

Background: Mitochondrial DNA deletions are prominent in human genetic disorders and cancer.
Results: Predicted mitochondrial G-quadruplex-forming sequences map in close proximity to known deletion breakpoints and form G-quadruplexes in vitro.
Conclusion: The mitochondrial replicative helicase Twinkle inefficiently unwinds intra- and intermolecular G-quadruplexes.
Significance: Mitochondrial G-quadruplexes are likely to cause genome instability by perturbing replication machinery.
Keywords: DNA Helicase, DNA Replication, G-quadruplex, Genetic Disease, Genomic Instability, Mitochondria, Twinkle
Abstract
Mitochondrial DNA deletions are prominent in human genetic disorders, cancer, and aging. It is thought that stalling of the mitochondrial replication machinery during DNA synthesis is a prominent source of mitochondrial genome instability; however, the precise molecular determinants of defective mitochondrial replication are not well understood. In this work, we performed a computational analysis of the human mitochondrial genome using the “Pattern Finder” G-quadruplex (G4) predictor algorithm to assess whether G4-forming sequences reside in close proximity (within 20 base pairs) to known mitochondrial DNA deletion breakpoints. We then used this information to map G4P sequences with deletions characteristic of representative mitochondrial genetic disorders and also those identified in various cancers and aging. Circular dichroism and UV spectral analysis demonstrated that mitochondrial G-rich sequences near deletion breakpoints prevalent in human disease form G-quadruplex DNA structures. A biochemical analysis of purified recombinant human Twinkle protein (gene product of c10orf2) showed that the mitochondrial replicative helicase inefficiently unwinds well characterized intermolecular and intramolecular G-quadruplex DNA substrates, as well as a unimolecular G4 substrate derived from a mitochondrial sequence that nests a deletion breakpoint described in human renal cell carcinoma. Although G4 has been implicated in the initiation of mitochondrial DNA replication, our current findings suggest that mitochondrial G-quadruplexes are also likely to be a source of instability for the mitochondrial genome by perturbing the normal progression of the mitochondrial replication machinery, including DNA unwinding by Twinkle helicase.
Introduction
The human mitochondrial genome consists of a 16,569-bp circular DNA molecule that encodes essential subunits of mitochondrial complexes I, II, III, IV, and V in oxidative phosphorylation, a process responsible for the generation of the majority of the cellular ATP pool (1). Therefore, energy production is intimately linked to an intact and correctly coding mitochondrial DNA (mtDNA) sequence. Because of the utmost importance of the mitochondrion, it naturally follows that the accumulation of mitochondrial DNA mutations leading to mitochondrial dysfunction are implicated in human genetic disorders (2), neurodegeneration (3), cancer (4), and aging (5). Understanding the molecular basis for mitochondrial DNA mutations has attracted considerable interest in the clinical and basic science fields.
mtDNA deletions are prominent in genetic disorders such as Pearson marrow-pancreas syndrome (PMPS),5 Kearns-Sayre syndrome (KSS), and progressive external ophthalmoplegia (PEO) as well as various cancers and age-related diseases (6). mtDNA deletions are often located in regions of the genome flanked by tandem repeat sequences. It is proposed that at least one mechanism likely to underlie the mitochondrial genomic instability that leads to deletions is the stalling of the mitochondrial DNA synthesis machinery during replication. Mitochondrial replication stalling can lead to replication slippage, aberrant DNA structures, and double strand breaks, which can in turn promote mtDNA deletions. Although this model is attractive, the molecular determinants of mitochondrial replication stalling are not well understood.
The mitochondrial genome consists of a heavy (H) strand, which is rich in guanine bases, and a complementary light (L) strand, which is rich in cytosine bases. Because of its chemical nature, the guanine-rich (G-rich) strand is prone to form G-quadruplex (G4) DNA, an alternative DNA structure characterized by planar stacks of four guanines interacting by nonconventional Hoogsteen hydrogen bonds. G4 DNA is stabilized by a monovalent cation (typically Na+ or K+) that resides in the central barrel of the G-tetrad. There has been growing interest in nuclear G-quadruplex DNA metabolism in recent years with increasing evidence that G4 exists in vivo (7, 8) (for review see Refs. 9–11). However, it has been only very recently that researchers have begun to focus on the mitochondrial genome and its propensity to form G-quadruplexes or other alternate DNA structures. The Zakian laboratory (12) performed the first analysis of G4 DNA motifs in yeast mitochondria, which indicates a 10-fold greater density of G4 motifs in mitochondrial versus nuclear genomes of Saccharomyces cerevisiae. Damas et al. (13) conducted a computational analysis of human mtDNA deletions and correlated these with non-B DNA conformations including hairpin, cruciform, and cloverleaf-like elements. Their findings suggest that non-B form conformations are likely to contribute to the formation of mitochondrial deletions. In 2013, Oliveira et al. (14) described their findings from meta-analysis of human mtDNA sequence leading them to propose that noncanonical DNA structures, including helix-distorting intrinsically curved regions and long G-quadruplex elements, are likely to drive mtDNA rearrangements.
In the current study, we employed “Pattern Finder,” a G4 predictor algorithm, to map potential G4-forming (G4P) sequences throughout the human mitochondrial genome and chart them to known mitochondrial deletion breakpoints. We utilized this information to correlate G4P sequences with deletions characteristic of representative mitochondrial genetic disorders and also those identified in various cancers and aging. We performed physical analysis of representative predicted G4-forming sequences residing adjacent to clinically relevant mitochondrial deletions to substantiate that they do indeed form G-quadruplexes. Finally, we tested G-quadruplex DNA substrates with various topologies including a unimolecular G4 derived from human mitochondrial sequence for unwinding by the human mitochondrial DNA helicase Twinkle. Our results provide insight into the hypothesis that mitochondrial G-quadruplexes are likely to be a source of instability for the mitochondrial genome by perturbing the normal progression of the mitochondrial replication machinery.
EXPERIMENTAL PROCEDURES
Mitochondrial G-quadruplex Motif Prediction and Correlation with Mitochondrial Deletions
Mitochondrial G4P sequences were searched using Pattern Finder software to predict G4 in a user-defined quadruplex pattern for a given input sequence (15). Human mtDNA sequences used for analysis were taken from accession number NC_012920 (formerly, AC_000021). Mitochondrial L-strand (C-rich) and H-strand (G-rich) DNA was subjected to Pattern Finder analysis to assign G4 motifs with a stem size of 2–5 nucleotides (nt) and loop size of 1–7 nt. The mitochondrial deletion breakpoints are reported based on L-strand numbering. The mitochondrial deletion data were obtained from MitoMap and MitoBreak.
Circular Dichroism Spectroscopy, UV Melting Curves, and Thermal Difference Spectra
Oligonucleotides used to prepare G4 DNA substrates for biophysical analysis were purchased from Eurogentec (Seraing, Belgium) without further purification. The oligonucleotides derived from human mitochondrial sequence were: KSS, 5′-GGGGAGGGGTGTTTAAGGGGTTGGCTAGGG-3′; human renal cell carcinoma (HRCC), 5′-GGGGGTTGGGTATGGGGAGGGGGG-3′; and PMPS, 5′-GGGACGCGGGCGGGGGATATAGGG-3′. Strand concentrations were determined by UV absorbance using the extinction coefficients provided by the manufacturer. All of the chemicals used were purchased from Sigma unless otherwise stated. The samples were heated to 95 °C for 5 min, cooled slowly to room temperature, and then stored at 4 °C before use unless otherwise stated (the concentration of each strand was 5 μm, and the buffer was prepared with 10 mm cacodylic acid buffered to pH 7.0 with LiOH containing 100 mm K+ or Na+).
Circular dichroism (CD) spectra were recorded on a Jasco J-815 equipped with a Peltier temperature control accessory (Jasco, Hachioji, Japan). Each CD spectrum was obtained by averaging five scans at a speed of 100 nm/min, and the collection temperature was set at 20 °C. UV absorbance measurements were performed on an Uvikon XS (Secomam) equipped with a circulating water bath. Temperature was measured with an inert glass sensor immersed into a quartz cell. UV absorbance was monitored at 295 nm with a temperature gradient of 0.5 °C/min. For each sample, the thermal difference spectrum was obtained by subtracting the absorbance spectrum at lower temperature from the one at higher temperature as described previously (16).
Recombinant Helicase Proteins
Recombinant human Twinkle protein (gene product of c10orf2) with a C-terminal hexahistidine tag was expressed in bacterial cells and purified as described previously (17). Recombinant human Fanconi anemia complementation group J (FANCJ) protein with a C-terminal FLAG tag was expressed in insect cells and purified as described previously (18). DnaB was purified from Escherichia coli as described previously (19).
DNA Helicase Assays
Gel-purified oligonucleotides used to prepare G4 DNA substrates for helicase assays were purchased from Lofstrand Labs Ltd. (Gaithersburg, MD) (Table 1). The four-stranded (tetramolecular) parallel TP-G4 substrate (20), two-stranded (bimolecular) anti-parallel OX-1-G2′ DNA substrate (20), four-stranded TelR2-G4 (20), four-stranded CEB1-G4 (21, 22), and unimolecular poly(A) Zic1-G4 substrate with 5′ single-stranded DNA (ssDNA) tail lengths of 20, 30, or 40 nt were prepared from gel-purified oligonucleotides as described previously (23). The 19-bp forked duplex DNA substrate was prepared by annealing DC26 and TSTEM25 as described earlier (24).
TABLE 1.
DNA substrates used in this study

Standard helicase reaction mixtures (20 μl) containing 5 fmol of the indicated TP-G4 or OX-1-G2′ DNA substrate or the forked duplex DNA substrate (0.25 nm DNA substrate concentration) and the indicated concentration of FANCJ (20) or DnaB hexamer (25) were as described previously unless stated otherwise. For the Twinkle hexamer, the reaction conditions for the forked duplex were as described previously (26) except for the inclusion of 25 mm KCl and the additional presence throughout the incubation period of a 100-fold excess of oligonucleotide of the same sequence as the labeled strand in the partial duplex substrate to serve as a displaced strand trap (17). Forked duplex helicase reactions were terminated as described previously and resolved on nondenaturing 12% polyacrylamide (19:1 acrylamide-bisacrylamide) gels (24). Twinkle reaction conditions for the intermolecular G4 substrates (TP-G4, OX-1-G2′, TelR2-G4, and CEB1-G4) were the same as for the forked duplex except there was no additional oligonucleotide present during the reaction. For the unimolecular poly(A) Zic1-G4 or HRCC-G4 DNA substrate (5 fmol), helicase reactions (20 μl) were performed in a reaction buffer containing a 20-fold excess of peptide-nucleic acid complementary oligonucleotide (23). G4 helicase reactions were terminated by the addition of 20 μl of stop buffer (74% glycerol, 0.01% xylene cyanol, 0.01% bromphenol blue, 10 mm KCl, and 20 mm EDTA). Reaction products for intermolecular and unimolecular G4 DNA substrates were resolved on 10 and 15% nondenaturing polyacrylamide (19:1 acrylamide-bisacrylamide) gels, respectively. Gels were scanned by PhosphorImager and quantitated as described previously (24).
RESULTS
Bioinformatic Analysis of Potential G-quadruplex-forming Sequences in the Human Mitochondrial Genome and Their Locations with Respect to Deletion Breakpoints
The human mitochondrial genome contains G-rich sequences predicted to form G-quadruplex DNA (Fig. 1); G4P sequences can be found in both the L-strand (top) and H-strand (bottom). However, a greater number of G4P sequences are present in the H-strand, which is also the G-rich strand. G4P sequences reside within the coding sequences of mitochondrial genes and also the tRNA genes. G4P sequences can also be found to a lesser extent in noncoding regions of the mitochondrial genome.
FIGURE 1.

Graphical representation of predicted G4 motifs and deletion breakpoints in the human mitochondrial genome coding and noncoding sequences. Mitochondrial genes, tRNA, and G-rich sequences predicted to form G4 are shown in the L-strand (top) and the H-strand (bottom). The 5′ and 3′ deletion breakpoints, collected from MitoMap and MitoBreak, residing within or adjacent to predicted G4-forming sequences, are shown. The number of guanines (2–5 G) and loop size (1–7 nt) were employed for search criteria using QuadBase Pattern Finder. The predicted G4-forming sequences with parameters ≥G3 and loop size 1–7 nt are also shown by bold dots. RNR1, 12 S ribosomal RNA; RNR2, 16 S ribosomal RNA; ND1, NADH dehydrogenase 1; ND2, NADH dehydrogenase 2; COX1, cytochrome c oxidase I; COXII, cytochrome c oxidase II; ATP8, ATP synthase 8; ATP6, ATP synthase 6; COXIII, cytochrome c oxidase III; ND3, NADH dehydrogenase 3; ND4L, NADH 4L dehydrogenase; ND4, NADH dehydrogenase 4; ND5, NADH dehydrogenase 5; ND6, NADH dehydrogenase 6; CYTB, cytochrome b.
We compared the G4P sequences in human mitochondrial DNA with other mammalian species to determine the degree of conservation. The number of G4 motifs in the mitochondrial genomes of mouse, rat, and monkey was markedly less than found in human (Table 2). The number of conserved G4P sequences identified by the Pattern Finder parameters between these species and humans was greatest in monkey followed by rat and then mouse.
TABLE 2.
Evolutionary conservation of G4 motifs in mitochondrial genomes of human and selected mammals
| Organisma | Mitochondrial genome | Sequence similarity | Total G4 motifsb | Conserved G4 motifsc |
|---|---|---|---|---|
| bp | % | |||
| Human | 16,569 | 100 | 270 | 270 |
| Monkey | 16,564 | 78 | 148 | 81 |
| Rat | 16,313 | 67 | 108 | 46 |
| Mouse | 16,299 | 66 | 59 | 34 |
a Accession numbers: human (NC_012920 AC_000021); monkey (NC_005943); rat (NC_001665); mouse (NC_005089).
b Total G4P motifs obtained based on stem size of 2–5 nt and loop size of 1–7 nt through Pattern Finder analysis.
c Number of predicted G4 motifs that are common in the indicated mammal and human.
The human mitochondrial genome deletions (collected from MitoMap and MitoBreak) that reside adjacent to the G4P sequences derived from our analysis are shown in Fig. 1. We identified both 5′ and 3′ deletion breakpoints that reside in the G4P sequence or adjacent (within 20 bp) to the G4P sequence. The mitochondrial genome deletion breakpoints were mapped proximal to G4P sequences located within the coding regions of mitochondrial genes and tRNA genes, as well as noncoding regions to a lesser extent.
To ascertain the relative abundance of deletion breakpoints in predicted mitochondrial G4-forming sequences, we compared the distribution and density of unique mitochondrial deletion breakpoints in G4P sequences, tRNA genes, and the D-loop region (Table 3). The total number of mitochondrial 5′ or 3′ deletion breakpoints was greater in G4P as compared with tRNA or the D-loop region. We next determined the deletion breakpoint density (deletions/kb) for each region of mitochondrial DNA. The 5′ mitochondrial deletion breakpoint density was ∼1.4- and 7.6-fold greater in G4P as compared with tRNA genes and the D-loop, respectively. The 3′ deletion breakpoint density of mitochondrial G4P was 1.4- greater and 0.8-fold less than that of tRNA genes and D-loop, respectively (Table 3).
TABLE 3.
Distribution and density of 5′ and 3′ deletion breakpoints
The number of unique deletion breakpoints = 620 5′ deletion breakpoints and 497 3′ deletion breakpoints (13).
| Deletion breakpoint | G4Pa |
tRNAb |
D-loop (AT-rich)c |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Deletion breakpoints |
Deletion breakpoints |
Deletion breakpoints |
|||||||
| Total no.d | %e | Density (deletions/kb)f | Total no.d | %e | Density (deletions/kb)f | Total no.d | %e | Density (deletions/kb)f | |
| 5′ deletion breakpoints | 194 | 31 | 38 | 57 | 9 | 28 | 6 | 1 | 5 |
| 3′ deletion breakpoints | 125 | 25 | 25 | 36 | 7 | 18 | 37 | 7 | 31 |
a Number of deletion breakpoints in G4P excluding those overlapping in tRNA and D-loop region.
b Number of deletion breakpoints in tRNA excluding those overlapping in G4P or D-loop region.
c Number of deletion breakpoints in D-loop excluding those overlapping in G4P or tRNA.
d Total number of unique deletion breakpoints in the case of G4P, tRNA, and D-loop.
e Number of deletion breakpoints in percentage (%) with respect to either 620 5′ deletions or 497 3′ deletions for G4P, tRNA, and D-loop.
f Deletion breakpoint density is based on 5071, 2017, and 1180 bp, respectively, for G4P, tRNA, and D-loop.
Association of Potential G-quadruplex-forming Sequences with Mitochondrial DNA Deletion Breakpoints Prevalent in Human Disease
Our computational analysis revealed that G4P sequences reside proximal to mtDNA deletions found in a number of genetic diseases (Table 4). Among the genetic disorders associated with mitochondrial deletions near G4P sequences is progressive external ophthalmoplegia. PEO is characterized by the adult onset of muscular deficiencies, including those of the external eye and skeletal system (27). G4P sequences were also located adjacent to mitochondrial deletion breakpoints associated with KSS, a clinical subgroup of mitochondrial encephalomyopathies associated with PEO and skeletal muscle defects (28).
TABLE 4.
Selected disease-associated mitochondrial deletion breakpoints in proximity to predicted G4-forming sequences
| Deletion breakpointa | G4P neighboring sequenceb | Genetic disease | References |
|---|---|---|---|
| 5′ 10190 | (10187) tata↓tcccccgcccgcgtcccttt (10210) | Pearson marrow-pancreas syndrome | (29, 84) |
| 3′ 13753 | (13751) tt↓tcccccgcatcccccttccaaacaacaatccccctcta (13790) | ||
| 5′ 12113 | (12081) tatcccccattctcctcctatccctcaaccccg↓ac (12115) | ||
| 5′ 12103 | (12078) cctatcccccattctcctcctatcc↓ctc (12106) | Kearns-Sayre syndrome, mitochondrial encephalomyopathy | (85–87) |
| 3′ 14414 | (14412) cc↓tcaacccctgacccccatgcc (14435) | ||
| 5′ 4398 | (4374) ccgtgccacctatcacaccccatcc↓ta (4400) | Diabetes mellitus | (88, 89) |
| 3′ 14822 | (14806) cctccccaccccatcc↓aacatctcc (14830) | ||
| 5′ 7974 | (7942) ctcctacatacttcccccattattcctagaacc↓agg (7977) | Kearns-Sayre syndrome, mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes syndrome | (90, 91) |
| 3′ 15496 | (15495) t↓aggcgacccagacaattataccctagccaaccccttaaacacccctccccacatc (15550) | ||
| 5′ 6325 | (6325) c↓ctccgtagacctaaccatcttctcct (6351) | Mitochondrial myopathy | (92) |
| 3′ 13989 | (13971) ccaaaacctgcccctact↓cctcctagacctaacc (14004) | ||
| 5′ 6329 | (6311) ctcccaccctggagcctc↓cgtagacc (6336) | Chronic progressive external ophthalmoplegia | (93) |
| 3′ 13994 | (13970) gccaaaacctgcccctactcctcc↓tagacctaacc (14004) | ||
| 5′ 6330 | (6311) ctcccaccctggagcctccg↓tagacctaacc (6341) | Kearns-Sayre syndrome, Pearson marrow-pancreas syndrome | (87) |
| 3′ 13994 | (13970) gccaaaacctgcccctactcctcc↓tagacctaacc (14004) | ||
| 5′ 7491 | (7365) accccccaaagctggtttcaagccaac↓cccatggcctcc (7503) | Kearns-Sayre syndrome, progressive external ophthalmoplegia | (85) |
| 3′ 11004 | (10995) caagccaac↓gccacttatccagtgaacc (11022) |
a Deletion breakpoints reside within the G4P sequence or ≤20 nt away from G4P. The breakpoint is indicated by a down arrow.
b L-strand sequence is shown (5′ to 3′). The predicted G4-forming sequence (G4P) is underlined.
Our analysis revealed that a G4P sequence is located in a breakpoint region of a mitochondrial DNA deletion associated with PMPS, a fatal disease of early infancy affecting the hematopoietic system and pancreas exocrine system (29) (Table 4). The molecular features of PMPS are attributed to a large-scale deficiency in oxidative phosphorylation. The site-specific mitochondrial deletions associated with PMPS and other mitochondrial disorders (KSS and PEO) are characterized by direct repeats of 8–13 bp present in the normal mitochondrial genome, which flank the deletion breakpoints (29–31). It has been proposed that homologous recombination may be responsible for the direct repeat signature pattern characteristic of PMPS and other sporadic mtDNA deletion disorders (32). The common 4977-bp deletion flanked by a 13-bp repeat in the normal mtDNA sequence frequently identified in sporadic PEO and other mitochondrial disorders (33) is characterized by a 5′ breakpoint at mtDNA position 8468 bp, which resides in a predicted G4-forming sequence according to the Pattern Finder algorithm. Unusual secondary DNA structures such as G-quadruplexes may be prone to form in the mitochondrial genome at loci such as the 5′ breakpoint of the common 4977-bp deletion and interfere with normal mitochondrial DNA metabolic processes such as replication or repair, leading to double strand breaks.
Association of Potential G-quadruplex-forming Sequences with Mitochondrial DNA Deletion Breakpoints Found in Cancer
We identified a number of cases in which G4P sequences flanked mtDNA deletion breakpoints reportedly associated with cancer (Table 5). G4P sequences flanking mtDNA deletions in thyroid tumors were prominent. G4P-associated mitochondrial genome instability may be relevant to previous observations that a subset of thyroid tumors display elevated mtDNA deletions (34). A relatively uncommon and small 264-bp deletion in the mitochondrial coding sequence for the first subunit (ND1) of NADH:ubiquinone oxidoreductase of the electron transport chain was reported in a HRCC (35). This deletion is flanked by G4P sequences according to our computational analysis (Table 5). Biophysical studies of the G4P flanking sequence confirmed its ability to form G4 (see Fig. 2A and “Biophysical Analysis of Selected G4P Mitochondrial Sequences” below). A 6-bp direct repeat flanked the deleted region (35), suggesting that replication slippage due to mtDNA synthesis stalling at G4 DNA may play a role.
TABLE 5.
Selected cancer-associated mitochondrial deletion breakpoints in proximity to predicted G4-forming sequences
| Deletion breakpointa | G4P neighboring sequenceb | Cancer | References |
|---|---|---|---|
| 5′ 3323 | (3310) cccatggccaacct↓cctactcc (3331) | Human renal cell carcinoma | (35) |
| 3′ 3588 | (3566) cccccctccccatacccaaccc↓cctggtcaaccct (3600) | ||
| 5′ 8290 | (8252) cccgtatttaccctatagcaccccctctaccccctctag↓agcccactgt (8300) | Thyroid tumor | (51) |
| 3′ 16275 | (16250) ccaaagccacccctcacccactagg↓ataccaacaaacctacccaccc (16296) | ||
| 5′ 8281 | (8262) ccctatagc accccctctac↓cccctctag agccc (8295) | ||
| 3′ 16371 | (16351) atcccttctc gtccccatgg↓atgacccccc (16380) | ||
| 3′ 13383 | (13364) ccgggtccatcatccacaa↓ccttaaca (13390) | ||
| 3′ 16280 | (16250) ccaaagccacccctcacccactaggatacc↓aacaaacctacccaccc (16296) | ||
| 5′ 8261 | (8252) cccgtattta↓ccctatagcaccccctctaccccctctagagccc (8295) | Colon adrenocarcinoma | (94) |
| 5′ 15591 | (15568) cctattcgcctacacaattctccg↓atccgtccc (15660) | Lung cancer | (52) |
| 5′ 8992 | (8983) ccaatagccc↓tggccgtacgcctaaccg (9010) | Hepatic tumor | (36) |
| 5′ 306 | (295) ccaccaaacccc↓ccctcccccgcttctggcc (325) | Hepatocellular carcoma | (95) |
| 3′ 16280 | (16250) ccaaagccacccctcacccactaggatacc↓aacaaacctacccaccc (16296) | Thyroid tumor | (96) |
a Deletion breakpoints reside within G4P sequence or ≤20 nt away from G4P. The breakpoint is indicated by a down arrow.
b L-strand sequence is shown (5′ to 3′). The predicted G4-forming sequence (G4P) is underlined.
FIGURE 2.
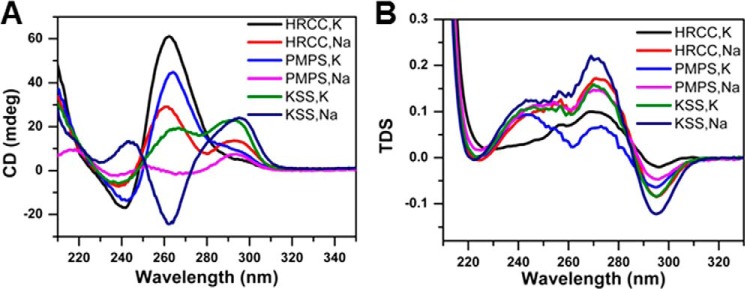
Circular dichroism and UV spectral analyses of G-rich human mitochondrial DNA sequences in the proximity of deletion breakpoints prevalent in human renal cell carcinoma, Pearson marrow-pancreas syndrome, and Kearns-Sayre syndrome. Oligonucleotide sequences used for biophysical analysis are listed under “Experimental Procedures.” A, CD spectra for 5 μm samples as indicated, in 10 mm lithium cacodylate buffer (pH 7.0) containing 100 mm Na+ or K+. The spectra were collected at 20 °C. B, thermal difference spectra (TDS) for 5 μm samples as indicated, in 10 mm cacodylate buffer (pH 7.0) containing 100 mm Na+ or K+.
Using the G4P algorithm, we determined that a G4P sequence flanked a 7079-bp mtDNA deletion site reported to exist in cirrhotic liver that surrounds hepatic tumors (36) (Table 5). This mtDNA deletion was not characterized by direct repeat flanking sequences, suggesting that a mechanism other than replication slippage may be involved. Interestingly, the 7079-bp mtDNA deletion was detected only in the cirrhotic liver surrounding the hepatic tumor but not in the tumor itself, suggesting that mitochondrial dysfunction contributes to liver cirrhosis, which in turn serves as a risk factor for liver cancer. Next generation sequencing of mtDNA from tumor-derived tissues should provide a more comprehensive analysis of the mtDNA mutation spectra, which may be informative for understanding the molecular mechanism underlying the origin of the mtDNA deletions associated with cancer and the potential role of G4P sequences.
Association of Potential G-quadruplex-forming Sequences with Mitochondrial DNA Deletion Breakpoints Observed in Aging
Using the computational analysis to map G4P sequences flanking mitochondrial deletion breakpoints, we sought to determine whether G4P sequences might play a role in the mitochondrial genome instability prevalent in aged tissues of the human body based on a survey of the literature. A comparison of the mitochondrial G4P sequences with reported mtDNA deletions associated with aging suggest this to be the case (Table 6). We will briefly discuss several examples to illustrate the potential importance of G4-forming sequences in mitochondrial DNA deletions found in aging tissues. An analysis of photo-aged tumor-free skin of nonmelanoma skin cancer patients reveals an increase in mitochondrial DNA deletions that correlates with patient age (37). Most of the identified mtDNA deletions contained repeat sequences, suggesting a replication slippage model. This would be consistent with the proposal that mtDNA photoadducts interfere with normal mtDNA replication; however, G4 DNA may also contribute to replication stalling and mitochondrial genomic instability. Understanding the potential role of G-quadruplexes in replication slippage or other aspects of replication mismanagement is an important area of study that deserves greater attention. Aberrant DNA structures such as G4 DNA, in addition to bulky adducts imposed by chronic UV exposure, may contribute to the accumulation of mtDNA deletions in skin during chronological aging.
TABLE 6.
Selected age-associated mitochondrial deletion breakpoints in proximity to predicted G4-forming sequences
| Deletion breakpointa | G4P neighboring sequenceb | Aging features | References |
|---|---|---|---|
| 5′ 540 | (535) ccccata↓ccccgaaccaaccaaaccccaaagacacccccc (573) | Heart tissue | (97) |
| 3′ 13764 | (13751) tttcccccgc atc↓ccccttcc ( 13771) | ||
| 5′ 5433 | (5421) gaacatacaaaa↓cccaccccattcctcccc (5450) | ||
| 3′ 13023 | (13010) cccaattaggtct↓ccacccctgactcccctcagcc (13044) | ||
| 5′ 1491 | (1482) cacaccgcccgtc↓accctcctc (1500) | Skin tissue | (37) |
| 3′ 5206 | (5191) cacccttaattccat↓ccaccctcctctccc (5220) | ||
| 5′ 6329 | (6311) ctcccaccctggagcctcc↓gtagacctaac catcttctcc (6350) | Skeletal muscle | (98) |
| 3′ 13994 | (13971) ccaaaacctgcccctactcctcc↓tagacctaacc (14004) | ||
| 5′ 6437 | (6420) ccccctgccataacccaa↓taccaaacgcccc (6450) | Substantia nigra neurons | (99) |
| 3′ 14077 | (14043) ccaaatct ccacctccat catcacctcaacccaa↓aaag (14080) | ||
| 5′ 6501 | (6463) ccgtcctaatcacagcagcctatctctcc↓cagtcctagc (6511) | Substantia nigra neurons | (38) |
| 3′ 13802 | (13781) tccccctctacctaaaactca↓cagccctcg (13810) | ||
| 3′ 16284 | (16250) ccaaagccacccctcacccactaggataccaaca↓aacctacccaccc (16296) | Cochlear tissue | (100) |
| 5′ 7409 | (7396) gccccccaccctac↓cacacattcga (7420) | Substantia nigra neurons of aged individual with Parkinson's disease | (101) |
| 3′ 5447 | (5333) cccaccccattcct↓ccccacactcatcgcccttaccac gctactccta cctatctcccc (5491) | Skeletal muscle | (102) |
a Deletion breakpoints reside within G4P sequence or ≤20 nt away from G4P. The breakpoint is indicated by a down arrow.
b L-strand sequence is shown (5′ to 3′). The predicted G4-forming sequence (G4P) is underlined.
A decline in mitochondrial function is prevalent in age-related disorders characterized by neurodegeneration (6). Kraytsberg et al. (38) found that aged human substantia nigra contain a great abundance of mitochondrial DNA deletions, some of which we determined to be in close proximity to G4P sequences (Table 6). It was observed that mtDNA deletions were enriched in cytochrome c oxidase-deficient neurons, suggesting that the mitochondrial genome instability was responsible for impaired cellular respiration and functional impairment of the aged neurons of the substantia nigra (38). Further work in this area to explore the potential role of G-quadruplexes in brain mtDNA deletions may be informative for age-related disorders such as Parkinson disease, which is characterized by primary neurodegeneration in the substantia nigra (39).
Biophysical Analysis of Selected G4P Mitochondrial Sequences
To determine whether representative mitochondrial G4P sequences residing adjacent to known deletion breakpoints form G-quadruplex structures, the classical spectroscopic methods of CD and UV light absorption were used. The oligonucleotides selected for physical analysis corresponded to G4P sequences flanking mitochondrial DNA deletion breakpoints detected in PMPS, KSS, and HRCC. CD spectral analysis demonstrated that HRCC and PMPS showed a positive peak around 265 nm and negative peak at 240 nm in the presence of 100 mm K+ (Fig. 2A), suggesting that parallel G-quadruplex structures were formed (40). However, PMPS and KSS exhibited positive and negative peaks around 295 and 260 nm, respectively, in the presence of 100 mm Na+, which is in agreement with the formation of nonparallel G-quadruplexes (40). Interestingly, both HRCC in Na+ and KSS in K+ displayed two positive peaks around 295 and 260 nm. These spectra are similar to those reported previously for the G-rich hTERT promoter, implying the coexistence of two different G-quadruplex conformations (41). Furthermore, the thermal difference spectra of all three sequences showed a negative peak around 295 nm and two positive peaks around 275 and 243 nm (Fig. 2B), which is a typical feature of G-quadruplex DNA (42). In addition, UV melting profiles of all these sequences exhibited a hypochromic transition at 295 nm, which is in agreement with the formation of G-quadruplex structures (42) (Fig. 3). Interestingly, Tm depends on the nature of the cation (compare left- and right-hand panels in Fig. 3), which is strong evidence for G-quadruplex formation (42).
FIGURE 3.

UV melting profiles of G-rich human mitochondrial DNA sequences (A–F). UV melting profiles at 295 nm of G4-prone sequences (5 μm) strand concentrations of G4-prone sequences in the presence of 100 mm Na+ or K+, as indicated in the respective panels. Arrows indicate direction of temperature changes.
Assessment of Twinkle Helicase to Unwind G-quadruplex DNA Substrates
Based on our computational analysis and biophysical studies, it is highly probable that human mitochondrial DNA forms G4 structures under physiological conditions. This led us to consider how the mitochondrial replication machinery might tolerate G-quadruplexes. The gene product of c10orf2, also known as Twinkle, is a DNA helicase required for the replication of human mitochondrial DNA (43). To acquire a better understanding of the potential role of Twinkle helicase in mitochondrial G-quadruplex DNA metabolism, we purified recombinant human Twinkle protein for biochemical assays with G4 DNA substrates. A representative Coomassie-stained polyacrylamide gel showing the purified recombinant Twinkle protein appears in Fig. 4A. Purified recombinant Twinkle protein unwound a forked duplex (19 bp) DNA substrate in a protein concentration-dependent manner (Fig. 4, B and C). DNA unwinding by Twinkle was ATP-dependent, as demonstrated by its poor activity in the absence of ATP or the presence of ATPγS (Fig. 4D).
FIGURE 4.

Purified recombinant Twinkle protein and its helicase activity on a forked duplex DNA substrate. A, Coomassie-stained denaturing SDS-polyacrylamide gel showing the purity of recombinant Twinkle protein (500 ng). kD (kilodalton), molecular mass protein standards. B, representative native polyacrylamide gel image showing helicase products from reaction mixtures containing the indicated concentration of purified recombinant Twinkle protein (hexamer) incubated with 19-bp forked duplex DNA substrate (0.25 nm) for 30 min at 30 °C as described under “Experimental Procedures.” Products were resolved on native 12% polyacrylamide gels, and a representative phosphorimage of a typical gel is shown. Triangle, heat-denatured DNA substrate control. Asterisk, denotes 5′-32P end label. C, quantitative analysis of data from Twinkle helicase reactions with forked duplex DNA substrate is shown. Data represent the mean of at least three independent experiments with S.D. indicated by error bars. D, Twinkle-catalyzed DNA unwinding is dependent on ATP. Twinkle (12.5 nm) was incubated with the 19-bp fork duplex substrate (0.25 nm) and the indicated nucleotide for 30 min at 30 °C and subsequently analyzed as described under “Experimental Procedures.”
We began tests for putative Twinkle G4 unwinding activity by assessing its ability to unwind previously characterized intermolecular G4 substrates with two distinct topologies: a four-stranded (tetramolecular) parallel TP-G4 substrate and a two-stranded (bimolecular) anti-parallel OX-1-G2′ substrate (Fig. 5). We monitored unwinding of the TP-G4 substrate with a 21-nt 5′ ssDNA tail (TP-G4–21) as a function of Twinkle helicase concentration (Fig. 5A). A low but detectable level of activity was observed, which reached 9% of the substrate unwound at 25 nm Twinkle hexamer concentration. In contrast, FANCJ unwound nearly all of the TP-G4–21 substrate at 3 nm protein monomer concentration (Fig. 5B). Twinkle (12.5 nm hexamer) inefficiently unwound the TP-G4 substrate with a 40-nt 5′ ssDNA tail TP-G4 (TP-G4–40) (Fig. 5C, lane 2), whereas the TP-G4–40 substrate was unwound efficiently by FANCJ (Fig. 5C, lane 4), as reported previously (20). We tested whether Twinkle would unwind the bimolecular OX-1-G2′ substrate with a 20-nt 5′ ssDNA tail (Fig. 5D). Twinkle showed little to no activity throughout the protein titration (Fig. 5D, lanes 2–7). In contrast, FANCJ (3 nm monomer) efficiently unwound OX-1-G2′ (Fig. 5D, lane 10), as reported previously (20). Quantitative analyses of Twinkle helicase activity on the TP-G4 or OX-1-G2′ substrates are shown in Fig. 5E. In contrast to the low unwinding activity on the G4 substrates, under identical reaction conditions Twinkle (12.5 nm hexamer) unwound to near completion the 20-bp forked duplex DNA substrate (Fig. 5F).
FIGURE 5.

Twinkle inefficiently unwinds intermolecular G4 DNA substrates TP-G4 and OX-1-G2′. Helicase reactions (20 μl) were performed at 30 °C for 30 min under standard helicase reaction conditions as described under “Experimental Procedures.” A, Twinkle hexamer titration in reaction mixtures containing 5 fmol of tetramolecular TP-G4 with a 21-nt 5′ ssDNA tail (TP-G4–21). B, FANCJ (3 nm monomer) efficiently unwinds 5′ ssDNA tail (TP-G4–21). C, Twinkle hexamer (12.5 nm) or FANCJ monomer (3 nm) was incubated with 5 fmol of TP-G4 with a 40-nt 5′ ssDNA tail (TP-G4–40). D, Twinkle hexamer titration on bimolecular G4 DNA (OX-1-G2′) with a 20-nt 5′ ssDNA tail. As a control, FANCJ (3 nm monomer) unwound OX-1-G2′ substrate. E, quantitative analysis of data from Twinkle helicase reactions with TP-G4–21 and OX-1-G2′ as shown in A and D, respectively. Data represent the mean of at least three independent experiments with S.D. indicated by error bars. F, Twinkle hexamer (12.5 nm) or FANCJ monomer (3 nm) unwound 20 bp forked duplex with 20 nt 5′ and 3′ ssDNA tails. Products were resolved on native 10% polyacrylamide gels, and representative phosphorimages of typical gels are shown. NE, no enzyme control; open triangle, heat-denatured DNA substrate control or ssDNA used as a marker.
To further investigate whether Twinkle was able to unwind G4 DNA with distinct sequence and topology, we tested its activity on two previously well characterized G4 substrates, one formed by a human telomeric sequence designated TelR2-G4–20 (20) and the other formed by a human CEB1 minisatellite G-rich sequence designated CEB1-G4–20 (21, 22). As reported previously (20), FANCJ efficiently unwound to near completion the TelR2-G4–20 substrate (Fig. 6A). We detected modest Twinkle helicase activity on the TelR2-G4–20 substrate, reaching a plateau of ∼22% of the substrate unwound even at the greatest Twinkle concentration tested, 125 nm hexamer (Fig. 6B). Twinkle-catalyzed unwinding of the TelR2-G4–20 substrate was dependent on hydrolysable ATP, as evident by the absence of helicase activity when the reactions were conducted in the presence of ATPγS (Fig. 6C). A quantitative assessment of Twinkle helicase activity on TelR2-G4–20 is shown in Fig. 6D. The G4 ligand telomestatin, shown previously to inhibit FANCJ unwinding of the TelR2-G4–20 substrate (20), also inhibited the modest DNA unwinding catalyzed by Twinkle in the presence of ATP (Fig. 6E) but did not affect Twinkle unwinding of a forked duplex DNA substrate (Fig. 6F).
FIGURE 6.

Twinkle shows modest activity for unwinding a G4 substrate formed by human telomeric G-rich sequence but is inactive on G4 formed by CEB1 minisatellite G-rich sequence. Helicase reactions (20 μl) were performed at 30 °C for 30 min with the indicated helicase and DNA substrate (5 fmol) under standard helicase reaction conditions as described under “Experimental Procedures.” A, FANCJ (3 nm monomer) was incubated with TelR2-G4–20 substrate. B, the indicated concentration of Twinkle hexamer was incubated with TelR2-G4–20 substrate. C, Twinkle hexamer was incubated with TelR2-G4–20 substrate as in B, except 4 mm ATPγS was substituted for ATP. D, quantitative analysis of data from Twinkle helicase reactions with TelR2-G4–20 substrate in the presence of ATP or ATPγS is shown. Data represent the mean of at least three independent experiments with S.D. indicated by error bars. E and F, the indicated concentrations of the G4 ligand telomestatin (TMS) was included in reaction mixtures containing 6.12 nm Twinkle hexamer and the TelR2-G4–20 DNA substrate (E) or a 19-bp forked duplex (F). G, FANCJ (3 nm) was incubated with CEB1-G4–20 substrate. H, the indicated concentrations of Twinkle hexamer were incubated with CEB1-G4–20 substrate. NE, no enzyme control or ssDNA used as a marker.
Next, we tested whether Twinkle helicase would unwind a G4 substrate formed by the CEB1 minisatellite G-rich sequence with a 20-nt 5′ single-stranded tail, designated CEB1-G4–20. This G4 substrate was reported previously to be unwound by the Pif1 helicase (21, 22). FANCJ (3 nm monomer) unwound the CEB1-G4–20 substrate to near completion (Fig. 6G). In contrast, we detected little to no activity of Twinkle on this substrate up to 140 nm hexamer concentration (Fig. 6H). Based on these results, we concluded that Twinkle did not unwind the CEB1-G4 substrate under the conditions in which it efficiently unwound a simple forked duplex substrate.
The relatively weak efficiency of Twinkle in unwinding the intermolecular G4 substrates raised the question of whether other helicases that share sequence homology with Twinkle are able to unwind G4 DNA. Therefore, we tested the E. coli replicative DnaB helicase, a member of superfamily 4 to which Twinkle belongs (44) (Fig. 7A). DnaB, like Twinkle, is a 5′-to-3′ hexameric DNA helicase (45). The results from helicase protein titrations demonstrated that DnaB unwound the OX-1-G2′ bimolecular G4 substrate in a protein concentration-dependent manner (Fig. 7B) similar to that observed for the forked duplex substrate (Fig. 7C), albeit with a slightly reduced efficiency (Fig. 7E). In contrast, DnaB failed to unwind the four-stranded parallel TP-G4 substrate (Fig. 7, D and E). DnaB unwound the OX-1-G2′ substrate in the presence of ATP, as evidenced by the product of the helicase reaction, which comigrated with the unannealed control radiolabeled oligonucleotide (Fig. 7, B and F). DnaB failed to unwind the G4 substrate in the absence of ATP or in the presence of ATPγS (Fig. 7F), indicating that DnaB-catalyzed G4 unwinding requires the hydrolysis of nucleoside triphosphate. Based on these results, we concluded that, unlike the sequence-related Twinkle helicase, DnaB is able to unwind the two-stranded anti-parallel G2′ substrate.
FIGURE 7.

DnaB unwinds anti-parallel bimolecular G4 DNA substrate. A, conserved helicase motifs in Homo sapiens Twinkle and E. coli DnaB proteins. B–D, DnaB was tested for helicase activity on various DNA substrates. The indicated concentration of DnaB hexamer was incubated at 37 °C for 15 min with 5 fmol of the OX-1-G2′ bimolecular G4 DNA substrate (B), a 19-bp forked duplex DNA substrate (C), or TP-G4–21 four-stranded parallel DNA substrate (D) under standard helicase conditions as described under “Experimental Procedures.” Lane 8, open triangle, heat-denatured DNA substrate control (C) or ssDNA as a marker control (B, D, and F). E, quantitative analysis of helicase activity on all DNA substrates is shown with S.D. indicated by error bars. Filled squares, 19-bp forked duplex; filled diamonds, OX-1-G2′; filled triangles, TP-G4–21. F, DnaB unwinding of OX-1-G2′ is ATP-dependent. Reactions were performed as described under “Experimental Procedures” in the presence or absence of ATP or the poorly hydrolyzable ATP analog ATPγS. Products were resolved on native 10% polyacrylamide gels, and representative phosphorimages of typical gels are shown.
We next assessed the ability of Twinkle helicase to unwind an entropically favored unimolecular G4 DNA substrate (poly(A) Zic1) that was shown previously to be unwound by FANCJ helicase (46). Twinkle was tested on a series of poly(A) Zic1-unimolecular G4 DNA substrates with increasing 5′ single-stranded DNA tail lengths of 20, 30, and 40 nt and was found to be completely inactive for unwinding (Fig. 8, A–C). In contrast, FANCJ efficiently unwound all three poly(A) Zic1 unimolecular G4 DNA substrates (Fig. 8, A–C). We also assessed the activity of Twinkle on a unimolecular G4 DNA substrate derived from a human mitochondrial sequence located in the vicinity of a deletion breakpoint observed in HRCC (Fig. 8, D and E). As described above, we determined by biophysical analysis that the HRCC G4P sequence incubated in the presence of potassium formed a G-quadruplex structure that conformed to the characteristic G4 parameters detected by CD and UV spectroscopy (Figs. 2 and 3). Twinkle failed to unwind the unimolecular HRCC G4 substrate flanked by a 20-nt 5′ ssDNA tail (HRCC-20) (Fig. 8D) or a 40-nt 5′ ssDNA tail (HRCC-40) (Fig. 8E). Moreover, FANCJ was able to efficiently unwind the HRCC-20 and HRCC-40 unimolecular G4 DNA substrates (Fig. 8, D and E).
FIGURE 8.

Twinkle inefficiently unwinds unimolecular G4 DNA substrates. Helicase reactions (20 μl) were performed at 30 °C for 30 min under standard helicase conditions in the presence of complementary PNA-oligonucleotide as described under “Experimental Procedures.” In contrast to helicase experiments with intermolecular G4 substrates, a PNA-conjugated complementary trap oligonucleotide was added to the reaction mixture to prevent refolding of the intramolecular G4 after helicase unwinding. A–C, Twinkle hexamer (12.5 nm) or FANCJ (0.5 nm) was incubated with 5 fmol of 5′ poly(A) Zic1 unimolecular G4 DNA substrate with increasing 5′ ssDNA tail lengths of 20 (A), 30 (B), or 40 nt (C). D and E, Twinkle (12.5 nm) or FANCJ (0.5 nm) was tested for unwinding of a unimolecular G4 DNA substrate derived from human mitochondrial DNA sequence flanked by a 20- or 40-nt ssDNA tail in HRCC-20 (D) and HRCC-40 (E), respectively. Helicase reaction products were resolved on native 15% polyacrylamide gels, and representative phosphorimages of typical gels are shown. F, increasing amounts of complementary PNA (HRCC-PNA) were included in reaction mixtures to trap transiently unfolded HRCC-40 G4 substrate. G, Twinkle shows modest helicase activity on intramolecular G4 substrate HRCC-40 (5 fmol) in reaction mixtures containing an excess of HRCC-PNA (1 pmol) to capture unfolded G4. H, a quantitative analysis of helicase data from experiments as shown in G is shown. Data represent the mean of at least three independent experiments with S.D. indicated by error bars.
In these helicase assays with a unimolecular G4 substrate, a peptide-nucleic acid (PNA) trap was used to capture the unwound radiolabeled complementary strand. The low but detectable unwinding by Twinkle of certain intermolecular G4 substrates raised the possibility that Twinkle might also unwind an intramolecular G4 substrate; however, due to rapid refolding the unfolded G4 failed to be captured. To investigate this possibility, we set out to test Twinkle unwinding of unimolecular G4 under conditions that would favor trapping of the unfolded G4 structure. Initially, we performed a titration of complementary PNA into Twinkle-free reaction mixtures containing the HRCC-40 G4 DNA substrate (5 fmol) and monitored G4 unfolding. As shown in Fig. 8F, a greater quantity of spontaneously unfolded HRCC-40 G4 substrate was captured by increasing the amount of PNA in the reaction mixture. We then tested for unwinding of the HRCC-40 G4 substrate by increasing the concentrations of Twinkle in the presence of a fixed level of PNA (1 pmol), which trapped ∼30% of the unfolded G4 structure in the absence of helicase. Under these conditions, Twinkle unwound a maximal 13% of the HRCC-40 G4 DNA substrate over background (Fig. 8, G and H). We performed a similar set of Twinkle helicase assays with the poly(A) Zic1 G4 substrate, which included the corresponding PNA (1 pmol) in the reaction mixture, but did not detect any Twinkle-catalyzed unfolded G4 product over background (data not shown). These results suggest that Twinkle cannot uniformly unwind all unimolecular G4 substrates, even in the presence of excess complementary PNA to capture the unfolded G4.
DISCUSSION
The source of mitochondrial DNA deletions and rearrangements is debated (47). A prominent hypothesis for a number of mitochondrion-based genetic disorders is that a significant fraction of mtDNA deletions are attributed to defective replication of the organelle's genome. Given the interest in and growing evidence that G-quadruplexes exist in vivo and can be sources of genomic instability, we sought to investigate their potential abundance in mitochondria by performing a meta-analysis of predicted G-quadruplex-forming human mitochondrial sequences. Using a G4 algorithm and information from mitochondrial deletion databases, we mapped predicted G-quadruplexes in the human mitochondrial genome to known mitochondrial deletion breakpoints and correlated this information with mitochondrial deletion breakpoints prevalent in human disease, cancer, and aging. As we used a conservative upper limit of 7 nt for all G4 loops, the number of G4-prone motifs found in mitochondrial human genome might be even larger (stable G4 may be formed with loops consisting of 8 or more nucleotides). A systematic bioinformatics and biophysical study of this genome using more relaxed parameters may yield interesting new correlations. Using biophysical approaches, we substantiated that selected predicted G4-forming human mitochondrial sequences indeed form G-quadruplexes in vitro. These results suggest that mitochondrial G-quadruplexes may be prevalent at sites of double strand breaks, which impose a source of mitochondrial genome instability observed in certain genetic mitochondrial disorders, cancer, and aging.
Mitochondrial G4 DNA and Genetic Disease
Mutations in several nuclear genes are associated with the mitochondrial DNA instability of PEO patients, including genes encoding the mitochondrial DNA polymerase γ and its accessory subunit, Twinkle/C10orf2 mitochondrial helicase, the RRM2B ribonucleotide reductase subunit, and the adenine nucleotide translocator (48). Interestingly, a recent study by Roos et al. (49) described a sibling pair with compound heterozygous POLG1 mutations that displayed PEO, cognitive impairment, and mitochondrial myopathy, characterized by multiple mitochondrial deletions. A subnormal level of pol γ in vitro resulted in reduced initiation of lagging strand synthesis, leading the authors to suggest uncoupled leading and lagging strand mitochondrial DNA synthesis and the prolonged presence of replication intermediates in which the H-strand is present in a non-double-stranded DNA conformation. It is plausible that delayed maturation of the G-rich template strand may contribute to prevalent mitochondrial genome deletions in the major arc between OH and OL and that this is exacerbated in this particular case of pol γ deficiency but perhaps also in other POLG1-associated multiple deletion syndromes. It is noteworthy that the density of deletion breakpoints within 20 bp of predicted G-quadruplexes in the major arc between OH and OL (33 deletions/kb) is considerably greater than the G4P-associated deletion breakpoints in the minor arc (5 deletions/kb), suggesting an alternative explanation for the prominence of mtDNA deletions in the major arc region.
Defects in nuclear genes encoding mitochondrial DNA replication proteins are responsible for mitochondrial multiple deletion syndromes. For example, the mitochondrial disorder Alpers syndrome is a progressive neurodevelopmental mitochondrial DNA depletion disease caused by mutations in polymerase γ (48). The prominent role of the mitochondrial DNA polymerase γ in mitochondrial DNA maintenance and human disease is attested to by the fact that ∼200 disease-causing mutations in POLG1 are reportedly responsible for clinical diseases characterized by mitochondrial depletion and/or deletions. Mutations in the mitochondrial DNA helicase Twinkle (c10orf2) are also prominently associated with instability of mitochondrial DNA in patients with PEO or encephalopathy (48). Wanrooij et al. (50) examined the consequences of POLG or Twinkle defects for the spectrum of multiple mtDNA deletions characteristic of PEO patients. Using the G4 Pattern Finder algorithm, we determined that of these reported deletion breakpoints, approximately one-half reside within 20 bp of predicted G4-forming sequences (Table 7). The frequency of the indicated 5′ or 3′ deletion breakpoint residing within ≤20 bp of the G4P sequence was calculated based on the number of specified deletion breakpoints per total number of mitochondrial deletions reported for the respective PEO patient. The most frequent deletion breakpoints residing in proximity to predicted G4 DNA were commonly found in more than one PEO patient. For example, 5′ deletion breakpoints, all occurring in the residue 7397–7401 window, were found in patient 1 (POLG), patient 2 (POLG), patient 3 (POLG), and patient 7 (Twinkle). Another common 5′ deletion breakpoint found in the residue 7397–7401 window appeared in patient 1 (POLG), patient 2 (POLG), patient 3 (POLG), and patient 7 (Twinkle). 3′ deletion breakpoints residing in close proximity to the G4P sequence were found only in patient 1 (POLG).
TABLE 7.
Mitochondrial deletion breakpoints in proximity to predicted G4-forming sequences from PEO patients with mutation in POLG or Twinkle

a The mitochondrial deletion breakpoints from PEO patients were collected from the supplemental data of Ref. 50.
b Deletion breakpoints mapped to within 20 bp of the predicted G4-forming sequences (shown) as determined by Pattern Finder analysis.
c Ratio of number of 5′ or 3′ deletion breakpoints per total number of mitochondrial deletions for respective PEO patient as reported in Ref. 50.
d L-strand sequence is shown (5′ to 3′).
e The predicted G4-forming sequence (G4P) is underlined.
A comparison of G4P-associated deletion breakpoint percentages with percentage values for tRNA genes or D-loop for the PEO patients with POLG or Twinkle defects, from the study by Wanrooij et al. (50), suggests that 5′ breakage in the vicinity of G4 motifs is associated with mtDNA instability (Table 8). All patients analyzed showed a greatly reduced percentage of 5′ deletion breakpoints in the tRNA genes or D-loop region compared with the G4P sequences. With the exception of patient 1, 3′ deletion breakpoints were not found in the G4P sequences, suggesting preferential breakage at the 5′ end of G4 motifs. For patient 1, there was a similar percentage of 3′ deletion breakpoints in the G4P sequence and the tRNA genes. Although the G4P algorithm predicts G4 motifs with a loop size of 1–7 nt, a greater loop size limit predicts that G4 structures may form at 3′ common deletion breakpoints (residues 16034–16035 and 16071–16075) found in the D-loop. Further analysis of mitochondrion-based genetic diseases should deepen our understanding of the relationship of mtDNA deletions to sequences that form G-quadruplexes or other alternative DNA structures and their clinical importance.
TABLE 8.
Distribution of mitochondrial deletion breakpoints in POLG or Twinkle patients
| Deletion breakpoint | Patient 1 PolG (64)a |
Patient 2 PolG (18)a |
Patient 3 PolG (17)a |
Patients 5 and 6 PolG (32)a |
Patient 7 Twinkle (11)a |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G4Pb | tRNAc | D-loopd | G4Pb | tRNAc | D-loopd | G4Pb | tRNAc | D-loopd | G4Pb | tRNAc | D-loopd | G4Pb | tRNAc | D-loopd | |
| % | % | % | % | % | |||||||||||
| 5′ deletion breakpoint | 31 | 5 | 0 | 72 | 0 | 0 | 47 | 0 | 0 | 59 | 0 | 0 | 64 | 9 | 0 |
| 3′ deletion breakpoint | 16 | 14 | 13 | 0 | 0 | 28 | 0 | 0 | 53 | 0 | 0 | 41 | 0 | 0 | 38 |
a The number of deletion breakpoints in the specified patients is indicated in parentheses.
b The percentage of deletion breakpoints in G4P, excluding those overlapping with tRNA or D-loop region.
c The percentage of deletion breakpoints in tRNA excluding those overlapping with G4P or D-loop region.
d The percentage of deletion breakpoints in D-loop excluding those overlapping with G4P or tRNA.
Mitochondrial G4 DNA and Cancer
Mitochondrial dysfunction is prevalent in tumor cells, and mitochondrial genomic instability is well documented in a wide spectrum of human cancers (4). The prevalence of mtDNA deletions in a variety of human malignancies has led researchers to explore the potential usefulness of cataloging mitochondrial deletions for personalized medicine in cancer diagnosis and treatment. The association of potential G-quadruplex-forming sequences with mtDNA deletion breakpoints associated with cancer raises the possibility that G4 DNA itself may serve as a predictive marker for cancer associated with abnormal mtDNA metabolism.
Rogounovitch et al. (51) observed a strong correlation between the number of mitochondrial deletions and the mtDNA content in radiation-associated human thyroid tumors, suggesting a useful biomarker for discerning the molecular differences between sporadic and radiation-induced tumors of the thyroid. However, the relationship between mtDNA and radiation-associated human thyroid diseases may be specific to random large-scale deletions flanked by 2–7 bp of microhomology versus the common 5000-bp deletion flanked by 13-bp direct repeats. A careful investigation of the potential role of G4P sequences or other alternative DNA structures contributing to the signature mtDNA deletions commonly found in thyroid tumors and other cancers is warranted.
In a 2012 study of a southwestern Chinese population, Zheng et al. (52) detected a defined 822-bp deletion in mtDNA designated as a cigarette smoking-associated risk factor for lung cancer. Our computational analysis predicts a G4P sequence adjacent to the 822-bp mtDNA deletion (Table 5). Further studies will be necessary to determine the putative role of the G4P flanking sequence in the origin of the mitochondrial genome instability and whether the characteristic small mtDNA deletion is associated with other cigarette smoking-associated diseases. The use of G4P sequences as biomarkers for various cancer types and risk factors may become more widely applicable in the future as our knowledge of G-quadruplex DNA metabolism in both the mitochondrial and nuclear genomes advances.
Mitochondrial G4 DNA and Aging
The mitochondrial theory of aging postulates that the accumulation of somatic mitochondrial mutations during the life span of an individual results in mitochondrial dysfunction, which contributes to the underlying pathological basis for age-related phenotypes of cells, tissues, and organs (53). An analysis of genetically inherited mtDNA disorders strongly suggests that defects in mtDNA replication make significant contributions to the decline of mitochondrial function, contributing to age-associated phenotypes (48). Moreover, mouse models that show an accumulation of mtDNA mutations recapitulate age-related phenotypes (54). Considerable evidence over the past 2 decades supports the general conclusion that mtDNA deletions accumulate with aging in various tissues. One recent model postulates that replication errors incurred in stem cells during development become clonally expanded, ultimately leading to mitochondrial dysfunction (5). Understanding the molecular-genetic basis for the accumulation of mtDNA mutations remains an active area of study. Two prominent models for the accumulation of mtDNA deletions with aging are: 1) replication slippage over repeated sequences (55) and 2) homologous recombination repair of double strand breaks (32). G-quadruplex DNA, which perturbs the progression of the mtDNA replication machinery, may contribute to either mechanism by causing stalling during DNA synthesis and/or double strand break formation during processing of replication intermediates. In addition, experimental evidence from Barros et al. (56) suggests a model in which recombination at the site of a double strand break may be followed by replication slippage of a G4-nested region during DNA synthesis, resulting in duplications.
Mitochondrial Deletions Associated with Potential G-quadruplex-forming Sequences as Polymorphic Anthropological Markers
There has been considerable interest in tracing the distant ancestry of an individual or population group by analyzing genetic markers (57). Different human populations with distinct mitochondrial genetic markers can be analyzed through generations to dissect their lineage and recreate past migration patterns. An excellent example of this strategy was provided by an analysis of an indigenous Alaskan population known as the Aleuts whose mtDNA variation from other native Americans suggested that multiple migrations occurred during the habitation of the New World (58). From the G4P computational analysis and a survey of the literature, we identified a reported 9-bp mitochondrial deletion used as a polymorphic anthropological marker for native North American groups (59) as residing within 20 bp of a G4P sequence (5′-GGGGGTAGAGGGGGTGCTATAGGGTAAATACGGG-3′ H-strand). The deletion has its highest frequency in the American Southwest and is absent in the Arctic and Subarctic regions (59). The distribution of the G4P-associated mitochondrial deletion marker has provided some insight into the migration patterns of East Asians to the New World. Genographic projects such as this will benefit from further genetic analysis of indigenous populations by using novel genetic markers as important tools. Understanding the role of G-quadruplex metabolism and other molecular mechanisms that affect chromosomal stability and mutations, thereby influencing the appearance of mitochondrial genetic polymorphisms, will help in deciphering the origin of polymorphic markers. These efforts will in turn be useful for anthropological studies such as the history of human migration patterns and other fields such as forensic science (60).
Implications of Twinkle G4 Studies for Mitochondrial DNA Replication
Because the Twinkle helicase is required for human mtDNA replication, we assessed whether Twinkle was able to unwind various uni-, bi-, and tetramolecular G4 DNA substrates. We found that the helicase is not efficient in unwinding the various topological forms of G4 DNA tested under the same conditions in which it efficiently unwound conventional forked duplex substrates. Therefore, it is reasonable to suggest that G4 DNA structures are likely to persist in the mitochondrial genome and may contribute to double strand breaks in regions nearby G4, ultimately leading to the mitochondrial deletions prevalent in human disease. Although this study does not definitively support the proposed model for a role of G4 DNA in mitochondrial genome instability, it sheds light on a novel aspect of mtDNA metabolism and provides a reasonable platform for its further study in biological systems.
An important biochemical observation made in this study is that Twinkle helicase poorly unwound all of the G-quadruplex substrates tested. This is paradoxical given that the human mitochondrial genome is very G4-prone. It was recently demonstrated that close coupling of T7 DNA polymerase with gp4 helicase during leading strand synthesis results in a synergistic interaction in which DNA synthesis drives fork unwinding (61). If Twinkle functions in a similar manner with the mitochondrial replicative polymerase γ, then the helicase might more readily melt G-quadruplex structures that impede mitochondrial DNA replication. Furthermore, auxiliary mitochondrial helicases are likely to assist in resolving G4 structures that impede mtDNA replication and other processes such as mtDNA repair. The most likely candidate for the role of G4 resolution in mitochondria is PIF1 helicase. Studies from the Nicolas and Zakian laboratories have implicated yeast PIF in G4 metabolism (21, 22, 62–66). Although the recombinant nuclear form of human Pif1 has been shown to unwind G4 DNA substrates in vitro (67), a definitive role for human PIF1 in mitochondrial G4 metabolism remains to be determined. Other DNA helicases with substantial residence time in mitochondria include the SUV3 helicase (68, 69) and DNA2 helicase-nuclease (70, 71), the latter of which has been shown to process G-quadruplex DNA in vitro (72, 73). Mutations in DNA2 were identified recently as associated with progressive myopathy in a multiple deletion syndrome (74).
In addition to helicases, a number of other G4 DNA-binding proteins are known to exist that may play a role in mitochondrial G4 metabolism (11). The demonstration that nuclear topoisomerase I interacts with G4 DNA structures (75) raises the possibility that the mitochondrion-specific topoisomerase I (76) plays a role in G-quadruplex metabolism as well. G4-binding and G4-metabolizing proteins may affect not only mtDNA replication during the elongation phase but also replication initiation in which G-quadruplexes have recently been implicated. Mitochondrial transcription creates primers in a CG-rich element, generally termed conserved sequence block II (CSBII), required for the initiation of leading strand DNA replication. G-quadruplexes formed by RNA result in transcription termination and dictate primer formation for DNA synthesis initiation (77). More recently, Wanrooij et al. (78) reported that the mitochondrial RNA primer required for the leading strand origin of mtDNA replication forms a hybrid G-quadruplex between RNA and DNA in CSBII and that this quadruplex formation plays a critical role in determining the rate of DNA synthesis initiation in human mitochondria by influencing the architecture and persistence of an RNA-DNA hybrid (R-loop) residing at the leading strand origin of DNA replication. Because CSBII-like sequences are widespread and found in both yeast and mammals (79) (but not, for example, in Drosophila), G-quadruplexes are now believed to play an important and often evolutionarily conserved role in mitochondrial replication initiation. Current models of mtDNA replication place the role of Twinkle helicase to unwind duplex DNA downstream of nascent DNA primer synthesized by polymerase γ (80). Thus, our experimental data suggest that Twinkle may be specialized to leave intact the RNA primer-associated G-quadruplex and focus its work on facilitating polymerase γ strand displacement DNA synthesis extension with the help of the mitochondrial ssDNA-binding protein.
Future Directions
The recent development of G4-specific probes (G4-directed antibodies (7, 8, 81) and ligands (82)) has made it more realistic to study the abundance and relevance of G4 mitochondrial structures in vivo. There has been a growing interest in nuclear G4 DNA metabolism to further the understanding of the biological roles of G4 structure, and it may also be a potential target for anticancer agents and therapies. This now applies to mitochondrial G4 metabolism as well. Recently, a discovery was made that guanine-rich oligonucleotides, which may be useful anti-cancer drugs, can be introduced into lung cancer cells where they persist as a topologically distinct class of G4 structures in the mitochondria (83). As these authors indicate, a more refined understanding of the cellular tracking and localization of guanine-rich oligonucleotides may provide greater insight into design optimization for cancer therapies (83). From a broader perspective, G-quadruplexes that form in the human mitochondrial genome are likely to have major consequences for mtDNA replication, transcription, and repair, impacting health and longevity.
This research was supported, in whole or in part, by the National Institutes of Health, NIA, Intramural Research Program.
- PMPS
- Pearson marrow-pancreas syndrome
- CSBII
- conserved sequence block II
- FANCJ
- Fanconi anemia complementation group J
- G4
- G-quadruplex
- G4P
- potential G4-forming (sequence)
- H
- heavy
- L
- light
- HRCC
- human renal cell carcinoma
- KSS
- Kearns-Sayre syndrome
- PEO
- progressive external ophthalmoplegia
- PNA
- peptide-nucleic acid
- ssDNA
- single-stranded DNA
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1. Anderson S., Bankier A. T., Barrell B. G., de Bruijn M. H., Coulson A. R., Drouin J., Eperon I. C., Nierlich D. P., Roe B. A., Sanger F., Schreier P. H., Smith A. J., Staden R., Young I. G. (1981) Sequence and organization of the human mitochondrial genome. Nature 290, 457–465 [DOI] [PubMed] [Google Scholar]
- 2. Wallace D. C. (1999) Mitochondrial diseases in man and mouse. Science 283, 1482–1488 [DOI] [PubMed] [Google Scholar]
- 3. Pinto M., Moraes C. T. (2013) Mitochondrial genome changes and neurodegenerative diseases. Biochim. Biophys. Acta 1842, 1198–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wallace D. C. (2012) Mitochondria and cancer. Nat. Rev. Cancer 12, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bratic A., Larsson N. G. (2013) The role of mitochondria in aging. J. Clin. Invest. 123, 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greaves L. C., Reeve A. K., Taylor R. W., Turnbull D. M. (2012) Mitochondrial DNA and disease. J. Pathol. 226, 274–286 [DOI] [PubMed] [Google Scholar]
- 7. Henderson A., Wu Y., Huang Y. C., Chavez E. A., Platt J., Johnson F. B., Brosh R. M., Jr., Sen D., Lansdorp P. M. (2014) Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 42, 860–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lam E. Y., Beraldi D., Tannahill D., Balasubramanian S. (2013) G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 4, 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bochman M. L., Paeschke K., Zakian V. A. (2012) DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 13, 770–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maizels N., Gray L. T. (2013) The G4 genome. PLoS Genet. 9, e1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu Y., Brosh R. M., Jr. (2010) G-quadruplex nucleic acids and human disease. FEBS J. 277, 3470–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Capra J. A., Paeschke K., Singh M., Zakian V. A. (2010) G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput. Biol. 6, e1000861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Damas J., Carneiro J., Gonçalves J., Stewart J. B., Samuels D. C., Amorim A., Pereira F. (2012) Mitochondrial DNA deletions are associated with non-B DNA conformations. Nucleic Acids Res. 40, 7606–7621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oliveira P. H., da Silva C. L., Cabral J. M. (2013) An appraisal of human mitochondrial DNA instability: new insights into the role of non-canonical DNA structures and sequence motifs. PLoS One 8, e59907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yadav V. K., Abraham J. K., Mani P., Kulshrestha R., Chowdhury S. (2008) QuadBase: genome-wide database of G4 DNA-occurrence and conservation in human, chimpanzee, mouse and rat promoters and 146 microbes. Nucleic Acids Res. 36, D381–D385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mergny J. L., Li J., Lacroix L., Amrane S., Chaires J. B. (2005) Thermal difference spectra: a specific signature for nucleic acid structures. Nucleic Acids Res. 33, e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sen D., Nandakumar D., Tang G. Q., Patel S. S. (2012) The human mitochondrial DNA helicase TWINKLE is both an unwinding and an annealing helicase. J. Biol. Chem. 287, 14545–14556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu Y., Sommers J. A., Loiland J. A., Kitao H., Kuper J., Kisker C., Brosh R. M. (2012) The Q motif of FANCJ DNA helicase regulates its dimerization, DNA binding, and DNA repair function. J. Biol. Chem. 287, 21699–21716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaplan D. L., O'Donnell M. (2002) DnaB drives DNA branch migration and dislodges proteins while encircling two DNA strands. Mol. Cell 10, 647–657 [DOI] [PubMed] [Google Scholar]
- 20. Wu Y., Shin-ya K., Brosh R. M., Jr. (2008) FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol. Cell. Biol. 28, 4116–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Piazza A., Boulé J. B., Lopes J., Mingo K., Largy E., Teulade-Fichou M. P., Nicolas A. (2010) Genetic instability triggered by G-quadruplex interacting Phen-DC compounds in Saccharomyces cerevisiae. Nucleic Acids Res. 38, 4337–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Piazza A., Serero A., Boulé J. B., Legoix-Né P., Lopes J., Nicolas A. (2012) Stimulation of gross chromosomal rearrangements by the human CEB1 and CEB25 minisatellites in Saccharomyces cerevisiae depends on G-quadruplexes or Cdc13. PLoS Genet. 8, e1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giri B., Smaldino P. J., Thys R. G., Creacy S. D., Routh E. D., Hantgan R. R., Lattmann S., Nagamine Y., Akman S. A., Vaughn J. P. (2011) G4 resolvase 1 tightly binds and unwinds unimolecular G4-DNA. Nucleic Acids Res. 39, 7161–7178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brosh R. M., Jr., Waheed J., Sommers J. A. (2002) Biochemical characterization of the DNA substrate specificity of Werner syndrome helicase. J. Biol. Chem. 277, 23236–23245 [DOI] [PubMed] [Google Scholar]
- 25. Suhasini A. N., Sommers J. A., Yu S., Wu Y., Xu T., Kelman Z., Kaplan D. L., Brosh R. M., Jr. (2012) DNA repair and replication fork helicases are differentially affected by alkyl phosphotriester lesion. J. Biol. Chem. 287, 19188–19198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Farge G., Holmlund T., Khvorostova J., Rofougaran R., Hofer A., Falkenberg M. (2008) The N-terminal domain of TWINKLE contributes to single-stranded DNA binding and DNA helicase activities. Nucleic Acids Res. 36, 393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Goethem G., Martin J. J., Van Broeckhoven C. (2003) Progressive external ophthalmoplegia characterized by multiple deletions of mitochondrial DNA: unraveling the pathogenesis of human mitochondrial DNA instability and the initiation of a genetic classification. Neuromolecular Med. 3, 129–146 [DOI] [PubMed] [Google Scholar]
- 28. Zeviani M., Moraes C. T., DiMauro S., Nakase H., Bonilla E., Schon E. A., Rowland L. P. (1988) Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 38, 1339–1346 [DOI] [PubMed] [Google Scholar]
- 29. Rötig A., Cormier V., Koll F., Mize C. E., Saudubray J. M., Veerman A., Pearson H. A., Munnich A. (1991) Site-specific deletions of the mitochondrial genome in the Pearson marrow-pancreas syndrome. Genomics 10, 502–504 [DOI] [PubMed] [Google Scholar]
- 30. Mita S., Rizzuto R., Moraes C. T., Shanske S., Arnaudo E., Fabrizi G. M., Koga Y., DiMauro S., Schon E. A. (1990) Recombination via flanking direct repeats is a major cause of large-scale deletions of human mitochondrial DNA. Nucleic Acids Res. 18, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Samuels D. C., Schon E. A., Chinnery P. F. (2004) Two direct repeats cause most human mtDNA deletions. Trends Genet. 20, 393–398 [DOI] [PubMed] [Google Scholar]
- 32. Chen X. J. (2013) Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol. Mol. Biol. Rev. 77, 476–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen T., He J., Huang Y., Zhao W. (2011) The generation of mitochondrial DNA large-scale deletions in human cells. J. Hum. Genet. 56, 689–694 [DOI] [PubMed] [Google Scholar]
- 34. Rogounovitch T., Saenko V., Yamashita S. (2004) Mitochondrial DNA and human thyroid diseases. Endocr. J. 51, 265–277 [DOI] [PubMed] [Google Scholar]
- 35. Horton T. M., Petros J. A., Heddi A., Shoffner J., Kaufman A. E., Graham S. D., Jr., Gramlich T., Wallace D. C. (1996) Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosomes Cancer 15, 95–101 [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto H., Tanaka M., Katayama M., Obayashi T., Nimura Y., Ozawa T. (1992) Significant existence of deleted mitochondrial DNA in cirrhotic liver surrounding hepatic tumor. Biochem. Biophys. Res. Commun. 182, 913–920 [DOI] [PubMed] [Google Scholar]
- 37. Eshaghian A., Vleugels R. A., Canter J. A., McDonald M. A., Stasko T., Sligh J. E. (2006) Mitochondrial DNA deletions serve as biomarkers of aging in the skin, but are typically absent in nonmelanoma skin cancers. J. Invest. Dermatol. 126, 336–344 [DOI] [PubMed] [Google Scholar]
- 38. Kraytsberg Y., Kudryavtseva E., McKee A. C., Geula C., Kowall N. W., Khrapko K. (2006) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 38, 518–520 [DOI] [PubMed] [Google Scholar]
- 39. Levy G. (2007) The relationship of Parkinson disease with aging. Arch. Neurol. 64, 1242–1246 [DOI] [PubMed] [Google Scholar]
- 40. Kypr J., Kejnovská I., Renciuk D., Vorlícková M. (2009) Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 37, 1713–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lim K. W., Amrane S., Bouaziz S., Xu W., Mu Y., Patel D. J,, Luu K. N., Phan A. T. (2009) Structure of the human telomere in K+ solution: a stable basket-type G-quadruplex with only two G-tetrad layers. J. Am. Chem. Soc. 131, 4301–4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mergny J. L., Phan A. T., Lacroix L. (1998) Following G-quartet formation by UV-spectroscopy. FEBS Lett. 435, 74–78 [DOI] [PubMed] [Google Scholar]
- 43. Rajala N., Gerhold J. M., Martinsson P., Klymov A., Spelbrink J. N. (2014) Replication factors transiently associate with mtDNA at the mitochondrial inner membrane to facilitate replication. Nucleic Acids Res. 42, 952–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matsushima Y., Farr C. L., Fan L., Kaguni L. S. (2008) Physiological and biochemical defects in carboxyl-terminal mutants of mitochondrial DNA helicase. J. Biol. Chem. 283, 23964–23971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Patel S. S., Picha K. M. (2000) Structure and function of hexameric helicases. Annu. Rev. Biochem. 69, 651–697 [DOI] [PubMed] [Google Scholar]
- 46. Bharti S. K., Sommers J. A., George F., Kuper J., Hamon F., Shin-ya K., Teulade-Fichou M. P., Kisker C., Brosh R. M., Jr. (2013) Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J. Biol. Chem. 288, 28217–28229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krishnan K. J., Reeve A. K., Samuels D. C., Chinnery P. F., Blackwood J. K., Taylor R. W., Wanrooij S., Spelbrink J. N., Lightowlers R. N., Turnbull D. M. (2008) What causes mitochondrial DNA deletions in human cells? Nat. Genet. 40, 275–279 [DOI] [PubMed] [Google Scholar]
- 48. Copeland W. C. (2012) Defects in mitochondrial DNA replication and human disease. Crit. Rev. Biochem. Mol. Biol. 47, 64–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roos S., Macao B., Fusté J. M., Lindberg C., Jemt E., Holme E., Moslemi A. R., Oldfors A., Falkenberg M. (2013) Subnormal levels of POL γ cause inefficient initiation of light-strand DNA synthesis and lead to mitochondrial DNA deletions and progressive external ophthalmoplegia. Hum. Mol. Genet. 22, 2411–2422 [DOI] [PubMed] [Google Scholar]
- 50. Wanrooij S., Luoma P., van Goethem G., van Broeckhoven C., Suomalainen A., Spelbrink J. N. (2004) Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 32, 3053–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rogounovitch T. I., Saenko V. A., Shimizu-Yoshida Y., Abrosimov A. Y., Lushnikov E. F., Roumiantsev P. O., Ohtsuru A., Namba H., Tsyb A. F., Yamashita S. (2002) Large deletions in mitochondrial DNA in radiation-associated human thyroid tumors. Cancer Res. 62, 7031–7041 [PubMed] [Google Scholar]
- 52. Zheng S., Qian P., Li F., Qian G., Wang C., Wu G., Li Q., Chen Y., Li J., Li H., He B., Ji F. (2012) Association of mitochondrial DNA variations with lung cancer risk in a Han Chinese population from southwestern China. PLoS One 7, e31322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Harman D. (1956) Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300 [DOI] [PubMed] [Google Scholar]
- 54. Trifunovic A., Wredenberg A., Falkenberg M., Spelbrink J. N., Rovio A. T., Bruder C. E., Bohlooly-Y M., Gidlöf S., Oldfors A., Wibom R., Törnell J., Jacobs H. T., Larsson N. G. (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 [DOI] [PubMed] [Google Scholar]
- 55. Madsen C. S., Ghivizzani S. C., Hauswirth W. W. (1993) In vivo and in vitro evidence for slipped mispairing in mammalian mitochondria. Proc. Natl. Acad. Sci. U.S.A. 90, 7671–7675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barros P., Boan F., Blanco M. G., Gomez-Marquez J. (2009) Effect of monovalent cations and G-quadruplex structures on the outcome of intramolecular homologous recombination. FEBS J. 276, 2983–2993 [DOI] [PubMed] [Google Scholar]
- 57. Rubicz R., Melton P. E., Crawford M. H. (2007) Molecular markers in anthropological genetic studies, in Anthropological Genetics, pp. 141–186, Cambridge University Press, Cambridge, UK [Google Scholar]
- 58. Rubicz R., Schurr T. G., Babb P. L., Crawford M. H. (2003) Mitochondrial DNA variation and the origins of the Aleuts. Hum. Biol. 75, 809–835 [DOI] [PubMed] [Google Scholar]
- 59. Lorenz J. G., Smith D. G. (1994) Distribution of the 9-bp mitochondrial DNA region V deletion among North American Indians. Hum. Biol. 66, 777–788 [PubMed] [Google Scholar]
- 60. Zapico S. C., Ubelaker D. H. (2013) mtDNA mutations and their role in aging, diseases and forensic sciences. Aging Dis. 4, 364–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pandey M., Patel S. S. (2014) Helicase and polymerase move together close to the fork junction and copy DNA in one-nucleotide steps. Cell Rep. 6, 1129–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lopes J., Piazza A., Bermejo R., Kriegsman B., Colosio A., Teulade-Fichou M. P., Foiani M., Nicolas A. (2011) G-quadruplex-induced instability during leading-strand replication. EMBO J. 30, 4033–4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Paeschke K., Capra J. A., Zakian V. A. (2011) DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145, 678–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Paeschke K., Bochman M. L., Garcia P. D., Cejka P., Friedman K. L., Kowalczykowski S. C., Zakian V. A. (2013) Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 497, 458–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ribeyre C., Lopes J., Boulé J. B., Piazza A., Guédin A., Zakian V. A., Mergny J. L., Nicolas A. (2009) The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 5, e1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sabouri N., McDonald K. R., Webb C. J., Cristea I. M., Zakian V. A. (2012) DNA replication through hard-to-replicate sites, including both highly transcribed RNA Pol II and Pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev. 26, 581–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sanders C. M. (2010) Human Pif1 helicase is a G-quadruplex DNA-binding protein with G-quadruplex DNA-unwinding activity. Biochem. J. 430, 119–128 [DOI] [PubMed] [Google Scholar]
- 68. Golik P., Szczepanek T., Bartnik E., Stepien P. P., Lazowska J. (1995) The S. cerevisiae nuclear gene SUV3 encoding a putative RNA helicase is necessary for the stability of mitochondrial transcripts containing multiple introns. Curr. Genet. 28, 217–224 [DOI] [PubMed] [Google Scholar]
- 69. Stepien P. P., Margossian S. P., Landsman D., Butow R. A. (1992) The yeast nuclear gene suv3 affecting mitochondrial post-transcriptional processes encodes a putative ATP-dependent RNA helicase. Proc. Natl. Acad. Sci. U.S.A. 89, 6813–6817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Duxin J. P., Dao B., Martinsson P., Rajala N., Guittat L., Campbell J. L., Spelbrink J. N., Stewart S. A. (2009) Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell. Biol. 29, 4274–4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zheng L., Zhou M., Guo Z., Lu H., Qian L., Dai H., Qiu J., Yakubovskaya E., Bogenhagen D. F., Demple B., Shen B. (2008) Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell 32, 325–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lin W., Sampathi S., Dai H., Liu C., Zhou M., Hu J., Huang Q., Campbell J., Shin-Ya K., Zheng L., Chai W., Shen B. (2013) Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 32, 1425–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Masuda-Sasa T., Polaczek P., Peng X. P., Chen L., Campbell J. L. (2008) Processing of G4 DNA by Dna2 helicase/nuclease and replication protein A (RPA) provides insights into the mechanism of Dna2/RPA substrate recognition. J. Biol. Chem. 283, 24359–24373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ronchi D., Di Fonzo A., Lin W., Bordoni A., Liu C., Fassone E., Pagliarani S., Rizzuti M., Zheng L., Filosto M., Ferrò M. T., Ranieri M., Magri F., Peverelli L., Li H., Yuan Y. C., Corti S., Sciacco M., Moggio M., Bresolin N., Shen B., Comi G. P. (2013) Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am. J. Hum. Genet. 92, 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Arimondo P. B., Riou J. F., Mergny J. L., Tazi J., Sun J. S., Garestier T., Hélène C. (2000) Interaction of human DNA topoisomerase I with G-quartet structures. Nucleic Acids Res. 28, 4832–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang H., Barceló J. M., Lee B., Kohlhagen G., Zimonjic D. B., Popescu N. C., Pommier Y. (2001) Human mitochondrial topoisomerase I. Proc. Natl. Acad. Sci. U.S.A. 98, 10608–10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wanrooij P. H., Uhler J. P., Simonsson T., Falkenberg M., Gustafsson C. M. (2010) G-quadruplex structures in RNA stimulate mitochondrial transcription termination and primer formation. Proc. Natl. Acad. Sci. U.S.A. 107, 16072–16077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wanrooij P. H., Uhler J. P., Shi Y., Westerlund F., Falkenberg M., Gustafsson C. M. (2012) A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop. Nucleic Acids Res. 40, 10334–10344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Xu B., Clayton D. A. (1995) A persistent RNA-DNA hybrid is formed during transcription at a phylogenetically conserved mitochondrial DNA sequence. Mol. Cell. Biol. 15, 580–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kasiviswanathan R., Collins T. R., Copeland W. C. (2012) The interface of transcription and DNA replication in the mitochondria. Biochim. Biophys. Acta 1819, 970–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schaffitzel C., Postberg J., Paeschke K., Lipps H. J. (2010) Probing telomeric G-quadruplex DNA structures in cells with in vitro generated single-chain antibody fragments. Methods Mol. Biol. 608, 159–181 [DOI] [PubMed] [Google Scholar]
- 82. Largy E., Granzhan A., Hamon F., Verga D., Teulade-Fichou M. P. (2013) Visualizing the quadruplex: from fluorescent ligands to light-up probes. Top. Curr. Chem. 330, 111–177 [DOI] [PubMed] [Google Scholar]
- 83. Tseng T. Y., Wang Z. F., Chien C. H., Chang T. C. (2013) In-cell optical imaging of exogenous G-quadruplex DNA by fluorogenic ligands. Nucleic Acids Res. 41, 10605–10618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rötig A., Bourgeron T., Chretien D., Rustin P., Munnich A. (1995) Spectrum of mitochondrial DNA rearrangements in the Pearson marrow-pancreas syndrome. Hum. Mol. Genet. 4, 1327–1330 [DOI] [PubMed] [Google Scholar]
- 85. Degoul F., Nelson I., Lestienne P., Francois D., Romero N., Duboc D., Eymard B., Fardeau M., Ponsot G., Paturneau-Jouas M. (1991) Deletions of mitochondrial DNA in Kearns-Sayre syndrome and ocular myopathies: genetic, biochemical, and morphological studies. J. Neurol. Sci. 101, 168–177 [DOI] [PubMed] [Google Scholar]
- 86. Harding A. E., Hammans S. R. (1992) Deletions of the mitochondrial genome. J. Inherit. Metab. Dis. 15, 480–486 [DOI] [PubMed] [Google Scholar]
- 87. Yamashita S., Nishino I., Nonaka I., Goto Y. (2008) Genotype and phenotype analyses in 136 patients with single large-scale mitochondrial DNA deletions. J. Hum. Genet. 53, 598–606 [DOI] [PubMed] [Google Scholar]
- 88. Ballinger S. W., Shoffner J. M., Hedaya E. V., Trounce I., Polak M. A., Koontz D. A., Wallace D. C. (1992) Maternally transmitted diabetes and deafness associated with a 10.4 kb mitochondrial DNA deletion. Nat. Genet. 1, 11–15 [DOI] [PubMed] [Google Scholar]
- 89. Gerbitz K. D., van den Ouweland J. M., Maassen J. A., Jaksch M. (1995) Mitochondrial diabetes mellitus: a review. Biochim. Biophys. Acta 1271, 253–260 [DOI] [PubMed] [Google Scholar]
- 90. Nakase H., Moraes C. T., Rizzuto R., Lombes A., DiMauro S., Schon E. A. (1990) Transcription and translation of deleted mitochondrial genomes in Kearns-Sayre syndrome: implications for pathogenesis. Am. J. Hum. Genet. 46, 418–427 [PMC free article] [PubMed] [Google Scholar]
- 91. Zupanc M. L., Moraes C. T., Shanske S., Langman C. B., Ciafaloni E., DiMauro S. (1991) Deletion of mitochondrial DNA in patients with combined features of Kearns-Sayre and MELAS syndromes. Ann. Neurol. 29, 680–683 [DOI] [PubMed] [Google Scholar]
- 92. Larsson N. G., Holme E. (1992) Multiple short direct repeats associated with single mtDNA deletions. Biochim. Biophys. Acta 1139, 311–314 [DOI] [PubMed] [Google Scholar]
- 93. Johns D. R., Rutledge S. L., Stine O. C., Hurko O. (1989) Directly repeated sequences associated with pathogenic mitochondrial DNA deletions. Proc. Natl. Acad. Sci. U.S.A. 86, 8059–8062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Savre-Train I., Piatyszek M. A., Shay J. W. (1992) Transcription of deleted mitochondrial DNA in human colon adenocarcinoma cells. Hum. Mol. Genet. 1, 203–204 [DOI] [PubMed] [Google Scholar]
- 95. Lee H. C., Li S. H., Lin J. C., Wu C. C., Yeh D. C., Wei Y. H. (2004) Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat. Res. 547, 71–78 [DOI] [PubMed] [Google Scholar]
- 96. Mkaouar-Rebai E., Chamkha I., Kammoun T., Chabchoub I., Aloulou H., Fendri N., Hachicha M., Fakhfakh F. (2010) A case of Kearns-Sayre syndrome with two novel deletions (9.768 and 7.253 kb) of the mtDNA associated with the common deletion in blood leukocytes, buccal mucosa and hair follicles. Mitochondrion 10, 449–455 [DOI] [PubMed] [Google Scholar]
- 97. Bodyak N. D., Nekhaeva E., Wei J. Y., Khrapko K. (2001) Quantification and sequencing of somatic deleted mtDNA in single cells: evidence for partially duplicated mtDNA in aged human tissues. Hum. Mol. Genet. 10, 17–24 [DOI] [PubMed] [Google Scholar]
- 98. Bua E., Johnson J., Herbst A., Delong B., McKenzie D., Salamat S., Aiken J. M. (2006) Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 79, 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Reeve A. K., Krishnan K. J., Elson J. L., Morris C. M., Bender A., Lightowlers R. N., Turnbull D. M. (2008) Nature of mitochondrial DNA deletions in substantia nigra neurons. Am. J. Hum. Genet. 82, 228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Markaryan A., Nelson E. G., Hinojosa R. (2008) Detection of mitochondrial DNA deletions in the cochlea and its structural elements from archival human temporal bone tissue. Mutat. Res. 640, 38–45 [DOI] [PubMed] [Google Scholar]
- 101. Bender A., Krishnan K. J., Morris C. M., Taylor G. A., Reeve A. K., Perry R. H., Jaros E., Hersheson J. S., Betts J., Klopstock T., Taylor R. W., Turnbull D. M. (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517 [DOI] [PubMed] [Google Scholar]
- 102. Katayama M., Tanaka M., Yamamoto H., Ohbayashi T., Nimura Y., Ozawa T. (1991) Deleted mitochondrial DNA in the skeletal muscle of aged individuals. Biochem. Int. 25, 47–56 [PubMed] [Google Scholar]