

Background: Kae1 proteins are universally conserved and are regulated by a variety of protein-protein interactions.
Results: P. falciparum expresses two Kae1 proteins with distinct localizations and atypical binding partners.
Conclusion: Kae1api is essential and a potential regulator of ribosome activity.
Significance: The Kae1 protein family likely has an expanded role outside of its known tRNA-modifying activity.
Keywords: Malaria, Nucleic Acid, Protein-Protein Interaction, Ribosome, Transfer RNA (tRNA)
Abstract
The universally conserved kinase-associated endopeptidase 1 (Kae1) protein family has established roles in N6-threonylcarbamoyl adenosine tRNA modification, transcriptional regulation, and telomere homeostasis. These functions are performed in complex with a conserved core of protein binding partners. Herein we describe the localization, essentiality, and protein-protein interactions for Kae1 in the human malaria parasite Plasmodium falciparum. We found that the parasite expresses one Kae1 protein in the cytoplasm and a second protein in the apicoplast, a chloroplast remnant organelle involved in fatty acid, heme, and isoprenoid biosynthesis. To analyze the protein interaction networks for both Kae1 pathways, we developed a new proteomic cross-validation approach. This strategy compares immunoprecipitation-MS data sets across different cellular compartments to enrich for biologically relevant protein interactions. Our results show that cytoplasmic Kae1 forms a complex with Bud32 and Cgi121 as in other organisms, whereas apicoplast Kae1 makes novel interactions with multiple proteins in the apicoplast. Quantitative RT-PCR and immunoprecipitation studies indicate that apicoplast Kae1 and its partners interact specifically with the apicoplast ribosomes and with proteins involved in ribosome function. Together, these data indicate an expanded, apicoplast-specific role for Kae1 in the parasite.
Introduction
Malaria represents a major threat to human health in many parts of the world, and it is estimated that the disease kills nearly 1,000,000 people annually (1). The causative agent of the most serious form of human malaria is Plasmodium falciparum. This parasite has a complex life cycle spanning an Anopheles mosquito vector and the human host. Clinical symptoms of the disease all arise from the intraerythrocytic development of the parasite. During this part of the life cycle, the parasite invades host red blood cells and replicates within a parasitophorous vacuole to produce 16–32 daughter merozoites. Following replication, the host cell is lysed, and the merozoite progeny reinvade new RBCs to begin another round of infection. In this study, we have characterized two members from the universally conserved Kae12 protein family that are expressed during blood stage growth.
Although the Kae1 proteins were described over 20 years ago (2, 3), the biological roles for this universally conserved family have only been elucidated recently. In 2006, two groups independently reported novel functions for Kae1 based on yeast suppressor screens. Kisseleva-Romanova et al. (4) utilized ChIP and qRT-PCR to demonstrate that Kae1 binds to specific gene sets in a transcription-dependent manner and that Kae1 is required for the recruitment of additional transcription factors. Downey et al. (5) showed that Kae1-null cells displayed shortened telomeres, although the biology underpinning this phenotype remains poorly understood. Kae1 was later shown to play an essential role in the threonylcarbamoyl 6-adenosine (t6A) tRNA pathway (6–8). t6A is a universal tRNA modification previously known to be important for efficient tRNA aminoacylation and translational fidelity. A subsequent series of studies elucidated the biochemical details of the t6A modification pathway, and it is now understood that a second protein called Sua5 or YrdC (in eukaryotes and bacteria, respectively) synthesizes carbamoylthreonine adenenylate, which is then appended to tRNA substrates by Kae1 (9–13).
Kae1 performs its function in complex with at least three other conserved proteins, although these partners differ between domains of life. In eukaryotes and Archaea, this functional unit is called endopeptidase, kinase, chromatin-associated (EKC; Kae1 was formerly thought to be an endopeptidase) (4) or kinase, endopeptidase, and other proteins of small size (KEOPS) (5) and comprises Kae1, the dimerization module Pcc1, the kinase/ATPase Bud32, and Cgi121, which is thought to allosterically regulate Bud32. The DEZ complex (YgjD-YjeE-YeaZ) is the prokaryotic counterpart to the EKC/KEOPS complex, and it is made up of Kae1, YrdC, and two proteins of unknown function called YeaZ and YjeE (9, 14). Interestingly, it was recently reported in yeast that the mitochondrial Kae1 homologue Qri7 can modify tRNA in the absence of protein binding partners (13).
In this study, we characterized the localization, essentiality, and protein interaction networks for the two Kae1 proteins encoded in the P. falciparum genome. We demonstrate that the parasite expresses both genes during intraerythrocytic development. One gene product localizes to the cytoplasm, and the other targets to the apicoplast, a non-photosynthetic plastid involved in heme, isoprenoid, and fatty acid biosynthesis. A mass spectrometry-driven proteomics approach was used to identify binding partners of both Kae1 proteins, and this approach was validated in an orthogonal immunoprecipitation assay. These data demonstrate that the cytoplasmic Kae1 protein forms a complex with homologues of Bud32 and Cgi121 similar to that seen in other eukaryotes. In contrast, the apicoplast-targeted Kae1 lacks predicted interaction partners, and multiple lines of data indicate that it instead makes atypical interactions with other apicoplast proteins and components of the apicoplast ribosome. These data suggest a novel role for Kae1 proteins in the regulation of ribosome function.
EXPERIMENTAL PROCEDURES
Sequence Alignment and Phylogenetic Analysis of Kae1 Proteins
All sequence alignments were done with ClustalW. Predicted Kae1 proteins were identified by BLAST search and aligned with known Kae1 proteins. Portions of the alignment were manually corrected as needed. Phylogenetic analysis was performed with 27 ASHKA (acetate and sugar kinase/heat shock protein 70/actin) ATPase superfamily sequences (18 Kae1 and nine diverse non-Kae1 proteins). Sequences were aligned and used to generate a phylogenetic tree in DNASTAR MegAlign by the neighbor-joining methodology and 1,000 bootstrap replicates. Distance values were calculated with the Kimura distance formula.
Generation of Parasite Integration and Overexpression Constructs
Integration constructs for P. falciparum Kae1cyto (PF3D7_1030600), Bud32 (PF3D7_0708300), Cgi121 (PF3D7_0511700), Sua5/YrdC (PF3D7_1203500), Kae1api (PF3D7_0408900.1), IspG (PF3D7_1022800), elongation factor Ts (EF-Ts) (PF3D7_0305000), and a putative RNA helicase (PF3D7_0504400) were made by PCR-amplifying the full-length gene sequence from genomic DNA. Note that the PF3D7_0408900 locus encodes two splice variants (PF3D7_0408900.1 and PF3D7_0408900.2) that differ by two amino acids. PCR products were ligated by In-Fusion cloning (Clontech) into the XhoI/AvrII restriction sites of the GDB vector in-frame with the C-terminal RFA tag (15). Kae1-GDB constructs were modified with the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) to generate allelic replacement constructs for testing Kae1 essentiality.
TyEOE overexpression constructs were made using a previously reported PiggyBac transposon-mediated plasmid with modifications (16). Briefly, the expression plasmid was modified to introduce the P. falciparum Hsp86 promoter and a downstream multicloning site containing XhoI and AvrII restriction sites. An in-frame 6xFLAG tag was placed 3′ to the AvrII site. Finally, the original human dihydrofolate reductase drug marker was replaced with a yeast dihydroorotate dehydrogenase selection cassette (17). Coding sequences for the target genes were amplified by RT-PCR from total parasite RNA. Amplification products were ligated into linearized TyEOE vector (XhoI and AvrII) in-frame with the C-terminal 6xFLAG tag.
Parasite Culture
Parasites were grown in human RBCs at 2% hematocrit in RPMI 1640 medium supplemented with 0.25% Albumax II (Invitrogen). Cultures were maintained at 37 °C under a 5% CO2 atmosphere.
Generation of Epitope-tagged Parasite Lines
Parasite lines were generated by transfecting uninfected RBCs followed by addition of 3D7-PM1 parasites (18) as described previously (19). Transfected RBCs were infected with asynchronous parasites, and subsequent drug selection was performed with 2.5 μg/ml blasticidin S and 5 μm trimethoprim (TMP). Parasites were enriched for integrants by alternating rounds of drug selection with 3-week periods in the absence of blasticidin S. Clones were isolated by limiting dilution.
For Southern blots, genomic DNA was isolated from clonal Kae1-RFA parasites and digested with SacI/ZraI (Kae1api) or AflII/HindIII (Kae1cyto). Probes against the first ∼500 bp of each gene were labeled using a AlkPhos Direct Labeling kit (GE Healthcare), and blots were visualized on BioMax light film (Carestream Health Inc., Rochester, NY).
Dual tagged RFA/FLAG parasite lines were made by transfecting 50 μg of the transposase expression plasmid (MRA-912) and 100 μg of TyEOE construct into the appropriate clonal RFA-tagged background (16). Transfections and parasite culture were performed as described above, and drug selection was performed in 2.5 μg/ml blasticidin S and 2 μm DSM-1.
Generation of Recombinant Overexpression Constructs
Codon-optimized (most abundant Escherichia coli codon for each amino acid) versions of the coding sequences for PF3D7_0408900.1 and PF3D7_1030600 were ordered from Integrated DNA Technologies, Inc. (Coralville, IA). The optimized sequences were amplified by PCR and ligated into linearized pET28B expression vector (NheI and XhoI) in-frame with the N-terminal His6 tag.
Electrophoretic Mobility Shift Assays
Electrophoretic mobility shift assays (EMSAs) were performed as described previously with modifications (20). Apicoplast DNA (PFC10_API_IRAB: 1–3000 and 2897–5979) and codon-optimized Kae1api sequences were PCR-amplified from total genomic DNA and gel-purified. pLEX vector was linearized with XhoI and gel-purified. Bovine serum albumin (BSA) or recombinant Kae1api was diluted in assay buffer (10 mm HEPES, pH 7.4, 5% glycerol, 5 mm MgCl2, 1 mm DTT) to a final volume of 10 μl and added to 150 ng of DNA. This solution was incubated at room temperature for 1 h before separation on a 0.8% agarose gel. Gels were visualized with ethidium bromide.
Immunofluorescence Microscopy
Immunofluorescence images were acquired using an Axio Imager.M1 epifluorescence microscope (Carl Zeiss Microimaging, Inc., Göttingen, Germany) equipped with a Hamamatsu ORCA-ER digital charge-coupled device camera and running Axiovision 4.8 software. Parasite cultures were incubated with 10 nm MitoTracker Red CM-H2XRos (Invitrogen) for 60 min then washed twice with PBS. Cells were fixed and stained for IFA as described previously (21). Primary antibody staining was performed in 3% BSA in PBS overnight at 4 °C with ab5450 goat α-GFP (1:500 dilution; Abcam, Cambridge, MA) and polyclonal rabbit α-acyl carrier protein (ACP) (Ref. 22; 1:1,000 dilution). Staining with secondary antibodies was carried out in 3% BSA in PBS for 45 min at a 1:500 dilution (Alexa Fluor 488-conjugated chicken α-goat, Invitrogen; IRDye 680RD-conjugated donkey α-rabbit, LI-COR Biosciences, Lincoln, NE). Stained cells were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen) to visualize nuclei.
Electron Microscopy
Parasites were fixed in 4% paraformaldehyde, 0.05% glutaraldehyde (Polysciences Inc., Warrington, PA) in 100 mm PIPES buffer. Samples were embedded in 10% gelatin and infiltrated overnight with 2.3 m sucrose, 20% polyvinyl pyrrolidone in PIPES at 4 °C. After freezing in liquid nitrogen, samples were sectioned with a cryo-ultramicrotome. Sections were probed with ab6556 rabbit α-GFP (1:1,000 dilution; Abcam) followed by secondary anti-rabbit antibody conjugated to 18-nm colloidal gold (Jackson ImmunoResearch Laboratories, West Grove, PA). Samples were then stained with uranyl acetate/methylcellulose and analyzed by transmission EM.
Immunoprecipitation and Mass Spectrometry
All buffers used during immunoprecipitation studies were kept at 4 °C and supplemented with Complete protease inhibitor mixture (Roche Applied Science) unless otherwise stated. Asynchronous parasites were grown to ∼10% parasitemia and resuspended in PBS with 0.05% saponin (Sigma-Aldrich). Parasites were released by gentle shaking on ice for 10 min, then collected by centrifugation (3400 × g for 30 min), and washed twice with PBS.
Cell pellets were lysed by sonication in 3 pellet volumes of radioimmune precipitation assay buffer. The lysate was cleared by centrifugation (16,100 × g for 30 min) and then precleared for 1 h by the addition of washed protein G Dynabeads (50-μl packed volume; Invitrogen). Precleared lysates were incubated with gentle mixing overnight with 10 μl of primary antibody (rabbit α-GFP ab6556). Washed protein G Dynabeads (50-μl packed volume) were then added for a final 2-h incubation at 4 °C. Beads were washed three times with radioimmune precipitation assay buffer and twice with PBS before boiling in loading buffer (LI-COR Biosciences) to release precipitated proteins. Protein samples were loaded onto a 10% acrylamide gel (ThermoFisher Scientific, Waltham, MA). After briefly running the gel, sample lanes were excised. Proteins were in-gel trypsin-digested and analyzed by MS-MS at the University of Dundee Fingerprints proteomics facility. Proteins failing to meet the Mascot significance threshold (p < 0.05) (23) or with <2 peptides were excluded from the raw data set.
Analysis of Mass Spectrometry Data Sets
MS data sets for each protein were obtained in two to three biological replicates and filtered against a series of negative control samples using Mathworks Matlab R2013b as described below.
Negative control samples were collected as described above in multiple parasite lines (3D7-PM1, ACP-GFP, PF3D7_1025200-RFA, and PF3D7_1311900-RFA). Hits identified in the 3D7-PM1 parental line were excluded from the analysis, and data sets were filtered based on bait protein localization. In this approach, hits appearing in the pooled apicoplast bait protein data set were subtracted from the hit list for each cytoplasmic bait protein and vice versa. Hits overlapping with the mitochondrial data set (PF3D7_1025200-RFA) were also removed. RFA tag interactions were identified by comparing IP samples in which Sua5 had been C-terminally tagged with either 3xHA or RFA. Hits appearing only in the RFA-tagged samples were removed. Remaining hits that were reproducible (detected in at least two biological replicates) were designated as true positives.
Cluster Analysis and Prediction of Cellular Localization
Housekeeping and apicoplast hit sets were analyzed based on GO annotation using the online DAVID Functional Annotation Tool (24). UniProt accession numbers for genes of interest were submitted as sample lists (david.abcc.ncifcrf.gov/tools.jsp). Analysis of hit lists was performed at default settings relative to the P. falciparum genome background. PlasmoAP (25) and PATS (26) were used to predict apicoplast localization, whereas the likelihood of mitochondrial targeting was determined by PlasMit (27).
Reciprocal Immunoprecipitation Studies
Dual tagged (RFA/6xFLAG) parasite lines were saponin-harvested and washed as described above. Cell pellets were lysed in Nonidet P-40 buffer (50 mm Tris, 150 mm NaCl, 1% Nonidet P-40, pH 7.4). Lysates were cleared by centrifugation (16,100 × g for 30 min) and precleared for 1 h by the addition of washed protein G Dynabeads (50-μl packed volume). 3 mm MgCl2 was added to all precleared lysates, and 2 units/μl Benzonase nuclease (Sigma-Aldrich) was used for nuclease-treated samples. Finally, rabbit α-GFP (1:500; ab6556) was added, and samples were incubated with gentle rocking overnight at 4 °C. 100 μl of washed protein G Dynabeads were added for an additional 2 h. Beads were washed and then boiled in loading buffer (LI-COR Biosciences) to release precipitated proteins. Proteins were separated by SDS-PAGE and blotted onto nitrocellulose membrane. Membranes were blocked for 1 h (blocking buffer, LI-COR Biosciences) and probed with mouse α-FLAG (1:2,000; Clontech) followed by IRDye 680RD donkey α-mouse (1:10,000; LI-COR Biosciences). Membranes were visualized on a LI-COR Biosciences Odyssey scanner.
RNA Sample Preparation
Bait proteins were immunoprecipitated as described for the mass spectrometry experiments but with the addition of 100 units/ml RNaseOUT (Invitrogen) in all buffers. For RNase I-treated samples, the IP beads were washed and then resuspended in room temperature PBS with 10 units/μl RNase I (Invitrogen). Samples were incubated at room temperature for 30 min with gentle rocking. Beads were washed twice with lysis buffer and twice with PBS. RNA was isolated from washed Dynabeads using the TRIzol method (Invitrogen), then DNase I-treated (Qiagen, Valencia, CA), and purified using an RNeasy Mini kit (Qiagen). RNA sample concentrations were measured by Qubit fluorometric quantitation (Invitrogen).
qRT-PCR Assay
6-FAM-labeled TaqMan minor groove binding probes and primer sequences were designed using Primer Express software (Invitrogen) and purchased from Invitrogen and Integrated DNA Technologies, Inc., respectively. Probe sequences were as follows: AGGAGTTGCAGGAGAT (actin), CGCATCCTAAAGCAG (apicoplast 23 S rRNA), TGGTGGAACATGTGGC (apicoplast 16 S rRNA), CGATGTCGGCTCTT (cytoplasmic 28 S rRNA), and CATGATTCATCAGATTGTG (cytoplasmic 18 S rRNA).
qRT-PCR was performed with TaqMan One-Step RT-PCR Master Mix Reagents kit (Invitrogen) on a ViiaA7 real time PCR system (Invitrogen). Relative abundance was determined for apicoplast rRNA (16 S (PFC10_API0002:rRNA) and 23 S (PFC10_API0010:rRNA)) as well as cytoplasmic rRNA (18 S (PF3D7_0531600) and 28 S (PF3D7_0532000)) and actin (PF3D7_1246200) mRNA. Relative enrichment values for 18 and 28 S rRNAs were determined for cytoplasmic proteins Kae1cyto, Bud32, Cgi121, and Sua5/YrdC relative to the untagged parental 3D7-PM1 control IP sample and normalized to apicoplast 16 and 23 S rRNA levels. -Fold enrichment for the apicoplast proteins Kae1api, IspG, EF-Ts, and the putative RNA helicase were determined relative to the ACP-GFP sample and normalized to nuclearly encoded cytoplasmic 18 and 28 S rRNA levels. Degradation of mRNA after RNase I treatment was confirmed by measurement of actin mRNA transcript levels. RNA levels were quantified based on the amplification Ct value.
RESULTS
Blood Stage Parasites Express Distinct Cytosolic and Apicoplast-targeted Kae1 Proteins
The predicted Kae1 genes PF3D7_1030600 and PF3D7_0408900 were identified based on sequence homology (Fig. 1A). Their identities were further confirmed by phylogenetic analysis, which shows that both genes cluster with known Kae1 members and not with other families of the ASHKA superfamily of ATPases (Fig. 1B). PF3D7_1030600 is of eukaryotic origin, whereas PF3D7_0408900 clusters most closely with prokaryotic Kae1 members. PF3D7_0408900 also displays conserved nucleotide-binding residues at positions 96 and 340, which are characteristic of a prokaryotic origin.
FIGURE 1.

Kae1 multiple sequence alignment and phylogenetic analysis. A, predicted Kae1 proteins were identified by BLAST search and aligned by ClustalW with established Kae1 proteins. Conserved metal-binding residues (red boxes) and nucleotide-binding residues (blue boxes) are shown. Two conserved nucleotide-binding residues at positions 96 and 340 (Kae1api numbering) differ between prokaryotes and eukaryotes/Archaea. Kae1api displays characteristic prokaryotic residues at both positions. B, phylogenetic analysis confirms that P. falciparum Kae1api and Kae1cyto are more closely related to Kae1 proteins than to other protein families of the ASHKA superfamily (shown in black). Kae1cyto clusters with eukaryotic and archaeal homologues (shown in red), whereas Kae1api groups with prokaryotic (bacterial or organellar) family members (shown in blue).
Both genes were RFA-tagged by single crossover homologous recombination at their endogenous loci in intraerythrocytic P. falciparum parasites. Integration was confirmed by PCR and Southern blotting (Fig. 2A). Western blot analysis confirmed that both proteins are expressed during intraerythrocytic development (Fig. 2B), and microscopy was performed to determine the localization of each protein. By IFA, PF3D7_0408900 (referred to hereafter as Kae1api) displayed an elongated, punctate signal overlapping precisely with that of the apicoplast-targeted ACP in trophozoites and early schizonts (Fig. 2C). Both signals were detected in a compartment adjacent to but distinct from the mitochondrion, consistent with apicoplast localization. Immuno-EM primarily detected Kae1api within structures bound by three to four membranes, further supporting that it is an apicoplast-targeted protein (Fig. 2D). In contrast, IFA detected a diffuse signal for PF3D7_1030600 (hereafter referred to as Kae1cyto) throughout the parasite, suggesting a cytoplasmic location (Fig. 2C). Consistent with previous reports in yeast (4), we also occasionally observed brighter foci overlapping with the nucleus, suggesting that Kae1cyto may target to both compartments.
FIGURE 2.

Expression and localization of Kae1api and Kae1cyto. A, a Southern blot confirms RFA tagging of the endogenous Kae1 loci. Kae1 genes were RFA-tagged at their 3′-ends by homologous recombination with an integration plasmid. Genomic DNA was isolated from clonal parasite lines and analyzed by Southern blotting. Bands corresponding to integrated loci are indicated by “i,” whereas plasmid bands are indicated by “p.” In Kae1api integrants, the parental locus band (predicted length of 7,925 bp) is replaced by a larger band matching the predicted size (11,263 bp) of the RFA-tagged gene. An additional band matching the predicted size of the plasmid (7,044 bp) is detected. The presence and relatively high signal of this band are due to concatamerization at the integrated locus, a common phenomenon in P. falciparum. In Kae1cyto integrants, both parental (3,299-bp) and RFA-tagged (4,323-bp) loci are detected, indicating the presence of multiple gene copies. A third band is detected at the predicted plasmid size (7,990 bp), and a fourth band at ∼7,000 bp is also detected. The fourth band is likely the result of a plasmid rearrangement event, which is not unusual in P. falciparum. B, RFA-tagged parasites were grown with or without TMP for 48 h, and Kae1 proteins were immunoprecipitated with an α-GFP antibody. An α-GFP Western blot confirms the expression of RFA-tagged Kae1 gene products in blood stage parasites, and a slight decrease in Kae1 protein levels is observed in the absence of TMP. Culture parasitemia was determined prior to immunoprecipitation to ensure equal loading. Note that IPs and Western blotting for all RFA-tagged proteins in this study were performed with identical antibodies and reagents and are used as negative controls for each other. C, representative images of trophozoites are shown. Kae1api (top row) co-localizes with an apicoplast marker (ACP), whereas Kae1cyto (bottom row) displays a diffuse signal throughout the parasite. Mitochondria and nuclei were visualized with MitoTracker and DAPI DNA stain, respectively. D, immuno-EM of Kae1api demonstrates localization to the apicoplast, bound by multiple membranes. Scale bar, 0.5 μm.
Kae1api Is Essential for Blood Stage Parasite Development, and Kae1cyto Is Present in Multiple Copies
The RFA tag comprises a GFP and a dihydrofolate reductase-based degradation domain, which misfolds in the absence of its stabilizing ligand TMP. This targets the RFA-tagged protein for degradation at the proteasome and allows protein levels to be controlled in a TMP-dependent manner, although the degree of knockdown is variable (15). We explored potential loss of function phenotypes by withdrawing TMP from cultured Kae1api-RFA and Kae1cyto-RFA parasites. Under these conditions, we observed a slight reduction in protein levels for both parasite lines (Fig. 2B). However, this relatively modest change did not result in detectable defects in morphology or growth rate (data not shown). We therefore set out to assess the essentiality of these genes through a series of allelic replacement experiments.
Kae1 function depends on a highly conserved histidine motif (HXXXH) that binds a metal cofactor at the central cleft (4, 28). We attempted to inactivate each Kae1 gene product by mutating both histidine residues to alanines by single crossover allelic replacement (Fig. 3A). Recombination at the endogenous locus can occur upstream or downstream of the target mutation, although only upstream crossover will result in incorporation of the mutation. To facilitate parasite screening, a unique synonymous restriction site (AgeI in the case of Kae1cyto and AciI for Kae1api) was introduced in each construct just upstream of the HXXXH mutation.
FIGURE 3.

Evaluation of Kae1 essentiality by allelic replacement. A, endogenous loci of the Kae1 genes are targeted by homologous recombination with an allelic replacement plasmid encoding either a synonymous or non-synonymous mutation to the essential metal-binding histidine motif (syn and non-syn lanes, respectively). Both mutations introduce a novel restriction site, and introduction of the mutation requires recombination upstream of the motif. Integration results in a second promoterless gene copy downstream of the RFA-tagged gene (not shown). B, PCR amplification of transfectant pools shows recombination at the target loci. Restriction digestion of PCR products with AciI (Kae1api) or AgeI (Kae1cyto) reveals the presence (no digest products) or absence (digest products indicated by *) of an intact metal-binding motif. Similar constructs were used to attempt to truncate the endogenous loci upstream of the metal-binding motif (trunc lanes). DHFRdd, dihydrofolate reductase-based degradation domain.
After transfecting with a non-synonymous (HXXXH → AXXXA) or a synonymous control (HXXXH → HXXXH) plasmid, the Kae1 loci of the integrant pools were PCR-amplified and screened for the presence of the target mutation by restriction digestion (Fig. 3B). In this assay, restriction endonuclease-sensitive PCR products are indicative of upstream crossover.
Allelic replacement of Kae1api resulted in mutation of the histidine codons in the synonymous control but not in the non-synonymous plasmid transfection (three independent transfections each), consistent with our inability to truncate the gene (Fig. 3B). Kae1api is therefore required for normal parasite growth, and its function requires an intact metal-binding motif. In contrast, we were readily able to truncate Kae1cyto and to introduce both synonymous and non-synonymous mutations at the metal binding site. However, RT-PCR of total parasite RNA indicated that the parasite still expresses wild-type Kae1cyto transcript even after truncation of the gene. Follow-up PCR and Southern blot studies indicated the presence of multiple copies of the gene (Fig. 2A); therefore it is unclear whether Kae1cyto is required for normal parasite growth.
Kae1api Binds DNA
Kae1api was expressed in E. coli using a codon-optimized plasmid in which the predicted bipartite targeting sequence was replaced with a His6 tag, and the protein was purified to homogeneity by nickel affinity and cation exchange chromatography (Fig. 4A).
FIGURE 4.

Recombinant Kae1api binds apicoplast DNA. A, P. falciparum sequences were aligned against Kae1 homologues to predict the mature sequence of Kae1api. The coding sequence for Kae1api was codon-optimized, and the predicted targeting sequence was replaced with a His6 tag. The final sequence was cloned into pET28B and expressed in E. coli, yielding an epitope-tagged protein of ∼70 kDa. B, purified Kae1api (right panels) was tested by EMSA for binding to 150 ng of PCR-amplified DNA. Results are shown for the following apicoplast genome sequences: PFC10_API_IRAB: 1–3000 (api1) and 2897–5979 (api2). Linearized pLEX vector and the codon-optimized coding sequence for Kae1api were also tested. BSA was tested with each DNA sequence as a negative control (left panels). Electrophoretic mobility of all tested PCR products decreased in a Kae1api protein concentration-dependent fashion.
Kae1api protein was incubated at different concentrations with a series of PCR-amplified sequences from the apicoplast genome in an EMSA (Fig. 4B). For each sequence tested, similar changes in electrophoretic mobility were observed at increasing protein concentrations. The progressive shifts in mobility at higher protein concentrations indicate that binding occurs at multiple sites on each PCR product. This indicates that there are either multiple copies of a Kae1api sequence recognition motif on each PCR product or more likely that the Kae1api binds DNA nonspecifically. It may be that Kae1api binds nucleic acids promiscuously in vivo or that specificity depends on regulatory partners or cofactors absent in our assay conditions.
Analysis of Protein-Protein Interaction Networks
We next set out to identify regulatory partners of both Kae1 pathways. Apicoplast Kae1 and cytoplasmic Kae1 were immunoprecipitated from RFA-tagged parasite cultures and analyzed by mass spectrometry. To identify false-positive hits due to high protein abundance or to nonspecific interactions, we performed control IPs in the untagged parental line 3D7-PM1. The top hits for Kae1api and Kae1cyto (discussed below) were RFA-tagged and tested by IP-MS.
IP-MS data were analyzed as shown in Fig. 5. Data sets were categorized based on the experimentally determined cellular localization of each bait protein (apicoplast, cytoplasm, or mitochondrion). To remove false-positive hits due to nonspecific interactions with the antibody or beads, hits identified in the untagged parental negative control were excluded.
FIGURE 5.
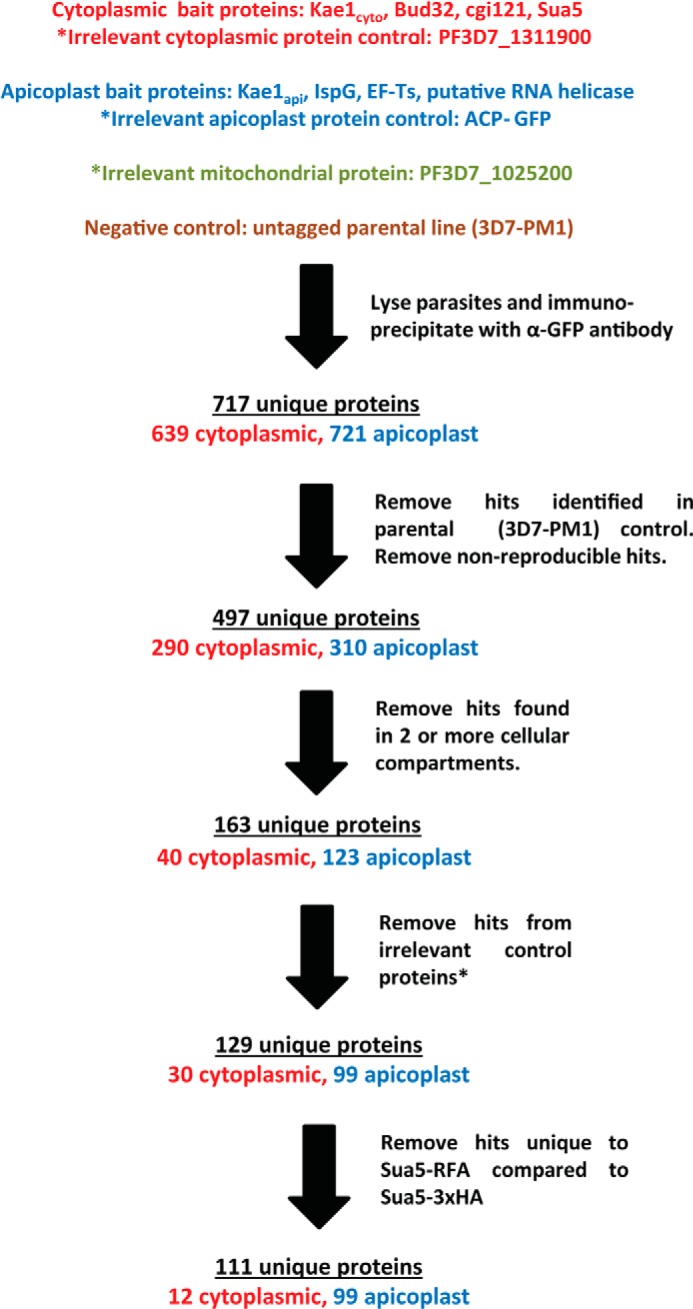
Mass spectrometry data flow. A series of control samples was used to filter out likely false-positives from the target bait protein hit lists. IP-MS data sets were classified as apicoplast, mitochondrial, or cytoplasmic based on the localization of each bait protein, and only reproducible hits were further considered. Proteins identified in the parental strain (3D7-PM1) negative control were removed from lists of potential hits. Proteins identified in bait proteins from different cellular compartments were also removed from consideration to focus on localization-specific protein interactions. Next, all proteins identified for any of the three irrelevant protein controls were excluded. Finally, IP-MS samples were compared between parasites in which Sua5 had been RFA- or 3xHA-tagged. Proteins found in only the RFA-tagged samples were removed from the analysis to account for interactions with the RFA tag itself.
To identify hits due to housekeeping interactions (protein expression, folding, degradation, etc.), data from each cellular compartment were pooled and compared with the other two compartments. Proteins targeted to different compartments such as the cytoplasm, apicoplast, or mitochondria are synthesized on a common set of machinery and then relocated to the sites where they perform their biological function. Therefore, hits common to bait proteins from different compartments are unlikely to be participants in their compartment-specific function. This category of proteins was therefore removed from lists of potential hits. Finally, to account for nonspecific interactions with the RFA tag, we compared IP samples from Sua5-RFA and Sua5–3xHA parasites. Hits detected in only the RFA-tagged line were excluded.
We performed GO annotation cluster analysis on hits found at the intersection of two or more cellular compartments. It is notable that this data set was strongly enriched for proteins involved in protein synthesis and protein folding (Table 1). In addition, hits at these intersections are similar regardless of the compartments being compared (i.e. hits at the intersection between the apicoplast and cytosol are similar to those common to the cytosol and mitochondria). Together, these findings indicate that our analysis based on cellular localization is an effective strategy for removing signals from housekeeping pathways that although biologically important can complicate the identification of more interesting protein-protein interactions. Finally, only nine proteins were excluded as hits based on the 3xHA/RFA tag comparison. This indicates that the upstream negative controls effectively remove false-positives due to interactions with the RFA tag.
TABLE 1.
GO annotation cluster analysis of pooled hits identified in two or more cellular compartments
348 of 388 hits were GO annotated and included in the analysis. Terms relating to the translational machinery and protein folding were significantly enriched in this data set relative to the parasite genome as a whole (background data set). adj., adjusted.
| Category | Term | No. proteins identified from hit list (out of 348) | Positive | p value | -Fold enrichment | Bonferroni adj. p value | Benjamini-Hochberg adj. p value | No. proteins identified from background |
|---|---|---|---|---|---|---|---|---|
| % | ||||||||
| KEGG_PATHWAY | pfa03010: ribosome | 43 | 12.4 | 1.12E-20 | 4.0 | 4.03E-19 | 4.03E-19 | 71 |
| SP_PIR_KEYWORDS | Ribosomal protein | 45 | 12.9 | 2.55E-19 | 4.7 | 2.42E-17 | 2.42E-17 | 129 |
| GOTERM_CC_FAT | GO:0005829, cytosol | 40 | 11.5 | 1.33E-16 | 4.0 | 1.79E-14 | 1.79E-14 | 90 |
| GOTERM_CC_FAT | GO:0033279, ribosomal subunit | 37 | 10.6 | 6.58E-14 | 3.7 | 1.06E-11 | 5.30E-12 | 90 |
| GOTERM_CC_FAT | GO:0005840, ribosome | 45 | 12.9 | 6.97E-14 | 3.1 | 1.12E-11 | 3.74E-12 | 130 |
| GOTERM_CC_FAT | GO:0022626, cytosolic ribosome | 27 | 7.8 | 3.55E-13 | 4.7 | 5.71E-11 | 1.43E-11 | 51 |
| GOTERM_CC_FAT | GO:0044445, cytosolic part | 30 | 8.6 | 3.74E-13 | 4.3 | 6.02E-11 | 1.20E-11 | 63 |
| GOTERM_MF_FAT | GO:0005198, structural molecule activity | 50 | 14.4 | 5.53E-13 | 2.8 | 1.19E-10 | 1.19E-10 | 136 |
| GOTERM_CC_FAT | GO:0043228, non-membrane-bounded organelle | 57 | 16.4 | 1.14E-11 | 2.3 | 1.83E-09 | 3.05E-10 | 221 |
| GOTERM_CC_FAT | GO:0043232, intracellular non-membrane-bounded organelle | 57 | 16.4 | 1.14E-11 | 2.3 | 1.83E-09 | 3.05E-10 | 221 |
| GOTERM_MF_FAT | GO:0003735, structural constituent of ribosome | 42 | 12.1 | 4.95E-11 | 2.8 | 1.07E-08 | 5.34E-09 | 113 |
| SP_PIR_KEYWORDS | nucleotide-binding | 49 | 14.1 | 2.02E-10 | 2.7 | 1.92E-08 | 9.60E-09 | 248 |
| GOTERM_CC_FAT | GO:0030529, ribonucleoprotein complex | 47 | 13.5 | 2.62E-10 | 2.4 | 4.22E-08 | 6.03E-09 | 172 |
| GOTERM_BP_FAT | GO:0006412, translation | 57 | 16.4 | 1.02E-09 | 2.2 | 3.48E-07 | 3.48E-07 | 202 |
| GOTERM_CC_FAT | GO:0022625, cytosolic large ribosomal subunit | 17 | 4.9 | 4.30E-09 | 5.3 | 6.93E-07 | 8.66E-08 | 29 |
| GOTERM_CC_FAT | GO:0015934, large ribosomal subunit | 23 | 6.6 | 6.99E-09 | 3.8 | 1.13E-06 | 1.25E-07 | 54 |
| SP_PIR_KEYWORDS | Chaperone | 16 | 4.6 | 2.61E-08 | 5.7 | 2.48E-06 | 8.27E-07 | 38 |
| SP_PIR_KEYWORDS | Cytoplasm | 15 | 4.3 | 6.33E-08 | 5.8 | 6.01E-06 | 1.50E-06 | 35 |
| SP_PIR_KEYWORDS | GTP binding | 17 | 4.9 | 5.07E-07 | 4.4 | 4.81E-05 | 9.63E-06 | 52 |
| SP_PIR_KEYWORDS | Ribonucleoprotein | 17 | 4.9 | 8.99E-07 | 4.2 | 8.54E-05 | 1.42E-05 | 54 |
| GOTERM_CC_FAT | GO:0015935, small ribosomal subunit | 14 | 4.0 | 5.30E-05 | 3.5 | 8.49E-03 | 8.52E-04 | 36 |
| SP_PIR_KEYWORDS | ATP binding | 34 | 9.8 | 6.17E-05 | 2.1 | 5.85E-03 | 8.37E-04 | 221 |
| INTERPRO | IPR001623: heat shock protein DnaJ, N-terminal | 14 | 4.0 | 1.31E-04 | 3.3 | 6.24E-02 | 6.24E-02 | 43 |
| SP_PIR_KEYWORDS | Stress response | 8 | 2.3 | 1.78E-04 | 6.0 | 1.68E-02 | 2.11E-03 | 18 |
| GOTERM_CC_FAT | GO:0022627, cytosolic small ribosomal subunit | 10 | 2.9 | 2.74E-04 | 4.1 | 4.31E-02 | 4.00E-03 | 22 |
| INTERPRO | IPR012335: thioredoxin fold | 11 | 3.2 | 3.31E-04 | 3.8 | 1.50E-01 | 7.79E-02 | 30 |
| GOTERM_MF_FAT | GO:0051082, unfolded protein binding | 17 | 4.9 | 3.40E-04 | 2.6 | 7.09E-02 | 2.42E-02 | 50 |
Kae1cyto Forms a Semiconserved KEOPS Complex
Sequence homology and cellular localization suggest that Kae1cyto is of eukaryotic origin, whereas Kae1api is of prokaryotic origin (Fig. 1, A and B). In eukaryotes, Kae1 functions within a highly conserved complex consisting of Kae1, Bud32, Cgi121, and the dimerization module Pcc1. Together, these act in concert downstream of Sua5 (YrdC in prokaryotes) to perform their tRNA modification function. Although there is no clear Pcc1 gene in P. falciparum, homologues of Bud32, Cgi121, and Sua5 are readily detectable. To confirm the expression of this pathway during blood stage development, we generated parasite lines in which the endogenous locus of each gene was RFA-tagged. Western blot analysis confirmed the expression of all three genes (Fig. 6A), and the localization of each protein was determined to be cytosolic by IFA (Fig. 6B). This is consistent with the idea that these proteins function in an intact t6A pathway with Kae1cyto.
FIGURE 6.

Expression and localization of Kae1cyto interaction partners and Sua5. A, an α-GFP Western blot confirms the expression of RFA-tagged Bud32, Cgi121, and Sua5 in blood stage parasites. B, representative IFA images from RFA-tagged parasites are shown. Localization for all three proteins is consistent with cytoplasmic localization, and Bud32 (top row), Cgi121 (middle row), and Sua5 (bottom row) show diffuse staining throughout the parasite. Nuclei were visualized with DAPI DNA stain.
We next analyzed the interaction networks of these proteins using the mass spectrometry approach described above. We demonstrated that Kae1cyto interacts with both Bud32 and Cgi121. Bud32 was reproducibly co-precipitated with Kae1cyto and Cgi121, and Kae1cyto was co-precipitated with both Bud32 and Cgi121. Bud32, but not Cgi121, was detected in Kae1cyto IP samples (Fig. 7A). These data most likely reflect the indirect nature of the Kae1cyto-Cgi121 interaction given that Kae1 and Bud32 interact independently of Cgi121 (28).
FIGURE 7.

Kae1cyto forms a partially conserved KEOPS complex. A, interaction map summarizing protein hits identified by mass spectrometry from immunoprecipitated Kae1cyto, Bud32, and Cgi121. Hits identified for the three bait proteins are indicated by colored lines as follows: Kae1cyto, gray; Bud32, red; Cgi121, orange. Hits without previously reported KEOPS interactions are boxed. B, summary of MS data comparing a typical eukaryotic KEOPS complex with the P. falciparum Kae1cyto complex.
No other binding partners were identified for Cgi121, and no hits common to all three proteins were found. This suggests that 1) Cgi121 may function solely within the EKC/KEOPS complex, and 2) there may be no functional homologue for Pcc1 in P. falciparum. Together, these data confirm the predicted eukaryotic origin of Kae1cyto and show that this protein forms a largely conserved protein complex with Bud32 and Cgi121. A total of seven other proteins with a variety of predicted activities were identified as hits for Kae1 and/or Bud32 (Fig. 7A), although the functional significance of these hits remains to be explored. As expected, the t6A pathway protein Sua5 did not interact with Kae1, Bud32, or Cgi121.
Kae1api Forms Non-canonical Interactions with Multiple Apicoplast Proteins
Initial mass spectrometry analyses of Kae1api IP samples identified the EF-Ts, a putative RNA helicase (PF3D7_0504400), the isoprenoid synthesis enzyme IspG, and a protein of unknown function (PF3D7_1025300) as the four top scoring hits (Table 2). Both isoforms of Kae1api (PF3D7_0408900.1 and PF3D7_0408900.2) were also detected. To further investigate these results, we successfully RFA-tagged three of the four candidate genes at their endogenous loci in blood stage parasites. The sole exception was PF3D7_1025300; we were unable to detect integration for this gene in multiple transfections.
TABLE 2.
Top Kae1api hits identified by mass spectrometry
Kae1api was immunoprecipitated from RFA-tagged parasites with α-GFP antibody and analyzed by MS. Hits were filtered against negative control samples and ranked by Mascot score. Data for top hits are shown from a representative replicate. PSMs, peptide-spectrum matches.
| Accession | Description | Score | Coverage | No. proteins | No. unique peptides | No. PSMs |
|---|---|---|---|---|---|---|
| % | ||||||
| PF3D7_0408900 | Product = peptidase, M22 family, putative | 1381.97 | 55.12 | 1 | 36 | 51 |
| PF3D7_1025300 | Product = conserved Plasmodium protein, unknown function | 1001.65 | 33.08 | 1 | 26 | 33 |
| PF3D7_1022800 | Product = 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase (GcpE) | 251.23 | 9.59 | 1 | 5 | 7 |
| PF3D7_0305000 | Product = EF-Ts | 195.21 | 6.92 | 1 | 2 | 4 |
| PF3D7_0504400 | Product = ATP-dependent helicase, putative | 160.04 | 5.71 | 2 | 3 | 4 |
Protein expression in blood stage parasites was verified by Western blotting (Fig. 8A), and IFA studies confirmed the apicoplast localization of all three proteins (Fig. 8B), consistent with the MS results. We next performed reciprocal IP experiments in which RFA-tagged IspG, EF-Ts, and the putative RNA helicase proteins were immunoprecipitated and analyzed by MS. The resulting data largely recapitulate the interactions detected in the Kae1api IP sample (Fig. 9A). All four bait proteins (Kae1api, IspG, EF-Ts, and RNA helicase) co-precipitated each other with the sole exception that Kae1api was not detected in the RNA helicase IP samples. It is possible that because of its higher abundance a relatively small percentage of total RNA helicase interacts with Kae1api. Alternatively, this interaction may be indirect, weak, or transient.
FIGURE 8.

Expression and localization of Kae1api interaction partners. A, an α-GFP Western blot confirms the expression of RFA-tagged IspG, EF-Ts, and a putative RNA helicase in blood stage parasites. B, representative IFA images from RFA-tagged parasites are shown. IspG (top row), EF-Ts (middle row), and a putative RNA helicase (bottom row) co-localize with an apicoplast marker (ACP) and not with mitochondria. Mitochondria and nuclei were visualized with MitoTracker and DAPI DNA stain, respectively.
FIGURE 9.

A, mass spectrometry reveals novel Kae1api protein interactions. An interaction map summarizes protein hits identified by mass spectrometry from immunoprecipitated Kae1api, IspG, EF-Ts, and the putative RNA helicase. Hits identified for the four bait proteins are indicated by colored lines as follows: Kae1api, gray; IspG, light blue; EF-Ts, dark blue; putative RNA helicase, purple. Hits not predicted to target to the apicoplast (as determined by PATS and PlasmoAP) are shown in orange. Genes predicted to function in apicoplast translation are underlined. B, distribution of hits predicted to function in translation and/or target to the apicoplast. api ribo, apicoplast ribosomal protein; INT, integrase; CMK, 4-diphosphocytidyl-2c-methyl-D-erythritol kinase; SPP, stromal processing peptidase; RAP, RNA-binding domain abundant in apicomplexans.
Several other proteins were reproducibly identified in the apicoplast IP data sets with varying degrees of overlap between the bait proteins (Fig. 9A). Remarkably, the vast majority of these proteins (96 of 99 hits) are known or predicted to be apicoplast-localized, and the large number of proteins identified in this screen are likely reflective of the interconnectedness between the biological pathways in this organelle. It is notable that we also identified the highly conserved interaction between EF-Ts and EF-Tu.
We next performed GO annotation cluster analysis on the pooled apicoplast hit list to better understand the functional implications of these results. As expected, there was strong enrichment for terms indicating apicoplast/plastid localization (Table 3). In addition, 23 of the 99 hits are predicted to directly participate in the translation or co-translational modification of apicoplast-expressed proteins (Fig. 9B). Together, these results suggest that Kae1api regulates ribosome activity, possibly at the level of protein translation.
TABLE 3.
GO annotation cluster analysis of pooled apicoplast hits
81 of 99 hits were GO annotated and included in the analysis. Translation and plastid localization terms were significantly enriched in this data set relative to the parasite genome as a whole (background data set). adj., adjusted.
| Category | Term | No. proteins identified from hit list (out of 81) | Positive | p value | -Fold enrichment | Bonferroni adj. p value | Benjamini-Hochberg adj. p value | No. proteins identified from background |
|---|---|---|---|---|---|---|---|---|
| % | ||||||||
| GOTERM_CC_FAT | GO:0020011, apicoplast | 64 | 79.0 | 6.07E-26 | 2.6 | 1.40E-24 | 1.40E-24 | 462 |
| GOTERM_CC_FAT | GO:0009536, plastid | 64 | 79.0 | 8.09E-26 | 2.6 | 1.86E-24 | 9.30E-25 | 464 |
| GOTERM_BP_FAT | GO:0006412, translation | 14 | 17.3 | 1.33E-04 | 3.1 | 9.13E-03 | 9.13E-03 | 202 |
Reciprocal IP Experiments in Dual Tagged Parasites Confirm Kae1api Protein Interactions
A series of parasite lines was generated to confirm the protein interactions observed by MS. In these experiments, RFA-tagged parasites (Kae1api, EF-Ts, RNA helicase, and IspG) were transfected with expression constructs encoding candidate interaction partners with a C-terminal 6xFLAG tag. We successfully obtained five parasite lines with different combinations of epitope-tagged proteins. Cell pellets from these parasites were collected, and RFA-tagged proteins were immunoprecipitated with α-GFP antibodies. The resulting samples were analyzed by Western blot and probed with α-FLAG antibodies (Fig. 10).
FIGURE 10.
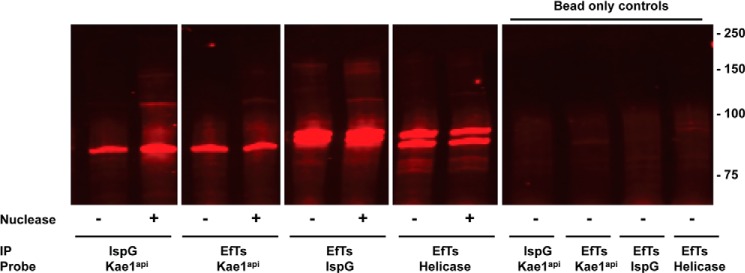
Reciprocal IP experiments validate MS approach and confirm protein-protein interactions. RFA-tagged proteins were immunoprecipitated with α-GFP antibody after treatment with or without Benzonase, a nonspecific nuclease. IP samples were blotted and visualized with α-FLAG antibody to assess protein-protein interactions. No difference was found between treated (+) and untreated (−) samples. To control for nonspecific interactions with beads, IPs were performed without α-GFP antibody (right panel).
These results confirm the interactions observed between Kae1api and IspG as well as the interactions observed between EF-Ts and the other three other apicoplast proteins (Kae1api, IspG, and the putative RNA helicase). These data further validate our MS-based approach and confirm that filtering MS data from complex samples against our array of negative controls enriches for bona fide biological interactions.
Apicoplast Protein Interactions Are Independent of Nucleic Acid Polymers
Kae1 proteins have established nucleic acid binding activity, the putative RNA helicase likely binds RNA, and EF-Ts interacts with the tRNA-binding protein EF-Tu. Together, these raise the possibility that the observed interactions may occur indirectly by binding to a common nucleic acid substrate or ribonucleoprotein complex (such as a ribosome). We directly tested this in the dual tagged parasite lines by performing co-IP experiments after treatment with Benzonase nuclease. Under these conditions, RNA and DNA were degraded to undetectable levels. There was no difference in precipitated protein signal between the two samples sets (Fig. 10), demonstrating that interactions between these proteins does not require DNA or RNA and is instead due to protein-protein interactions.
Kae1api and Its Partners Play a Potential Role in Ribosome Function
We next set out to quantitatively measure the apicoplast ribosome interactions observed in the IP-MS experiments. We immunoprecipitated RFA-tagged Kae1api, EF-Ts, IspG, RNA helicase, Kae1cyto, Bud32, Cgi121, and Sua5/YrdC and isolated total RNA from each sample. Negative control samples from 3D7-PM1 or ACP-GFP parasite lines were collected in parallel, and qRT-PCR was performed to test for enrichment for apicoplast or cytoplasmic rRNA.
Although no enrichment was observed for any of the cytoplasmic bait protein samples (data not shown), dramatic enrichment for apicoplast 16 and 23 S rRNA was observed for IspG, EF-Ts, and RNA helicase samples (Fig. 11). Kae1api displayed modest but reproducible 2–3-fold enrichment for both apicoplast rRNAs compared with the ACP-GFP control. To test the possibility that bait proteins interact with rRNA through binding of a common mRNA partner (i.e. bait proteins may bind actively translated mRNA), we treated IP samples with RNase I to specifically degrade single-stranded mRNA. Enrichment for apicoplast rRNA was essentially identical for all four bait proteins under treated and untreated conditions, suggesting that Kae1api and its partners directly bind rRNA or ribosomal proteins.
FIGURE 11.

Kae1api and its partners specifically interact with apicoplast rRNA. Enrichment for apicoplast rRNA relative to the ACP-GFP control was determined (nuclearly encoded cytoplasmic rRNAs were used as internal standards for total RNA abundance). Triplicate data are shown from a representative experiment. Error bars represent one standard deviation from the mean.
DISCUSSION
Mass spectrometry is a valuable tool for studying the biology of protein complexes. The high sensitivity of IP-MS assays allows for the detection of weak or low abundance interactions, although this sensitivity also makes such assays highly prone to false-positives. Although false-positives are often due to abundant or nonspecifically binding (i.e. sticky) proteins, a greater challenge can be eliminating the myriad housekeeping interactions that occur with the protein of interest. These interactions are biologically real and important for proteostasis of the target protein (i.e. synthesis, modification, folding, and degradation), although they often hamper efforts to identify interactions relevant to the specific functions of a protein. To address this issue, we developed a novel data filtering approach based on the cellular localization of multiple control proteins in P. falciparum. This was then used to identify and exclude the vast majority of housekeeping interactions, and we utilized this strategy to characterize the protein-protein interaction networks of Kae1api and Kae1cyto.
This approach successfully identified all predicted interactions for the bait proteins in our test set. Our data confirm the predicted Kae1cyto-Bud32-Cgi121 complex in the cytoplasm as well as the highly conserved interaction between EF-Tu and EF-Ts in the apicoplast. In addition, we identified a novel set of interacting partners for Kae1 in the apicoplast. These findings were confirmed in an orthogonal reciprocal IP assay and validate the effectiveness of our data analysis strategy. This approach has potential advantages over other MS-based analyses. Although other techniques such as tandem affinity purification tagging remove background signal by utilizing multiple purification steps that can lead to dissociation of weakly binding partners, we reduced background by comparison with a stringent series of negative control samples. We have demonstrated that this effectively reduces signal from housekeeping pathways without removing predicted conserved interactions with our set of bait proteins.
The Kae1 protein family has been implicated in a wide range of biological processes since its initial discovery over 20 years ago. Several recent studies have established the requirement for Kae1 in the t6A modification of ANN-decoding tRNAs, and mechanistic details of this pathway are emerging. In addition to its clear tRNA modification function, Kae1 is recruited to specific gene sets in Saccharomyces cerevisiae where it appears to regulate transcription by recruitment of various co-activators. Kae1 is also required for telomere homeostasis, although whether this function is distinct from its role in t6A metabolism is unclear. Our findings suggest an expanded role for Kae1api as well as new mechanisms of transcriptional and translational co-regulation.
Kae1api binds DNA in vitro, and it is tempting to speculate that it may regulate gene expression in the P. falciparum apicoplast. Interestingly, the association of Kae1api with IspG may suggest a role in transcriptional regulation. In prokaryotes, IspG participates in the late stages of non-mevalonate isoprenoid biosynthesis and catalyzes transformation of the small molecule intermediate 2-C-methyl-d-erythritol 2,4-cyclodiphosphate to 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate. Intriguingly, flux through this pathway is coupled to transcription in Chlamydia trachomatis via 2-C-methyl-d-erythritol 2,4-cyclodiphosphate levels (29). Increased levels of 2-C-methyl-d-erythritol 2,4-cyclodiphosphate are thought to disrupt histone-DNA interactions, leading to transcriptional activation and cellular differentiation. The physical association between Kae1 and the 2-C-methyl-d-erythritol 2,4-cyclodiphosphate-producing enzyme IspG raises the possibility that transcription may be similarly regulated in the apicoplast. This is especially notable as isoprenoid biosynthesis is proposed to be the essential function of the apicoplast in intraerythrocytic parasites (30).
Alternatively, Kae1api nucleic acid binding may simply reflect its predicted tRNA-modifying activity. The novel interactions of Kae1api with IspG, EF-Ts, and a predicted RNA helicase suggest that Kae1 function is modulated by protein-protein interactions even outside the context of a typical EKC/KEOPS complex. The unexpected nature of these interactions suggests a more complex regulation of Kae1 than previously thought, and their functional implications warrant further investigation. EF-Ts has a well known role in recycling EF-Tu after peptide bond formation at the ribosome, whereas the putative RNA helicase may act on rRNA or mRNA substrates to facilitate ribosome biogenesis or translation. The physical association of Kae1api with these proteins suggests a regulatory role in protein synthesis and is further supported by interactions of these proteins with ribosomal components as demonstrated by MS and qRT-PCR. In certain archaeal genomes, the KEOPS protein Pcc1 is fused to the ribosome maturation factor RimI, indicating a possible link between Kae1 and ribosome maturation (31).
Although cytosolic Kae1 appears to be a canonical tRNA modification enzyme, its apicoplast-targeted counterpart likely has additional functions. Further study of Kae1api associations with the DNA, ribosomes, and isoprenoid biosynthesis pathway of the apicoplast will yield further insight into the biology of this essential and unusual organelle.
Acknowledgments
We thank the following people for generously sharing reagents: John Adams (MRA-912 plasmid), Akhil Vaidya (DSM-1), and Geoff McFadden (α-ACP antibodies). We thank Kenneth Beattie and Douglas Lamont at the Dundee Fingerprints Proteomics facility for help and feedback. We also thank Edwin Chen for feedback on recombinant Kae1 expression, Nicholas Szrama for help on data analysis in MatLab, and Erica Siebrasse for assistance with qRT-PCR assay development. Finally, we thank Eva Istvan, Vasant Muralidharan, and Sergei Djuranovic for insights and helpful discussions.
Footnotes
- Kae1
- kinase-associated endopeptidase 1
- qRT-PCR
- quantitative RT-PCR
- IP
- immunoprecipitation
- t6A
- threonylcarbamoyl 6-adenosine
- EKC
- endopeptidase, kinase, chromatin-associated
- KEOPS
- kinase, endopeptidase, and other proteins of small size
- EF
- elongation factor
- RFA
- regulatable fluorescent affinity
- TMP
- trimethoprim
- IFA
- immunofluorescence assay
- ACP
- acyl carrier protein
- GO
- gene ontology.
REFERENCES
- 1. World Health Organization (2013) World Malaria Report 2013, World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Abdullah K. M., Lo R. Y., Mellors A. (1991) Cloning, nucleotide sequence, and expression of the Pasteurella haemolytica A1 glycoprotease gene. J. Bacteriol. 173, 5597–5603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abdullah K. M., Udoh E. A., Shewen P. E., Mellors A. (1992) A neutral glycoprotease of Pasteurella haemolytica A1 specifically cleaves O-sialoglycoproteins. Infect. Immun. 60, 56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kisseleva-Romanova E., Lopreiato R., Baudin-Baillieu A., Rousselle J. C., Ilan L., Hofmann K., Namane A., Mann C., Libri D. (2006) Yeast homolog of a cancer-testis antigen defines a new transcription complex. EMBO J. 25, 3576–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Downey M., Houlsworth R., Maringele L., Rollie A., Brehme M., Galicia S., Guillard S., Partington M., Zubko M. K., Krogan N. J., Emili A., Greenblatt J. F., Harrington L., Lydall D., Durocher D. (2006) A genome-wide screen identifies the evolutionarily conserved KEOPS complex as a telomere regulator. Cell 124, 1155–1168 [DOI] [PubMed] [Google Scholar]
- 6. Srinivasan M., Mehta P., Yu Y., Prugar E., Koonin E. V., Karzai A. W., Sternglanz R. (2011) The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 30, 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. El Yacoubi B., Hatin I., Deutsch C., Kahveci T., Rousset J. P., Iwata-Reuyl D., Murzin A. G., de Crécy-Lagard V. (2011) A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J. 30, 882–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Daugeron M. C., Lenstra T. L., Frizzarin M., El Yacoubi B., Liu X., Baudin-Baillieu A., Lijnzaad P., Decourty L., Saveanu C., Jacquier A., Holstege F. C., de Crécy-Lagard V., van Tilbeurgh H., Libri D. (2011) Gcn4 misregulation reveals a direct role for the evolutionary conserved EKC/KEOPS in the t6A modification of tRNAs. Nucleic Acids Res. 39, 6148–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deutsch C., El Yacoubi B., de Crécy-Lagard V., Iwata-Reuyl D. (2012) Biosynthesis of threonylcarbamoyl adenosine (t6A), a universal tRNA nucleoside. J. Biol. Chem. 287, 13666–13673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lauhon C. T. (2012) Mechanism of N6-threonylcarbamoyladenonsine (t(6)A) biosynthesis: isolation and characterization of the intermediate threonylcarbamoyl-AMP. Biochemistry 51, 8950–8963 [DOI] [PubMed] [Google Scholar]
- 11. Perrochia L., Crozat E., Hecker A., Zhang W., Bareille J., Collinet B., van Tilbeurgh H., Forterre P., Basta T. (2013) In vitro biosynthesis of a universal t6A tRNA modification in Archaea and Eukarya. Nucleic Acids Res. 41, 1953–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perrochia L., Guetta D., Hecker A., Forterre P., Basta T. (2013) Functional assignment of KEOPS/EKC complex subunits in the biosynthesis of the universal t6A tRNA modification. Nucleic Acids Res. 41, 9484–9499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wan L. C., Mao D. Y., Neculai D., Strecker J., Chiovitti D., Kurinov I., Poda G., Thevakumaran N., Yuan F., Szilard R. K., Lissina E., Nislow C., Caudy A. A., Durocher D., Sicheri F. (2013) Reconstitution and characterization of eukaryotic N6-threonylcarbamoylation of tRNA using a minimal enzyme system. Nucleic Acids Res. 41, 6332–6346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Handford J. I., Ize B., Buchanan G., Butland G. P., Greenblatt J., Emili A., Palmer T. (2009) Conserved network of proteins essential for bacterial viability. J. Bacteriol. 191, 4732–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muralidharan V., Oksman A., Iwamoto M., Wandless T. J., Goldberg D. E. (2011) Asparagine repeat function in a Plasmodium falciparum protein assessed via a regulatable fluorescent affinity tag. Proc. Natl. Acad. Sci. U.S.A. 108, 4411–4416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Balu B., Shoue D. A., Fraser M. J., Jr., Adams J. H. (2005) High-efficiency transformation of Plasmodium falciparum by the lepidopteran transposable element piggyBac. Proc. Natl. Acad. Sci. U.S.A. 102, 16391–16396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ganesan S. M., Morrisey J. M., Ke H., Painter H. J., Laroiya K., Phillips M. A., Rathod P. K., Mather M. W., Vaidya A. B. (2011) Yeast dihydroorotate dehydrogenase as a new selectable marker for Plasmodium falciparum transfection. Mol. Biochem. Parasitol. 177, 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu J., Gluzman I. Y., Drew M. E., Goldberg D. E. (2005) The role of Plasmodium falciparum food vacuole plasmepsins. J. Biol. Chem. 280, 1432–1437 [DOI] [PubMed] [Google Scholar]
- 19. Deitsch K., Driskill C., Wellems T. (2001) Transformation of malaria parasites by the spontaneous uptake and expression of DNA from human erythrocytes. Nucleic Acids Res. 29, 850–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rodgers J. T., Patel P., Hennes J. L., Bolognia S. L., Mascotti D. P. (2000) Use of biotin-labeled nucleic acids for protein purification and agarose-based chemiluminescent electromobility shift assays. Anal. Biochem. 277, 254–259 [DOI] [PubMed] [Google Scholar]
- 21. Tonkin C. J., van Dooren G. G., Spurck T. P., Struck N. S., Good R. T., Handman E., Cowman A. F., McFadden G. I. (2004) Localization of organellar proteins in Plasmodium falciparum using a novel set of transfection vectors and a new immunofluorescence fixation method. Mol. Biochem. Parasitol. 137, 13–21 [DOI] [PubMed] [Google Scholar]
- 22. Waller R. F., Keeling P. J., Donald R. G., Striepen B., Handman E., Lang-Unnasch N., Cowman A. F., Besra G. S., Roos D. S., McFadden G. I. (1998) Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 95, 12352–12357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 24. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 25. Foth B. J., Ralph S. A., Tonkin C. J., Struck N. S., Fraunholz M., Roos D. S., Cowman A. F., McFadden G. I. (2003) Dissecting apicoplast targeting in the malaria parasite Plasmodium falciparum. Science 299, 705–708 [DOI] [PubMed] [Google Scholar]
- 26. Zuegge J., Ralph S., Schmuker M., McFadden G. I., Schneider G. (2001) Deciphering apicoplast targeting signals—feature extraction from nuclear-encoded precursors of Plasmodium falciparum apicoplast proteins. Gene. 280, 19–26 [DOI] [PubMed] [Google Scholar]
- 27. Bender A., van Dooren G. G., Ralph S. A., McFadden G. I., Schneider G. (2003) Properties and prediction of mitochondrial transit peptides from Plasmodium falciparum. Mol. Biochem. Parasitol. 132, 59–66 [DOI] [PubMed] [Google Scholar]
- 28. Mao D. Y., Neculai D., Downey M., Orlicky S., Haffani Y. Z., Ceccarelli D. F., Ho J. S., Szilard R. K., Zhang W., Ho C. S., Wan L., Fares C., Rumpel S., Kurinov I., Arrowsmith C. H., Durocher D., Sicheri F. (2008) Atomic structure of the KEOPS complex: an ancient protein kinase-containing molecular machine. Mol. Cell 32, 259–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grieshaber N. A., Fischer E. R., Mead D. J., Dooley C. A., Hackstadt T. (2004) Chlamydial histone-DNA interactions are disrupted by a metabolite in the methylerythritol phosphate pathway of isoprenoid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 101, 7451–7456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yeh E., DeRisi J. L. (2011) Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 9, e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoshikawa A., Isono S., Sheback A., Isono K. (1987) Cloning and nucleotide sequencing of the genes rimI and rimJ which encode enzymes acetylating ribosomal proteins S18 and S5 of Escherichia coli K12. Mol. Gen. Genet. 209, 481–488 [DOI] [PubMed] [Google Scholar]