systems (0.150 mm concentration of E1o-ec active centers; 0.2% occupancy), and (c) ∼1.3 μm for the OGDHc-h assembled from E1o-h, E2o-h, and E3 components (0.222 mm concentration of E1o-h active centers; 0.59% occupancy). It is evident that less than 1% of the active centers of E1o-h and E1o-ec are occupied by the radical species.
The ThDP-bound Intermediate Preceding the Thiamin-Enamine Radical
Assignment of the Positive CD Band Centered near 300 nm to the Second Postdecarboxylation Intermediate
Because both the E1o-h and E1o-ec display the same CD behavior with OG (Fig. 2), and given the greater stability of the intermediates on E1o-ec, this latter enzyme was used to characterize the ThDP-bound intermediate responsible for the newly observed CD/UV-visible signal. Upon the incremental addition of OG to E1o-ec, there developed two positive CD signals, one at ∼300 and the other at ∼350 nm (Fig. 2, bottom). The positive band near 300 nm could pertain to either pre- or postdecarboxylation ThDP-bound intermediate with C2α-tetrahedral substitution (55).
To resolve this ambiguity, a gCHSQC NMR experiment was carried out by using [C2,C6′-13C2]ThDP-labeled E1o-ec similar to that reported recently for E1p-ec (43). As was reported earlier, the reaction of pyruvate with E1p-ec produces C6′H resonances at 7.34 ppm for C2α-hydroxyethyl-ThDP and at 7.36, 7.37, and 8.62 ppm for the keto, hydrate, and carbinolamine forms of 2-acetyl-ThDP (43, 65, 66) when DCPIP is present. The 1H NMR spectrum of [C2,C6′-13C2]ThDP-labeled E1o-ec with OG revealed a chemical shift of C6′H at 7.37 ppm (Fig. 5, left), corresponding to a C2α-hydroxyalkyl-ThDP-like compound, C2-(α-hydroxy)-γ-carboxypropyl-ThDP, according to the Tittman-Hübner method (65). In the presence of the alternate enamine acceptor DCPIP, an external oxidizing agent leading to oxidation to 2-succinyl-ThDP, 1H resonances were observed at 7.37 ppm for the C2α-hydroxyalkyl-ThDP-like compound and at 7.47 ppm for the keto/hydrate form of 2-succinyl-ThDP, probably overlapping with another signal (67) (Fig. 5, right). No resonance for the tricyclic carbinolamine form of 2-succinyl-ThDP could be observed (Fig. 5, right), suggesting that the keto/enol form is more stabilized on E1o-ec, as also observed with another ThDP-dependent enzyme pyruvate oxidase (67). At later times (above 0.20 s), there is a decrease in the area of the resonances assigned to 2-succinyl-ThDP, presumably due to hydrolysis of succinyl-ThDP, as reported for hydrolysis of 2-acetyl-ThDP (43).
FIGURE 5.

Distribution of ThDP-bound covalent intermediates in the reaction of E1o-ec. gCHSQC NMR spectra of the supernatant after acid quench of the E1o-ec reaction and removal of protein from the reaction. The [C2,C6′-13C2]ThDP-labeled E1o-ec was incubated with OG for 50 ms in the absence (left) and in the presence of DCPIP (right). The C6′H chemical shift of 8.01, 7.37, and 7.47 ppm corresponds to ThDP, C2-(α-hydroxy)-γ-carboxypropyl-ThDP, and succinyl-ThDP, respectively. The 7.36–7.37 ppm resonance has contributions from both species.
Formation of C2-(α-Hydroxy)-γ-carboxypropyl-ThDP was further confirmed by carrying out the reverse reaction. Succinic semialdehyde (SSA) is the expected product of non-oxidative decarboxylation of OG in the absence of enamine oxidation. The [C2,C6′-13C2]ThDP-labeled E1o-ec was reacted with SSA, followed by acid quench and produced a resonance at 7.37 ppm (not shown), consistent with that observed in the forward direction and assigned to the second postdecarboxylation product (C2-(α-hydroxy)γ-carboxypropyl-ThDP; Scheme 1). The presence of C2-(α-hydroxy)-γ-carboxypropyl-ThDP in the NMR spectrum even after a 3-min incubation of E1o-ec with OG suggests stability of the second postdecarboxylation intermediate and correlates well with the CD results (stability of the positive 300 nm CD band).
Assignment of the Positive CD Band at 350 nm to the ThDP-Enamine Intermediate
Evidence is presented below consistent with the assignment of this band to the enamine, the experiments being necessitated by our own earlier observations, where generation of the enamine derived from pyruvate decarboxylation was modeled and gave a UV spectrum with λmax of 290–295 nm (68), at a much shorter wavelength than here seen for OG. Pre-steady-state rates of formation of ThDP-bound intermediates on E1o-ec with OG were determined by a stopped-flow CD experiment. To determine which of the species giving rise to the CD bands at 300 and 350 nm is formed first, the E1o-ec (38 μm concentration of active centers) from one syringe was mixed in a reaction cell with OG (4 mm) from the second syringe, and the reaction was monitored for 50 s at 297 nm or at 365 nm (two different experiments; the optimal intensity of the lamp in the instrument dictates these wavelengths as giving the strongest lines nearest to the wavelength of interest) (Fig. 6). The observed rate of formation of the second postdecarboxylation intermediate (C2-(α-hydroxy)γ-carboxypropyl-ThDP in Scheme 1) at 297 nm is 0.78 ± 0.02 s−1 (Fig. 6A), whereas at 365 nm, the rates are k1 = 2.8 ± 0.1 s−1 and k′1 = 0.3 ± 0.01 s−1 for the biphasic buildup of the enamine intermediate and k2 = 0.011 ± 0.008 s−1 for its depletion (Fig. 6B). These results suggest that formation of the 350-nm species is 3–4 times faster than that of the now proven second postdecarboxylation intermediate at 300 nm, further suggesting that the species represented by the CD band at 350 nm is formed before the second (C2α-tetrahedral) postdecarboxylation intermediate, consistent with the assignment of the 350 nm band to the enamine intermediate. This positive CD band has never been observed before with either E1o or any other ThDP enzymes.
FIGURE 6.

Pre-steady-state kinetics of E1o-ec with OG. A, time dependence of build-up of the C2-(α-hydroxy)-γ-carboxypropyl-ThDP intermediate (second postdecarboxylation intermediate in Scheme 1). The data points were fit using a single exponential equation (CD297(t) = CD1·e−k1t + c), and the line is a regression fit trace. B, time dependence of enamine formation on E1o-ec. The data were fitted using the triple exponential equation (CDλ(t) = CD1·e−k1t + CD2·e−k1′t − CD2·e−k2t + c), and the line is the regression fit trace. k1, k−1, and k2 are the apparent rate constants; c and CDmaxλ in the exponential rise to maximum model. All stopped-flow CD experiments were performed in 20 mm KH2PO4 (pH 7.0) with 0.5 mm ThDP and 2 mm MgCl2 at 20 °C.
Additional Evidence Supporting the Assignment of the Positive CD Band at 350 nm to the Enamine Intermediate
One could hypothesize three plausible candidates for this transient species produced upon the addition of OG to E1o-h or to E1o-ec (Scheme 1, bottom right): the enamine intermediate; the one-electron oxidation product of the enamine, C2-(α-hydroxy)-γ-carboxypropylidene-ThDP radical; or the two-electron oxidation product, 2-succinyl-ThDP (see Scheme 1 for an explanation of intermediates). The following arguments support the formation of the enamine intermediate. (a) Observation of the ThDP-enamine radical with less than 1% active center occupancy (i.e. submicromolar concentration) rules out the radical because the CD instrumentation employed is incapable of observing such low amounts by either steady-state of time-resolved methods. (b) The recent report of the observation of a CD band near 390–395 nm for 2-acetyl-ThDP tends to rule out 2-succinyl-ThDP because it should have a characteristic CD signal near that of 2-acetyl-ThDP (69). (c) The 1H NMR spectrum of [C2,C6′-13C2]ThDP-labeled E1o-ec with OG revealed a chemical shift of C6′H at 7.37 ppm (Fig. 5, left) corresponding to a C2α-hydroxyalkyl-ThDP-like compound, C2-(α-hydroxy)-γ-carboxypropyl-ThDP (second postdecarboxylation product), a result of C2α protonation of the enamine (Scheme 1, right). (d) We then hypothesized that the presence of an alternate electrophilic acceptor for the putative enamine would change the kinetics of build-up and/or depletion of the CD band corresponding to the intermediate. The following potential enamine acceptors were tested: (i) as a mimic of the physiological reaction, the E2o-ec-derived didomain was used (E2o-ec(1–176), comprising the N terminus, the lipoyl domain, the peripheral subunit binding domain, and linkers); (ii) DCPIP was used as an external oxidizing agent; and finally (iii) glyoxylate was used as a known enamine acceptor in a so-called “carboligase” reaction (33, 70, 71). As control, a stopped-flow CD experiment of E1o-ec with OG was carried out and displayed relatively good stability of the 365 nm signal (Fig. 7a). It is evident from comparing the data in Fig. 7 (a–d) that the three acceptors (DCPIP, glyoxylate, and E2o-ec(1–176) didomain) shortened the lifetime of the species corresponding to the 365 nm CD band, consistent with it being pertinent to the ThDP-enamine. (e) A UV-visible stopped-flow photodiode array (PDA) experiment was carried out by mixing E1o-ec with OG (Fig. 8) and resolved several issues. (i) From the experience gained in CD studies of 12 ThDP enzymes so far, when there is a strong UV-visible signal observable along with the CD band at the same wavelength, a conjugated system is the source of the electronic transition rather than a charge transfer band, whose λmax is not predictable with current theory. (ii) Unexpectedly, the PDA experiment could resolve two species with λmax at 348 and 361 nm and a clean isosbestic point (Fig. 8, top); with time, the species monitored at 348 nm diminishes, whereas the one at 361 nm increases in amplitude (Fig. 8, bottom). Recognizing that these species correspond to a postdecarboxylation intermediate(s), two possible explanations could be envisioned once more: (i) perhaps the ThDP-enamine and the ThDP-enamine radical (Scheme 1) have uncannily similar λmax values, where the λmax at 348 nm corresponds to the former, which is converted to the radical with longer λmax, or (ii) both species are ThDP-enamines, perhaps in different conformations/configurations. Several arguments favor the second explanation. The species at 348 nm is fully formed at the first time point (Fig. 8, bottom), consistent with the time-resolved CD behavior, whereas the formation of the species at 361 nm has a significant lag time. Also, the recently published x-ray structure of 2-oxoglutarate decarboxylase (72) from Mycobacterium smegmatis with OG (this enzyme is sometimes referred to as E1o in M. smegmatis) and 2-hydroxy-3-ketoadipate synthase in Mycobacterium tuberculosis (66) suggested the existence of two enamines (postdecarboxylation intermediates), which the authors termed “early” and “late” (72), the early one converted with time to the late one. (f) Although OG added to E1o-ec gave rise to this CD band near 350 nm, the related 2-oxo acids with no side chain carboxyl groups, 2-oxovalerate and 2-oxoisovalerate, did not (data not shown) (70).
FIGURE 7.

Effect of different enamine acceptors on the CD at 365 nm. a, stopped-flow CD behavior at 365 nm, where E1o-ec from one syringe was mixed with 100 μm OG from the second syringe at 20 °C. In the other traces, E1o (38 μm active site concentration) from one syringe was mixed with the enamine acceptor (concentration indicated in the figure) premixed with OG in the second syringe. b, E2o-ec(1–176) didomain (DD). c, DCPIP. d, glyoxylate.
FIGURE 8.
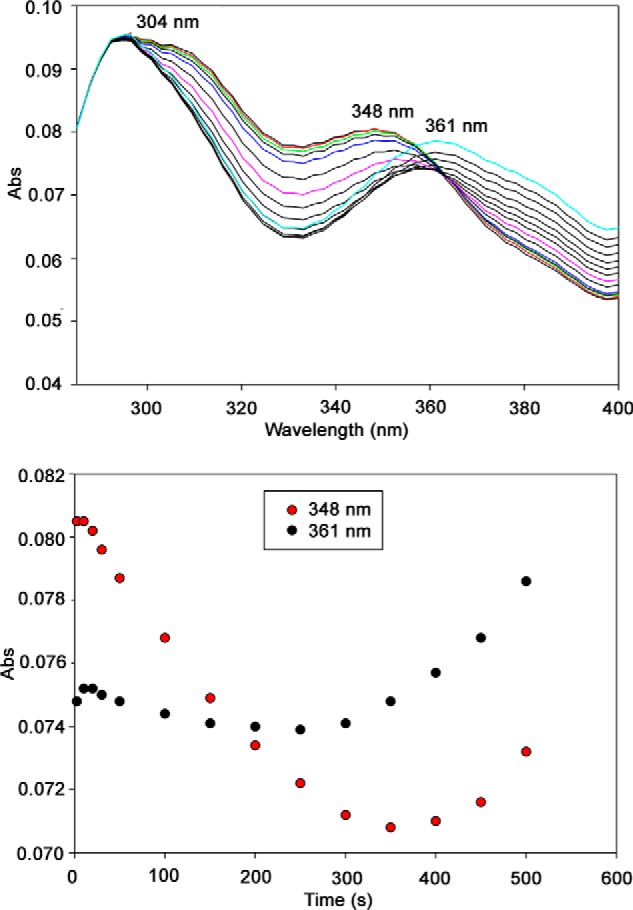
Reaction of E1o-ec with OG monitored by stopped-flow PDA. Top, E1o-ec spectra with 10 mm OG showing formation of ThDP-enamines, perhaps in different conformations/configurations. E1o-ec (40 μm concentration of active centers) was mixed with an equal volume of 10 mm OG in the same buffer at 25 °C. The reaction was monitored in the 280–400-nm wavelength range for 500 s. Bottom, time course of the formation of ThDP-enamines at the indicated wavelength.
The cumulative evidence points to the ThDP-enamine as the species responsible for the newly identified electronic transition near 350 nm produced by OG on E1o-h and E1o-ec. Possible explanations of the λmax observed are shown in Scheme 2, but whatever the correct explanation is, it must involve a longer conjugated system than present in the ThDP-enamine derived from pyruvate.
SCHEME 2.
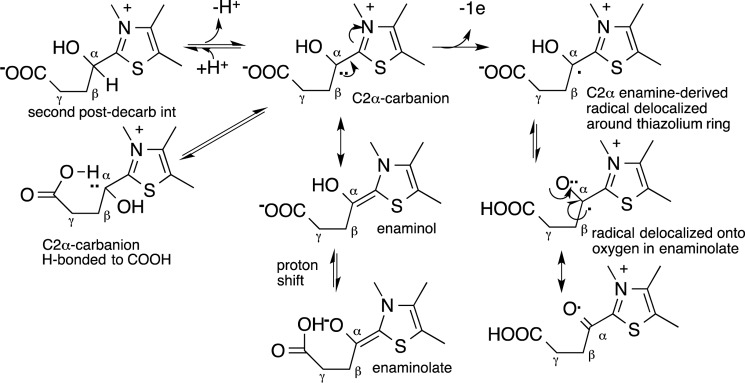
Possible role of side chain carboxylate in stabilizing ThDP-bound enamine and radical.
Evidence for Reactive Oxygen Species Production by E1o-h
Generation of ROS by E1o-h was detected by two independent and widely used methods: the Amplex Red fluorescence assay for detection of H2O2 and the acetylated cyt c reduction assay for detection of superoxide anion. The assays verified superoxide and H2O2 generation by E1o-h and exhibited quantitatively the same order of magnitude specific activity (Fig. 9). An E1o-h specific activity of 4.45 ± 0.41 nmol·min−1 mg E1o-h−1 (n = 3) (pH 7.3) and 4.05 ± 0.10 nmol·min−1·mg E1o-h−1 (n = 3) (pH 6.3) was determined for H2O2 generation, with no statistically significant difference detected between the results at the two pH values. The E1o-h specific activity for superoxide generation was 2.06 ± 0.07 nmol·min−1·mg E1o-h−1 (n = 5) (pH 6.3). No statistically significant difference in activity was found when the pH was changed from 7.3 to 6.3 (measured in the Amplex Red assay). These values were compared with the specific activity of the E1o-h as an isolated component or as part of OGDHc assembled from individual components (Table 1). The comparison shows that the ROS generation capacity by E1o-h is 0.23% for H2O2 and 0.12% for superoxide as compared with the E1o-h specific activity measured in the DCPIP assay.
FIGURE 9.
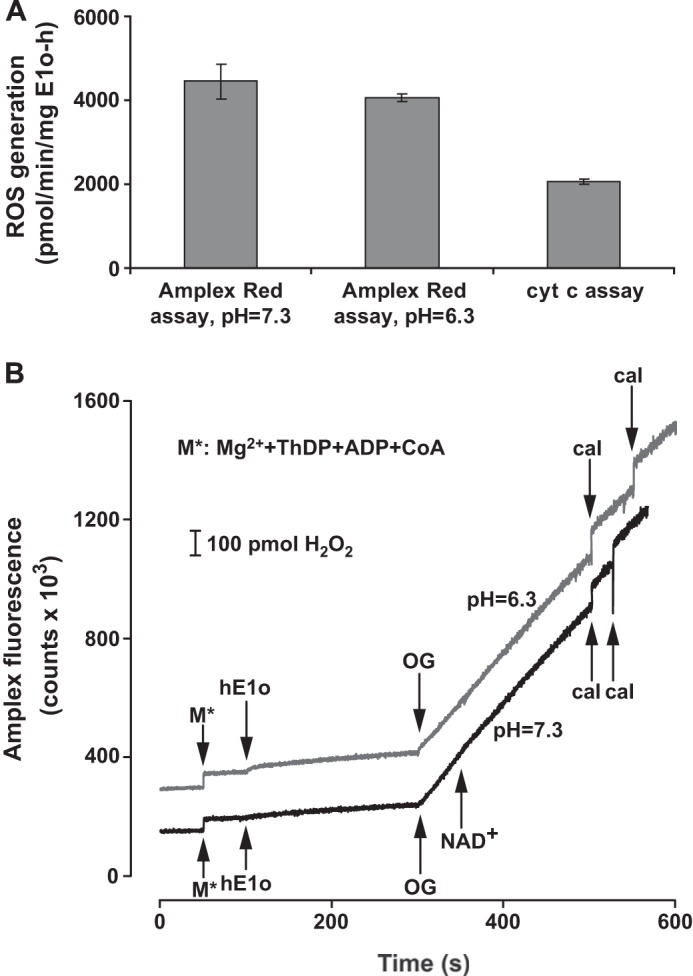
Superoxide/H2O2 production by E1o-h. A, superoxide/H2O2 production by E1o-h as measured by the cyt c or the Amplex Red assay and expressed as the amount of superoxide or H2O2 produced (pmol)·min−1·mg E1o-h−1. Linear fitting to initial velocity data points generated always an R2 > 0.982 in the cyt c assay or greater in the Amplex Red assay. The primary data were corrected with the average slopes of the blanks (for both assays). Error bars, S.E. values for five (cyt c assay) or three (Amplex Red assay) parallel measurements. No statistically significant difference was found between the rates of H2O2 production measured at two different pH values by the Amplex Red assay. For experimental conditions, see “Experimental Procedures.” B, representative Amplex Red fluorescence traces for H2O2 generation by E1o-h at different pH values with additions indicated. For experimental conditions, see “Experimental Procedures.” Curves have been offset for clarity. For calibration of all experiments (designated as cal at the bottom), 100 pmol of H2O2 was applied in duplicates. 1 mm NAD+ was added to the reaction assay at pH 7.3.
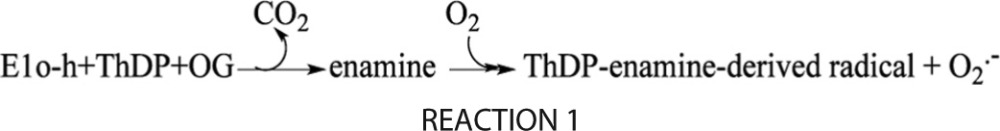
Because the primary product is proposed to be superoxide (a one-electron reduction of O2 to O2⨪ according to Reaction 2), the H2O2 present in the assay could be considered to be the product of spontaneous dismutation (disproportionation) of superoxide (6, 21, 23, 73), similar to that reported for other enzymes (74), rather than two-electron reduction of O2 to H2O2 according to Reaction 3.
 |
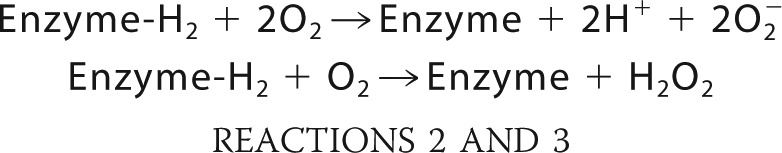 |
In support of this conclusion we present the following. (a) Because one-electron enamine oxidation on E1o-h leading to enamine-derived radical was detected by the EPR experiment, a one-electron reduction of O2 leading to superoxide is logical. (b) The production of superoxide by E1o-h was directly detected by the cyt c assay. (c) In a control experiment, the superoxide dismutase, when added in the cyt c assay, completely eliminated the signal by trapping superoxide produced by E1o-h. (d) When catalase was added to the Amplex Red assay in a similar control experiment, the signal was also eliminated, confirming H2O2 accumulation in the Amplex Red assay.
Additional control experiments were carried out to confirm ROS generation by E1o-h. (a) In the Amplex Red assay, neither OG nor E1o-h revealed H2O2 production by itself in the presence of Mg2+, ThDP, ADP, and CoA. Further, the order of addition of OG or E1o-h did not change the experimental results beyond experimental error. (b) The Amplex Red assay was also tested using commercial porcine heart OGDHc (Sigma) and the protocol reported earlier (6), leading to the observation that the rate of H2O2 generation by porcine heart OGDHc was in good accord with that reported earlier. Nevertheless, a direct quantitative comparison was not reliable because of the impurities present in the commercial OGDHc preparations.
Next, we estimated the rate of spontaneous dismutation reaction under the assay conditions as follows. (a) With E1o-h activity of 2.06 nmol·min−1·mg E1o−1 for superoxide production, a kcat = 0.0076 s−1 (first order rate constant) and an O2⨪ concentration of ∼1 × 10−5 m could be estimated. (b) If no cyt c is present in the assay medium, a second order rate constant for spontaneous dismutation of O2⨪ of 2 × 105 m−1 s−1 (pH 7.3) was estimated based on a rate constant of 2 × 105 m−1·s−1 (pH 7.4) reported in the literature (73), yielding a first order rate constant of 2 s−1. (iii) When cyt c is present in the assay, using a k of 3.5 × 105 m−1 s−1 (pH ∼7.0) (74), for the same concentration of O2⨪, the value of 3.5 s−1 is estimated.
From these simple estimates we conclude that superoxide formation by E1o-h is the rate-limiting step (7.6 × 10−3 s−1), whereas spontaneous dismutation to H2O2 (2 s−1) and reaction of HO2/O2⨪ with acetylated cyt c (3.5 s−1) are both much faster and have nearly equal probability.
At present, the functional relevance of ROS generation by OGDHc-h under various pathological conditions has been proven and is discussed in detail in the literature, including by the Semmelweis group and others (4–6, 19–22, 30, 31, 75–78). In theory, E1o-h could generate the same amount of superoxide as the entire OGDHc functioning in the mitochondrion based on data presented in this paper and in the literature. However, a direct comparison of ROS production by OGDHc on isolated mitochondria with data presented in this paper on E1o-h is not possible because these values were calculated per total protein but not per amount of OGDHc content. A reasonably relevant comparison is with OGDHc isolated from pig heart, providing a superoxide-producing activity of 1 nmol·min−1·mg protein−1 in both the physiological reaction (with OG and CoA) and in the reverse reaction with NADH. This reaction was attributed to E3-h in the OGDHc and constituted 0.3–0.4% of the overall OGDHc reaction (20).
Recently, the relative maximum capacities for O2⨪/H2O2 generation were compared for NADH/NAD+-dependent enzyme complexes in intact skeletal muscle mitochondria. The studies revealed that under optimal conditions, the OGDHc produced superoxide/H2O2 at about twice the rate of PDHc and at 4 times the rate of BCOADHc (when oxidizing their specific 2-oxo acid substrates in the physiological reaction) and at 8 times the rate of respiratory complex site IF, indicating that OGDHc has the highest capacity in this group (19).
Conclusions
The first successful expression and purification of active, full-length E1o and E2o components of the human 2-oxoglutarate dehydrogenase multienzyme complex enabled this collaborative effort to undertake studies directed to an elucidation of the fundamental reactions carried out by this enzyme complex. (a) Formation of a ThDP-enamine radical was demonstrated by EPR spectroscopy generated from OG for both the human and E. coli E1o components and for OGDHc-h reconstituted from the three component enzymes, as shown for the E1o-ec earlier (18). How and whether this radical, formed by reaction of dioxygen with the enamine, receives special stabilization remains to be determined. (b) Upon the addition of OG to E1o-h and E1o-ec, a new electronic transition was observed centered at 350 nm by both UV-visible and CD spectroscopy. Multiple experiments are suggestive of the transition being pertinent to the ThDP-enamine intermediate, the first ThDP-bound postdecarboxylation intermediate in Scheme 1. The stability of the ThDP-enamine, compared with that derived from pyruvate, is more than likely due to some stabilization afforded by the side chain C5 carboxylate group (Scheme 2), perhaps of the enaminolate oxyanionic forming an intramolecular hydrogen bond or of the lone pair of electrons at C2α in the enamine forming an intramolecular hydrogen bond. Either scenario is difficult to model, unlike the enaminol form derived from pyruvate, where it was modeled by replacing an OH group by an OCH3 group (68). (c) Of greatest import to human physiology and pathology, the E1o-h component now joins the E3-h component as being able to generate the superoxide anion and thence hydrogen peroxide, probably by dismutation, shown here for the first time. The specific activity on this pathway is 0.1–0.2% of the physiological activity, certainly significant. Here both products formed in Reaction 1 are clearly demonstrated for E1o-h, adding to the important differences in the aerobic behavior between the pyruvate and 2-oxoglutarate dehydrogenase complexes. The importance of superoxide formation from E1o-h under physiological conditions remains to be demonstrated in the human OGDHc.
Finally, the ability to assess the fractional occupancy of active sites by the ThDP-enamine radical, as well as the fractional specific activity of ROS formation compared with NADH production, allows us to conclude that the O2-induced oxidation-reduction chemistry in Reaction 1 is “off-pathway” at much less than 1% of the “on-pathway” reactivity of the enamine toward reductive succinylation of E2o and thence succinyl-CoA formation. Therefore, there is little likelihood that the principal pathway follows a radical mechanism for the reductive acylation of the lipoyl-E2. The low occupancy of E1o active centers by the thiamin enamine radical suggests that the on-pathway mechanism produces succinyl-CoA via 2-electron enamine oxidation (ionic mechanism) proposed in a model reaction (79).
Supplementary Material
Acknowledgment
We thank Dr. Anand Balakrishnan for help with NMR sample preparation using the quench flow instruments.
This work was supported, in whole or in part, by National Institutes of Health Grants GM097499 (to G. J. G.) and GM-050380. This work was also supported by Hungarian Academy of Sciences MTA Grant 02001 (to A.-V. V.), Hungarian Scientific Research Fund (OTKA) Grants 85082 and 81983 to A.-V. V.), Hungarian Brain Research Program Grant KTIA_13_NAP-A-III/6 (to A.-V. V.), Bolyai and Fulbright fellowships (to A. A.), and National Science Foundation Grant CHE 1213550 (to G. J. G.).

This article contains supplemental Tables S1 and S2.
- PDHc
- pyruvate dehydrogenase complex
- ThDP
- thiamin diphosphate
- DCPIP
- 2,6-dichlorophenolindophenol
- OGDHc
- 2-oxoglutarate dehydrogenase multienzyme complex
- E1o
- 2-oxoglutarate decarboxylase
- E1o-h
- human E1o-h
- E1o-ec
- E. coli E1o
- E2o
- dihydrolipoyl succinyltransferase
- E2o-h
- human E2o
- E2o-ec
- E. coli E2o-ec
- E3
- dihydrolipoyl dehydrogenase
- E3-h
- human E3
- E3-ec
- E. coli E3
- OG
- 2-oxoglutarate
- ROS
- reactive oxygen species
- IP form
- 1′,4′-iminopyrimidine tautomer IP form of ThDP-bound intermediate
- gCHSQC
- gradient Carbon Heteronuclear Single Quantum Coherence NMR experiment
- SSA
- succinic semialdehyde
- PDA
- UV-visible stopped-flow photodiode array spectroscopy
- cyt c
- cytochrome c.
REFERENCES
- 1. Perham R. N. (2000) Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu. Rev. Biochem. 69, 961–1004 [DOI] [PubMed] [Google Scholar]
- 2. Reed L. J. (2001) A trial of research from lipoic acid to α-keto acid dehydrogenase complexes. J. Biol. Chem. 276, 38329–38336 [DOI] [PubMed] [Google Scholar]
- 3. Perham R. N. (1991) Domains, motifs, and linkers in 2-oxo acid dehydrogenase multienzyme complexes: a paradigm in the design of a multifunctional protein. Biochemistry 30, 8501–8512 [DOI] [PubMed] [Google Scholar]
- 4. Gibson G. E., Park L. C. H., Sheu K. F. R., Blass J. P., Calingasan N. Y. (2000) The α-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochem. Int. 36, 97–112 [DOI] [PubMed] [Google Scholar]
- 5. Starkov A. A., Fiskum G., Chinopoulos C., Lorenzo B. J., Browne S. E., Patel M. S., Beal M. F. (2004) Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 24, 7779–7788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tretter L., Adam-Vizi V. (2004) Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. J. Neurosci. 24, 7771–7778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ciszak E. M., Makal A., Hong Y. S., Vettaikkorumakankauv A. K., Korotchkina L. G., Patel M. S. (2006) How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J. Biol. Chem. 281, 648–655 [DOI] [PubMed] [Google Scholar]
- 8. Brautigam C. A., Wynn R. M., Chuang J. L., Machius M., Tomchick D. R., Chuang D. T. (2006) Structural insight into interactions between dihydrolipoamide dehydrogenase (E3) and E3 binding protein of human pyruvate dehydrogenase complex. Structure 14, 611–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brautigam C. A., Chuang J. L., Tomchick D. R., Machius M., Chuang D. T. (2005) Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J. Mol. Biol. 350, 543–552 [DOI] [PubMed] [Google Scholar]
- 10. Brautigam C. A., Wynn R. M., Chuang J. L., Naik M. T., Young B. B., Huang T. H., Chuang D. T. (2011) Structural and thermodynamic basis for weak interactions between dihydrolipoamide dehydrogenase and subunit-binding domain of the branched-chain alpha-ketoacid dehydrogenase complex. J. Biol. Chem. 286, 23476–23488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandrasekhar K., Wang J., Arjunan P., Sax M., Park Y. H., Nemeria N. S., Kumaran S., Song J. Y., Jordan F., Furey W. (2013) Insight to the interaction of the dihydrolipoamide acetyltransferase (E2) core with the peripheral components in the Escherichia coli pyruvate dehydrogenase complex via multifaceted structural approaches. J. Biol. Chem. 288, 15402–15417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ambrus A., Adam-Vizi V. (2013) Molecular dynamics study of the structural basis of dysfunction and the modulation of reactive oxygen species generation by pathogenic mutants of human dihydrolipoamide dehydrogenase. Arch. Biochem. Biophys. 538, 145–155 [DOI] [PubMed] [Google Scholar]
- 13. Frank R. A. W., Price A. J., Northrop F. D., Perham R. N., Luisi B. F. (2007) Crystal structure of the E1 component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex. J. Mol. Biol. 368, 639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ragsdale S. W. (2003) Pyruvate ferredoxin oxidoreductase and its radical intermediate. Chem. Rev. 103, 2333–2346 [DOI] [PubMed] [Google Scholar]
- 15. Mansoorabadi S. O., Seravalli J., Furdui C., Krymov V., Gerfen G. J., Begley T. P., Melnick J., Ragsdale S. W., Reed G. H. (2006) EPR spectroscopic and computational characterization of the hydroxyethylidene-thiamin pyrophosphate radical intermediate of pyruvate:ferredoxin oxidoreductase. Biochemistry 45, 7122–7131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tittmann K., Wille G., Golbik R., Weidner A., Ghisla S., Hübner G. (2005) Radical phosphate transfer mechanism for the thiamin diphosphate- and FAD-dependent pyruvate oxidase from Lactobacillus plantarum. Kinetic coupling of intercofactor electron transfer with phosphate transfer to acetyl-thiamin diphosphate via a transient FAD semiquinone/hydroxyethyl-ThDP radical pair. Biochemistry 44, 13291–13303 [DOI] [PubMed] [Google Scholar]
- 17. Tittmann K. (2009) Reaction mechanisms of thiamin diphosphate enzymes:redox reactions. FEBS J. 276, 2454–2468 [DOI] [PubMed] [Google Scholar]
- 18. Frank R. A., Kay C. W., Hirst J., Luisi B. F. (2008) Off-pathway, oxygen-dependent thiamin radical in the Krebs cycle. J. Am. Chem. Soc. 130, 1662–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quinlan C. L., Goncalves R. L. S., Hey-Mogensen M., Yadava N., Bunik V. I., Brand M. D. (2014) The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 289, 8312–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bunik V. I., Sievers C. (2002) Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur. J. Biochem. 269, 5004–5015 [DOI] [PubMed] [Google Scholar]
- 21. Ambrus A., Tretter L., Adam-Vizi V. (2009) Inhibition of the α-ketoglutarate dehydrogenase-mediated reactive oxygen species generation by lipoic acid. J. Neurochem. 109, 222–229 [DOI] [PubMed] [Google Scholar]
- 22. Ambrus A., Torocsik B., Tretter L., Ozohanics O., Adam-Vizi V. (2011) Stimulation of reactive oxygen species generation by disease-causing mutations of lipoamide dehydrogenase. Hum. Mol. Genet. 20, 2984–2995 [DOI] [PubMed] [Google Scholar]
- 23. Bando Y., Aki K. (1991) Mechanisms of generation of oxygen radicals and reductive mobilization of ferritin iron by lipoamide dehydrogenase. J. Biochem. 109, 450–454 [DOI] [PubMed] [Google Scholar]
- 24. Gazaryan I. G., Krasnikov B. F., Ashby G. A., Thorneley R. N., Kristal B. S., Brown A. M. (2002) Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J. Biol. Chem. 277, 10064–10072 [DOI] [PubMed] [Google Scholar]
- 25. Cruts M., Backhovens H., Van Gassen G., Theuns J., Wang S. Y., Wehnert A., van Duijn C. M., Karlsson T., Hofman A., Adolfsson R. (1995) Mutation analysis of the chromosome 14q24.3 dihydrolipoyl succinyltransferase (dlst) gene in patients with early-onset Alzheimer-disease. Neurosci. Lett. 199, 73–77 [DOI] [PubMed] [Google Scholar]
- 26. Nakano K., Takase C., Sakamoto T., Nakagawa S., Inazawa J., Ohta S., Matuda S. (1994) Isolation, characterization and structural organization of the gene and pseudogene for the dihydrolipoamide succinyltransferase component of the human 2-oxoglutarate dehydrogenase complex. Eur. J. Biochem. 224, 179–189 [DOI] [PubMed] [Google Scholar]
- 27. Shi Q., Chen H. L., Xu H., Gibson G. E. (2005) Reduction in the E2k subunit of the α ketoglutarate dehydrogenase complex has effects independent of complex activity. J. Biol. Chem. 280, 10888–10896 [DOI] [PubMed] [Google Scholar]
- 28. Gibson G. E., Zhang H., Sheu K. F., Bogdanovich N., Lindsay J. G., Lannfelt L., Vestling M., Cowburn R. F. (1998) α-Ketoglutarate dehydrogenase in Alzheimer brains bearing the APP670/671 mutation. Ann. Neurol. 44, 676–681 [DOI] [PubMed] [Google Scholar]
- 29. Dumont M., Ho D. J., Calingasan N. Y., Xu H., Gibson G., Beal M. F. (2009) Mitochondrial dihydrolipoyl succinyltransferase deficiency accelerates amyloid pathology and memory deficit in a transgenic mouse model of amyloid deposition. Free Radic. Biol. Med. 47, 1019–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adam-Vizi V., Starkov A. A. (2010) Calcium and mitochondrial reactive oxygen species generation: how to read the facts. J. Alzhheimers Dis. 20, Suppl. 2, S413–S426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gibson G. E., Starkov A., Blass J. P., Ratan R. R., Beal M. F. (2010) Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim. Biophys. Acta 1802, 122–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakano K., Takase C., Sakamoto T., Ohta S., Nakagawa S., Ariyama T., Inazawa J., Abe T., Matuda S. (1993) An unspliced cDNA for human dihydrolipoamide succinyltransferase: characterization and mapping of the gene to chromosome-14q24.2-q24.3. Biochem. Biophys. Res. Commun. 196, 527–533 [DOI] [PubMed] [Google Scholar]
- 33. Shim da J., Nemeria N. S., Balakrishnan A., Patel H., Song J., Wang J., Jordan F., Farinas E. T. (2011) Assignment of function to histidines 260 and 298 by engineering the E1 component of the Escherichia coli 2-oxoglutarate dehydrogenase complex: substitutions that lead to acceptance of substrates lacking the 5-carboxyl group. Biochemistry 50, 7705–7709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Song J., Park Y. H., Nemeria N. S., Kale S., Kakalis L., Jordan F. (2010) Nuclear magnetic resonance evidence for the role of the flexible regions of the E1 component of the pyruvate dehydrogenase complex from Gram-negative bacteria. J. Biol. Chem. 285, 4680–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu T.-C., Korotchkina L. G., Hyatt S. L., Vettakkorumakankav N. N., Patel M. S. (1995) Spectroscopic studies of the characterization of recombinant human dihydrolipoamide dehydrogenase and its site-directed mutants. J. Biol. Chem. 270, 15545–15550 [DOI] [PubMed] [Google Scholar]
- 36. Khaĭlova L. S., Bernkhardt R., Khiubner G. (1977) Study of the kinetic mechanism of pyruvate-2,6-dichlorophenolindophenol reductase activity of muscle pyruvate dehydrogenase. Biokhimiia 42, 113–117 [PubMed] [Google Scholar]
- 37. Mayo L. A., Curnutte J. T. (1990) KInetic microplate assay for superoxide production by neutrophils and other phagocytic cells. Methods Enzymol. 186, 567–575 [DOI] [PubMed] [Google Scholar]
- 38. Azzi A., Montecucco C., Richter C. (1975) Use of acetylated ferricytochrome c for detection of superoxide radicals produced in biological membranes. Biochem. Biophys. Res. Commun. 65, 597–603 [DOI] [PubMed] [Google Scholar]
- 39. McCord J. M., Fridovich I. (1968) The reduction of cytochrome c by milk xantine oxidase. J. Biol. Chem. 243, 5753–5760 [PubMed] [Google Scholar]
- 40. Rosen G. M., Finkelstein E., Rauckman E. J. (1982) A method for the detection of superoxide in biological systems. Arch. Biochem. Biophys. 215, 367–378 [DOI] [PubMed] [Google Scholar]
- 41. Bradford M. M. (1976) Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 42. Mohanty J. G., Jaffe J. S., Schulman E. S., Raible D. G. (1997) A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J. Immunol. Methods 202, 133–141 [DOI] [PubMed] [Google Scholar]
- 43. Balakrishnan A., Nemeria N. S., Chakraborty S., Kakalis L., Jordan F. (2012) Determination of pre-steady-state rate constants on the Escherichia coli pyruvate dehydrogenase complex reveals that loop movement controls the rate-limiting step. J. Am. Chem. Soc. 134, 18644–18655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burghaus O., Rohrer M., Götzinger T., Plato M., Möbius K. (1992) A novel high-field/high-frequency EPR and ENDOR spectrometer operating at 3 mm wavelength. Meas. Sci. Technol. 3, 765–774 [Google Scholar]
- 45. Gerfen G. J., Bellew B. F., Griffin R. G., Singel D. J., Ekberg C. A., Whittaker J. W. (1996) High-frequency electron paramagnetic resonance spectroscopy of the apogalactose oxidase radical. J. Phys. Chem. 100, 16739–16748 [Google Scholar]
- 46. Armstrong C. T., Anderson J. L. R., Denton R. M. (2014) Studies on the regulation of the human E1 subunit of the 2-oxoglutarate dehydrogenase complex, including the identification of a novel calcium-binding site. Biochem. J. 459, 369–381 [DOI] [PubMed] [Google Scholar]
- 47. Bunik V. I., Denton T. T., Xu H., Thompson C. M., Cooper A. J. L., Gibson G. E. (2005) Phosphonate analogues of α-ketoglutarate inhibit the activity of the α-ketoglutarate dehydrogenase complex isolated from brain and in cultured cells. Biochemistry 44, 10552–10561 [DOI] [PubMed] [Google Scholar]
- 48. Lawlis V. B., Roche T. (1981) Regulation of bovine kidney α-ketoglutarate dehydrogenase complex by calcium ion and adenine nucleotides. Effects on S0.5 for α-ketoglutarate. Biochemistry 20, 2512–2518 [DOI] [PubMed] [Google Scholar]
- 49. Hamada M., Koike K., Nakaula Y., Hiraoka T., Koike M. (1975) A kinetic study of the α-keto acid dehydrogenase complexes from pig heart mitochondria. J. Biochem. 77, 1047–1056 [DOI] [PubMed] [Google Scholar]
- 50. McMinn C. L., Ottaway J. H. (1977) Studies on the mechanism and kinetics of the 2-oxoglutarate dehydrogenase system from pig heart. Biochem. J. 161, 569–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gupta S. C., Dekker E. E. (1980) Evidence for the identity and some comparative properties of α-ketoglutarate and 2-keto-4-hydroxyglutarate dehydrogenase activity. J. Biol. Chem. 255, 1107–1112 [PubMed] [Google Scholar]
- 52. Strumilo S. A., Taranda N. I., Senkevich S. B., Vinogradov V. V. (1981) 2-Oxoglutarate dehydrogenase complex from bovine adrenal-cortex mitochondria. Purification and partial characterization. Acta Biol. Med. Ger. 40, 257–264 [PubMed] [Google Scholar]
- 53. Kiselevsky Y. V., Ostrovtsova S. A., Strumilo S. A. (1990) Kinetic characterization of the pyruvate and oxoglutarate dehydrogenase complexes from human heart. Acta Biochim. Pol. 37, 135–139 [PubMed] [Google Scholar]
- 54. Hirashima M., Hayakawa T., Koike M. (1967) Mammalian α-keto acid dehydrogenase complexes. J. Biol. Chem. 242, 902–907 [PubMed] [Google Scholar]
- 55. Nemeria N., Baykal A., Joseph E., Zhang S., Yan Y., Furey W., Jordan F. (2004) Tetrahedral intermediates in thiamin diphosphate-dependent decarboxylations exist as a 1′,4′-imino tautomeric form of the coenzyme, unlike the Michaelis complex or the free coenzyme. Biochemistry 43, 6565–6575 [DOI] [PubMed] [Google Scholar]
- 56. Kluger R., Pike D. C. (1977) Active site generated analogues of reactive intermediates in enzymic reactions. Potent inhibition of pyruvate dehydrogenase by a phosphonate analogue of pyruvate. J. Am. Chem. Soc. 99, 4504–4506 [DOI] [PubMed] [Google Scholar]
- 57. Kluger R., Pike D. C. (1979) Chemical synthesis of a proposed enzyme-generated “reactive intermediate analogue” derived from thiamin diphosphate. Self-activation of pyruvate dehydrogenase by conversion of the analogue to its components. J. Am. Chem. Soc. 101, 6425–6428 [Google Scholar]
- 58. Biryukov A. I., Bunik V. I., Zhukov Y. N., Khurs E. N., Khomutov R. M. (1996) Succinyl phosphonate inhibits α-ketoglutarate oxidative decarboxylation, catalyzed by α-ketoglutarate dehydrogenase complexes from E. coli and pigeon breast muscle. FEBS Lett. 382, 167–170 [DOI] [PubMed] [Google Scholar]
- 59. O'Brien T. A., Kluger R., Pike D. C., Gennis R. B. (1980) Phosphonate analogues of pyruvate. Probes of substrate binding to pyruvate oxidase and other thiamin pyrophosphate-dependent decarboxylases. Biochim. Biophys. Acta 613, 10–17 [DOI] [PubMed] [Google Scholar]
- 60. Schönbrunn-Hanebeck E., Laber B., Amrhein N. (1990) Slow-binding inhibition of the Escherichia coli pyruvate dehydrogenase multienzyme complex by acetylphosphinate. Biochemistry 29, 4880–4885 [DOI] [PubMed] [Google Scholar]
- 61. Arjunan P., Sax M., Brunskill A., Chandrasekhar K., Nemeria N., Zhang S., Jordan F., Furey W. (2006) A thiamin-bound, pre-decarboxylation reaction intermediate analogue in the pyruvate dehydrogenase E1 subunit induces large scale disorder-to-order transformations in the enzyme and reveals novel structural features in the covalently bound adduct. J. Biol. Chem. 281, 15296–15303 [DOI] [PubMed] [Google Scholar]
- 62. Nemeria N. S., Korotchkina L. G., Chakraborty S., Patel M. S., Jordan F. (2006) Acetylphosphinate is the most potent mechanism-based substrate-like inhibitor of both the human and Escherichia coli pyruvate dehydrogenase components of the pyruvate dehydrogenase complex. Bioorg. Chem. 34, 362–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baillie A. C., Wright K., Wright B. J., Earnshaw C. G. (1988) Inhibitors of pyruvate dehydrogenase as herbicides. Pestic. Biochem. Physiol. 30, 103–112 [Google Scholar]
- 64. Nemeria N., Chakraborty S., Baykal A., Korotchkina L. G., Patel M. S., Jordan F. (2007) The 1′,4′-iminopyrimidine tautomer of thiamin diphosphate is poised for catalysis in asymmetric active centers on enzymes. Proc. Natl. Acad. Sci. U.S.A. 104, 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tittmann K., Golbik R., Uhlemann K., Khailova L., Schneider G., Patel M., Jordan F., Chipman D. M., Duggleby R. G., Hübner G. (2003) NMR analysis of covalent intermediates in thiamin diphosphate enzymes. Biochemistry 42, 7885–7891 [DOI] [PubMed] [Google Scholar]
- 66. Balakrishnan A., Jordan F., Nathan C. F. (2013) Influence of allosteric regulators on individual steps in the reaction catalyzed by Mycobacterium tuberculosis 2-hydroxy-3-oxoadipate synthase. J. Biol. Chem. 288, 21688–21702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wille G., Meyer D., Steinmetz A., Hinze E., Golbik R., Tittmann K. (2006) The catalytic cycle of a thiamin diphosphate enzyme examined by cryocrystallography. Nat. Chem. Biol. 2, 324–328 [DOI] [PubMed] [Google Scholar]
- 68. Jordan F., Kudzin Z. H., Rios C. B. (1987) Generation and physical properties of enamines related to the key intermediate in thiamin diphosphate dependent enzymatic pathways. J. Am. Chem. Soc. 109, 4415–4416 [Google Scholar]
- 69. Patel H., Nemeria N. S., Andrews F. H., McLeish M. J., Jordan F. (2014) Identification of charge transfer transitions related to thiamin-bound intermediates on enzymes provides a plethora of signatures useful in mechanistic studies. Biochemistry 53, 2145–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Patel H. (2014) Ph.D. dissertation, Rutgers University, Newark, NJ [Google Scholar]
- 71. Patel H., Shim D. J., Farinas E. T., Jordan F. (2013) Investigation of the donor and acceptor range for chiral carboligation catalyzed by the E1 component of the 2-oxoglutarate dehydrogenase complex. J. Mol. Catal. B Enzym. 98, 42–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wagner T., Barilone N., Alzari P. M., Bellinzoni M. (2014) A dual conformation of the post-decarboxylation intermediate is associated with distinct enzyme states in mycobacterial KGD (α-ketoglutarate decarboxylase). Biochem. J. 457, 425–434 [DOI] [PubMed] [Google Scholar]
- 73. Fridovich I. (1975) Superoxide dismutases. Annu. Rev. Biochem. 44, 147–159 [DOI] [PubMed] [Google Scholar]
- 74. Bielski B. H. J., Cabelli D. E., Arudi R. L., Ross A. B. (1985) Reactivity of HO2/O2⨪ radicals in aqueous solution. J. Phys. Chem. Ref. Data 14, 1041–1100 [Google Scholar]
- 75. Tretter L., Adam-Vizi V. (2005) α-Ketoglutarate dehydrogenase: a target and generation of oxidative stress. Phil. Trans. R. Soc. B Biol. Sci. 360, 2335–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Adam-Vizi V., Tretter (2013) The role of mitochondrial dehydrogenases in the generation of oxidative stress. Neurochem. Int. 62, 757–763 [DOI] [PubMed] [Google Scholar]
- 77. Adam-Vizi V. (2005) Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid. Redox. Signal. 7, 1140–1149 [DOI] [PubMed] [Google Scholar]
- 78. Starkov A. A. (2013) An update on the role of mitochondrial α-ketoglutarate dehydrogenase in oxidative stress. Mol. Cell. Neurosci. 55, 13–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pan K., Jordan F. (1998) dl-S-Methyllipoic acid methyl ester, a kinetically viable model for S-protonated lipoic acid as the oxidizing agent in reductive acyl transfers catalyzed by the 2-oxoacid dehydrogenase multienzyme complex. Biochemistry 37, 1357–1364 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.