

Background: Recent studies suggest that Mincle expression is induced by Dectin-3-mediated signaling in response to TDM stimulation.
Results: Deficiency in Dectin-3 and the CARD9-Bcl10-Malt1 complex are defective for TDM-induced NF-κB activation and Mincle.
Conclusion: Dectin-3- and CARD9/Bcl10/Malt1-dependent NF-κB activation plays an essential role for TDM-induced Mincle expression.
Significance: This study provides the molecular insight for designing adjuvants that stimulate the immune system.
Keywords: Innate Immunity, Lectin, NF-kappa B (NF-κB), NF-κB Transcription Factor, Signal Transduction
Abstract
Previous studies indicate that both Dectin-3 (also called MCL or Clec4d) and Mincle (also called Clec4e), two C-type lectin receptors, can recognize trehalose 6,6′-dimycolate (TDM), a cell wall component from mycobacteria, and induce potent innate immune responses. Interestingly, stimulation of Dectin-3 by TDM can also induce Mincle expression, which may enhance the host innate immune system to sense Mycobacterium infection. However, the mechanism by which Dectin-3 induces Mincle expression is not fully defined. Here, we show that TDM-induced Mincle expression is dependent on Dectin-3-mediated NF-κB, but not nuclear factor of activated T-cells (NFAT), activation, and Dectin-3 induces NF-κB activation through the CARD9-BCL10-MALT1 complex. We found that bone marrow-derived macrophages from Dectin-3-deficient mice were severely defective in the induction of Mincle expression in response to TDM stimulation. This defect is correlated with the failure of TDM-induced NF-κB activation in Dectin-3-deficient bone marrow-derived macrophages. Consistently, inhibition of NF-κB, but not NFAT, impaired TDM-induced Mincle expression, whereas NF-κB, but not NFAT, binds to the Mincle promoter. Dectin-3-mediated NF-κB activation is dependent on the CARD9-Bcl10-MALT1 complex. Finally, mice deficient for Dectin-3 or CARD9 produced much less proinflammatory cytokines and keyhole limpet hemocyanin (KLH)-specific antibodies after immunization with an adjuvant containing TDM. Overall, this study provides the mechanism by which Dectin-3 induces Mincle expression in response to Mycobacterium infection, which will have significant impact to improve adjuvant and design vaccine for antimicrobial infection.
Introduction
The C-type lectin receptors (CLRs)3 are a large superfamily of proteins that contain a carbohydrate recognition domain and calcium-binding sites on their extracellular domains (1–3). The Dectin-2 family of CLRs encoded by the natural killer gene cluster is composed of Dectin-2, Dectin-3 (originally named murine macrophage C-type lectin, MCL, and also classified as CLECSF8 or Clec4d), BDCA-2 (blood dendritic cell antigen 2), Mincle, and DCIR (dendritic cell immunoreceptor) (4, 5). These CLRs are type II transmembrane receptors and share a common structure consisting of a single extracellular C-type lectin-like domain, a stalk region of varying length, and a transmembrane region (6).
Interestingly, Mincle is a recently characterized receptor for macrophage recognition of Mycobacterium tuberculosis, which recognizes the Mycobacterium cord factor, trehalose-6,6′-dimycolate (TDM) (7–11). Mincle is important for macrophage-associated innate immune response (12, 13), as well as in vitro macrophage activation (14). Furthermore, Mincle-deficient mice show defective adaptive immune responses to immunization with a synthetic TDM analog (11, 15, 16).
Emerging evidence indicate that upon ligand binding, CLRs, such as Dectin-1, Dectin-2, Mincle, DCAR, and BDCA-2, induce multiple signal transduction cascades through their own immunoreceptor tyrosine-based activation motifs or interacting with immunoreceptor tyrosine-based activation motif-containing adaptor proteins such as FcRγ (5, 17). These CLR-induced signaling cascades lead to the activation of nuclear factor κ B (NF-κB) family of transcriptional factors through a Syk- and CARD9-dependent pathway (9, 18, 19). The activation of NF-κB plays a critical role in the induction of innate immune and inflammatory responses following microbial infection and tissue damages (18, 20, 21). Dectin-3, which has a short cytoplasmic tail without any signaling motif and is presumably associated with other signaling adaptors, is the least characterized member of this family (22, 23). We have recently demonstrated that Dectin-3 forms a heterodimeric pattern-recognition receptor with Dectin-2 for sensing fungal infection through activation of NF-κB (24).
In a recent study, Dectin-3/MCL was identified as another receptor for TDM (25). Moreover, Lobato-Pascual et al. found that Mincle can form a receptor complex with Dectin-3 and FcϵRI-γ in a rat system (26). It is interesting to know whether Dectin-3 and Mincle are functionally linked for the recognition of TDM. In this study, we report that Dectin-3-mediated NF-κB activation is critical for TDM-induced Mincle expression, which is dependent on the CARD9-Bcl10-MALT1 complex. Although Dectin-3 has been found to form a heterodimeric complex with Dectin-2, only Dectin-3, but not Dectin-2, is required for induction of Mincle. In addition, Dectin-3 neither form a heterodimeric complex nor synergistically induce NF-κB activation with Mincle. Instead, it only serves as a sensor for induction of Mincle. Functionally, we showed that Dectin-3-deficient mice, as CARD9-deficient mice, produce much less cytokines and antigen-specific antibodies when using the adjuvant containing TDM.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies against phospho-p38 (4631), phospho-ERK (9101), phospho-JNK (9251), phospho-IKK (2697), total p38 (9212), and total JNK (9252) were purchased from Cell Signaling Technology; antibodies against p65 (sc-8008), proliferating cell nuclear antigen (sc-56), NFAT-c1 (sc-7294), ERK (sc-154), IKKα (sc-7218), IκBα (sc-371), FLAG (sc-807), and tubulin (sc-8035) were from Santa Cruz Biotechnology. TDM (catalog no. T3034) and TPCA-1 (T1452) were from Sigma. In all experiments, TDM was coated in six-well plate with 20 μg/ml concentration at 4 °C for overnight. NFAT inhibitor 11R-VIVIT (catalog no. 13855) was from Cayman Chemical. Dectin-2 and Dectin-3 monoclonal antibodies (IgG1 and IgG2a) were generated by using the extracellular domain of Dectin-2 or Dectin-3 as immunogens that were described previously (24). Monoclonal antibodies against Mincle were generated by using the extracellular domain of Mincle as immunogens. Fluorescence-conjugated monoclonal antibodies CD11b (catalog no. 45-0112-82) and F4/80 (catalog no. 123108) were purchased from eBioscience and Biolegend. FITC-conjugated goat anti-mouse IgG (H+L) secondary antibody was purchased from Invitrogen (catalog no. 62-6511).
Plasmid Construction
Human Dectin-2, Dectin-3, and Mincle were amplified by PCR using full-length cDNA of human peripheral blood cells as a template (24). All PCR amplifying fragments including FLAG-encoding DNA sequence were inserted into the SalI and BglII digested site of pRV3 (a lentiviral vector).
Mice
Dectin-3-knock-out (Clec4d−/−) mice, CARD9-knock-out (Card9−/−) mice, Bcl10 knock-out (Bcl10−/−) mice and Malt1 knock-out (Malt1−/−) mice have been described previously (20, 24, 27, 28). All mice were housed in the specific pathogen-free animal facility at MD Anderson Cancer Center. 8- to 16-week-old mice were used in the experiments. All animal experiments were performed in compliance with institutional guidelines and according to the protocol approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center.
Immunization of mice was conducted as described previously (29–31). Briefly, CARD9- and Dectin-3-deficient mice and their controls (7-week-old female mice) were immunized with keyhole limpet hemocyanin (KLH, 0.5 mg/ml) emulsified in complete Freund's adjuvant (0.5 mg/ml) at the base of the tail (100 μl for each mouse). Forty-eight hour later, blood was collected from the tail and measured for cytokine production. Seven days after immunization, mice were sacrificed, and serum samples were subjected to immunoglobulin analysis by ELISA. In brief, different dilutions of serum were added into wells of plates precoated with KLH protein (10 μg/ml), and antigen-specific antibodies were detected with biotinylated anti-mouse IgG, HRP-conjugated goat anti-mouse IgM, goat anti-mouse IgG1-HRP, and goat anti-mouse IgG2a-HRP antibodies (Southern Biotechnology Associates).
BMDM Preparation
Primary cultures of bone marrow-derived macrophages (BMDMs) from C57BL/6J mice were prepared as described previously (24, 32). Briefly, bone marrow cells were harvested from the femurs and tibias of mice. Erythrocytes were removed from cells by using a hypotonic solution. Cells were cultured for 7 days in Dulbecco's modified Eagle's medium containing 20% fetal bovine serum, 55 μm β-mercaptoethanol, streptomycin (100 μg/ml), penicillin (100 units/ml), and 30% conditioned medium from L929 cells expressing macrophage colony-stimulating factor. Non-adherent cells were removed, and cells were passed every 3 days. After 1 week of culturing, flow cytometry analysis indicated that the harvested cell population contained above 97% CD11b+ F4/80+cells.
Immunoblotting and Immunoprecipitation
BMDMs were serum-starved overnight, stimulated, and lysed in lysis buffer (150 mm NaCl, 50 mm HEPES (pH 7.4), 1 mm EDTA, 1% Nonidet P-40, protease inhibitors). Nuclear extracts or total cell lysates were immunoprecipitated with antibody-conjugated agarose. The resulting immunoprecipitates and lysates were subjected to SDS-PAGE and then blotted using antibodies.
Quantitative PCR
Total RNA was isolated using RNeasy kit (Qiagen) and reverse transcribed using SuperScriptIII (Invitrogen). Quantitative PCR was performed in triplicates using Power SYBR Green PCR Master Mix (Applied Biosystems). The amounts of transcript were normalized to GAPDH. Melting curves were run to ensure amplification of a single product. Primers were as follows: Mincle, 5′-AGTGCTCTCCTGGACGATAG-3′ (forward) and 5′-CCTGATGCCTCACTGTAGCAG-3′ (reverse); GAPDH, 5′-AACAGCAACTCCCACTCTTC-3′ (forward) and 5′-CCTGTTGCTGTAGCCGTATT-3′ (reverse).
ChIP Assay
The ChIP assay was performed with a ChIP kit (Upstate Biotechnology) as described previously (33, 34). In brief, after cross-linking with 1% formaldehyde for 10 min at room temperature, cells were lysed in SDS buffer (50 mm Tris-Cl (pH 8.1), 10 mm EDTA, 1% SDS, and protease inhibitors). Lysates were sonicated using Bioruptor and diluted with a ChIP dilution buffer (20 mm Tris-Cl (pH 8.1), 1 mm EDTA, 150 mm NaCl, and 0.3% Triton X-100). Lysates were then precleared with protein G beads and incubated with p65, NFAT-c1, or control rabbit IgG at 4 °C overnight. Protein G agarose was added to the lysates and incubated for 2 h at 4 °C. Ab-protein-DNA complexes were eluted and incubated at 65 °C overnight to reverse formaldehyde cross-linking, and DNA was recovered by a PCR purification kit (Clontech). DNA fragments were amplified by RT-PCR with specific primers. The antibodies used for ChIP are as follows: NFκB-p65 (sc-109) and NFATc1 (sc-7294). The ChIP PCR primers were as follows: 5′-CAGTGACAACTCAACCAAGC-3′ (forward) and 5′-GAGGTTATGAGTGTCAGGAG-3′ (reverse); 5′-ATGGCTCCTGCCACATATGT-3′ (forward) and 5′-ACTCCATTCCCTCACTACTC-3′ (reverse).
Luciferase Reporter Assay
The Mincle promoter was amplified by PCR using the genome DNA of wild type C57BL/6 mice as a template. PCR primers were as follows: 1783F, 5′-GACTGACGCGTGGTTTGCAGCCCCATAGGAG-3′ (forward); 1207F, 5′-GCGAGACGCGTGGTTTGGATCTTGGAAATGATG-3′ (forward); 835F, 5′-TTGTGACGCGTATGTGTAGCACTTCTATCTCCTGAC-3′ (forward); and 5′-GATAAAGATCTAAGAGCCCCTGGAAAGTGAGT-3′ (reverse). The two NF-kB-binding sites were mutated by site-directed mutagenesis. The primers are as follows: m1, 5′-TGAGCACTAGAGTGAATCTGTGGAAATAATAGAAAGTTTCCTTGAGGCA-3′(forward) and 5′-TCCACAGATTCACTCTAGTGCTCAGAACTTTTTATTTTACAGACACGTGGAT-3′ (reverse); m2, 5′-TCATCAAACACCACTAGAGTGAAGTCATATGTGTAGCACTTCTATCTCCTGAC-3′ (forward) and 5′-TGACTTCACTCTAGTGGTGTTTGATGAGCCAGATTTCCTATGTGCTAC-3′ (reverse). All of the reporter constructs were cloned into PGL3 luciferase reporter plasmid. RAW264.7 cells were transfected with different luciferase reporter constructs together with a Dectin-3 expression plasmid (pRV3-Dectin-3) and a control Renilla luciferase reporter plasmid by using a NeofectTM DNA transfection reagent Kit (catalog no. TF201201) according to manufacturer's instructions. The Dual-Luciferase reporter system (Promega) was used to examine firefly and Renilla luciferase activity. Renilla luciferase was used to normalize transfection efficiency and luciferase activity.
Cytokine Measurements
TNF-α, IL-6, IL-1β, and IL-10 levels in the supernatant of the cells were measured with Ready-SET-GO enzyme-linked immunosorbent assay (ELISA) kits (eBioscience). All samples were measured in triplicate according to the manufacturer's protocol.
Statistical Analysis
At least two biological replicates were performed for all experiments unless otherwise indicated. Student's t test for paired observations was used for statistical analyses. Statistical significance was set at a p value of <0.05 or 0.01.
RESULTS
Dectin-3 Is Necessary for TDM-induced Mincle Expression
Recent studies suggest that Dectin-3/MCL functions as a pattern recognition receptor for TDM, and stimulation of Dectin-3 by TDM induces the expression of Mincle (25). To confirm this result, BMDMs isolated from wild type or Dectin-3 knock-out mice were stimulated with plate-bound TDM, and then Mincle expression was determined. Consistent with a recent study, the TDM-induced Mincle expression both at protein (Fig. 1A) and mRNA levels (Fig. 1B) was impaired in Dectin-3-deficient cells. Because our recent studies suggest that Dectin-3 and Dectin-2 form a heterodimeric receptor for sensing fungal infection (24), we examined whether Dectin-2/Dectin-3 heterodimers were involved in sensing TDM. BMDMs were pretreated with neutralizing antibodies against Dectin-3 or Dectin-2 to block the expression of Dectin-3 or Dectin-2 (24). Consistent with what we observed in Dectin-3 knock-out BMDMs, Dectin-3-blocking antibodies effectively blocked TDM-induced Mincle expression (Fig. 1C). However, Dectin-2 antibodies were not able to block this induction of Mincle expression both at mRNA and protein level (Fig. 1, D and E), although Dectin-2 expression was blocked very efficiently (Fig. 1E). These results indicate that TDM only activates receptor containing Dectin-3 but not both Dectin-2 and Dectin-3, suggesting that the Dectin-3, but not Dectin-2/Dectin-3 heterodimer, is a pattern-recognition receptor for TDM.
FIGURE 1.

Dectin-3 is required for TDM-induced Mincle expression. A and B, BMDMs from WT and Dectin-3 KO mice were stimulated with 20 μg/ml plate-coated TDM for 3, 6, 9, and 12 h. A, cell lysates were analyzed by immunoblotting with the indicated antibodies. B, total mRNA was analyzed by RT-PCR for Mincle expression. C–E, BMDMs from WT mice were pretreated with control (Ctr) IgG or blocking antibodies to Dectin-3 (C) or Dectin-2 (D and E) for 45 min and then stimulated with plate-coated TDM for indicated time points. C, D, total RNA was prepared from these cells, and real-time PCR was performed to quantify Mincle mRNA. E, cell lysates were analyzed by immunoblotting with the indicated antibodies.
Dectin-3 Is Required for TDM-induced NF-κB Activation
To determine the role of Dectin-3 after TDM stimulation, we examined the impact of Dectin-3 deficiency on TDM-induced signaling. We found that TDM-induced IKK phosphorylation, IκBα degradation (Fig. 2A), and p65 subunit nuclear translocation were defective in Dectin-3-deficient BMDMs following stimulation by TDM (Fig. 2B). Moreover, there is also a significant reduction of nuclear NFAT-c1 (Fig. 2B). However, TDM-induced activation of p38, ERK, and JNK were comparable in WT and Dectin-3-deficient BMDMs (Fig. 2C), indicating that these kinases can be activated by other receptors on BMDMs in response to TDM stimulation. Consistently, when Dectin-3 expression was blocked with neutralizing antibody, both α-mannan and TDM stimulation cannot induce p65 subunit nuclear translocation. However, blocking of Dectin-2 expression impaired the p65 translocation only when stimulated with α-mannan, but not in the TDM stimulation case (Fig. 2D). This indicates that TDM only activates Dectin-3, whereas Dectin-2 is not necessary for NF-κB activation. Moreover, the expression levels of proinflammatory cytokines including TNF-α, IL-6, and IL-1β in Dectin-3-deficient BMDMs were also significantly reduced in comparison with those in WT cells (Fig. 2E). Taken together, these results demonstrate that Dectin-3 mediates TDM-induced NF-κB activation.
FIGURE 2.

Dectin-3 is required for TDM-induced NF-κB activation. A–C, BMDMs from WT and Dectin-3 KO mice were either untreated or stimulated with plate-coated TDM for indicated time points. A, total cell lysates were prepared from these cells and then subjected to immunoblotting analysis using indicated antibodies. B, nuclear extracts were prepared and analyzed by immunoblotting with p65, NFAT-c1, and proliferating cell nuclear antigen antibodies. C, immunoblotting with the indicated antibodies were done with the total cell lysates. D, BMDMs from WT mice were pretreated with Dectin-2 or Dectin-3 blocking antibodies for 45 min. Resulting cells were stimulated with plate-coated α-mannan (man) or TDM for 1 h. Nuclear extracts were prepared and analyzed by immunoblotting with p65 and proliferating cell nuclear antigen (PCNA) antibodies. Total cell lysates were immunoblotting with Dectin-2 or Dectin-3 antibodies. E, ELISA results for TNF-α, IL-6, and IL-1β in supernatants of BMDMs derived from WT or Dectin-3 KO mice, which were stimulated with plate-coated TDM for 8 h. Data shown are representative of three independent experiments. S.D. is indicated. **, p < 0.01 (t test). Ctr, control.
NF-κB, but Not NFAT, Is Required for Mincle Induction
To determine whether Dectin-3-induced Mincle expression is through NF-κB-dependent mechanism, we used TPCA-1, an inhibitor for IKK, to inhibit NF-κB activation (Fig. 3, A and C) and then examined the expression level of Mincle. We found that Mincle expression level is significantly reduced with this inhibitor both at protein (Fig. 3A) and mRNA level (Fig. 3C). Because NFAT-c1 is also affected in Dectin-3-deficient cells, we used NFAT inhibitor, 11R-VIVIT, with TDM stimulation. However, we found that inhibition of NFAT did not affect Mincle expression (Fig. 3, B and C). Consistently, we found that TPCA-1 inhibited TDM-induced expression of TNFα and IL-6, but not IL-10 expression (Fig. 3D). In contrast, NFAT inhibitor 11R-VIVIT could significantly reduce IL-10 expression (Fig. 3D) as expected, but slightly influenced TNFα and IL-6 production. These results show that NF-κB but not NFAT activation is essential for Mincle induction.
FIGURE 3.

NF-κB but not NFAT activation is essential for Mincle induction. A–C, BMDMs from WT mice were stimulated with plate-coated TDM for indicated time points. TPCA-1 (NF-κB inhibitor, 1 μm) or 11R-VIVIT (NFAT inhibitor, 10 μm) was added to cells when they were stimulated. A and B, cell lysates were prepared and analyzed by immunoblotting with the indicated antibodies. C, total mRNA was analyzed by RT-PCR for Mincle expression. D, BMDMs were treated as described in A–C by plate-coated TDM for 8 h, and supernatants were subjected to ELISA analysis for TNF-α, IL-6, and IL-10. Data shown are representative of three independent experiments. S.D. is indicated. **, p < 0.01 (t test). Ctr, control.
As three predicted NF-κB-binding sites were found on Mincle promoter: −1589, −941, and −851 (Fig. 4A), we hypothesize that NF-κB may directly regulate Mincle expression through binding to its promoter following TDM stimulation. To test this hypothesis, we performed ChIP assay and found that there was a strong binding of NF-κB p65 subunit on Mincle promoter in control cells after 2 hour stimulation with TDM. However, there is almost no p65 binding in Dectin-3-deficient cells. Moreover, NFAT-c1 cannot bind with Mincle promoter after TDM stimulation neither in control cells nor in Dectin-3-deficient cells (Fig. 4B). To further test whether NF-κB dictates transcription of Mincle genes, luciferase reporter assay and NF-κB binding sites mutation was performed. The Mincle promoter activation exhibited substantially increase whether the −1589 site was present or not (Fig. 4C). However, mutation of either −941 or −851 site severely impaired Mincle promoter activation, which is consistent with the impaired activation by deletion of all three NF-κB binding sites (Fig. 4C). These results clearly demonstrate that NF-κB regulates Mincle expression through directly activating its promoter, and both NF-κB binding site −941 and −851 are necessary. Together, our results indicate that Dectin-3-mediated NF-κB activation is necessary for TDM-induced Mincle expression.
FIGURE 4.

NF-κB p65 subunit binds with Mincle promoter to regulate Mincle expression. A, the structure for Mincle gene promoter region. −1589, −941, and −851 are the three predicted NF-κB-binding sites. Different luciferase reporter constructs are also shown. B, BMDMs were stimulated with plate-coated TDM for 2 h or left untreated (0h), followed by ChIP assay with the indicated antibodies and real-time PCR analysis for Mincle promoter. Results are presented as means plus S.D. after normalization to input. C, RAW264.7 cells were transfected with different luciferase reporter constructs together with Dectin-3 expression plasmid. The luciferase activities of the Mincle promoter were measured. Two putative NF-κB-binding sites were mutated and the mutant reporter was designated as −941m and −851m. Data shown are representative of two independent experiments. S.D. is indicated. **, p < 0.01 (t test). Ctr, control.
Dectin-3 and Mincle Neither Form a Heterodimeric Receptor Nor Synergistically Induce Signaling
Our recent studies have found that Dectin-3 forms heterodimers with Dectin-2, another C-type lectin receptor, which provides a high sensitivity for recognition of the cell wall component of Candida albicans (24). Because both Dectin-3 and Mincle can respond to TDM stimulation, we examine whether Dectin-3 and Mincle can form a heterodimer and function synergistically. To examine whether Dectin-3 and Mincle form heterodimers, RAW264.7 cells stably expressing Mincle, Dectin-3, or both were stimulated with TDM and then immunoprecipitated with Dectin-3 or Mincle. In contrast with Dectin-2 and Dectin-3, Dectin-3 and Mincle were not co-immunoprecipitated when they were co-expressed in RAW264.7 cells (Fig. 5A). These results indicate that Dectin-3 and Mincle are not physically linked.
FIGURE 5.

Dectin-3 neither forms heterodimer nor synergistically induces signaling together with Mincle. A, RAW264. 7 cells stably expressing human Mincle (M), Dectin-3 (D3), or both (M + D3) were stimulated with plate-coated TDM for indicated time points and then lysed. Cell lysates were immunoprecipitated with anti-Dectin-3 or anti-Mincle antibodies (5 ug), and then the immunoprecipitated (IP) and lysate fractions were analyzed by immunoblotting with the indicated antibodies. B, RAW264.7 cells stably expressing human Dectin-3, Mincle, both (D3 + M), or control vector (Mock) were stimulated with plate-coated TDM for 60 min. Nuclear extracts (top and middle panel) and cytoplasmic fractions (bottom panel) of these cells were prepared and subjected to immunoblotting analysis. C, ELISA results for TNF-α and IL-6 in supernatants of RAW264.7 cells stimulated as described in B for 6 h, cells treated only with the solvent for TDM were used as unstimulated control (Unsti). Data shown are representative of three independent experiments. PCNA, proliferating cell nuclear antigen.
To determine whether Dectin-3 and Mincle can work synergistically to induce NF-κB activation and cytokine expression, RAW264.7 cells stably expressing Mincle, Dectin-3, or both were stimulated with TDM, and then p65 nuclear translocation was examined. We found that there was no obvious difference for p65 nuclear translocation no matter Dectin-3 and Mincle were co-expressed or not (Fig. 5B), and the expression of proinflammatory cytokines such as TNFα and IL-6 did not have significantly difference between Dectin-3 or Mincle alone and Mincle/Dectin-3 co-expression (Fig. 5C). Taken together, these results indicate that unlike Dectin-2/Dectin-3 heterodimers, Dectin-3 and Mincle neither form heterodimers nor work synergistically to activate NF-κB.
Dectin-3 Serves as a Sensor for Mincle Induction
To determine the role of Dectin-3 for sensing TDM, we treated BMDMs with Dectin-3 antibodies, which can induce Dectin-3 endocytosis (24), before (Fig. 6A, Pre+a-D3) or after TDM treatment for 3 h (Fig. 6A, 3 h+a-D3). As expected, pretreatment with Dectin-3 antibodies could sufficiently impair Mincle induction (Fig. 6B). However, after TDM stimulation for 3 h, blocking Dectin-3 no longer affected Mincle induction at all (Fig. 6B), which indicates that Mincle was already induced in the first 3-h treatment, and TDM could induce Mincle expression through stimulating Mincle itself. Consistent with this result, TNFα and IL-6 production were not affected when we blocked Dectin-3 expression 3 h after TDM stimulation (Fig. 6, C and D). Together, these results indicate that Dectin-3 serves as a sensor for TDM when Mincle level is low. However, once Mincle is induced, Dectin-3 is not required for further expression of Mincle and cytokines.
FIGURE 6.

Dectin-3 serves as the sensor for induction of Mincle expression. A and B, BMDMs were incubated with Dectin-3-blocking antibodies for 30 min at 37 °C and then stimulated with TDM (pre+α-D3) or stimulated with TDM for 3 h and then Dectin-3-blocking antibodies (3hr+α-D3). After another 6-h incubation, all cells were collected and stained with Dectin-3 antibody (A) or Mincle antibody (B), followed by FITC-labeled goat anti-mouse secondary antibodies. Samples were then examined by flow cytometry. C and D, the indicated cell supernatant were collected and measured by ELISA for TNFα and IL-6 production. Data shown are representative of three independent experiments. S.D. is indicated. **, p < 0.01 (t test). Ctr, control.
CARD9, Bcl10, and Malt1 Are Required for Mincle Induction Following TDM Stimulation
It has been shown that the CARD9-Bcl10-Malt1 complex is necessary for NF-κB activation upon different stimulation. Recent study shows that CARD9-dependent pathway plays important roles in TDM-induced inflammation and Th17 response (15). Therefore, we examined whether the CARD9-Bcl10-Malt1 complex is required for TDM-induced Mincle expression. To test this hypothesis, CARD9-deficient BMDMs were prepared from CARD9 knock-out mice. Then, these cells were stimulated with TDM for different time points. We found that although wild type and CARD9-deficient cells expressed comparable level of Dectin-3, Mincle induction was severely impaired in CARD9-deficient cells at both protein and mRNA level (Fig. 7, A and C). We then checked whether Bcl10 or Malt1 is required for the Mincle induction using BMDMs from Bcl10 or Malt1 knock-out mice and found that both Bcl10 and Malt1 were required for TDM-induced Mincle expression (Fig. 7, B and D). Moreover, TDM-induced proinflammatory cytokine, TNF, IL-6, and IL-1β were also significantly reduced in these CARD9-, Bcl10-, or Malt1-deficient cells (Fig. 7E). These results demonstrate that NF-κB activation is necessary for TDM-induced Mincle expression in a CARD9/Bcl10/Malt1-dependent manner.
FIGURE 7.

CARD-9, Bcl10, and Malt1 are required for Mincle induction. A and B, BMDMs from WT and CARD9-deficient (KO) mice (A) or from WT, Bcl10-deficient (KO) and Malt1-deficient (KO) mice (B) were stimulated with plate-coated TDM for different time points. Cell lysates were prepared and analyzed by immunoblotting with the indicated antibodies. C and D, total mRNA from above stimulated cells was prepared and analyzed by RT-PCR for Mincle expression. E, BMDMs from different mice were stimulated with or without plate-coated TDM for 8 h. Supernatants from these cells were subjected to ELISA analysis for the level of TNF-α, IL-6, and IL-1β. Data shown are representative of three independent experiments. S.D. is indicated. **, p < 0.01 (t test). Ctr, control.
Dectin-3 and CARD9 Deficiency Significantly Affect TDM-mediated Adjuvant Function
TDM, also known as cord factor, is a Mycobacterium glycolipid with potent adjuvant activity. To determine whether Dectin-3 and CARD9 could account for the adjuvant activity, complete Freund's adjuvant a water-in-oil emulsion containing heat-killed Mycobacterium tuberculosis, together with an immune antigen, KLH, were used to immunize Dectin-3 or CARD9 knock-out mice. Forty-eight hours after immunization, serum was taken to measure the cytokine production. TNF and IL-6 concentration both were significant lower in Dectin-3 and CARD9 knock-out mice (Fig. 8A). Seven days after immunization, KLH-specific antibody production was determined. We found that Dectin-3 and CARD9 knock-out mice showed significant less IgG (both IgG1 and IgG2a) and IgM production (Fig. 8B). Together, these results indicated that Dectin-3 and CARD9 are required for adaptive immune response when TDM-containing adjuvant is used.
FIGURE 8.

Dectin-3- and CARD9-deficient mice produce much less cytokine and antibodies upon immunization using adjuvant containing TDM. CARD9- and Dectin-3-deficient mice and their controls were immunized with KLH emulsified in complete Freund's adjuvant at the base of the tail. A, forty-eight hours later, blood was collected from the tail and measured with ELISA for TNFα and IL-6 production. **, p < 0.01 (t test). B, seven days after immunization, sera from the indicated immunized mice were subjected to serial dilutions, and the concentrations of KLH-specific IgG and IgM antibodies were analyzed by ELISA and averaged for each group. Graphs show the means ± S.D. from three mice of each genotype.
DISCUSSION
Dectin-3, a CLR, has been shown to function as a pattern recognition receptor for Mycobacterium glycolipid, TDM, and stimulation of Dectin-3 by TDM induces the expression of another CLR, Mincle (25). It has been shown that Mincle also plays a critical role for TDM-induced proinflammatory and adjuvant responses (7, 8). Therefore, it is critical to determine the molecular mechanism of the functional connection between Dectin-3 and Mincle. In this study, we have shown that Dectin-3 serves as a molecular sensor for recognition of TDM, which leads to induction of Mincle expression (Fig. 9). The Dectin-3-induced Mincle expression is directly regulated by transcription factor NF-κB that is activated through a signaling pathway dependent on CARD9, Bcl10, and Malt1. Consistently, we have found that Dectin-3- and CARD9-deficient mice produce much less amount of proinflammatory cytokines and antigen-specific antibodies when immunized with adjuvant containing TDM. Together, our study provides the molecular mechanism by which Dectin-3 functions as a key regulator for the adjuvant activity of TDM.
FIGURE 9.
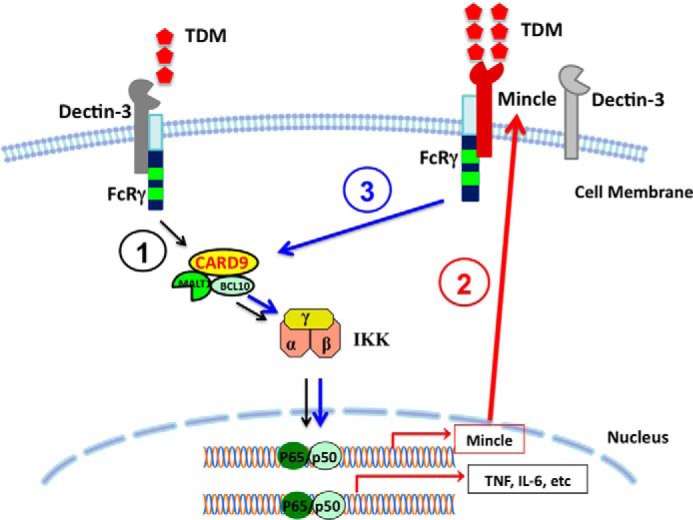
Proposed model for the mechanism by which Dectin-3 induces Mincle expression. Step 1, Dectin-3 functions as an initial sensor for detecting low level of TDM and triggers an intracellular signaling pathway leading to activation of NF-κB in a CARD9-Bcl10-Malt1-dependent manner. Step 2, The activated NF-κB induces the expression of Mincle gene, which results in significantly higher amount of Mincle expressed on the cell surface. Step 3, The high amount of Mincle further activates by TDM to induce the expression of proinflammatory cytokines.
A recent report by Lobato-Pascual et al. (26) described a receptor complex formed by Mincle with Dectin-3 and FcϵRI-γ, and this association enhanced phagocytosis of antibody-coated beads. However, in our study, we could not detect the heterodimeric complex of Dectin-3 and Mincle when they were co-expressed in mouse macrophage-like cells, RAW264.7 cells. The most possible explanation for the discrepancy between Lobato-Pascual's results and our results could be due to the expression level of Dectin-3 and Mincle in rat cells used by Lobato-Pascual et al. (26) versus mouse cells used in our study. However, co-expression of Dectin-3 and Mincle cannot synergistically induce NF-κB activation and cytokine expression, which was found upon co-expression of heterodimeric complexes of Dectin-2 and Dectin-3 (24). Moreover, there is no evidence that endogenous Dectin-3 and Mincle can form heterodimeric complexes (26).
TDM promotes both normal innate and acquired immunity and contributes to immune disorders such as granuloma formation (2) and experimental autoimmune encephalomyelitis (15, 16). Consistently, Dectin-3-deficient mice have defect in both innate and adaptive immune response (4, 24, 25). However, TDM-induced adaptive immune response was almost completely dependent on Dectin-3, but not Mincle (25), demonstrating that Dectin-3 plays important roles for TDM-induced innate immune response.
We have shown that Dectin-3 induces NF-κB activation in response to C. albicans hyphae challenging (24) and have found that a portion of Dectin-3 forms heterodimers with another CLR Dectin-2, which has a higher affinity recognizing α-mannan on the cell wall of C. albicans, and potently induces proinflammatory response to the fungal infection (24). However, unlike Dectin-2/Dectin-3 heterodimers, we have found that Dectin-3 and Mincle neither form heterodimers nor synergistically regulate NF-κB activation together with Dectin-3 upon TDM stimulation. Instead, Dectin-3 and Mincle function in a sequential cascade to provide an amplification loop to enhance TDM-induced proinflammatory response (Fig. 9). In this case, Dectin-3 is only necessary for the initial induction of Mincle expression. After the initial stimulation of Dectin-3 and induction of Mincle expression, blocking Dectin-3 no longer influences the further induction of Mincle expression. These results indicate that Decin-3 serves as the initial sensing molecule for recognition of TDM and induces expression of Mincle through activation of NF-κB. Once Mincle level is induced, Mincle can further regulate its own expression (Fig. 9).
NFAT is another transcription factor that may be involved in regulation of Dectin-3-induced Mincle expression because there is defect for NFAT-c1 translocation in Dectin-3-deficient cells upon TDM stimulation (25). However, treatment of BMDMs with 11R-VIVIT, a well characterized NFAT inhibitor did not affect TDM-induced Mincle expression, although this inhibitor could efficiently block NFAT-c1 translocation (data not shown) and subsequently IL-10 production. Consistently, we were unable to detect the binding of NFAT-c1 to Mincle promoter as NF-κB does. Together, these results indicate that Dectin-3-induced Mincle expression is regulated by transcription factor NF-κB but not NFAT.
In summary, our study provides the genetic and biochemical evidence for the molecular link between Dectin-3 and Mincle in response to TDM stimulation. We have demonstrated that Dectin-3 functions as a sensing molecule for detecting low level of TDM molecule, which leads to the induction of Mincle expression by activating NF-κB through CARD9-Bcl10-Malt1 complex-dependent mechanism. The induced Mincle can also recognize TDM and further amplifies the response to induce strong innate immune and inflammatory response.
Acknowledgment
We thank Shilei Zhang for technical assistance and helpful discussion.
This work was supported in part by The Shanghai Basic Research Program Grant 12JC1408401 (to X. M. J.) and National Institutes of Health Grants R01 AI050848 and R01 GM065899 and a pilot grant from the Center for Inflammation and Cancer of the MD Anderson Cancer Center (to X. L.).
- CLR
- C-type lectin receptor
- BMDM
- bone marrow-derived macrophage
- TDM
- trehalose-6,6′-dimycolate.
REFERENCES
- 1. Netea M. G., Brown G. D., Kullberg B. J., Gow N. A. (2008) An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 6, 67–78 [DOI] [PubMed] [Google Scholar]
- 2. Hardison S. E., Brown G. D. (2012) C-type lectin receptors orchestrate antifungal immunity. Nat. Immunol. 13, 817–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vautier S., MacCallum D. M., Brown G. D. (2012) C-type lectin receptors and cytokines in fungal immunity. Cytokine 58, 89–99 [DOI] [PubMed] [Google Scholar]
- 4. Kerscher B., Willment J. A., Brown G. D. (2013) The Dectin-2 family of C-type lectin-like receptors: an update. Int. Immunol. 25, 271–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Graham L. M., Brown G. D. (2009) The Dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine 48, 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kingeter L. M., Lin X. (2012) C-type lectin receptor-induced NF-κB activation in innate immune and inflammatory responses. Cell. Mol. Immunol. 9, 105–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ishikawa E., Ishikawa T., Morita Y. S., Toyonaga K., Yamada H., Takeuchi O., Kinoshita T., Akira S., Yoshikai Y., Yamasaki S. (2009) Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 206, 2879–2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsunaga I., Moody D. B. (2009) Mincle is a long sought receptor for mycobacterial cord factor. J. Exp. Med. 206, 2865–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schoenen H., Bodendorfer B., Hitchens K., Manzanero S., Werninghaus K., Nimmerjahn F., Agger E. M., Stenger S., Andersen P., Ruland J., Brown G. D., Wells C., Lang R. (2010) Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J. Immunol. 184, 2756–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feinberg H., Jégouzo S. A., Rowntree T. J., Guan Y., Brash M. A., Taylor M. E., Weis W. I., Drickamer K. (2013) Mechanism for recognition of an unusual mycobacterial glycolipid by the macrophage receptor mincle. J. Biol. Chem. 288, 28457–28465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Werninghaus K., Babiak A., Gross O., Hölscher C., Dietrich H., Agger E. M., Mages J., Mocsai A., Schoenen H., Finger K., Nimmerjahn F., Brown G. D., Kirschning C., Heit A., Andersen P., Wagner H., Ruland J., Lang R. (2009) Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRγ-Syk-Card9-dependent innate immune activation. J. Exp. Med. 206, 89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Behler F., Steinwede K., Balboa L., Ueberberg B., Maus R., Kirchhof G., Yamasaki S., Welte T., Maus U. A. (2012) Role of Mincle in alveolar macrophage-dependent innate immunity against mycobacterial infections in mice. J. Immunol. 189, 3121–3129 [DOI] [PubMed] [Google Scholar]
- 13. Lang R. (2013) Recognition of the mycobacterial cord factor by Mincle: relevance for granuloma formation and resistance to tuberculosis. Front. Immunol. 4, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsumoto M., Tanaka T., Kaisho T., Sanjo H., Copeland N. G., Gilbert D. J., Jenkins N. A., Akira S. (1999) A novel LPS-inducible C-type lectin is a transcriptional target of NF-IL6 in macrophages. J. Immunol. 163, 5039–5048 [PubMed] [Google Scholar]
- 15. Shenderov K., Barber D. L., Mayer-Barber K. D., Gurcha S. S., Jankovic D., Feng C. G., Oland S., Hieny S., Caspar P., Yamasaki S., Lin X., Ting J. P., Trinchieri G., Besra G. S., Cerundolo V., Sher A. (2013) Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through mincle/CARD9 signaling and the inflammasome. J. Immunol. 190, 5722–5730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Desel C., Werninghaus K., Ritter M., Jozefowski K., Wenzel J., Russkamp N., Schleicher U., Christensen D., Wirtz S., Kirschning C., Agger E. M., Prazeres da Costa C., Lang R. (2013) The Mincle-activating adjuvant TDB induces MyD88-dependent Th1 and Th17 responses through IL-1R signaling. PLoS One 8, e53531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamasaki S., Ishikawa E., Sakuma M., Hara H., Ogata K., Saito T. (2008) Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat. Immunol. 9, 1179–1188 [DOI] [PubMed] [Google Scholar]
- 18. Marakalala M. J., Graham L. M., Brown G. D. (2010) The role of Syk/CARD9-coupled C-type lectin receptors in immunity to Mycobacterium tuberculosis infections. Clin. Dev. Immunol. 2010, 567571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drummond R. A., Saijo S., Iwakura Y., Brown G. D. (2011) The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur. J. Immunol. 41, 276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsu Y. M., Zhang Y., You Y., Wang D., Li H., Duramad O., Qin X. F., Dong C., Lin X. (2007) The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 8, 198–205 [DOI] [PubMed] [Google Scholar]
- 21. Strasser D., Neumann K., Bergmann H., Marakalala M. J., Guler R., Rojowska A., Hopfner K. P., Brombacher F., Urlaub H., Baier G., Brown G. D., Leitges M., Ruland J. (2012) Syk kinase-coupled C-type lectin receptors engage protein kinase C-σ to elicit Card9 adaptor-mediated innate immunity. Immunity 36, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arce I., Martínez-Muñoz L., Roda-Navarro P., Fernández-Ruiz E. (2004) The human C-type lectin CLECSF8 is a novel monocyte/macrophage endocytic receptor. Eur. J. Immunol. 34, 210–220 [DOI] [PubMed] [Google Scholar]
- 23. Graham L. M., Gupta V., Schafer G., Reid D. M., Kimberg M., Dennehy K. M., Hornsell W. G., Guler R., Campanero-Rhodes M. A., Palma A. S., Feizi T., Kim S. K., Sobieszczuk P., Willment J. A., Brown G. D. (2012) The C-type lectin receptor CLECSF8 (CLEC4D) is expressed by myeloid cells and triggers cellular activation through Syk kinase. J. Biol. Chem. 287, 25964–25974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu L. L., Zhao X. Q., Jiang C., You Y., Chen X. P., Jiang Y. Y., Jia X. M., Lin X. (2013) C-type lectin receptors dectin-3 and dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 39, 324–334 [DOI] [PubMed] [Google Scholar]
- 25. Miyake Y., Toyonaga K., Mori D., Kakuta S., Hoshino Y., Oyamada A., Yamada H., Ono K., Suyama M., Iwakura Y., Yoshikai Y., Yamasaki S. (2013) C-type lectin MCL is an FcRγ-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity 38, 1050–1062 [DOI] [PubMed] [Google Scholar]
- 26. Lobato-Pascual A., Saether P. C., Fossum S., Dissen E., Daws M. R. (2013) Mincle, the receptor for mycobacterial cord factor, forms a functional receptor complex with MCL and FϵRI-γ. Eur. J. Immunol. 43, 3167–3174 [DOI] [PubMed] [Google Scholar]
- 27. Ruland J., Duncan G. S., Wakeham A., Mak T. W. (2003) Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity 19, 749–758 [DOI] [PubMed] [Google Scholar]
- 28. Xue L., Morris S. W., Orihuela C., Tuomanen E., Cui X., Wen R., Wang D. (2003) Defective development and function of Bcl10-deficient follicular, marginal zone and B1 B cells. Nat. Immunol. 4, 857–865 [DOI] [PubMed] [Google Scholar]
- 29. Blonska M., Joo D., Nurieva R. I., Zhao X., Chiao P., Sun S. C., Dong C., Lin X. (2013) Activation of the transcription factor c-Maf in T cells is dependent on the CARMA1-IKKβ signaling cascade. Sci. Signal. 6, ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nurieva R. I., Chung Y., Martinez G. J., Yang X. O., Tanaka S., Matskevitch T. D., Wang Y. H., Dong C. (2009) Bcl6 mediates the development of T follicular helper cells. Science 325, 1001–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nurieva R. I., Podd A., Chen Y., Alekseev A. M., Yu M., Qi X., Huang H., Wen R., Wang J., Li H. S., Watowich S. S., Qi H., Dong C., Wang D. (2012) STAT5 protein negatively regulates T follicular helper (Tfh) cell generation and function. J. Biol. Chem. 287, 11234–11239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gorjestani S., Darnay B. G., Lin X. (2012) Tumor necrosis factor receptor-associated factor 6 (TRAF6) and TGFbeta-activated kinase 1 (TAK1) play essential roles in the C-type lectin receptor signaling in response to Candida albicans infection. J. Biol. Chem. 287, 44143–44150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blonska M., Joo D., Zweidler-McKay P. A., Zhao Q., Lin X. (2012) CARMA1 controls Th2 cell-specific cytokine expression through regulating JunB and GATA3 transcription factors. J. Immunol. 188, 3160–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu X., Li H., Zhong B., Blonska M., Gorjestani S., Yan M., Tian Q., Zhang D. E., Lin X., Dong C. (2013) USP18 inhibits NF-κB and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB1 complex. J. Exp. Med. 210, 1575–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]