

Background: IL-6/Stat3 promote breast cancer metastasis through regulation of the fascin gene.
Results: In addition to IL-6, TNF-α induces binding of a Stat3·NFκB complex to the fascin promoter to induce transcription.
Conclusion: Both NFκB and Stat3 are required for cytokine-induced fascin expression and cell migration.
Significance: Identification of proteins critical for breast cancer metastasis will reveal drug targets.
Keywords: Breast Cancer; Stat3; Transcription Regulation; Tumor Metastasis; Tumor Necrosis Factor (TNF); Fascin, NF KappaB
Abstract
IL-6 mediated activation of Stat3 is a major signaling pathway in the process of breast cancer metastasis. One important mechanism by which the IL-6/Stat3 pathway promotes metastasis is through transcriptional regulation of the actin-bundling protein fascin. In this study, we further analyzed the transcriptional regulation of the fascin gene promoter. We show that in addition to IL-6, TNF-α increases Stat3 and NFκB binding to the fascin promoter to induce its expression. We also show that NFκB is required for Stat3 recruitment to the fascin promoter in response to IL-6. Furthermore, Stat3 and NFκB form a protein complex in response to cytokine stimulation. Finally, we demonstrate that an overlapping STAT/NFκB site in a highly conserved 160-bp region of the fascin promoter is sufficient and necessary to induce transcription in response to IL-6 and TNF-α.
Introduction
The STAT (signal transducers and activators of transcription) proteins are a family of transcription factors that play various roles in cellular processes, including immune response, apoptosis, and oncogenesis (1–4). STATs are members of the JAK-STAT signaling pathway, which is activated by growth factors and cytokines (1, 2). In response to cytokine binding to its cell surface receptor, the receptor dimerizes and activates its associated Janus kinase (JAK). The JAK phosphorylates the cell surface receptor, which serves as a docking site for the STATs. Upon recruitment of the STATs to the phosphorylated receptor, STATs become phosphorylated by JAKs on a conserved tyrosine near the Src homology 2 domain of STAT (1, 2). The phosphorylated STATs dimerize and translocate to the nucleus where they bind to the promoters of STAT target genes to form enhancersomes with other transcriptional co-activators to activate transcription (5, 6).
Stat3 is one member of the STAT family that functions in a wide range of cellular processes, including wound healing, postnatal survival, stem cell renewal, and tumorigenesis (7–12). Stat3 is activated by cytokines that bind to the gp130 receptor, including interleukin-6 (IL-6) and oncostatin M (13, 14). Activated Stat3 has been associated with several cancers, including head and neck and breast cancer (7, 15). In breast cancer, increased levels of IL-6 and activated Stat3 promote not only tumor growth but also metastasis (7, 16, 17). Recently, we have shown that one possible mechanism by which IL-6 and Stat3 promote metastasis is through transcriptional induction of the fascin gene (18).
Fascin is a highly conserved actin-bundling protein that localizes to microspikes and filopodia with functions in cell adhesion and motility (19). In mammalian cells, three isoforms of fascin exist (20). Fascin-1 (herein referred to as fascin) is expressed during embryonic development and in some normal adult cell types, including dendritic cells (20). Fascin-2 is retinal-specific, and fascin-3 is only found in the testes (21). Fascin expression increases significantly in many cancers, including metastatic gastric, colon, and breast cancer (19, 20). Fascin is a key regulator of breast cancer metastasis. Knocking down fascin or inhibition of fascin with small chemical compounds block breast cancer metastasis (22). The mechanism by which fascin is up-regulated in cancer and the signaling pathways that regulate transcription of the fascin gene are not well understood, although the β-catenin pathway and the transcription factor cAMP-responsive element-binding protein (CREB) have been shown to be involved in controlling fascin expression in colon cancer (23–25).
The transcription factor NFκB2 has also been associated with tumorigenesis, apoptosis, inflammation, and cancer cell migration (26, 27). NFκB activity is controlled through shuttling between the cytoplasm and the nucleus. In response to stimulus by inflammatory factors such as tumor necrosis factor (TNF)-α, increased levels of NFκB accumulate in the nucleus (27–29). Interactions between Stat3 and NFκB have been demonstrated previously (30, 31). It has been shown that unphosphorylated Stat3 can be recruited to promoters through its interaction with NFκB (30). The fascin promoter contains a 160-bp conserved region that contains overlapping STAT and NFκB binding sites (18). We have shown previously that in response to IL-6 or oncostatin M treatment, Stat3 and NFκB are recruited to the fascin promoter to increase transcription of the fascin gene and promote migration of breast cancer cells (18). Knocking down Stat3 or NFκB blocks IL-6 or oncostatin M-mediated transcriptional expression of fascin. Furthermore, Stat3 is also required for breast cancer cell migration in response to IL-6 or oncostatin M treatment (18).
In this report, we further analyze the role of Stat3 and NFκB in transcriptional regulation of the fascin promoter. We demonstrate that in addition to IL-6, TNF-α induces Stat3 and NFκB binding to the fascin promoter in the MDA-MB-231 human breast cancer cells. NFκB is necessary for TNF-α-induced fascin expression and cell migration. In response to IL-6 treatment, binding of Stat3 to the fascin promoter requires NFκB. Furthermore, we show that treatment of human breast cancer cells with IL-6 and TNF-α leads to the formation of a complex of Stat3, NFκB p50, and NFκB p65. Finally, we demonstrate that the 160-bp conserved region in the fascin promoter containing the overlapping STAT and NFκB sites is sufficient to activate transcription in response to IL-6 and TNF-α, and transcription driven by this conserved region is dependent on specific nucleotides shared by both the STAT and NFκB binding sites.
EXPERIMENTAL PROCEDURES
Cell Culture, Reagents, and Plasmids
MDA-MB-231 cells were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS. The Stat3, NFκB p50, and p65 antibodies for ChIP and co-immunoprecipitation experiments were from Santa Cruz Biotechnology. Antibodies for Western blot analysis were anti-Stat3 (BD Transduction Laboratories), anti-tubulin (Sigma), anti- NFκB p50 and p65, and anti-fascin (Santa Cruz Biotechnology). Ligands were used at the following concentrations: 40 ng/ml TNF-α (Millipore) and 20 ng/ml IL-6 combined with 200 ng/ml IL-6 Rα (R&D Systems). PCR reactions for cloning used Pfu DNA polymerase (Agilent). The plasmid FasP-luc was constructed by amplifying a 160-bp conserved region with the primers 5′-ggggatccagaccttgtgggcagcctgt and 5′-ggggatccgggacattccctgcagacaccac from ChIP DNA. It was cloned into the BamHI site of pBluescript (Addgene). The fascin promoter conserved region was then PCR-amplified with the primers 5′-ggggatccaccttgtgggcagcctgt and 5′-atgcgctcgaggggacattccctgcagacaccac and cloned into the BamHI/XhoI site of the plasmid tkluc.
Chromatin Immunoprecipitations (ChIP) Assays
ChIP assays were performed as described previously (32–34). MDA-MB-231 cells were treated with IL-6 for 30 min, and the resulting cell lysates (input) were subjected to immunoprecipitations with 2.5 μg of indicated antibodies. ChIP primers for the human fascin promoter were 5′-accttgtgggcagcctgt and 5′-attccctgcagacaccacct.
Quantitative Real-time Reverse Transcription (RT)-PCR
RT-PCR analyses were performed as described previously (18, 32, 33). RNA was extracted using the TRIzol method (Invitrogen). RNA was reverse-transcribed using qScript cDNA SuperMix (Quanta Biosciences) and subjected to quantitative real-time PCR using PerfeCTa SYBR Green FastMix, ROX (Quanta Biosciences). RT-PCR primers for human fascin were 5′-aaaagtgtgccttccgtacc and 5′-cccattcttcttggaggtca.
RNAi
RNAi was performed with NFκB p50 siRNA oligonucleotides from Qiagen (catalogue nos. SI02662618 and SI00300958), NFκB p65 siRNA from Integrated DNA Technologies (catalogue nos. HSC.RNAI.N001145138.12.1 and HSC.RNAI.N001145138.12.2), or a negative control siRNA (Qiagen, catalogue no. 1022563). Cells were transfected using HiPerFect transfection reagent (Qiagen).
Co-immunoprecipitation Assays
MDA-MB-231 cells were treated with IL-6 and TNF-α for 30 min. Nuclear extracts were prepared and dialyzed overnight at 4 °C in 20 mm Tris, pH 8.0, 0.5 mm EDTA, 0.1 m KCl, 10 mm Na3VO4, and 10% glycerol. Dialyzed extracts (50 μg) were subjected to immunoprecipitation (IP) against Stat3, NFκB p50, NFκB p65, or rabbit polyclonal IgG. IP reactions were performed with 5 μg of antibody in 20 mm Hepes, pH 8.0, 10 mm KCl, 140 mm NaCl, 1 mm EDTA, 0.1 mm Na3VO4, 0.1% Nonidet P-40, and 13% glycerol at 4 °C overnight. IP reactions were subjected to Western blot analysis with antibodies against Stat3, NFκB p50, or p65.
Cell Migration Assays
Wound-healing assays were performed as described previously (26). Cells transfected with siRNAs were cultured to confluence in RPMI 1640 medium supplemented with 10% FBS in 24-multiwell plates. Wounds were made with a sterile pipette tip, and cells were washed with PBS and then cultured in indicated media for 24 h.
Luciferase Assays
Luciferase assays were performed using the Dual-Luciferase Reporter System (Promega). Luciferase results were represented as relative luciferase units, which are the luciferase activities standardized to Renilla luciferase (Promega).
RESULTS
Both IL-6 and TNF-α Induce Fascin mRNA Expression through Stat3 and NFκB
The actin-bundling protein fascin is up-regulated in some cancers and promotes metastasis (19, 20). We have previously shown that in response to IL-6, Stat3 and NFκB are recruited to the fascin promoter, and fascin expression increases to promote breast cancer cell migration (18). However, the role of TNF-α and NFκB in fascin transcriptional regulation has not been fully analyzed. To determine whether TNF-α induces NFκB to bind to the fascin promoter, ChIP analyses were performed in the human breast cancer cell line MDA-MB-231. In response to TNF-α treatment, there was an increase in NFκB p50 and p65 binding to the fascin promoter compared with untreated cells (Fig. 1A, lanes 1 and 3). TNF-α treatment also caused an increase in Stat3 binding to the promoter (Fig. 1A, lanes 1 and 3). There was a stronger signal in NFκB p50, p65, and Stat3 bound to the promoter in cells treated with IL-6 and TNF-α together (Fig. 1A, lanes 1 and 2).
FIGURE 1.
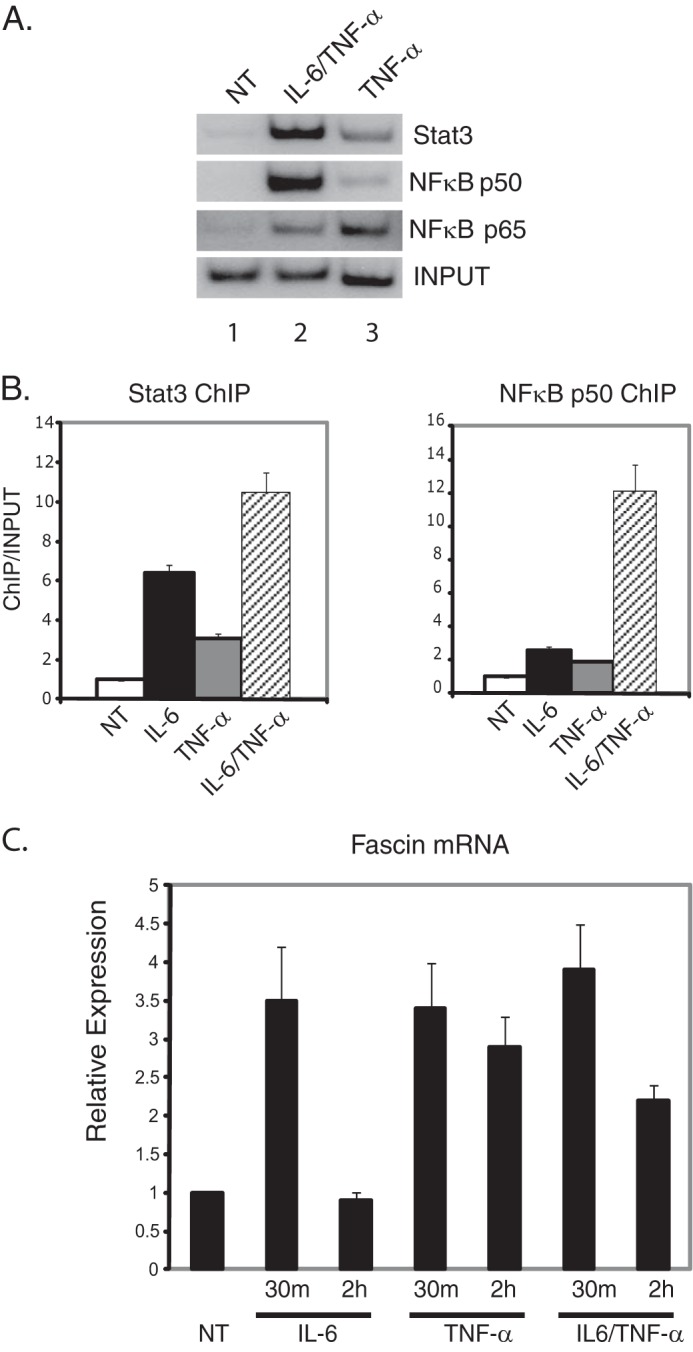
Stat3 and NFκB induce fascin expression in response to IL-6 and TNF-α. A, MDA-MB-231 cells were treated with TNF-α alone or IL-6 and TNF-α combined for 30 min, and ChIP analyses were performed with antibodies against Stat3, NFκB p50, or p65. NT, no treatment. B, quantitation of ChIP assays for MDA-MB-231 cells treated with IL-6 alone, TNF-α alone, or IL-6 and TNF-α combined for 30 min. ChIP analyses were performed for Stat3 or NFκB p50. Gels were quantitated using a phosphorimaging device and expressed as the intensities of ChIP/input with untreated values (NT) set at 1. Results represent the averages and S.D. of at least two experiments. C, cells were serum-starved for 48 h and treated with IL-6, TNF-α, or IL-6 plus TNF-α for 30 min or 2 h. RNA was extracted and subjected to quantitative real-time RT-PCR analyses for fascin. Results are standardized to GAPDH with untreated samples (NT) set at 1. Results represent the averages and S.D. of at least three independent experiements performed in triplicate.
ChIP results of Stat3 and NFκB p50 for IL-6, TNF-α, and both TNF-α and Il-6 together were quantitated for comparison (Fig. 1B) (18). For Stat3 binding, IL-6 induced a stronger signal than TNF-α (Fig. 1B). IL-6 and TNF-α together further increased the binding of Stat3 to the fascin promoter (Fig. 1B). For NFκB p50, either IL-6 or TNF-α alone induced its binding to the fascin promoter; however, IL-6 and TNF-α together enhanced p50 binding significantly (Fig. 1B). These ChIP results demonstrate that either IL-6 or TNF-α induce Stat3 and NFκB binding to the fascin promoter. However, simultaneous treatment of IL-6 and TNF-α can induce much stronger binding of these proteins to the promoter, possibly through increased stabilization of the complex.
To see whether these cytokines induced fascin mRNA expression, quantitative real-time RT-PCR analyses were performed for cells treated with TNF-α. As demonstrated previously (18), IL-6 treatment alone induced fascin expression almost 4-fold after 30 min of treatment. Treatment of MDA-MB-231 cells with TNF-α alone induced fascin mRNA expression 3-fold after 30 min of treatment (Fig. 1C). Combined treatment of IL-6 plus TNF-α showed a similar increase in fascin expression to treatment with either cytokine alone at the 30-min time point. However, after 2 h of treatment with TNF-α, either alone or with IL-6, fascin mRNA levels were higher compared with untreated or IL-6 treated cells (Fig. 1C). These results demonstrate that either TNF-α or IL-6 can induce expression of fascin through recruitment of Stat3 and NFκB. However, TNF-α can induce a longer and more sustained transcriptional activation of the fascin gene.
NFkB Is Necessary for Fascin Expression and Binding of Stat3 to the Fascin Promoter in Response to Cytokines
We have shown previously that recruitment of NFκB to the fascin promoter in response to IL-6 requires Stat3 (18). To determine whether Stat3 binding to the fascin promoter requires NFκB, we performed ChIP assays in NFκB siRNA knockdown cells. Two siRNAs targeting separate regions of NFκB p50 were described previously (18). NFκB p50 was efficiently knocked down in MDA-MB-231 cells compared with cells transfected with a control siRNA (Fig. 2A). In cells with NFκB p50 knockdown, no fascin mRNA was induced by IL-6 or TNF-α treatment (data not shown) (18) and fascin protein was not induced by TNF-α (Fig. 2B). To see whether there is any Stat3 binding at the fascin promoter in NFκB p50 knockdown cells, ChIP analyses were performed in both NFκB p50 knockdown and control cells. In response to IL-6, there was a significant increase in Stat3 and NFκB p50 binding to the fascin promoter in control cells (Fig. 2C, lanes 1 and 2). In contrast, there was a significant decrease in Stat3 binding to the promoter in NFκB p50 knockdown cells (Fig. 2C, lanes 3 and 4). There was no binding of NFκB p50 in the knockdown cells (Fig. 2C, lanes 3 and 4).
FIGURE 2.
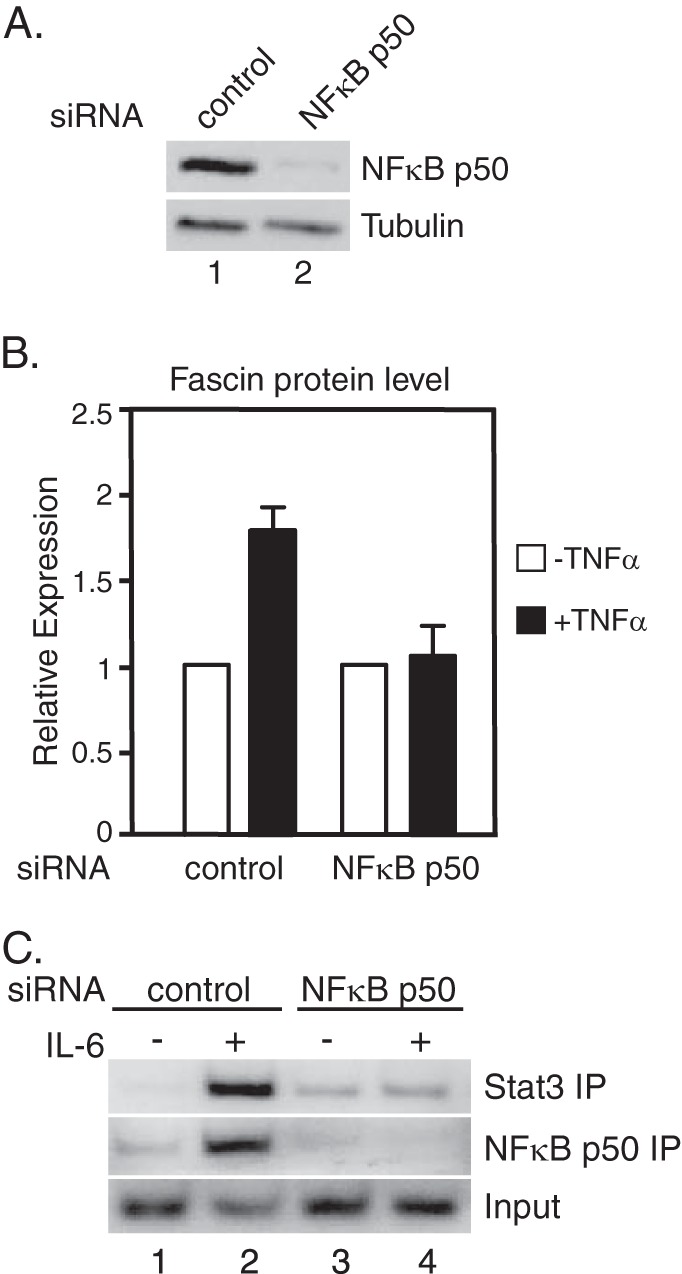
NFκB p50 is required for fascin protein expression and for Stat3 binding to the fascin promoter. A, MDA-MB-231 cells were transfected with either a control siRNA or NFκBp50 siRNAs and cultured for 4 days. Western blot analyses were performed with antibodies against NFκB p50 or tubulin. B, two rounds of RNAi-mediated knockdown of NFκBp50 was performed (days 1 and 4). On day 7, cells were treated with TNF-α for 3 h followed by Western blot analyses. Autoradiographs of Western blotting analyses were quantitated by ImageJ software, and the results of three experiments + S.D. are shown. C, RNAi-mediated knockdown of NFκB p50 was performed as described in A. Cells were treated with IL-6 for 30 min. ChIP analyses were performed with antibodies against Stat3 or NFκB p50. Results represent two independent experiments.
To further demonstrate that NFκB is necessary for fascin induction, NFκB p65 was knocked down in MDA-MB-231 cells. In cells transfected with control siRNA, both fascin mRNA and protein were induced by TNF-α, whereas in cells transfected with NFκB p65 siRNA, fascin mRNA and protein were not induced in response to TNF-α (Fig. 3, A–C). Altogether, these results demonstrate that NFκB is required for fascin expression and Stat3 recruitment to the fascin promoter in response to cytokine treatment.
FIGURE 3.
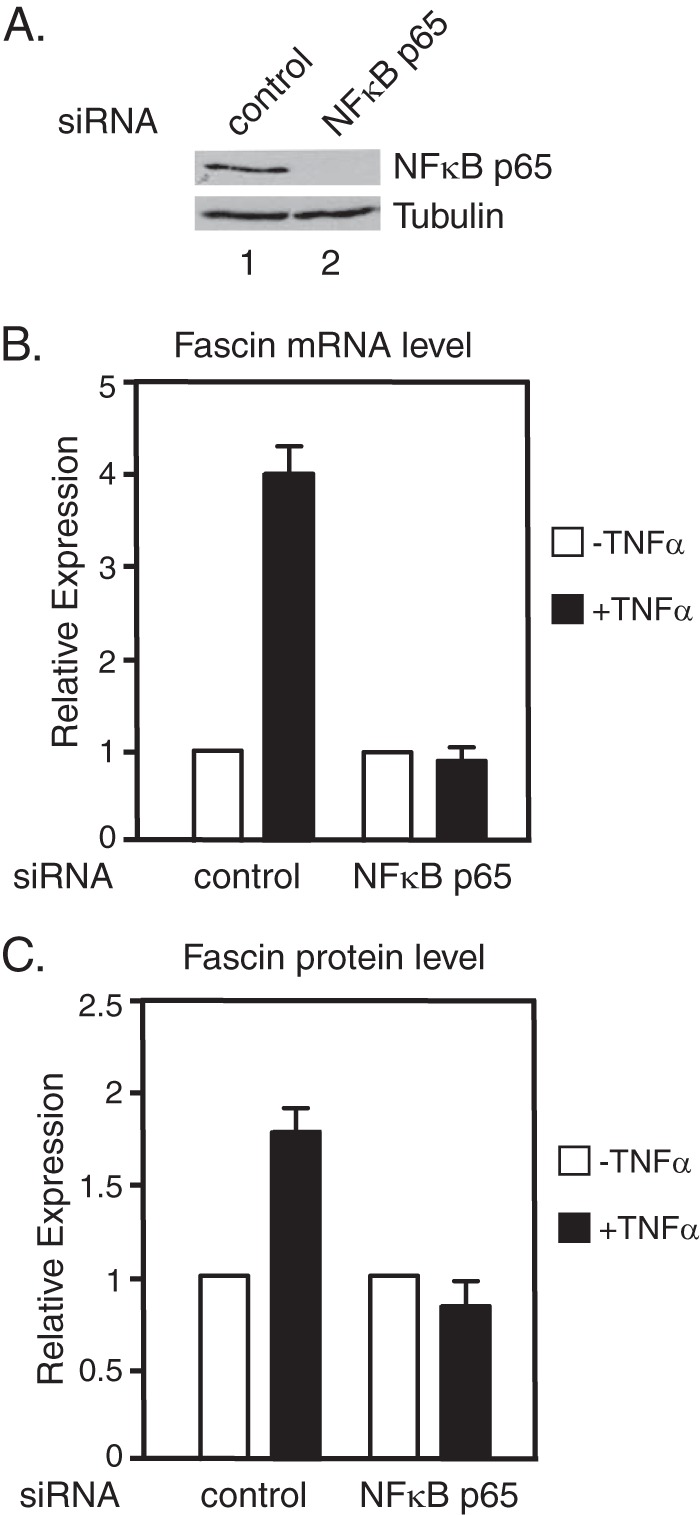
NFκB p65 is required for fascin expression. MDA-MB-231 cells were transfected with either a control siRNA or NFκBp65 siRNAs and cultured for 4 days. A, Western blot analyses of whole cell lysates were performed with antibodies against NFκB p65 or tubulin. B, cells were treated with TNF-α for 30 min followed by real-time RT-PCR analyses. C, cells were treated with TNF-α for 3 h followed by Western blot analyses. Autoradiographs of Western blotting analyses were quantitated by ImageJ software, and the results of three experiments + S.D. are shown.
NFκB Is Required for IL-6 and TNF-α-induced Cell Migration of MDA-MB-231 Cells
To determine whether NFκB is also required for cytokine-induced cell migration, two rounds of siRNA knockdown of NFκB p50 were performed to maintain the knock-down of fascin protein in the cells (Fig. 4). The cells transfected with control siRNA or NFκB p50 siRNA were serum-starved for 10 h followed by wound healing assays. Cells did not migrate in medium without serum (Fig. 4, i and vi). In medium containing serum, both cells transfected with control siRNA or NFκB p50 siRNA migrated well (Fig. 4, ii and vii). Cells transfected with control siRNA migrated efficiently in response to treatment with IL-6, TNF-α, or IL-6 plus TNF-α (Fig. 4, iii–v). However, the cells transfected with NFκB p50 siRNA did not migrate in any cytokine treated medium (Fig. 4, viii–x). Similar results were obtained with NFκB p65 knockdown. These results demonstrate that NFκB is necessary for MDA-MB-231 cells to migrate in response to IL-6 and TNF-α signaling.
FIGURE 4.
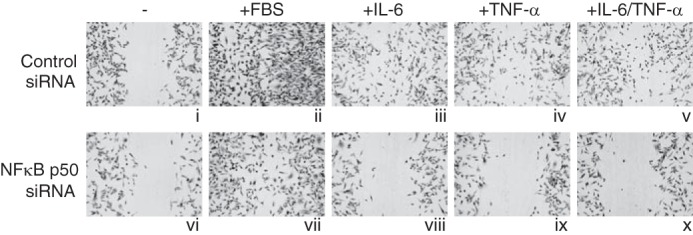
NFκB is required for cell migration in response to IL-6 and TNF-α. MDA-MB-231 cells were transfected with either a control siRNA or NFκB p50 siRNAs on days 1 and 4. On day 7, the cells were starved for 10 h, and wound healing assays were performed in RPMI 1640 medium (i and v), RPMI 1640 medium supplemented with 10% FBS (ii and vii), IL-6 (iii and viii), TNF-α (iv and ix), or IL-6 plus TNF-α (v and x). Cells were cultured in the indicated conditions for 24 h.
Stat3, NFκB p50, and NFκB p65 Form a Complex
We have shown that in response to IL-6 and TNF-α, Stat3 and members of the NFκB complex interact with the fascin promoter using ChIP analyses. To further analyze the interactions between Stat3 and NFκB in vivo, co-immunoprecipitation experiments were performed on nuclear extracts from cells treated with both IL-6 and TNF-α. Co-immunoprecipitation experiments with the Stat3 antibody showed that NFκB p50 and p65 co-immunoprecipitated with Stat3 (Fig. 5, lane 2). The NFκB p50 antibody co-immunoprecipitated both Stat3 and NFκB p65 (Fig. 5, lane 3), and the NFκB p65 antibody co-immunoprecipitated both Stat3 and NFκB p50 (Fig. 5, lane 4). An IgG antibody was used as a negative control (Fig. 5, lane 5). Stat3 and NFκB also can be co-immunoprecipitated in untreated MDA-MB-231 cells; however, the interactions were much weaker because NFκB was present at lower levels in untreated nuclear extracts (data not shown). Altogether, these results demonstrate that Stat3, NFκB p50, and NFκB p65 form a protein complex in the nucleus of MDA-MB-231 cells treated with IL-6 and TNF-α.
FIGURE 5.
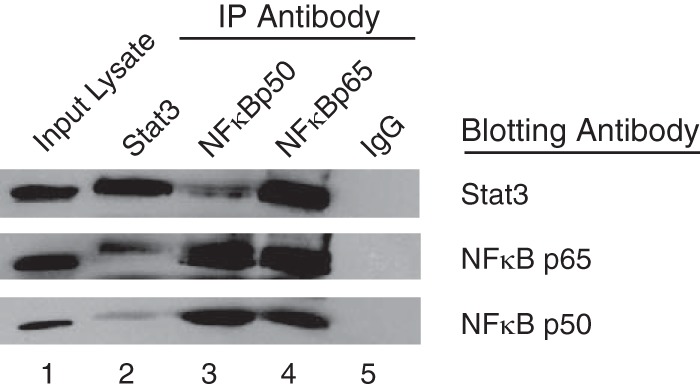
Stat3, NFκB p50, and NFκB p65 form a complex. MDA-MB-231 cells were treated with IL-6 and TNF-α for 30 min. Nuclear extracts were subjected to immunoprecipitation with antibodies against Stat3, NFκB p50, NFκB p65, or rabbit polyclonal IgG. Immunopreciptiations were analyzed by Western blot with antibodies against Stat3, NFκB p50, and NFκB p65. Input lysate (10%) is shown as a control.
A Conserved Region in the Fascin Promoter Containing the STAT/NFκB Sites Is Sufficient to Induce Transcription in Response to IL-6 and TNF-α
There is an evolutionarily conserved 160-bp region in the fascin promoter that contains overlapping STAT and NFκB sites (Fig. 6A) (18). To further analyze the role of this conserved region in transcriptional activation, we subcloned it into a luciferase reporter (FasP-luc) (Fig. 6A). Luciferase assays were performed in MDA-MB-231 cells transiently transfected with either the empty luciferase vector (tkluc) as a negative control, M67 (which contains four Stat3-binding sites (35)) as a positive control, and FasP-luc. In response to IL-6 treatment, there was an ∼8-fold induction of relative luciferase units for M67 compared with untreated cells (Fig. 6B). IL-6 treatment did not cause a significant increase in relative luciferase units for the empty vector tkluc (Fig. 6B). Cells transfected with the FasP-luc contruct showed a ∼10-fold increase in relative luciferase units compared with untreated cells (Fig. 6B).
FIGURE 6.

A 160-bp conserved region of the fascin promoter is sufficient to induce luciferase expression in response to IL-6. A, a 160-bp conserved region of the human fascin promoter containing an overlapping STAT site and NFκB site was subcloned into the luciferase reporter tkluc to generate FasP-luc. B, luciferase assays were performed in MDA-MB-231 cells transfected for 24 h with empty vector tkluc (negative control), M67 (positive control), which contains four Stat3 binding sites, and FasP-luc. Cells were treated with IL-6 for 6 h. C, MDA-MB-231 cells were transfected with tkluc or FasP-luc and treated with IL-6, TNF-α, or IL-6 and TNF-α together for 6 h. Results represent the averages and S.D. of at least three independent experiments.
Once it was determined that IL-6 could activate the FasP-luc reporter containing the conserved 160-bp region, it was necessary to determine whether TNF-α could activate transcription of the FasP-luc reporter. Cells transfected with the empty vector tkluc did not show any transcriptional activation when treated with IL-6, TNF-α, or both IL-6 and TNF-α together (Fig. 6C). In response to TNF-α treatment, there was a significant induction of luciferase in cells transfected with the FasP-luc reporter compared with untreated cells (Fig. 6C). Luciferase levels were similar to those induced by IL-6 alone (Fig. 6C). When cells transfected with FasP were treated with both cytokines together, luciferase was induced at levels similar to treatment with either cytokine alone (Fig. 6C). These results demonstrate that a 160-bp conserved region of the fascin promoter is sufficient to activate transcription in response to IL-6 or TNF-α.
The Overlapping STAT/NFκB site in the FasP-luc Contruct Is Necessary for Transcriptional Activation in Response to IL-6 and TNF-α
To determine whether the overlapping sequences of the STAT/NFκB site are required for cytokine-induced transcriptional activation, point mutations of nucleotides important for Stat3 and NFκB binding were introduced into the FasP-luc contruct (Fig. 7A). Luciferase assays were performed for MDA-MB-231 cells transiently transfected with tkluc, FasP-luc, and the constructs containing the indicated point mutations. Cells transfected with the wild-type FasP-luc construct showed a significant increase in relative luciferase units (∼8-fold) compared with untreated cells (Fig. 7B). In contrast, the constructs containing the point mutations G1086A and G1085A showed an induction of only ∼2-fold compared with untreated cells (Fig. 7B). The construct containing the point mutation G1084A showed an induction of ∼4-fold, whereas the AA1082CC mutation did not show significant induction in response to IL-6 (Fig. 7B).
FIGURE 7.

IL-6- and TNF-α-induced luciferase expression is dependent on the STAT/NFκB site. A, point mutations of the STAT/NFκB site were generated in FasP-luc. Luciferase assays were performed for the empty vector (tkluc), FasP-luc (wild-type), and the mutant constructs, G1086A, G1085A, G1084A, and AA1082CC in response to IL-6 (B), TNF-α (C), or both IL-6 and TNF-α combined (D). Results represent the averages and S.D. of three independent experiments.
Luciferase assays were performed to determine whether these mutations had an effect in response to TNF-α treatment. When the cells were treated with TNF-α alone, there was a significant increase in luciferase expression for the FasP-luc construct compared with untreated cells (Fig. 7C). There was no induction in response to TNF-α for the G1086A, G1085A, or AA1082CC mutations (Fig. 7C). The G1084A mutation did not significantly affect luciferase expression (Fig. 7C).
Luciferase asssays were performed to determine whether combined treatment with IL-6 plus TNF-α would restore expression of any of the mutants. Treatment with both cytokines induced luciferase expression of the FasP-luc contruct to levels similar to treatment with either cytokine alone (Fig. 7D). Treatment with both IL-6 and TNF-α together showed some induction with the G1086A and G1085A constructs that was similar to results obtained with IL-6 treatment alone (Fig. 7D). For the G1084A construct, treatment with both cytokines induced luciferase expression levels similar to the wild-type FasP-luc construct (Fig. 7D). Although the AA1082CC construct was not induced with either cytokine alone, there was some induction of luciferase expression with both together (Fig. 7D). These results, taken together, show that the overlapping sequences in the STAT and NFκB sites in the 160-bp conserved region of the fascin promoter are required for efficient transcriptional activation by IL-6 and TNF-α.
DISCUSSION
The cytokine IL-6 has long been associated with inflammation and metastatic cancers including breast cancer (36–39). IL-6 induces activation of Stat3, which is also associated with breast cancer metastasis (16, 17). We have shown previously that through induction by IL-6, Stat3 along with NFκB bind to the fascin promoter to increase its expression and promote breast cancer cell migration (18). In this work, we further explored the roles of Stat3, NFκB, and the TNF-α signaling pathway in fascin gene regulation.
Similar to IL-6/Stat3, TNF-α/NFκB has been linked to inflammation and cancer (28). This study demonstrates that in response to TNF-α, NFκB binds to the fascin promoter and increases its expression. Compared with IL-6 induction of fascin, TNF-α induces a longer, more sustained induction of the fascin gene. It is interesting that IL-6 and TNF-α treatment together does not lead to an additive effect of both cytokines. Despite an increase in Stat3 and NFκB binding to the fascin promoter based on ChIP analyses, treatment with both cytokines induced mRNA expression levels similar to either cytokine alone. It has been shown that MDA-MB-231 cells express and constitutively secrete IL-6 and TNF-α (40), which probably results in a high basal level of fascin because there is always a presence of both cytokines in the media. If these cytokines were not secreted into the medium, a more dramatic induction of fascin expression in response to simultaneous treatment of IL-6 and TNF-α might be observed. However, further treatment with high levels of cytokines can induce more cell migration, which is dependent on both Stat3 (18) and NFκB (Fig. 4), suggesting that in vivo, local inflammation caused by the growth of a tumor may result in high levels of inflammatory cytokines such as IL-6 and TNF-α in the tumor micro-environment, which would then lead to tumor cell migration and metastasis.
NFκB is required for Stat3 binding to the fascin promoter in response to IL-6 treatment (Fig. 2), and NFκB binding requires Stat3 as shown in our previous studies (18). Neither transcription factor can bind to the fascin promoter separately. Co-immunoprecipitation data demonstrates that in response to IL-6 and TNF-α, Stat3, NFκB p50, and NFκB p65 form a complex in the nucleus (Fig. 5). This suggests that for either Stat3 or NFκB to bind to the fascin promoter, the formation of the Stat3·NFκB complex is a prerequisite step. Therefore, Stat3 and NFκB are co-dependent in their binding to the fascin promoter in response to cytokine treatment.
We had previously identified a 160-bp region of the fascin promoter that is highly conserved in both the human and mouse genes (18). Furthermore, this conserved region contains overlapping STAT and NFκB sites. Using a luciferase reporter, we isolated this region and showed that it is sufficient for transcriptional activation in response to IL-6 and TNF-α. We further demonstrated that the overlapping sequences are required for IL-6 and TNF-α induced transcription. It is interesting to note that the STAT site deviates from the consensus at the first three nucleotides (TTC to TGT) (41). It is possible that binding of NFκB to the overlapping NFκB site serves as a stabilizer for Stat3 binding to the sequence, as well as recruitment of other co-activators in the transcriptional complex.
Cancer metastasis is a multistep process that involves many cellular factors and signaling pathways (42). Fascin has been shown to be a key regulator of breast cancer metastasis (22). Identification of Stat3 and NFκB as two important regulators of IL-6 and TNF-α-induced fascin expression is a major step in understanding the process of metastasis. Further analysis of the interaction of these two transcription factors could potentially lead to identification of drug targets to block breast cancer metastasis.
Acknowledgments
We thank all laboratory members for helpful discussions and the Department of Microbiology and Immunology at Weill Cornell Medical College for the use of the Applied Biosystems PRISM 7900HT Sequence Detection Equipment.
This work was supported by National Institutes of Health Grant CA136837 (to X. Y. H.).
- NFκB
- nuclear factor κB
- luc
- luciferase.
REFERENCES
- 1. Levy D. E., Darnell J. E., Jr. (2002) Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662 [DOI] [PubMed] [Google Scholar]
- 2. O'Shea J. J., Gadina M., Schreiber R. D. (2002) Cytokine signaling in 2002: new surprises in the Jak/STAT pathway. Cell 109, S121–S131 [DOI] [PubMed] [Google Scholar]
- 3. Akira S. (1999) Functional roles of STAT family proteins: lessons from knockout mice. Stem Cells 17, 138–146 [DOI] [PubMed] [Google Scholar]
- 4. Santos C. I., Costa-Pereira A. P. (2011) Signal transducers and activators of transcription-from cytokine signalling to cancer biology. Biochim. Biophys. Acta 1816, 38–49 [DOI] [PubMed] [Google Scholar]
- 5. Zhang X., Wrzeszczynska M. H., Horvath C. M., Darnell J. E., Jr. (1999) Interacting regions in Stat3 and c-Jun that participate in cooperative transcriptional activation. Mol. Cell. Biol. 19, 7138–7146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lerner L., Henriksen M. A., Zhang X., Darnell J. E., Jr. (2003) STAT3-dependent enhanceosome assembly and disassembly: synergy with GR for full transcriptional increase of the α2-macroglobulin gene. Genes Dev. 17, 2564–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bromberg J. F., Wrzeszczynska M. H., Devgan G., Zhao Y., Pestell R. G., Albanese C., Darnell J. E., Jr. (1999) Stat3 as an oncogene. Cell 98, 295–303 [DOI] [PubMed] [Google Scholar]
- 8. Shen Y., Schlessinger K., Zhu X., Meffre E., Quimby F., Levy D. E., Darnell J. E., Jr. (2004) Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol. Cell. Biol. 24, 407–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Akira S. (2000) Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 19, 2607–2611 [DOI] [PubMed] [Google Scholar]
- 10. Raz R., Lee C. K., Cannizzaro L. A., d'Eustachio P., Levy D. E. (1999) Essential role of STAT3 for embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. U.S.A. 96, 2846–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Niwa H., Burdon T., Chambers I., Smith A. (1998) Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 12, 2048–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu H., Pardoll D., Jove R. (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 9, 798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akira S., Nishio Y., Inoue M., Wang X. J., Wei S., Matsusaka T., Yoshida K., Sudo T., Naruto M., Kishimoto T. (1994) Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63–71 [DOI] [PubMed] [Google Scholar]
- 14. Heinrich P. C., Horn F., Graeve L., Dittrich E., Kerr I., Müller-Newen G., Grötzinger J., Wollmer A. (1998) Interleukin-6 and related cytokines: effect on the acute phase reaction. Z Ernahrungswiss 37, 43–49 [PubMed] [Google Scholar]
- 15. Grandis J. R., Drenning S. D., Zeng Q., Watkins S. C., Melhem M. F., Endo S., Johnson D. E., Huang L., He Y., Kim J. D. (2000) Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 97, 4227–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barbieri I., Pensa S., Pannellini T., Quaglino E., Maritano D., Demaria M., Voster A., Turkson J., Cavallo F., Watson C. J., Provero P., Musiani P., Poli V. (2010) Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 70, 2558–2567 [DOI] [PubMed] [Google Scholar]
- 17. Ling X., Arlinghaus R. B. (2005) Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 65, 2532–2536 [DOI] [PubMed] [Google Scholar]
- 18. Snyder M., Huang X. Y., Zhang J. J. (2011) Signal transducers and activators of transcription 3 (STAT3) directly regulates cytokine-induced fascin expression and is required for breast cancer cell migration. J. Biol. Chem. 286, 38886–38893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams J. C. (2004) Roles of fascin in cell adhesion and motility. Curr. Opin. Cell Biol. 16, 590–596 [DOI] [PubMed] [Google Scholar]
- 20. Machesky L. M., Li A. (2010) Fascin: invasive filopodia promoting metastasis. Commun. Integr. Biol. 3, 263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kureishy N., Sapountzi V., Prag S., Anilkumar N., Adams J. C. (2002) Fascins, and their roles in cell structure and function. Bioessays 24, 350–361 [DOI] [PubMed] [Google Scholar]
- 22. Chen L., Yang S., Jakoncic J., Zhang J. J., Huang X. Y. (2010) Migrastatin analogues target fascin to block tumour metastasis. Nature 464, 1062–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vignjevic D., Schoumacher M., Gavert N., Janssen K. P., Jih G., Laé M., Louvard D., Ben-Ze'ev A., Robine S. (2007) Fascin, a novel target of β-catenin-TCF signaling, is expressed at the invasive front of human colon cancer. Cancer Res. 67, 6844–6853 [DOI] [PubMed] [Google Scholar]
- 24. Hashimoto Y., Loftis D. W., Adams J. C. (2009) Fascin-1 promoter activity is regulated by CREB and the aryl hydrocarbon receptor in human carcinoma cells. PLoS One 4, e5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bros M., Ross X. L., Pautz A., Reske-Kunz A. B., Ross R. (2003) The human fascin gene promoter is highly active in mature dendritic cells due to a stage-specific enhancer. J. Immunol. 171, 1825–1834 [DOI] [PubMed] [Google Scholar]
- 26. DiDonato J. A., Mercurio F., Karin M. (2012) NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 246, 379–400 [DOI] [PubMed] [Google Scholar]
- 27. Carbone C., Melisi D. (2012) NF-κB as a target for pancreatic cancer therapy. Expert Opin. Ther. Targets 16, S1–S10 [DOI] [PubMed] [Google Scholar]
- 28. Ben-Neriah Y., Karin M. (2011) Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 12, 715–723 [DOI] [PubMed] [Google Scholar]
- 29. Ramadoss S., Li J., Ding X., Al Hezaimi K., Wang C. Y. (2011) Transducin β-like protein 1 recruits nuclear factor κB to the target gene promoter for transcriptional activation. Mol. Cell. Biol. 31, 924–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang J., Liao X., Agarwal M. K., Barnes L., Auron P. E., Stark G. R. (2007) Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 21, 1396–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang J., Stark G. R. (2008) Roles of unphosphorylated STATs in signaling. Cell. Res. 18, 443–451 [DOI] [PubMed] [Google Scholar]
- 32. Snyder M., Huang X. Y., Zhang J. J. (2008) Identification of novel direct Stat3 target genes for control of growth and differentiation. J. Biol. Chem. 283, 3791–3798 [DOI] [PubMed] [Google Scholar]
- 33. Snyder M., Huang X. Y., Zhang J. J. (2010) Stat3 directly controls the expression of Tbx5, Nkx2.5, and GATA4 and is essential for cardiomyocyte differentiation of P19CL6 Cells. J. Biol. Chem. 285, 23639–23646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Snyder M., Huang X. Y., Zhang J. J. (2011) Stat3 is essential for neuronal differentiation through direct transcriptional regulation of the Sox6 gene. FEBS Lett. 585, 148–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Besser D., Bromberg J. F., Darnell J. E., Jr., Hanafusa H. (1999) A single amino acid substitution in the v-Eyk intracellular domain results in activation of Stat3 and enhances cellular transformation. Mol. Cell. Biol. 19, 1401–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knüpfer H., Preiss R. (2007) Significance of interleukin-6 (IL-6) in breast cancer (review). Breast Cancer Res. Treat. 102, 129–135 [DOI] [PubMed] [Google Scholar]
- 37. Sullivan N. J., Sasser A. K., Axel A. E., Vesuna F., Raman V., Ramirez N., Oberyszyn T. M., Hall B. M. (2009) Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 28, 2940–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walter M., Liang S., Ghosh S., Hornsby P. J., Li R. (2009) Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene 28, 2745–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sasser A. K., Sullivan N. J., Studebaker A. W., Hendey L. F., Axel A. E., Hall B. M. (2007) Interleukin-6 is a potent growth factor for ER-α-positive human breast cancer. FASEB J. 21, 3763–3770 [DOI] [PubMed] [Google Scholar]
- 40. Faggioli L., Costanzo C., Merola M., Bianchini E., Furia A., Carsana A., Palmieri M. (1996) Nuclear factor kappa B (NF-κB), nuclear factor interleukin-6 (NFIL-6 or C/EBP β) and nuclear factor interleukin-6 beta (NFIL6-β or C/EBPδ) are not sufficient to activate the endogenous interleukin-6 gene in the human breast carcinoma cell line MCF-7. Comparative analysis with MDA-MB-231 cells, an interleukin-6-expressing human breast carcinoma cell line. Eur. J. Biochem. 239, 624–631 [DOI] [PubMed] [Google Scholar]
- 41. Ehret G. B., Reichenbach P., Schindler U., Horvath C. M., Fritz S., Nabholz M., Bucher P. (2001) DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J. Biol. Chem. 276, 6675–6688 [DOI] [PubMed] [Google Scholar]
- 42. Said N. A., Williams E. D. (2011) Growth factors in induction of epithelial-mesenchymal transition and metastasis. Cells Tissues Organs 193, 85–97 [DOI] [PubMed] [Google Scholar]