

Background: Membrane attachment of PrP via a GPI anchor is critical for conversion into PrPSc.
Results: Semisynthetic membrane-anchored PrP variants are generated and analyzed in different lipid environments.
Conclusion: A membrane anchor drastically changes PrP interactions with membranes and enables pore formation.
Significance: Membrane-induced conformational changes and pore formation of PrP play a role in the conversion into PrPSc and its toxic effects.
Keywords: Liposome, Prion, Protein Aggregation, Protein Chemistry, Protein Conformation, lipid-Protein Interactions, Membrane Anchor, Pore Formation, Protein Semisynthesis
Abstract
Membrane attachment via a C-terminal glycosylphosphatidylinositol anchor is critical for conversion of PrPC into pathogenic PrPSc. Therefore the effects of the anchor on PrP structure and function need to be deciphered. Three PrP variants, including full-length PrP (residues 23–231, FL_PrP), N-terminally truncated PrP (residues 90–231, T_PrP), and PrP missing its central hydrophobic region (Δ105–125, ΔCR_PrP), were equipped with a C-terminal membrane anchor via a semisynthesis strategy. Analyses of the interactions of lipidated PrPs with phospholipid membranes demonstrated that C-terminal membrane attachment induces a different binding mode of PrP to membranes, distinct from that of non-lipidated PrPs, and influences the biochemical and conformational properties of PrPs. Additionally, fluorescence-based assays indicated pore formation by lipidated ΔCR_PrP, a variant that is known to be highly neurotoxic in transgenic mice. This finding was supported by using patch clamp electrophysiological measurements of cultured cells. These results provide new evidence for the role of the membrane anchor in PrP-lipid interactions, highlighting the importance of the N-terminal and the central hydrophobic domain in these interactions.
Introduction
Prion diseases are fatal neurodegenerative disorders characterized by the conformational conversion of the normal, α-helical, cellular prion protein (PrPC)3 into the toxic, β-sheet-enriched isoform called scrapie PrP (PrPSc) (1–4). PrPC is a glycosylphosphatidylinositol (GPI)-anchored protein that is found predominantly at the outer leaflet of the neuronal cell membrane (5). Although the biological function of PrPC, especially within a membrane environment, is not fully understood, the C-terminal membrane attachment of PrPC via a GPI anchor appears to play a key role in the conversion of PrPC into PrPSc (6–9). Indeed, several studies provide evidence that the conversion depends on posttranslational GPI anchor attachment of PrP to the outer leaflet of the cell membrane. Methods to release PrPC from the cell surface or to disrupt the transport of PrPC to the plasma membrane are shown to prevent or significantly reduce conversion (10–12). Baron and Caughey and co-workers (13, 14) found further proof that a membrane environment as well as a GPI anchor on PrP is required for conversion using cell-free conversion conditions. Recently, by developing a new cellular system in which epitope-tagged PrPC was expressed in a PrP knockdown neuronal cell line and then exposed to the RML (Rocky Mountain Laboratory) prion (15), Goold et al. (16) found that the conversion occurred primarily on the plasma membrane, especially in GM1-enriched lipid microdomains. At the same time the infection of such cells exposed to the RML prion was extremely rapid. Moreover, lipids are also one of the factors currently believed to be essential for efficient conversion in vitro and in vivo (17–19). In this context, studying the interaction of PrP carrying a membrane anchor with membranes under fully controlled conditions is of paramount importance, e.g. to identify the effects of membrane composition on protein conformation and conversion. However, GPI-anchored PrPC isolated from neuronal cells is typically heterogeneous with respect to N-glycosylation and the molecular structure of the GPI anchor. Furthermore, contamination with other compounds such as lipids, carbohydrates, and even misfolded PrP cannot be excluded.
Thus, generating membrane-anchored PrP using recombinant expression in combination with selective chemical modifications is a viable alternative, which has been used by several groups so far (9, 20, 21). Here, we utilize a strategy developed previously in our group that relies on expressed protein ligation to prepare PrP containing a C-terminal membrane anchor that mimics a GPI anchor and conveys strong membrane attachment (22). We demonstrated previously that PrP with the GPI anchor mimic performs similarly to recombinant PrP equipped with a synthetic GPI anchor in comparative aggregation assays (23). This finding implies that the effects of both membrane anchors on PrP conversion are very similar and that the lipidated peptide constitutes a useful and easily accessible GPI anchor mimic.
Here we used expressed protein ligation to produce three membrane-anchored PrP constructs: full-length PrP (residues 23–231, FL_PrP-MA), PrP missing its central hydrophobic region (residues 23–104-126–231, ΔCR_PrP-MA) and N-terminally truncated PrP (residues 90–231, T_PrP-MA). The conformational and biochemical properties of the resulting PrP variants within different membrane environments as well as the influence of PrP-lipid interactions on the integrity of membranes are investigated.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
PrP-Mxe intein fusion proteins were produced and purified according to the procedure described previously (22, 24). The plasmids (Fig. 1A) were constructed and transformed into Escherichia coli chemical-competent cells (BL21(DE3)RIL or Rosetta 2 (Invitrogen) for expression of T_PrP or FL_PrP and ΔCR_PrP, respectively). The cells were cultured in 2YT medium containing 100 μg/ml ampicillin and 30 μg/ml chloramphenicol at 37 °C and 200 rpm with shaking. Expression was induced at A600 nm = 1.0 by the addition of 1 mm IPTG to the medium and continued for another 4 h. Cells were harvested, dissolved in TBS-EDTA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.5 mm EDTA), lysed with a cell disruption system (Constant Systems, TS series), and then centrifuged at 50,000 × g for 30 min at 4 °C to collect the pellet. The pellet was washed with TBS-EDTA buffer containing 0.5% (v/v) Triton X-100 and centrifuged at 50,000 × g for 20 min a 4 °C. Washing and centrifugation steps were repeated three times with TBS-EDTA buffer. Next, the pellet was dissolved in 8 m Gnd-HCl, 50 mm Tris-HCl, pH 8.0, by stirring at room temperature overnight. The solubilized pellet solutions were loaded on nickel-nitrilotriacetic acid resin equilibrated with 8 m GndHCl, 50 mm Tris-HCl, pH 8.0. The resin slurry was washed with 8 m GndHCl, 50 mm Tris-HCl, pH 8.0. Finally, PrP-Mxe intein proteins were eluted from the resin with 8 m GndHCl, 50 mm Tris-HCl, 250 mm imidazole, pH 8.0. The PrP-Mxe intein proteins were transferred into 8 m urea, 50 mm Tris-HCl, pH 8.0, using a PD-10 column (GE Healthcare). Purified PrP-Mxe intein fusion proteins in 8 m urea buffer were slowly diluted to 4 m urea with 50 mm Tris-HCl, pH 8.0, and then sodium 2-mercaptoethanesulfonate (Sigma-Aldrich) was added to reach a final concentration of 500 mm, and the mixture was gently stirred at room temperature overnight (Fig. 1B). The cleavage mixtures containing PrP-Cα-thioester proteins were loaded on a preparative C4 column (22 mm diameter, 250 mm length, Grace Vydac) equilibrated with 5% (v/v) of buffer B (acetonitrile and 0.08% (v/v) TFA) mixed with buffer A (dH2O and 0.1% (v/v) TFA) at a flow rate of 10 ml/min for 20 min. Next, a linear gradient of 30 to 60% (v/v) of buffer B mixed with buffer A over 60 min was applied to elute PrP-Cα-thioesters from the column (Fig. 1C). Fractions containing PrP-Cα-thioester proteins were pooled and analyzed by MALDI-MS and SDS-PAGE (Fig. 1D). Pure fractions were lyophilized and used for native chemical ligation (NCL) reactions.
FIGURE 1.

Preparation of PrP constructs containing C-terminal α-thioesters for expressed protein ligation. A, plasmid maps of FL_PrP or ΔCR_PrP (top) and T_PrP (bottom) C-terminally fused to Mxe intein. B, cleavage reactions of PrP-intein fusion proteins. C and D, SDS-PAGE (C) and MALDI-MS (D) analyses of purified PrP-Cα-thioesters.
Synthesis of Membrane Anchor
An Fmoc-based solid phase peptide synthesis was conducted according to the strategy previously described (22). The peptide was purified by RP-HPLC using a semipreparative C4 column (10 mm diameter, 250 mm length, Grace Vydac) with an elution gradient of 30 to 90% (v/v) of buffer B mixed with buffer A at a flow rate of 3 ml/min for 60 min. Pure fractions were identified by ESI-MS and checked by analytical HPLC (Fig. 2).
FIGURE 2.

Chemical synthesis of the GPI anchor-mimicking peptide (membrane anchor) using Fmoc-based solid phase peptide synthesis. peptide carries two palmitoyl moieties, a polymer tag to enhance solubility, and an N-terminal cysteine for the NCL reaction. A, structure and representation of membrane anchor. B, analytical RP-HPLC chromatogram. C, ESI-MS spectrum and deconvoluted mass of the purified membrane anchor.
Native Chemical Ligation
0.5 μmol of PrP-Cα-thioester proteins and 2.5 μmol of membrane anchor were dissolved in 500 μl of degased NCL buffer (6 m GndHCl, 300 mm Na2HPO4, pH 7.8), and then 2% (v/v) thiophenol was added and mixed vigorously under an argon atmosphere. The NCL reactions were incubated at 37 °C for 24 h (Fig. 3B). The reactions were stopped by adding 15% (v/v) β-mercaptoethanol. Reaction yields were checked by SDS-PAGE; ligation products (lipidated PrPs) were purified by the same HPLC procedure used above and lyophilized (Fig. 3, C and D).
FIGURE 3.

Semisynthesis of PrP variants with membrane anchors. A, scheme of the semisynthesis strategy. B and C, SDS-PAGE analyses of ligation reactions (B) and purified PrP constructs (C) carrying MAs. D and E, MS analysis (D) and CD measurements (E) of purified PrPs with membrane anchor.
Folding of Lipidated PrPs
The lyophilized lipidated PrPs were dissolved in 8 m GndHCl, 50 mm Tris-HCl pH 8.5, 1 mm EDTA, and 3.0/0.3 mm GSH/GSSG. A stepwise dilution strategy was applied to dilute Gdn-HCl from 8 to 2.5 m using dilution buffer containing 20 mm sodium acetate, pH 5.0, 20 mm N-octyl-β-d-glucopyranoside (SERVA), and 0.3/3 mm GSSG/GSH on ice. Folding reactions were incubated at 10 °C for at least 48 h to prevent precipitation in the following step in which lipidated PrPs were exchanged into storage buffer (20 mm sodium acetate, pH 5.0, and 20 mm N-octyl-β-d-glucopyranoside) using D-tube dialyzers (Novagen). Ultracentrifugation at 100,000 × g for 1 h at 4 °C was applied, and the supernatant was collected for CD spectrometry analysis (Fig. 3E). A similar folding protocol was also applied for PrPs without membrane anchors.
Preparation of Phospholipid Liposomes
10 mg of phospholipids was dissolved in 1 ml of chloroform in a 25-ml round-bottom flask, and chloroform was evaporated by nitrogen flow to form a thin lipid film. The film was dried in a vacuum desiccator for at least 2 h or overnight. Hydration of the film was performed in 20 mm sodium phosphate, pH 7.0, for 1 h, with vortexing every 10 min in between to obtain an opalescent solution (large multilamellar liposomes were formed in this step). The multilamellar liposome suspension was frozen in dry ice-methanol for 5 s and then vortexed rapidly to form a thin frozen layer of phospholipid on the wall of the tube. Next, the frozen phospholipid layer was thawed by warming up the tube under cool tap water and then under warm (42 °C) water for 30 s. The freeze/thaw cycle was repeated five times. The lipid solution was passed 20 times through a mini-extruder with a 100-nm polycarbonate membrane (Avanti Polar Lipids). The resulting liposomes were collected and kept under argon at 4 °C for the next steps. Liposomes were produced using non-charged phospholipid DPPC or DOPC, anionic phospholipid POPG, or phospholipid mixtures mimicking neuronal membrane (NM). These NM mixtures consisted of PC:PE:PI:SM:Chol:POPG (30:30:2.5:5:22.5:10) for NM-PG and PC:PE:PI:SM:Chol:POPS (30:30:2.5:5:22.5:10) for NM-PS (25).
Flotation Assays
These assays were carried out as described previously (26). Briefly, PrP proteins and liposomes were mixed and incubated for 30 min at room temperature. The mixtures were loaded under a discontinuous iodixanol (Optiprep, Sigma-Aldrich) gradient containing, sequentially, 1 ml of 36% (v/v), 1 ml of 31%, and 0.4 ml of 5% iodixanol in a centrifuge tube. The gradient was centrifuged at 200,000 × g at 4 °C for 3 h (MLS-50 rotor, Optima ultracentrifugation system, Beckman Coulter Inc.). Next, 200-μl fractions were collected from top to bottom of the centrifuge tube, and PrP in each fraction was precipitated by TCA and dissolved in 1× SDS loading buffer for SDS-PAGE and Western blot analysis with polyclonal anti-PrP antibody M20 (Santa Cruz Biotechnology) (Fig. 4, C and D).
FIGURE 4.

Strong and specific binding of PrPs to liposomes via their C-terminal membrane anchors. The interactions of anchorless PrPs (black) and PrPs with a membrane anchor (red) with DPPC liposomes (A) and POPG liposomes (B) were analyzed by flotation assays in combination with Western blots. C and D, The original Western blots are depicted in C for experiments with DPPC liposomes and in D for POPG liposomes. POPG liposomes loaded with PrP variants were treated with a solution of 10 mm NaOH to disrupt electrostatic interactions. Quantification of all blots was performed by using ImageJ software.
Proteinase K (PK) Resistance Assay
20 μl of PrP-phospholipid mixtures were subjected to PK digestions at 37 °C for 30 min with the indicated PK:PrP molar ratios of 1:4, 1:8, and 1:16. Reactions were stopped by adding 5 mm PMSF and kept on ice for 15 min. The mixtures were subjected to SDS-PAGE and Western blot analysis as described above.
Calcein Release
The assay was performed as described previously (27) with minor modifications. The release of calcein from the calcein-containing liposomes was monitored by calcein fluorescence at 512 nm over 120 min after mixing five parts of the liposomes and one part of protein solution (5:1 (v/v)). The excitation wavelength was 490 nm. Experiments were carried out at room temperature. The concentration of liposomes was 50 μm, and protein concentrations varied from 10 to 500 nm. The percentage of calcein release (Rf) was calculated based on Equation 1,
where Ft is the fluorescence intensity recorded at time t after adding PrP to liposomes, Fo is fluorescence intensity without PrP proteins, and Ftot is the total fluorescence intensity recorded after total release of calcein by disrupting the liposomes with 1% (v/v) Triton X-100. Each measurement was repeated three times and measured in 96-well plates with a microplate reader (BioTek Synergy Mx).
NBD Fluorescence Quenching
NBD-phosphatidylethanolamine (PE) (Avanti Polar Lipids) was dissolved in chloroform and added to phospholipids at 1 mole % and dried together under nitrogen flow to form thin films. The films were used to generate the NBD-liposomes as described above. Samples for fluorescence measurements were prepared by adding PrP at a concentration of 1 μm, or using buffers only (blank run), and 200 μm NBD-liposomes to 20 mm sodium phosphate, pH 7.0. Fluorescence quenching measurements were initiated by adding the quencher sodium dithionite at 10 mm (Merck) from a 130 mm stock solution freshly prepared in 50 mm Tris-base buffer, ∼pH 11. NBD fluorescence kinetics were recorded on a Fluoromax 4 system (Horiba) for 900 s at 22 °C using an excitation wavelength of 467 nm and an emission wavelength of 535 nm. The NBD fluorescence decay was calculated as a percentage of the initial NBD fluorescence measured before adding sodium dithionite (28).
Circular Dichroism (CD) Spectroscopy
Far-UV CD spectra were recorded at 22 °C on an Applied Photophysics Chirascan Plus system. PrP proteins were diluted in 20 mm sodium acetate, pH 5.0, to final concentration of 0.1–0.2 mg/ml. CD spectra were acquired at a speed of 20 nm/min, a 1-nm bandwidth, and a response time of 4 s. All spectra were recorded in a 0.1-cm quartz cuvette with a wavelength ranging between 195 and 260 nm. Each spectrum was recorded 10 times and subsequently averaged.
Tryptophan Fluorescence Measurements
Tryptophan fluorescence measurements were performed by mixing the protein solution with an equal volume of lipid liposomes. After equilibration for 3 min, four fluorescence spectra from 300 to 450 nm were recorded and averaged on a Fluoromax 4 system (Horiba) with an excitation wavelength of 295 nm (2 nm bandwidth) at 22 °C. Fluorescent background was also recorded and subtracted from the final spectra (27). Tryptophan fluorescence quenching measurements were performed by adding freshly prepared aliquots of 1 m acrylamide stock solution in 20 mm sodium phosphate, pH 7.0, to PrP in the presence or absence of liposomes (DOPC or POPG) at concentrations of 3 μm PrP and 3 mm lipid liposomes. Tryptophan fluorescence spectra for each concentration of the quencher acrylamide were recorded and corrected (29). The fluorescence quenching data were analyzed by using the Stern-Volmer equation for collisional quenching as given in Equation 2,
where Fo is the initial fluorescence, F is the fluorescence at the acrylamide concentration [Q], and KSV is the Stern-Volmer constant. A plot of Fo/F versus [Q] gives a linear plot that yields the KSV value (59).
Cryo-electron Microscopy (EM) Measurements
Two samples including POPG liposomes alone and POPG liposomes mixed with lipidated ΔCR_PrP were prepared and visualized using a urea system (FEI Tecnai F30 Helium “Polara”). The resulting images were processed by using ImageJ software.
Electrophysiology
The pore-forming activity of FL_PrP and ΔCR_PrP with and without a membrane anchor was measured in HEK293 cells using patch clamping in the whole-cell configuration. HEK293 cells were purchased from American Type Cell Culture (ATCC CRL-1573). Cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 units/ml penicillin, and 100 μg/ml streptomycin (Sigma-Aldrich).
Whole-cell currents were acquired from HEK293 cells at room temperature (19–22 °C) 24–48 h after plating using an Axopatch 200B amplifier and Digidata 1440A with pCLAMP10.2 software (Axon Instruments). Currents were recorded at 10 kHz and filtered at 5 kHz. Glass coverslips with the cultured cells were transferred to a chamber of 1 ml capacity, mounted on the stage of an inverted microscope (Zeiss, Axiovert 100). After gigaohm seal formation, the equilibration period of 5 min was followed by recordings at a holding potential (Eh) of −100 mV unless stated otherwise. During a control period of 5 min, no ion current activity was observed. Aliquots of indicated prion proteins were thawed on ice and added into the bathing solution, resulting in the desired concentrations. Subsequent membrane current activities were recorded for up to 15 min. Capacity transients were cancelled, and series resistance was compensated. Access resistance was monitored and 75% compensated. Data acquisition and storage were performed directly on a PC. Gigaseals were monitored continuously during the experiments to avoid inadequate voltage clamp. Current densities were calculated by dividing the total current generated in a given cell by the membrane capacitance of the cell.
For studying pore-induced divalent cation current, the extracellular bath solution contained 20 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, and 140 mm choline-Cl titrated to pH 7.4 with methane sulfonic acid. The borosilicate glass patch pipettes (Harvard Apparatus, Holliston, MA) with resistances of 2–4 MΩ were pulled and polished using a DMZ universal puller (Zeitz Instruments, Martinsried, Germany) and were filled with pipette solution containing 145 mm CsCl, 3 mm MgCl2,10 mm HEPES and 10 mm EGTA, titrated to pH 7.25 with CsOH. The composition of the bath solution for the measurement of monovalent cation currents was 145 mm NaCl, 2.7 mm KCl, 1.8 mm CaCl2, 2 mm MgCl2, 5.5 mm glucose, and 10 mm HEPES, adjusted to pH 7.4 with 1 m NaOH solution. The pipette solution contained 135 mm potassium methanesulfonate, 10 mm KCl, 6 mm NaCl, 1 mm Mg2ATP, 2 mm Na3ATP, 10 mm HEPES, and 0.5 mm EGTA, adjusted to pH 7.2 with 1 m KOH solution.
RESULTS
Semisynthesis of Lipidated PrP Variants
Three PrP constructs including full-length PrP (residues 23–231, FL_PrP), N-terminally truncated PrP (residues 90–231, T_PrP), and PrP missing its central hydrophobic region (Δ105–125, ΔCR_PrP) were equipped with a C-terminal, chemically synthesized membrane anchor (MA) (22, 24). Briefly, PrP constructs were C-terminally fused to Mxe intein to generate recombinantly expressed PrPs containing a C-terminal α-thioester (Fig. 1) for NCL with a synthetic membrane anchor carrying an N-terminal cysteine residue (Fig. 2) (30). The resulting PrPs with a C-terminal membrane anchor were purified from NCL reactions (Fig. 3) and folded to obtain predominantly α-helical proteins (Fig. 3E) typical for cellular PrPC (31, 32).
Strong and Specific Binding of PrP Variants to Phospholipid Liposomes via Their C-terminal Membrane Anchors
To assess the binding of prion proteins to liposomes, a flotation assay in a discontinuous iodixanol gradient (36, 31, and 5% (v/v) iodixanol) was used (26, 33). Two kinds of phospholipid liposomes, DPPC (a temperature-dependent gel-phase, non-charged phospholipid) and POPG (an anionic phospholipid), were mixed with PrP constructs and loaded underneath the iodixanol gradient. In the case of flotation assays using the non-charged phospholipid DPPC, PrPs without a membrane anchor were deposited at the bottom of the centrifuge tube (Fig. 4A, black). In contrast, PrPs with a C-terminal membrane anchor displayed strong binding to DPPC liposomes. These were found in fractions 1, 2, and 3 from the upper volume of the centrifuge tube (Fig. 4A, red). All PrPs carrying the membrane anchor analyzed here bind to liposomes, as demonstrated previously for T_PrP-MA (22). The high affinity binding of prion protein to vesicles made of the anionic phospholipid POPG has been reported previously to induce structural changes from α-helical into β-sheet-rich structures (26, 27, 34). Therefore, we analyzed the interaction of PrPs with POPG liposomes in greater detail. In contrast to the behavior with DPPC liposomes described above, all PrP variants (with and without membrane anchor) interacted with POPG liposomes (Fig. 4B). However, there was a pronounced difference between PrP variants with and without membrane anchor after treatment of PrP-POPG liposomes with 10 mm NaOH, which can partially extract PrP from POPG liposomes by disrupting weak electrostatic interactions without denaturing the protein (26). For PrPs without a membrane anchor, the interactions with POPG liposomes were disrupted by NaOH, and large amounts of protein were deposited at the bottom of the iodixanol gradient (Fig. 4B, black). In contrast, the interactions of PrPs with membrane anchors and POPG liposomes were stable under NaOH extraction conditions, as indicated by PrPs present mainly in the top fractions of the iodixanol gradient (Fig. 4B, red). These results clearly demonstrated that all PrP variants interact with negatively charged liposomes but that only the C-terminal membrane anchor imparts a strong interaction.
Conformation of PrP Constructs in the Presence of Phospholipid Membranes
Far-UV CD spectroscopy was used to investigate whether PrP variants with and without a membrane anchor undergo conformational changes during interaction with phospholipid liposomes. PrP variants were incubated with liposomes at 25 °C, and CD spectra were recorded. The CD data depicted in Fig. 5A and Table 1 for PrPs without a membrane anchor indicate that PrP mixed with non-charged DOPC liposomes (green dots) did not undergo a change in their predominantly α-helical conformation and were almost indistinguishable from PrPs in the absence of lipids (black dots). In contrast, incubation with anionic POPG liposomes leads to a significant change in the secondary structure of PrP, from a typically α-helical to a β-sheet-rich structure (Fig. 5A, red), accompanied by an increase in random coil content as well (Table 1). The conformational changes of FL_PrP and T_PrP without a membrane anchor upon interaction with POPG liposomes found here are consistent with previous reports (26, 27). In addition, we found the same effect for ΔCR_PrP without a membrane anchor. Surprisingly, PrP variants carrying a membrane anchor did not convert into β-sheet-rich structures during interaction with POPG liposomes. In the case of T_PrP-MA and ΔCR_PrP-MA, their secondary structures remained mostly α-helical (Fig. 5A), without any significant changes in secondary structure parameters (Table 1). For FL_PrP-MA attached to POPG liposomes, a slight loss of secondary structure was observed, whereas more β-sheet was observed for FL_PrP without membrane anchor (Fig. 5A and Table 1). This finding suggests that the secondary structure of PrP attached to the lipid membrane via a membrane anchor differs considerably from that of PrP without a membrane anchor (Table 1). This possibly accounts for the distinct differences in biochemical characteristics such as PK resistance described below.
FIGURE 5.

Biochemical and structural properties of PrP constructs within membrane environments. A, CD analyses of PrPs without membrane anchor (top row) and PrPs with membrane anchor (bottom row) in solution (black) or in the presence of DOPC (green) and POPG (red) liposomes. B, PK digestions of FL_PrP constructs after incubation at 37°C for 1 h with buffer alone (left) or POPG liposomes (right) are shown in each panel; the results of PrPs with and without a membrane anchor are presented in the left- and right-hand panels, respectively. The PK-to-PrP molar ratios were 1:16, 1:8, and 1:4.
TABLE 1.
Secondary structure parameters of PrP variants in buffer only and upon interaction with DOPC or POPG liposomes
The CD spectra depicted in Fig. 5A were analyzed and deconvoluted using CDNN software.
| Sample | α-Helices | β-Sheets | Turns | Random coils |
|---|---|---|---|---|
| % | % | % | % | |
| FL_PrP | 43.68 | 13.97 | 16.28 | 26.10 |
| FL_PrP+DOPC | 32.74 | 17.42 | 17.02 | 32.84 |
| FL_PrP+POPG | 11.78 | 28.30 | 17.48 | 42.48 |
| FL_PrP-MA | 37.63 | 16.03 | 16.85 | 29.52 |
| FL_PrP-MA+DOPC | 39.97 | 15.28 | 16.65 | 28.14 |
| FL_PrP-MA+POPG | 33.75 | 18.20 | 17.48 | 30.55 |
| ΔCR_PrP | 34.77 | 16.66 | 16.90 | 31.67 |
| ΔCR_PrP+DOPC | 32.07 | 17.66 | 17.07 | 33.13 |
| ΔCR_PrP+POPG | 24.03 | 21.14 | 17.55 | 37.25 |
| ΔCR_PrP-MA | 32.13 | 17.70 | 17.10 | 33.20 |
| ΔCR_PrP-MA+DOPC | 34.25 | 17.22 | 16.82 | 31.68 |
| ΔCR_PrP-MA+POPG | 34.36 | 18.01 | 17.40 | 30.26 |
| T_PrP | 36.00 | 15.94 | 16.44 | 31.60 |
| T_PrP+DOPC | 35.28 | 16.31 | 16.62 | 31.79 |
| T_PrP+POPG | 17.59 | 24.60 | 17.17 | 40.62 |
| T_PrP-MA | 33.75 | 15.87 | 15.34 | 35.00 |
| T_PrP-MA + DOPC | 35.58 | 15.04 | 16.96 | 32.42 |
| T_PrP-MA + POPG | 36.27 | 15.63 | 16.07 | 32.23 |
Proteinase K Resistance of PrP Constructs within a Membrane Environment
PK resistance is one of the typical characteristics of PrPSc and distinguishes it from PrPC, which can be digested completely. Here PK digestion was used to evaluate whether the binding of PrPs to anionic POPG liposomes, as demonstrated above, can lead to PK resistance. PK digestion was carried out with PK to PrP molar ratios of 1:16, 1:8, and 1:4, after which samples were analyzed by Western blot. One sample without PK treatment was always included as a control. FL_PrP without a membrane anchor showed PK-resistant bands after mixing with POPG liposomes, whereas FL_PrP incubated in the absence of POPG liposomes remained PK-sensitive (Fig. 5B, upper left). This result is in good agreement with a previous study by Wang et al. (26).
In contrast, FL_PrP-MA incubated with POPG liposomes still remained PK-sensitive under similar conditions (Fig. 5B, lower left). This observation supports our CD data, which indicates that binding of FL_PrP-MA to POPG liposomes differs from binding of FL_PrP without a membrane anchor. This different binding prevents formation of PK-resistant PrP. For T_PrP and ΔCR_PrP no PK resistance was observed, either for variants with or without membrane anchor (Fig. 5B). These variants remained PK-sensitive and even though changes in secondary structure were observed, no fibril formation occurred (Fig. 5). This finding is consistent with the idea that both electrostatic (weak in the case of T_PrP due to the missing, positively charged N-terminal region) and hydrophobic (lacking in ΔCR_PrP) interactions play important roles in the interaction of PrP with anionic POPG liposomes (27, 33). To investigate this point further, we analyzed the interaction of different PrP variants with membranes in more detail using tryptophan fluorescence quenching.
Characterizing the Interactions of PrP Variants with Membranes via Tryptophan Fluorescence Quenching
Tryptophan fluorescence quenching by water-soluble quenchers such as acrylamide can provide information about the location of tryptophan residues in the complex structure of membrane-bound proteins (35). In the case of PrP, seven tryptophan residues are present in the unstructured N-terminal region (residues 23–99), which is supposed to play a key role in interaction of prion proteins and phospholipid membranes (26, 27). Linear Stern-Volmer plots were created for the quenching of tryptophan fluorescence of anchorless FL_PrP in phosphate buffer and under denaturing conditions after the addition of 8 m urea (Fig. 6A). The resulting Stern-Volmer constants (KSV) were found to be 5.96 ± 0.18 m−1 and 6.16 ± 0.26 m−1, respectively (Table 2). The good agreement between both values suggests that tryptophan residues of folded and unfolded FL_PrP without a membrane anchor are equally accessible to the quencher in solution.
FIGURE 6.

Stern-Volmer plots of tryptophan fluorescence quenching. A, FL_PrP without a membrane anchor in a phosphate buffer (red) or in the presence of 8 m urea (black). B–D, PrPs with and without a membrane anchor in solution (black and green) or bound to POPG liposomes (red and blue) or DOPC liposomes (cyan and magenta) for T_PrP, ΔCR_PrP and FL_PrP, respectively.
TABLE 2.
Stern-Volmer constant (Ksv) values of PrP variants in solution or mixed with POPG and DOPC liposomes
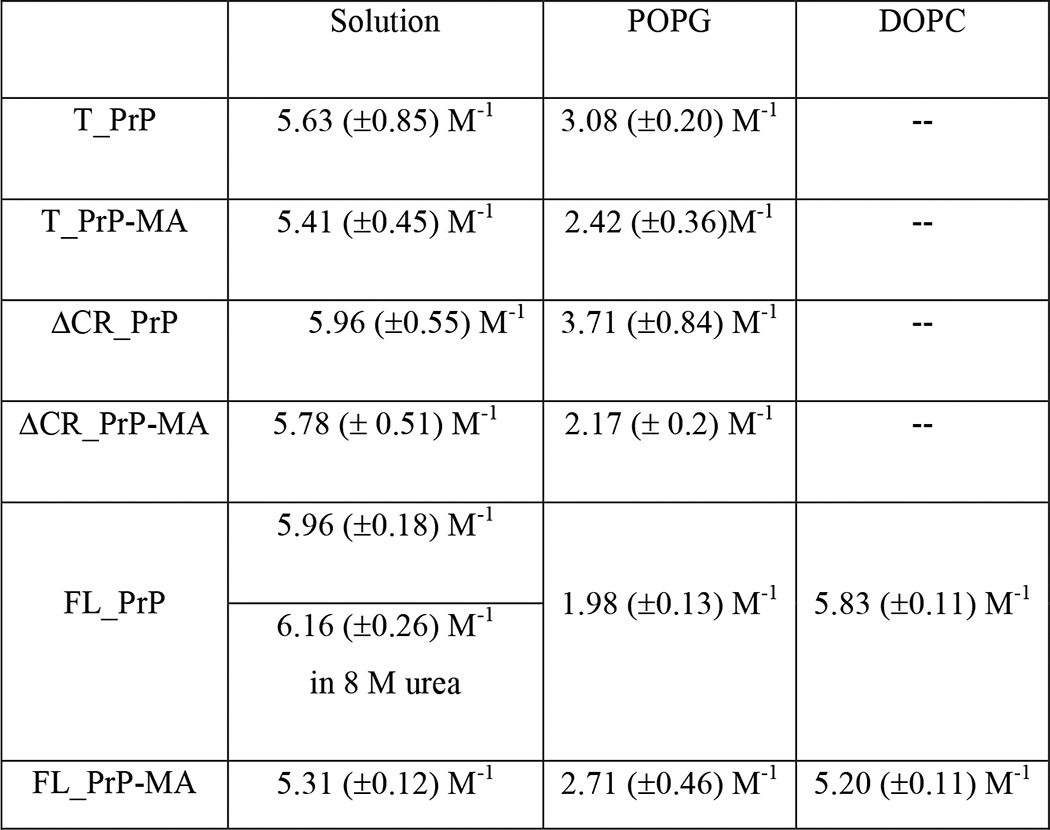
Next, linear Stern-Volmer plots corresponding to the quenching of the tryptophan fluorescence of each PrP variant in solution and mixed with liposomes were measured (Fig. 6, B–D). The resulting KSV values are summarized in Table 2. KSV values for PrPs without a membrane anchor in the presence of POPG liposomes were found to be significantly lower than those of PrPs in solution, indicating that the tryptophan residues have become less accessible to the quencher acrylamide due to their binding to POPG liposomes. This suggests either close contact with or even insertion of tryptophan residues into the lipid membrane (27). To further illuminate the insertion behavior, the same experimental procedure was applied to FL_PrP with and without a membrane anchor mixed with non-charged DOPC liposomes. The resulting KSV values were found to be 5.83 ± 0.11 m−1 and 5.20 ± 0.11 m−1, respectively, which is very close to those values measured for PrPs in solution (Table 2). The close agreement of KSV values measured in the presence and absence of DOPC liposomes indicates little protection of tryptophan residues from quencher. This is due to absence of or very weak binding of the N-terminal region to DOPC liposomes and is in good agreement with data from calcein release and NBD fluorescence quenching assays shown below. Thus the C-terminal membrane anchor attachment has no significant impact on the interaction of PrP N termini with membranes composed of DOPC.
In contrast to these results, the interactions of PrP variants with anionic POPG liposomes are strongly influenced by the membrane anchor, as indicated by KSV values. For T_PrP and ΔCR_PrP constructs, the interaction of variants with a membrane anchor and POPG liposomes resulted in far lower KSV values than those found for variants without a membrane anchor (Table 2). Thus, the C-terminal membrane anchor enhanced the contact and/or insertion of tryptophan residues of T_PrP and ΔCR_PrP into lipid bilayers of POPG liposomes.
Surprisingly, the membrane anchor limited the protection of tryptophan residues from FL_PrP by anionic POPG liposomes. The KSV values indicated that the tryptophan residues from FL_PrP-MA were more accessible to the polar quencher acrylamide than those from FL_PrP without a membrane anchor. These results suggest that the contact/insertion of N-terminal tryptophan residues of FL_PrP without a membrane anchor into the POPG bilayer is stronger than that of FL_PrP-MA. Overall the C-terminal membrane anchor influences the binding of the N-terminal region of PrP variants to POPG liposomes. Having analyzed the impact of the membrane anchor on protein conformation and flexibility, we also wanted to investigate the effect of PrP variants on membrane integrity.
Calcein Release from Liposomes
To investigate whether the binding of prion proteins can affect the stability of lipid membranes, a well established method based on the release of calcein from liposomes was applied. The amounts of calcein released from DOPC liposomes upon interaction with PrPs with and without a membrane anchor showed no significant difference compared with background controls (BG) in the absence of any PrP (Fig. 7A). This result suggested minimal binding of PrPs without a membrane anchor to DOPC liposomes, as already deduced from flotation assays. Interestingly the already established interactions of PrPs with a membrane anchor with DOPC liposomes also had no influence on the release of calcein. This result implies that the insertion of the C-terminal membrane anchor into liposomes has no destabilizing effect per se.
FIGURE 7.

Calein release assays. The influence of PrP-lipid interaction on the integrity of lipid membranes was analyzed by calcein release assays using concentrations of 100 nm of different PrP variants and 200 μm of calcein-loaded vesicles DOPC (A) and POPG (B) and phospholipid mixtures mimicking neuronal membranes with anionic phospholipids POPG (NM-PG) (C) and POPS (NM-PS) (D). One sample containing only calcein-loaded vesicles was included as the background control (BG).
In contrast to the interaction of PrPs with non-charged DOPC liposomes, the interaction of PrPs with anionic POPG liposomes leads to a distinct release of calcein when compared with background control (Fig. 7B). The binding of PrP variants to POPG liposomes has a destabilizing effect on the membrane and supports the fact that electrostatic interactions are a prerequisite for binding of PrP to lipid membranes. To test more physiologically relevant membranes using the calcein release assay, liposomes consisting of a phospholipid mixture that mimics neuronal membranes were produced. Two kinds of anionic phospholipids (POPG and POPS) were used to analyze whether the difference in the anionic phospholipid component can affect the interaction of PrPs with membranes. As shown in Fig. 7, C and D, no significant difference of calcein release was observed for binding of PrP variants to NM-PG and NM-PS liposomes. One interesting observation is that the total amount of calcein released from liposomes containing PrPs with a membrane anchor is always higher than that from liposomes interacting with PrPs without a membrane anchor. Only in the case of T_PrP is no significant difference observed (Fig. 7). These results suggest that attaching PrP constructs via their C-terminal membrane anchor helps the N-terminal polybasic region of PrP (residues 23–27), which is lacking in the T_PrP construct, to enhance the electrostatic interaction of PrP with anionic phospholipid membranes. Indeed, Wang et al. (33) already determined that this region initiates (electrostatic) interaction between PrP and anionic phospholipids. Moreover, Turnbaugh et al. (36) recently found that this polybasic region dictates the efficiency of prion propagation by binding to PrPSc. A deletion of this region can dramatically reduce susceptibility of prion protein to prion infection and significantly elongate the survival of transgenic (Δ23–31 PrP) mice after scrapie inoculation (36).
The interactions of ΔCR_PrP equipped with a membrane anchor with POPG, NM-PG, and NM-PS liposomes revealed the highest extent of calcein release followed by FL-PrP-MA (Fig. 7, B–D). Such an effect is likely explained by the formation of discrete membrane pores if gross destabilization and rupture of liposomes can be excluded as in this case (see cryo-EM data in Fig. 9B). In previous experiments based on electrophysiological measurements with neuronal cells expressing ΔCR_PrP, Solomon et al. (37, 38) already surmised that ΔCR_PrP might form ion channel-like assemblies.
FIGURE 9.

Electrophysiological and cryo-EM analysis. A, ΔCR_PrP with a membrane anchor induces spontaneous inward currents in HEK293 cells. Representative whole-cell patch clamp recordings are shown. HEK293 cells were clamped from a holding potential of 0 mV to a test potential of −100 mV for 10 s every 30 s. After a control phase, the indicated proteins were added directly to the bath solution. In the presence of FL_PrP-MA (left) and ΔCR_PrP without a membrane anchor (middle), no or only weak pore forming activity was observed, whereas ΔCR_PrP-MA induced large sodium (top right) and calcium (bottom right) inward currents. B, cryo-EM images of POPG vesicles (arrows) mixed with ΔCR_PrP-MA (left) and POPG vesicles alone (right). The scale bar is 100 nm.
NBD Fluorescence Quenching
To further understand the effects of PrP binding and the role of the membrane anchor on the integrity of lipid bilayers, NBD fluorescence quenching was used (Fig. 8A). With this approach we detected how much and how fast the quencher sodium dithionite could penetrate into PrP-loaded vesicles and quench the NBD fluorescence. Fluorescently labeled PE (NBD-PE) with the fluorophore attached to the head group of phosphatidylethanolamine was incorporated into the liposomes at a ratio of labeled to unlabeled lipids of 1 mole %. NBD is localized close to the head group of the lipids and thus is a sensitive probe to detect the interaction of proteins/peptides with the lipid bilayer. In addition, the accessibility of the dithionite quencher to NBD moieties can also be correlated with bilayer perforation or disruption (39, 40). Fig. 8B depicts dithionite-induced fluorescence quenching of NBD-PE incorporated into DOPC (left) and POPG (right) after these liposomes were mixed with different PrP constructs. Fig. 8C shows similar quenching experiments with NM-PG (left) and NM-PS (right) liposomes. The control (SUVs only) is based on incubation of the liposomes with dithionite in the absence of any PrP. In all cases the addition of any PrP variant led to accelerated fluorescence quenching.
FIGURE 8.

Fluorescence quenching assays for PrP variants mixed with different NBD-labeled vesicles. A, explains the principle setup and control experiments with vesicles in the absence of protein. The green trace indicates stable fluorescence during measurements. i, dithionite quenching of NBD lipids on the outside of intact vesicles (blue trace, negative control); ii, dithionite quenching of NBD fluorescence on triton-disrupted vesicles (red trace, positive control). B, results for PrP variants on NBD-DOPC (left) and NBD-POPG vesicles (right). C, results for PrP variants on vesicles mimicking neuronal membranes NBD-NM-PG (left) and NBD-NM-PS (right). Graphs on the left show NBD fluorescence decay recorded for 900 s. After 800 s, NBD fluorescence was averaged and summarized in a bar graph shown on the right. D, ΔCR_PrP-MA causes the influx of the quencher dithionite into NBD-POPG vesicles in a concentration-dependent manner. Concentrations of ΔCR_PrP-MA from 0 to 1.0 μm were mixed with NBD-POPG vesicles (200 μm lipids), and 10 mm dithionite was added (left side). After 800 s (vertical line) the remaining fluorescence was measured. Averages of two independent measurements were plotted (right panel).
However, in the case of PrPs mixed with NBD-DOPC liposomes, no distinguishable difference in NBD fluorescence quenching was recorded for any of the PrP variants (Fig. 8B). This observation indicates that PrP variants with and without a membrane anchor have no significant effect on NBD-DOPC liposomes, similar to the results obtained in our calcein release assays. The interactions of T_PrP and FL_PrP with NBD-POPG liposomes also did not show any significant differences between proteins with and without a membrane anchor (Fig. 8B). Therefore the binding of these variants to NBD-POPG liposomes does not destabilize or permeabilize liposome membranes, even though a clear interaction was demonstrated by floatation and tryptophan quenching assays.
In contrast, the interaction of ΔCR_PrP-MA with NBD-POPG liposomes led to a very significant quenching effect (Fig. 8B, right panel, cyan), which provides additional evidence that ΔCR_PrP-MA induces pores in POPG liposomes. Moreover, ΔCR_PrP without a membrane anchor did not permeabilize NBD-POPG liposomes (Fig. 8B, right panel, blue), indicating that the C-terminal membrane anchor is a crucial factor contributing to pore formation. Fluorescence quenching assays were performed with different concentrations (from 0 to 1 μm) of ΔCR_PrP-MA. The quenching rate has a fast and a slow component, with the fast one corresponding to quenching of fluorophores immediately accessible to the quencher on the outer surface of all liposomes. The slower component represents the average rate of penetration of the quencher through the bilayer, where it quenches fluorophores on the inner leaflet (Fig. 8D). Langner and Hui (41) suggest a single exponential function that combines both components of the quenching rate. Here, the plot of concentrations of ΔCR_PrP-MA versus NBD fluorescence was fitted with an exponential function to clearly demonstrate that the extent of NBD fluorescence quenching increased with the amount of ΔCR_PrP-MA added (Fig. 8D). In an extension of this assay, liposomes mimicking neuronal membranes (NM-PG and NM-PS, with the phospholipid components as described above) including 1 mole % NBD-PE were prepared and mixed with PrPs with and without membrane anchors. Although NM-PG and NM-PS liposomes were made of different anionic phospholipid components (POPG and POPS, respectively), the NBD fluorescence quenching data depicted in Fig. 8C for the interactions of both liposomes with PrPs reveal a close similarity and are in very good agreement with previous results obtained from calcein release assays (Fig. 8A). However, NM-PG and NM-PS liposomes were less stable than POPG liposomes (as seen during the preparation of calcein-loaded liposomes), and thus the fluorescence quenching processes of NM-PG and NM-PS for all samples including controls (Fig. 8C, SUVs only (black)) are much faster and show higher background reaction. In addition, the fluorescence quenching results in Fig. 8C for NBD-NM-PG and NBD-NM-PS liposomes indicated that the C-terminal membrane anchor increased the extent of NBD fluorescence quenching in liposomes mimicking neuronal membranes. This observation most likely reflects a preferred localization of PrPs with membrane anchor at the lipid-water interface, where the NBD probe attached to the hydrophilic head group of PE also localizes. Interestingly, similar to the observation from NBD-POPG liposomes, the interactions of ΔCR_PrP-MA with NBD-NM-PS or NBD-NM-PG liposomes also resulted in the highest extent of NBD fluorescence quenching (Fig. 7D, cyan). Therefore the results from NBD fluorescence quenching strongly support the hypothesis that the membrane-anchored versions of ΔCR_PrP and, to a lesser extent, of FL_PrP induce the formation of pores in liposomes containing anionic phospholipids, as found in our previous assays and in vivo.
Cryo-EM
Cryo-electron microscopy of SUV samples in the presence and absence of ΔCR_PrP-MA was carried out to ensure integrity of POPG liposomes under all conditions used here and to prove that no disruption or visible damage of vesicles occurred during our measurements. Two samples, POPG liposomes only and POPG liposomes plus ΔCR_PrP-MA, were prepared and visualized with a transmission electron microscopy (TEM) system. The resulting images (Fig. 9B) clearly show that ΔCR_PrP-MA bound to POPG liposomes has no visible influence on the integrity of liposomes. Therefore the efflux of calcein and influx of acrylamide or dithionite did not occur due to nonspecific damage or disruption of liposomes by ΔCR_PrP-MA. The formation of pores by ΔCR_PrP-MA is a viable explanation for the observed effects. To further support this hypothesis, we performed patch clamp experiments on whole cells to detect ion flow through such pores.
Lipidated ΔCR_PrP Induces Spontaneous Inward Currents in HEK293 Cells
Whole-cell currents from HEK293 were recorded to determine the pore forming activity of ΔCR_PrP, with and without membrane anchor, and of FL_PrP-MA. As shown in Fig. 9A, neither ΔCR_PrP without a membrane anchor nor FL_PrP-MA induced currents with a monovalent cation (Na+) or a divalent cation (Ca2+). ΔCR_PrP only infrequently generated small currents in the pA range (95 ± 10 pA/pF, n = 3). In contrast, the addition of ΔCR_PrP-MA up to a concentration of 500 nm induced a large spontaneous inward current. ΔCR_PrP-MA-induced pores conducted Na+ (2.1 ± 0.1 nA/pF, n = 5) and Ca2+ (1.9 ± 0.5 nA/pF, n = 5) cations in all tested cells after a wash-in period of 5 to 7 min, similar to the known pore-forming toxin Panton-Valentine leukocidin used as a positive control here (data not shown).
DISCUSSION
The C-terminal Membrane Anchor Strongly Influences the Biochemical and Conformational Characteristics of PrP Variants in Different Membrane Environments
PrP is a GPI-anchored protein that is localized at the outer leaflet of the plasma membrane (11), and its close association with this membrane already suggests that the conversion of PrPC into PrPSc may be influenced by the lipid bilayer as demonstrated previously (14–16). Several studies of the interactions of PrP with phospholipid membranes have been carried out, mostly by using bacterially expressed PrP constructs without any posttranslational modifications (7, 26, 27, 33, 34, 42). We have successfully extended our previously developed expressed protein ligation strategy to generate multimilligram amounts of three different PrP variants. Our previous results demonstrate that homogeneous, folded PrPs equipped with a membrane anchor behave similarly to GPI-modified PrP in vitro (23), and they have allowed us to address several questions about the membrane interaction and conformational changes of PrPs.
As demonstrated above, the presence of a membrane anchor dramatically influences the behavior of PrP when it is “tethered” to membranes, in comparison with free, soluble PrP. In our flotation assays, weak (alkaline-sensitive) binding of non-modified PrPs to anionic POPG liposomes was observed, as described previously by others (26, 27, 33, 43). This interaction nevertheless induces conversion of the typical α-helical structure of PrPs into β-sheet-enriched structures for PrPs without membrane anchor, which agrees well with earlier findings (26, 27, 33, 34, 42). The results from these assays also revealed that PrPs with a membrane anchor bind very strongly to negatively charged liposomes, mainly via their C-terminal anchors. However, PrPs with a membrane anchor did not undergo a similar conformational change into β-sheet-rich structures. T_PrP-MA and ΔCR_PrP-MA still retained a predominantly α-helical structure, and FL_PrP-MA partially converted into a random coil. These findings are summarized in Scheme 1 and provide an overview of the changes in secondary structure measured by CD and the functional consequences discussed in more detail below. In general, we concluded that the membrane anchor modulates the interaction of PrP variants with negatively charged membranes in a way that inhibits conformational changes into β-sheet-enriched structures, thereby slowing PrP aggregation and fibril formation as demonstrated previously (22). Such processes seem to be accelerated in the absence of the membrane anchor, as can be caused by enzymatic GPI anchor removal in vivo. However, we could not exclude the early formation of smaller aggregates.
SCHEME 1.
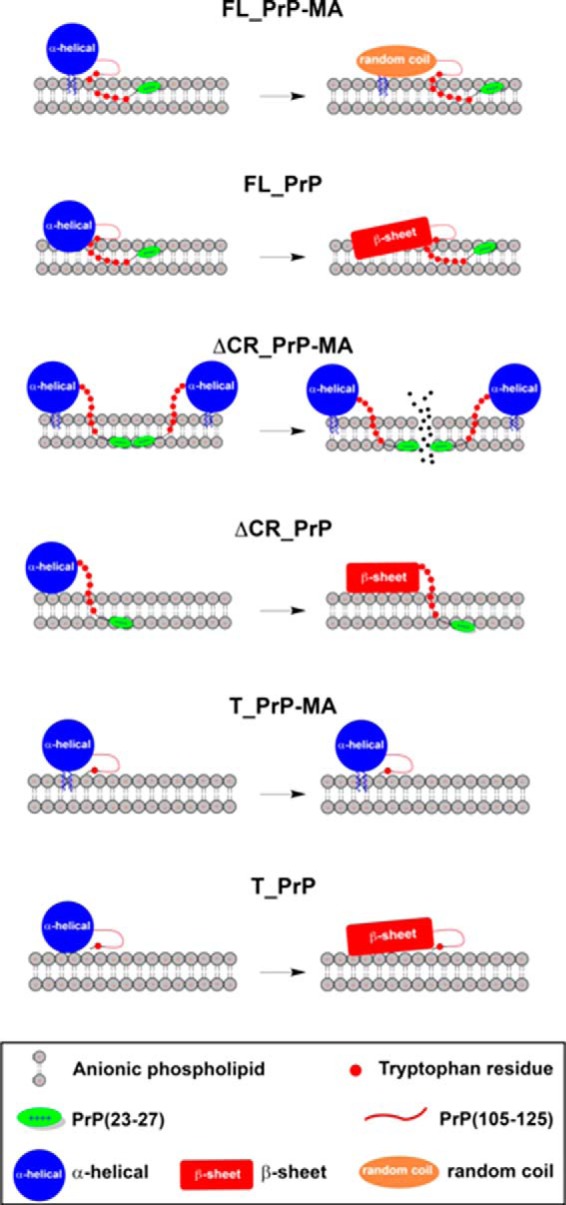
Schematic illustration of how the PrP variants FL_PrP, ΔCR_PrP, and T_PrP with or without an MA may interact with anionic phospholipid membranes. The scheme illustrates a distinct binding mode of predominantly α-helical PrP constructs (large blue circle) carrying a membrane anchor to POPG vesicles when compared with PrP constructs without a membrane anchor. Tryptophan residues (small red circles), the N-terminal polybasic region (residues 23–27, green oval) and central hydrophobic region (residues 105–125, red line) are indicated. The position of the latter region has not been determined by our experiments and is therefore not accurately depicted in this scheme. In addition, the conformational changes of the globular domain of PrP (residues 125–231) from mainly α-helical (large blue circle) to random coil (orange oval) or β-sheet (red rectangle) are highlighted.
The conformational changes in FL_PrP without a membrane anchor in the presence of anionic POPG liposomes were also found to lead to increased PK resistance (26), one of the typical biochemical characteristics of PrPSc. A similar behavior in the presence of POPG liposomes has already been described by Wang et al. (17, 26). However, T_PrP and ΔCR_PrP remained PK-sensitive even though a conversion into β-sheet-rich conformations occurred. An explanation of this apparently inconsistent behavior is based on the missing protein domains in both variants. T_PrP is missing the basic N-terminal region and has much weaker electrostatic interactions with POPG; ΔCR_PrP lacking the core hydrophobic region cannot engage in hydrophobic interactions with the membrane. We suggest that these differences are sufficient to maintain the PK sensitivity of T_PrP and ΔCR_PrP on the time scale of our experiments (up to 12 h). A straightforward interpretation of these findings is that the interactions of both of the latter constructs with anionic POPG are much weaker than that of FL_PrP, which can be protected from PK by its strong association with POPG liposomes. The addition of Triton X-100 to disrupt the liposomes countermands these effects and allows PK to digest FL-PrP as well (data not shown). Tryptophan fluorescence quenching experiments also support this interpretation. FL_PrP-MA, as well as the other two PrP variants with a membrane anchor, bound to POPG vesicles was sensitive to PK, suggesting a different binding mode that keeps the protein fully exposed to PK.
A suitable method for studying the binding of tryptophan-rich domains, as found in the N terminus of PrP, to membranes is by monitoring the changes in intrinsic tryptophan fluorescence and the response to a fluorescence quencher. Our tryptophan fluorescence experiments show that the N-terminal region of PrP (residues 23–99) binds to anionic POPG liposomes but not to non-charged DOPC liposomes, as already observed by others (27). In general this effect does not depend on the presence or absence of a membrane anchor, as a similar effect was found for all PrP variants in interactions with POPG liposomes. Based on this observation a direct interaction of the PrP N-terminal region with phospholipid membranes always occurs, as described for other lipid-anchored proteins such as Src proteins and HIV-1 Gag protein (27, 44). However, tryptophan fluorescence quenching occurred to a different extent for PrPs with a membrane anchor compared with PrPs without a membrane anchor. In the case of the T_PrP and ΔCR_PrP constructs, the comparison of KSV values between PrPs with and without a membrane anchor revealed that the membrane anchor enhances the insertion of tryptophan residues into the bilayer. In contrast, the membrane anchor limits the extent of binding for FL_PrP. Scheme 1 provides a simplified illustration of the different conformational effects of binding of PrP variants with or without a membrane anchor to negatively charged liposomes. Similar conformational effects have been described previously, e.g. for the sensitivity of PrPC to treatment with GPI-specific phospholipase when compared with PrPSc. Whereas PrPC was easily cleaved by GPI-specific phospholipase and removed from the membrane surface, PrPSc was retained on the membrane under similar conditions (45).
Pore Formation by ΔCR_PrP with Membrane Anchor
Spontaneous neurodegenerative illness has been observed in transgenic mice expressing PrP molecules carrying any one of several different internal deletions (46–48). The most severe phenotype is produced by the ΔCR mutation, which consists of deletion of 21 amino acids in the central, primarily hydrophobic region of PrPC (47). Transgenic mice expressing the ΔCR_PrP mutant in the absence of endogenous, wild-type PrP suffered from lethality during the neonatal period. Interestingly, ΔCRPrP was found to contain the full set of posttranslational modifications (N-glycosylations and GPI attachment) typical of wild-type PrPC (49). Expression of ΔCRPrP in transfected cell lines was found to induce spontaneous ionic currents, and the same was true for PrPs harboring several point mutations in the central hydrophobic region that are linked to familial prion diseases in humans (37, 38). These ionic currents can be silenced by co-expression of wild-type PrPC. Relying on biophysical results from cell-based assays Biasini et al. (50, 51) suggested two possible explanations for these ionic currents: either ΔCR_PrP and the familial mutants in the CR influence endogenous ion channel(s), or these PrP constructs themselves form an ion channel or a pore in the cellular membrane.
To follow up on these studies and make use of our unique membrane-anchored PrP variants, we carried out calcein release assays from POPG liposomes. As shown above the largest amount of calcein was released upon binding of ΔCR_PrP-MA to POPG liposomes. Gross liposome disruption by ΔCR_PrP was ruled out by analyzing liposome integrity using cryo-EM. This was necessary because such membrane-disrupting effects have been described for other transmembrane proteins or peptides (52–54). Additional support for the formation of pores within the lipid bilayer was obtained by NBD fluorescence quenching assays. Here binding of ΔCR_PrP-MA to liposomes also induced significant quenching of NBD fluorescence, much higher than observed for all other PrP variants with or without a membrane anchor. Comparative electrophysiological measurements with different cations (Ca2+ and Na+) on HEK cells incubated with ΔCR_PrP-MA were carried out, and strong inward currents were observed. This finding is compelling evidence for pore formation by ΔCR_PrP-MA and may explain the dramatic neurotoxicity that this mutant produces in transgenic mice (47). Based on our results, we hypothesized critical roles for both C-terminal membrane attachment and the N-terminal domain of PrP in pore formation, suggesting that this latter region dips into the lipid bilayer to create a pore (Scheme 1). Consistent with this idea, the N terminus of PrPC contains a positively charged region (residues 23–31) that is reminiscent of “protein transduction domains” capable of penetrating the lipid bilayer and inducing ion channels (55, 56). We have shown previously that these nine amino acids are essential for the neurotoxic and neuroprotective activities of PrP (57, 58). We surmise that the N terminus of membrane-anchored wild-type PrPC also interacts with the lipid bilayer and that this may play an important role in the physiological activity of the protein.
Acknowledgments
We thank Katja Baeuml for help with peptide synthesis, Manuel Brehs for figure design, Aleksandr Kravchuk for assistance with mass spectrometry, and Guenter Resch (Electron Microscopy, Campus Science Support Facilities GmbH, Vienna, Austria) for performing cryo-EM measurements.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 NS065244 (to D. A. H.). This work was also supported by Deutsche Forschungsgemeinschaft (DFG) Grant BE3270/6-1 (to C. F. W. B.).
- PrPc
- cellular prion protein
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DPPC
- 1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- Gdn-HCl
- guanidine hydrochloride
- GPI
- glycosylphosphatidylinositol
- MA
- membrane anchor
- NBD
- 4-chloro-7-nitrobenzofurazan
- NCL
- native chemical ligation
- NM
- neuronal membrane
- PE
- phosphatidylethanolamine
- PK
- proteinase K
- POPG
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- PrP
- prion protein
- PrPSc
- scrapie PrP
- SUV
- small unilammelar vesicle.
REFERENCES
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cobb N. J., Surewicz W. K. (2009) Prion diseases and their biochemical mechanisms. Biochemistry 48, 2574–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caughey B., Baron G. S., Chesebro B., Jeffrey M. (2009) Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 78, 177–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aguzzi A., Heppner F. L. (2000) Pathogenesis of prion diseases: a progress report. Cell Death Differ. 7, 889–902 [DOI] [PubMed] [Google Scholar]
- 5. Linden R., Martins V. R., Prado M. A., Cammarota M., Izquierdo I., Brentani R. R. (2008) Physiology of the prion protein. Physiol. Rev. 88, 673–728 [DOI] [PubMed] [Google Scholar]
- 6. Kim J. I., Surewicz K., Gambetti P., Surewicz W. K. (2009) The role of glycophosphatidylinositol anchor in the amplification of the scrapie isoform of prion protein in vitro. FEBS Lett. 583, 3671–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Critchley P., Kazlauskaite J., Eason R., Pinheiro T. J. (2004) Binding of prion proteins to lipid membranes. Biochem. Biophys. Res. Commun. 313, 559–567 [DOI] [PubMed] [Google Scholar]
- 8. Priola S. A., McNally K. L. (2009) The role of the prion protein membrane anchor in prion infection. Prion 3, 134–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hicks M. R., Gill A. C., Bath I. K., Rullay A. K., Sylvester I. D., Crout D. H., Pinheiro T. J. (2006) Synthesis and structural characterization of a mimetic membrane-anchored prion protein. FEBS J. 273, 1285–1299 [DOI] [PubMed] [Google Scholar]
- 10. Borchelt D. R., Taraboulos A., Prusiner S. B. (1992) Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 267, 16188–16199 [PubMed] [Google Scholar]
- 11. Caughey B., Raymond G. J. (1991) The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 266, 18217–18223 [PubMed] [Google Scholar]
- 12. Gilch S., Winklhofer K. F., Groschup M. H., Nunziante M., Lucassen R., Spielhaupter C., Muranyi W., Riesner D., Tatzelt J., Schätzl H. M. (2001) Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease. EMBO J. 20, 3957–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baron G. S., Wehrly K., Dorward D. W., Chesebro B., Caughey B. (2002) Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J. 21, 1031–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baron G. S., Caughey B. (2003) Effect of glycosylphosphatidylinositol anchor-dependent and -independent prion protein association with model raft membranes on conversion to the protease-resistant isoform. J. Biol. Chem. 278, 14883–14892 [DOI] [PubMed] [Google Scholar]
- 15. Goold R., Rabbanian S., Sutton L., Andre R., Arora P., Moonga J., Clarke A. R., Schiavo G., Jat P., Collinge J., Tabrizi S. J. (2011) Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goold R., McKinnon C., Rabbanian S., Collinge J., Schiavo G., Tabrizi S. J. (2013) Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J. Cell Sci. 126, 3552–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang F., Wang X., Yuan C. G., Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deleault N. R., Harris B. T., Rees J. R., Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deleault N. R., Piro J. R., Walsh D. J., Wang F., Ma J., Geoghegan J. C., Supattapone S. (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc .Natl. Acad. Sci. U.S.A. 109, 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eberl H., Tittmann P., Glockshuber R. (2004) Characterization of recombinant, membrane-attached full-length prion protein. J. Biol. Chem. 279, 25058–25065 [DOI] [PubMed] [Google Scholar]
- 21. Breydo L., Sun Y., Makarava N., Lee C. I., Novitskaia V., Bocharova O., Kao J. P., Baskakov I. V. (2007) Nonpolar substitution at the C terminus of the prion protein, a mimic of the glycosylphosphatidylinositol anchor, partially impairs amyloid fibril formation. Biochemistry (Mosc) 46, 852–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Olschewski D., Seidel R., Miesbauer M., Rambold A. S., Oesterhelt D., Winklhofer K. F., Tatzelt J., Engelhard M., Becker C. F. (2007) Semisynthetic murine prion protein equipped with a GPI anchor mimic incorporates into cellular membranes. Chem. Biol. 14, 994–1006 [DOI] [PubMed] [Google Scholar]
- 23. Becker C. F., Liu X., Olschewski D., Castelli R., Seidel R., Seeberger P. H. (2008) Semisynthesis of a glycosylphosphatidylinositol-anchored prion protein. Angew. Chem. Int. Ed. Engl. 47, 8215–8219 [DOI] [PubMed] [Google Scholar]
- 24. Chu N. K., Becker C. F. (2009) Semisynthesis of membrane-attached prion proteins. Methods Enzymol. 462, 177–193 [DOI] [PubMed] [Google Scholar]
- 25. Mingeot-Leclercq M. P., Lins L., Bensliman M., Van Bambeke F., Van Der Smissen P., Peuvot J., Schanck A., Brasseur R. (2002) Membrane destabilization induced by beta-amyloid peptide 29–42: importance of the amino terminus. Chem. Phys. Lipids 120, 57–74 [DOI] [PubMed] [Google Scholar]
- 26. Wang F., Yang F., Hu Y., Wang X., Wang X., Jin C., Ma J. (2007) Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry 46, 7045–7053 [DOI] [PubMed] [Google Scholar]
- 27. Sanghera N., Pinheiro T. J. (2002) Binding of prion protein to lipid membranes and implications for prion conversion. J. Mol. Biol. 315, 1241–1256 [DOI] [PubMed] [Google Scholar]
- 28. Katz M., Tsubery H., Kolusheva S., Shames A., Fridkin M., Jelinek R. (2003) Lipid binding and membrane penetration of polymyxin B derivatives studied in a biomimetic vesicle system. Biochem. J. 375, 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raja S. M., Rawat S. S., Chattopadhyay A., Lala A. K. (1999) Localization and environment of tryptophans in soluble and membrane-bound states of a pore-forming toxin from Staphylococcus aureus. Biophys. J. 76, 1469–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muir T. W. (2003) Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 72, 249–289 [DOI] [PubMed] [Google Scholar]
- 31. Pan K. M., Baldwin M., Nguyen J., Gasset M., Serban A., Groth D., Mehlhorn I., Huang Z., Fletterick R. J., Cohen F. E., et al. (1993) Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U.S.A. 90, 10962–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hornemann S., Korth C., Oesch B., Riek R., Wider G., Wüthrich K., Glockshuber R. (1997) Recombinant full-length murine prion protein, mPrP(23–231): purification and spectroscopic characterization. FEBS Lett. 413, 277–281 [DOI] [PubMed] [Google Scholar]
- 33. Wang F., Yin S., Wang X., Zha L., Sy M. S., Ma J. (2010) Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry 49, 8169–8176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanghera N., Correia B. E., Correia J. R., Ludwig C., Agarwal S., Nakamura H. K., Kuwata K., Samain E., Gill A. C., Bonev B. B., Pinheiro T. J. (2011) Deciphering the molecular details for the binding of the prion protein to main ganglioside GM1 of neuronal membranes. Chem. Biol. 18, 1422–1431 [DOI] [PubMed] [Google Scholar]
- 35. Eftink M. R. (1991) Fluorescence techniques for studying protein structure. Methods Biochem. Anal. 35, 127–205 [DOI] [PubMed] [Google Scholar]
- 36. Turnbaugh J. A., Unterberger U., Saá P., Massignan T., Fluharty B. R., Bowman F. P., Miller M. B., Supattapone S., Biasini E., Harris D. A. (2012) The N-terminal, polybasic region of PrP(C) dictates the efficiency of prion propagation by binding to PrP(Sc). J. Neurosci. 32, 8817–8830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Solomon I. H., Huettner J. E., Harris D. A. (2010) Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J. Biol. Chem. 285, 26719–26726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Solomon I. H., Khatri N., Biasini E., Massignan T., Huettner J. E., Harris D. A. (2011) An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J. Biol. Chem. 286, 14724–14736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Halevy R., Rozek A., Kolusheva S., Hancock R. E., Jelinek R. (2003) Membrane binding and permeation by indolicidin analogs studied by a biomimetic lipid/polydiacetylene vesicle assay. Peptides 24, 1753–1761 [DOI] [PubMed] [Google Scholar]
- 40. McIntyre J. C., Sleight R. G. (1991) Fluorescence assay for phospholipid membrane asymmetry. Biochemistry 30, 11819–11827 [DOI] [PubMed] [Google Scholar]
- 41. Langner M., Hui S. W. (1993) Dithionite penetration through phospholipid bilayers as a measure of defects in lipid molecular packing. Chem. Phys. Lipids 65, 23–30 [DOI] [PubMed] [Google Scholar]
- 42. Morillas M., Swietnicki W., Gambetti P., Surewicz W. K. (1999) Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem. 274, 36859–36865 [DOI] [PubMed] [Google Scholar]
- 43. Robinson P. J., Pinheiro T. J. (2010) Phospholipid composition of membranes directs prions down alternative aggregation pathways. Biophys. J. 98, 1520–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gerlach H., Laumann V., Martens S., Becker C. F., Goody R. S., Geyer M. (2010) HIV-1 Nef membrane association depends on charge, curvature, composition and sequence. Nat. Chem. Biol. 6, 46–53 [DOI] [PubMed] [Google Scholar]
- 45. Stahl N., Borchelt D. R., Prusiner S. B. (1990) Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry 29, 5405–5412 [DOI] [PubMed] [Google Scholar]
- 46. Baumann F., Tolnay M., Brabeck C., Pahnke J., Kloz U., Niemann H. H., Heikenwalder M., Rülicke T., Bürkle A., Aguzzi A. (2007) Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 26, 538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li A., Christensen H. M., Stewart L. R., Roth K. A., Chiesa R., Harris D. A. (2007) Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 26, 548–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shmerling D., Hegyi I., Fischer M., Blättler T., Brandner S., Götz J., Rülicke T., Flechsig E., Cozzio A., von Mering C., Hangartner C., Aguzzi A., Weissmann C. (1998) Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93, 203–214 [DOI] [PubMed] [Google Scholar]
- 49. Christensen H. M., Harris D. A. (2009) A deleted prion protein that is neurotoxic in vivo is localized normally in cultured cells. J. Neurochem. 108, 44–56 [DOI] [PubMed] [Google Scholar]
- 50. Biasini E., Turnbaugh J. A., Unterberger U., Harris D. A. (2012) Prion protein at the crossroads of physiology and disease. Trends Neurosci. 35, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Biasini E., Unterberger U., Solomon I. H., Massignan T., Senatore A., Bian H., Voigtlaender T., Bowman F. P., Bonetto V., Chiesa R., Luebke J., Toselli P., Harris D. A. (2013) A mutant prion protein sensitizes neurons to glutamate-induced excitotoxicity. J. Neurosci. 33, 2408–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Butko P., Huang F., Pusztai-Carey M., Surewicz W. K. (1996) Membrane permeabilization induced by cytolytic δ-endotoxin CytA from Bacillus thuringiensis var. israelensis. Biochemistry 35, 11355–11360 [DOI] [PubMed] [Google Scholar]
- 53. Garg P., Nemec K. N., Khaled A. R., Tatulian S. A. (2013) Transmembrane pore formation by the carboxyl terminus of Bax protein. Biochim. Biophys. Acta 1828, 732–742 [DOI] [PubMed] [Google Scholar]
- 54. Vogt T. C., Bechinger B. (1999) The interactions of histidine-containing amphipathic helical peptide antibiotics with lipid bilayers: the effects of charges and pH. J. Biol. Chem. 274, 29115–29121 [DOI] [PubMed] [Google Scholar]
- 55. Herce H. D., Garcia A. E. (2007) Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. U.S.A. 104, 20805–20810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wadia J. S., Schaller M., Williamson R. A., Dowdy S. F. (2008) Pathologic prion protein infects cells by lipid-raft dependent macropinocytosis. PLoS One 3, e3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Westergard L., Turnbaugh J. A., Harris D. A. (2011) A nine amino acid domain is essential for mutant prion protein toxicity. J. Neurosci. 31, 14005–14017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Turnbaugh J. A., Westergard L., Unterberger U., Biasini E., Harris D. A. (2011) The N-terminal, polybasic region is critical for prion protein neuroprotective activity. PLoS One 6, e25675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stern O., Volmer M. (1919) On the quenching time of fluorescence. Phys. Z. 20, 183–188 [Google Scholar]