

Background: Mogat1 inhibition improves glucose metabolism in obese mice, but its effects on liver injury are unknown.
Results: Mogat1 knockdown improved hepatic metabolic abnormalities but did not reduce liver inflammation or injury.
Conclusion: Hepatic injury and metabolic abnormalities are separable disease entities.
Significance: Attention to liver injury markers should be made when evaluating metabolic therapies.
Keywords: Diacylglycerol, Fatty Acid Metabolism, Inflammation, Insulin Resistance, Liver, Nonalcoholic Steatohepatitis
Abstract
Abnormalities in hepatic lipid metabolism and insulin action are believed to play a critical role in the etiology of nonalcoholic steatohepatitis. Monoacylglycerol acyltransferase (MGAT) enzymes convert monoacylglycerol to diacylglycerol, which is the penultimate step in one pathway for triacylglycerol synthesis. Hepatic expression of Mogat1, which encodes an MGAT enzyme, is increased in the livers of mice with hepatic steatosis, and knocking down Mogat1 improves glucose metabolism and hepatic insulin signaling, but whether increased MGAT activity plays a role in the etiology of nonalcoholic steatohepatitis is unclear. To examine this issue, mice were placed on a diet containing high levels of trans fatty acids, fructose, and cholesterol (HTF-C diet) or a low fat control diet for 4 weeks. Mice were injected with antisense oligonucleotides (ASOs) to knockdown Mogat1 or a scrambled ASO control for 12 weeks while remaining on diet. The HTF-C diet caused glucose intolerance, hepatic steatosis, and induced hepatic gene expression markers of inflammation, macrophage infiltration, and stellate cell activation. Mogat1 ASO treatment, which suppressed Mogat1 expression in liver and adipose tissue, attenuated weight gain, improved glucose tolerance, improved hepatic insulin signaling, and decreased hepatic triacylglycerol content compared with control ASO-treated mice on HTF-C chow. However, Mogat1 ASO treatment did not reduce hepatic diacylglycerol, cholesterol, or free fatty acid content; improve histologic measures of liver injury; or reduce expression of markers of stellate cell activation, liver inflammation, and injury. In conclusion, inhibition of hepatic Mogat1 in HTF-C diet-fed mice improves hepatic metabolic abnormalities without attenuating liver inflammation and injury.
Introduction
Nonalcoholic fatty liver disease (NAFLD)3 is one of the most common types of chronic liver disease. NAFLD is extremely prevalent in obese individuals, and rates of NAFLD are rising with the increased incidence of obesity. The term “NAFLD” encompasses a spectrum of disorders that ranges from simple steatosis to steatohepatitis (NASH) and cirrhosis, although only a small percentage of patients with NAFLD are classified to have NASH (1). Moreover, it is not clear whether simple steatosis progresses to NASH or whether these are two separate entities. Several genetic, dietary, and environmental factors have been implicated as risk factors for NASH (2–4). In addition, evidence has emerged that hepatic insulin resistance and inflammation, probably caused by accumulation of toxic lipids, may play a role in the development and progression of NASH. However, much remains unclear regarding the pathogenic mechanisms of NASH development, and the dynamic interplay among abnormal intermediary metabolism, insulin resistance, inflammation, and liver injury is unclear.
The hallmark of NAFLD is accumulation of neutral lipids, primarily triacylglycerol (TAG) by mass, in hepatocytes. Mammalian cells synthesize TAG by two convergent pathways: the glycerol-3-phosphate and the monoacylglycerol acyltransferase (MGAT) pathways. MGAT enzymes catalyze the acylation of MAG to DAG, which is the direct precursor of TAG. In higher organisms, a family of three related genes (Mogat1, Mogat2, and Mogat3) encoding enzymes with MGAT activity has been identified, but the mouse Mogat3 is a pseudogene (5). MGATs have been most thoroughly studied in intestinal enterocytes, where they play important roles in mediating dietary fat absorption and chylomicron secretion (6, 7). MGAT activity may also be important for TAG recycling by re-esterifying fatty acids to lipolytic remnants (8, 9).
MGAT activity in human liver is substantial (10), and MGAT expression is strikingly increased in NAFLD (10–13). Previous work using antisense oligonucleotides (ASOs) and RNAi approaches have shown that short term hepatic suppression of Mogat1 led to a significant improvement in hepatic insulin signaling and whole-body glucose homeostasis (12, 13). The improved glucose tolerance after ASO-mediated knockdown was associated with improved insulin signaling in liver but not other tissues and was not associated with enhanced insulin secretion (13). Although both previous studies demonstrated a profound insulin-sensitizing effect, neither study examined markers of liver injury, inflammation, or fibrosis after knockdown of Mogat1.
In this study, we characterized the transcriptional effects of inhibiting Mogat1 in diet-induced obese (DIO) mice. The surprising finding was that Mogat1 knockdown by ASO for 3 weeks actually exacerbated expression of markers of oxidative stress and inflammatory signaling in mice with marked improvements in glucose homeostasis and hepatic insulin signaling. Therefore, we also evaluated the effects of prolonged inhibition of Mogat1 in liver and adipose tissue by ASO injection in a mouse model of NASH provoked by feeding a diet enriched with trans fat, fructose, and cholesterol (14, 15). Suppression of hepatic and adipose tissue Mogat1 attenuated weight gain, reduced hepatic TAG content, and markedly improved glucose tolerance in mice fed this diet. However, Mogat1 inhibition ultimately did not reduce hepatocyte ballooning, NAFLD scoring, or expression of gene markers of inflammation, macrophage infiltration, and stellate cell activation. These data suggest a disconnect between the beneficial metabolic effects of Mogat1 inhibition, hepatic inflammation, and the pathogenesis of NASH in a mouse model. This study also aids in the understanding of the difference between the benign entity of fat accumulation in the liver and hepatic injury, inflammation, and fibrosis.
EXPERIMENTAL PROCEDURES
Animal Study Design
For data shown in Fig. 1, C57BL/6J male mice were fed chow providing 60% of calories from fatty acids (D12492, Research Diets Inc.) starting at 6 weeks of age. Age-matched mice were maintained on a matched 10% fat chow (D12450B, Research Diets Inc.). Mice received intraperitoneal injections of ASO directed against Mogat1 or a scrambled control ASO (25 mg/kg body weight; ISIS Pharmaceuticals, Carlsbad, CA) twice a week for 3 weeks. Treatments were initiated after 14 weeks of high fat diet feeding as described (13). Mice were sacrificed after 3 weeks of injections with ASOs, and tissues were harvested, frozen in liquid nitrogen, and stored at −80° for further analyses.
FIGURE 1.

Hepatic gene expression in DIO mice after Mogat1 inhibition. A, pathway analyses of hepatic gene expression array studies of DIO mice treated with control or Mogat1 ASO. B, hepatic expression of the indicated genes in livers of lean and DIO mice treated with control or Mogat1 ASOs. *, p < 0.05 versus lean controls; **, p < 0.05 versus lean and DIO controls. Error bars, S.E. AU, arbitrary units.
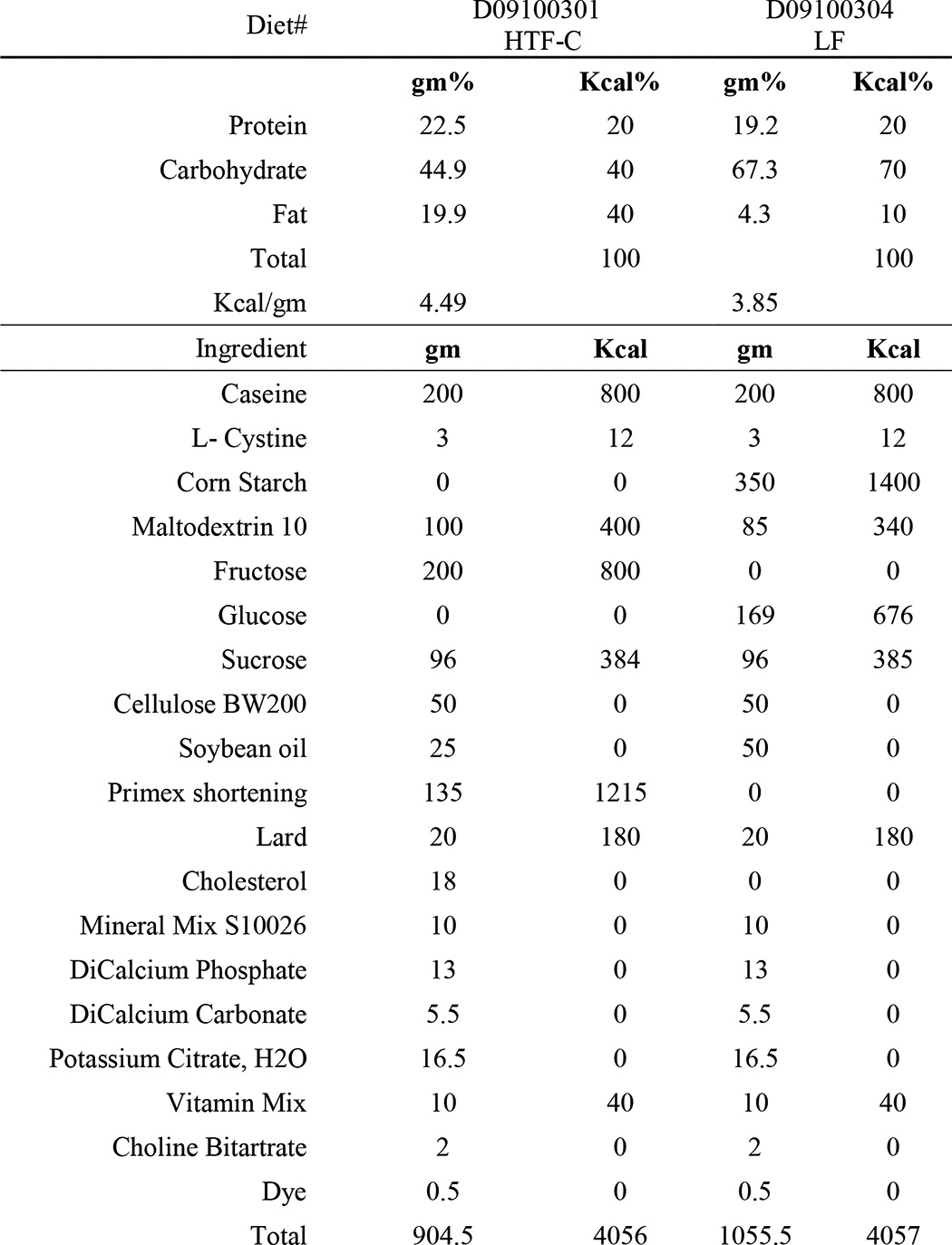
In a second study, 7-week-old C57BL/6J male mice purchased from Jackson Laboratories (Bar Harbor, ME) were placed on a diet enriched with fat (40% kcal, mainly trans fat; trans-oleic and trans-linoleic acids), cholesterol (2% weight), and fructose (22% weight) (HTF-C diet) (D09100301 Research Diets Inc.) that has been described to cause hepatic injury and inflammation (14). Age- and strain-matched mice were fed a matched low fat (10% kcal) control diet that was not supplemented with fructose or cholesterol (LF diet) (D09100304, Research Diets Inc.). Diet compositions can be found in Table 1. After remaining on the designated diets for 4 weeks, the mice received intraperitoneal injections of ASO directed against Mogat1 or scrambled control ASO (25 mg/kg body weight; ISIS Pharmaceuticals, Carlsbad, CA). Injections were given twice a week for 2 weeks and then once a week for 10 weeks. Body weight was checked weekly. Mice were sacrificed, and tissues were harvested at the end of week 16 of the study after a 4-h fast. Liver, gonadal, and subcutaneous fat tissue samples were frozen in liquid nitrogen and stored at −80 °C. Animal studies were approved by the institutional animal use and care committees of Washington University School of Medicine and fulfilled National Institutes of Health requirements for humane care.
TABLE 1.
HTF-C and control LF diet composition
Glucose Tolerance Test
At week 14 of the study, two mice were fasted for 6 h and then injected with a 10% d-glucose solution (1 g/kg). Tail blood glucose was measured at 0, 30, 60, and 120 min after injection using a One-Touch Ultra glucometer (Life Scan, Inc.). Total area under the curve was calculated using the trapezoidal rule.
Hepatocyte Isolation and Metabolic Studies
Primary mouse hepatocytes were isolated and cultured as described previously (16). All metabolic experiments were carried out 4–5 h after the hepatocytes were plated. Palmitate oxidation rates were assessed using [3H]palmitate as described previously (16, 17). De novo lipogenesis was measured by determining the incorporation rate of [14C]acetate in cellular fatty acids following a 2-h incubation period (18). TAG synthesis rates were quantified by using [3H]glycerol in the presence of 0.2 mm BSA-conjugated oleic acid as described previously (19), and TAG turnover was estimated by counting the intracellular [3H]TAG content 14 h after [3H]glycerol pulse.
Western Blot Analyses of Insulin Signaling
After a 4-h attachment period, hepatocytes were incubated in serum-free DMEM for 1 h. Protein extracts were obtained from hepatocytes 5 min after stimulation with 100 nm insulin. Antibodies to Akt, Ser(P)-473 Akt, and Thr(P)-308 Akt (Cell Signaling, Danvers, MA) or tubulin (Sigma) were used according to the manufacturer's instructions for Western blotting analyses.
mRNA Isolation and Quantitative RT-PCR
Total liver mRNA was extracted with RNA Bee (Isotex Diagnostics, Friendswood, TX) based on the manufacturer's instructions. cDNA was synthesized using Vilo reagents. Quantitative real-time PCR was performed with Power SYBR Green and using an ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster City, CA). Arbitrary units of mRNA of the genes of interest were corrected to the level of the 36B4 mRNA quantified by using Δ/ΔCt. Primers were obtained through Integrated DNA Technologies (Coralville, IA). The sequences of primers used in this paper are available upon request.
Microarray Study and Pathway Analysis
Total liver mRNA was used for Illumina Mouse 6 whole microarrays (version 2). Parametric analysis of gene set enrichment was performed as described previously (20, 21). In brief, gene sets were obtained from the Molecular Signatures Database (gene ontology gene sets, C5 collection), and Z scores and p values were calculated for each gene set. A p value of <0.05 was considered significantly changed. All data were analyzed by the R statistical software package (available at the Bioconductor Web site). All microarray data used in this study have been deposited into the NCBI GEO database (accession number GSE60349).
Histology
Harvested liver tissue samples for histology were fixed in 10% formalin for 24 h and then embedded in paraffin. Sections of the samples were stained by hematoxylin and eosin (H&E) and Mason's trichrome stains and assessed by a single pathologist blinded to the groups to which the samples belonged. Specific histological features (steatosis, inflammation, hepatocyte ballooning, and fibrosis) were evaluated and scored according to NAFLD activity score (NAS) (22, 23). Steatosis grading was done on low to medium power evaluation of parenchymal involvement by steatosis (0 for <5%, 1 for 5–33%, 2 for 33–66%, and 3 for more than 66%). Inflammation was graded by overall assessment of all inflammatory foci on a ×200 field (0 for no foci, 1 for <2, 2 for 2–4 foci, and 3 for >4 foci). The ballooning score was 0 for no ballooned cells per field, 1 for few, and 2 for many. Fibrosis was graded 0 for no fibrosis, 1 for perisinusoidal or periportal/portal, 2 for perisinusoidal and periportal/portal, 3 for bridging fibrosis, and 4 for cirrhosis. NAS is the sum of steatosis, inflammation, and ballooning scores.
Plasma Aspartate Aminotransferase (AST) and Alanine Aminotransferase (ALT) Concentrations
Plasma AST and ALT concentrations were measured by using a commercially available colorimetric kinetic assay (Teco Diagnostics, Anaheim, CA) and plasma collected at sacrifice.
Tissue Lipid Measurement
Hepatic TAG content was measured after homogenizing the liver samples in PBS, solubilizing the lipids in the samples by adding 100 μl of 1% sodium deoxycholate to 100 μl of the liver homogenate, and then incubating for 5 min at 37 °C. The TAG content was then determined using an enzymatic assay kit from Thermo Scientific following the manufacturer's instructions. DAG, free cholesterol, and free fatty acid (FFA) content was determined by mass spectrometry as described previously (24–26).
Statistical Analyses
Results are presented as the means ± S.E. Statistical significance was calculated using an unpaired Student's t test, with a statistically significant difference defined as a p value of ≤0.05.
RESULTS
Improvements in Glucose Homeostasis in Diet-induced Obese Mice by Mogat1 Knockdown Are Associated with Enhanced Hepatic Inflammation
We have recently shown that ASO-mediated knockdown of Mogat1 in mice fed a 60% fat diet markedly improves hepatic insulin signaling and systemic glucose metabolism (13). To further characterize the Mogat1 inhibition-induced global transcriptional changes in the liver of these mice, we performed microarray analyses and conducted parametric analysis of gene set enrichment using these gene expression data and gene ontology gene sets. Mogat1 inhibition suppressed several pathways composed of genes encoding proteins involved in fat synthesis, storage, and trafficking (Fig. 1A, blue highlighted pathways). As reported (13), many of these genes are known to be targets of PPARγ (Cfd, Fsp27, and Plin4) and SREBP1 (Cidea and Elovl3) (data not shown). Interestingly, pathway analysis of the regulated genes also suggested that a number of genes encoding markers of increased oxidative stress, chemokines, and inflammatory mediators were increased, rather than reduced, by Mogat1 knockdown (Fig. 1A, red highlighted pathways). For example, glutathione S-transferase α1 (Gsta1) was significantly increased by Mogat1 ASO (Fig. 1B). Serum amyloid A family (Saa2 and Saa3) expression was increased by high fat diet feeding but further increased by Mogat1 knockdown. Finally, expression of a number of chemokines (Ccl2, Ccl5, Ccl7, Cxcl9, and Cxcl10) and proinflammatory cytokines (Il1a and Il1b) were increased or tended to be increased by Mogat1 inhibition. These findings suggest a disconnect between hepatic inflammation and metabolic improvements and suggest that the inflammation that occurs in response to this diet was actually accentuated by Mogat1 inhibition.
Inhibition of Mogat1 Expression in Liver and Adipose Tissue Leads to Decreased Weight Gain and Adiposity
To properly examine the effects of Mogat1 knockdown on hepatic inflammatory correlates of NASH, mice were fed a diet rich in trans fatty acids and fructose with supplemented cholesterol (HTF-C diet). Interestingly, the HTF-C diet was not obesogenic compared with the LF diet because weight gain of mice on LF and HTF-C diets injected with control ASO was not different (Fig. 2A). Mogat1 ASO administration reduced weight gain compared with both the HTF-C and LF control ASO groups at weeks 13, 15, and 16 of the study (Fig. 2A). Decreased adiposity as evidenced by reduced fat pad weight of both the subcutaneous and epididymal fat compartments was noted (Fig. 2B).
FIGURE 2.

Mogat1 ASO treatment improved glucose tolerance. A, Mogat1 inhibition led to decreased weight gain. B, reduced body weight after Mogat1 inhibition is associated with diminished weight of subcutaneous (SQ) and epididymal (EPI) white adipose tissue (WAT) depots. C, results of GTT studies in mice fed an LF or HTF-C diet and treated with control or Mogat1 ASOs. The bar graph at the right represents area under the curve values among the groups. n = 10 mice for all groups. *, p < 0.05 versus LF control and HTF-C Mogat1; **, p < 0.05 versus LF and HTF-C control. D, insulin-stimulated phosphorylation of Akt in hepatocytes isolated from mice treated with LF or HTF-C diet and control or Mogat1 ASO. Error bars, S.E.
Mogat1 Suppression Improved Glucose Tolerance
Glucose tolerance tests done after 10 weeks of ASO administration showed that HTF-C led to a significant worsening of glucose tolerance compared with LF controls in mice injected with control ASO (Fig. 2C), and Mogat1 ASO treatment significantly improved glucose tolerance in HTF-C-fed mice (Fig. 2C). Insulin-stimulated Akt phosphorylation (Ser-473 and Thr-308), which was reduced in hepatocytes from mice fed HTF-C compared with LF diet, was increased by Mogat1 knockdown (Fig. 2D). These data are consistent with our recent work showing that knocking down Mogat1 in livers of mice fed a 60% fat diet markedly improves glucose tolerance and hepatic insulin signaling (13).
Mogat1 Knockdown in Liver and Adipose Tissue Reduced Hepatic TAG Content
Consistent with our previous observations using a 60% fat diet, hepatic expression of Mogat1 was increased in HTF-C diet-fed C57BL/6 mice compared with low fat (LF) diet controls (Fig. 3A). The HTF-C diet also tended to increase Mogat2 expression. Mogat1 ASO administration inhibited hepatic and epididymal adipose tissue Mogat1 mRNA expression. The expression of Mogat2 was not significantly affected by Mogat1 ASO (Fig. 3A). Mogat1 expression in the small intestine was not affected by Mogat1 ASO (Fig. 3A). The HTF-C diet significantly increased liver weight and liver/body ratio compared with LF diet controls in mice treated with control ASO (Fig. 3B). Mogat1 ASO treatment did not affect absolute liver weight or the liver weight/body weight ratio. A 6-fold increase in hepatic TAG content was detected in HTF-C compared with LF mice injected with control ASO (Fig. 3C). Mogat1 ASO administration reduced hepatic TAG content compared with HTF-C control mice, although TAG was still significantly increased compared with LF controls (Fig. 3C). Liver free cholesterol was increased in mice fed the HTF-C diet compared with the LF diet (Fig. 3D). However, there was no difference between the control and Mogat1 ASO groups.
FIGURE 3.
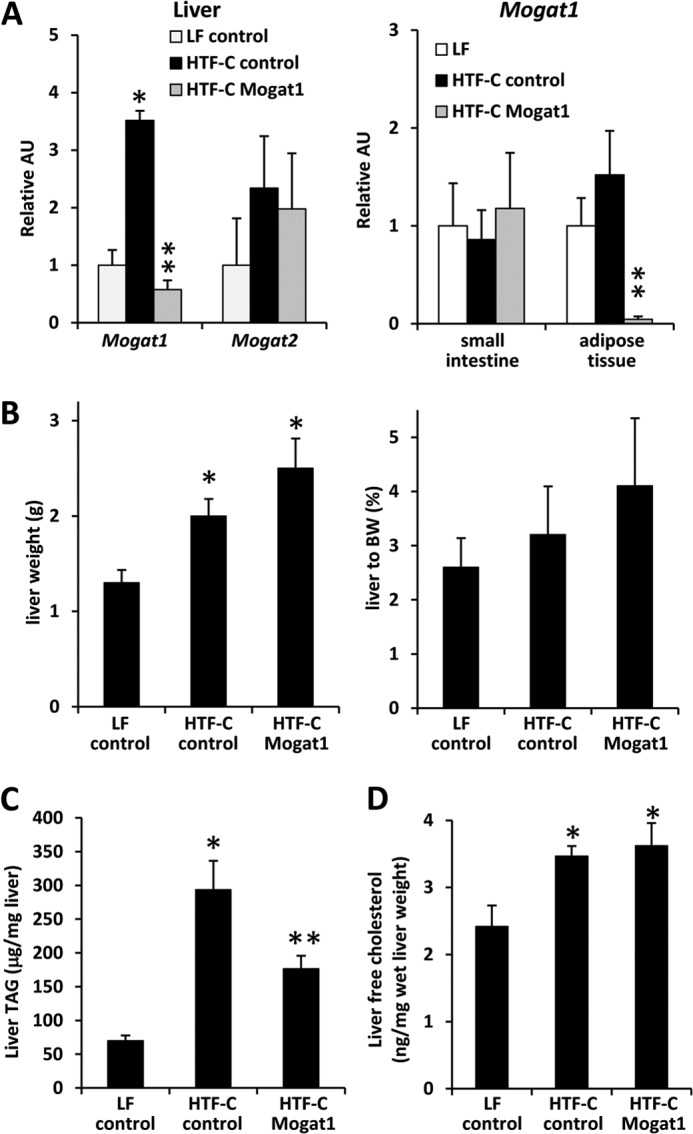
Mogat1 inhibition reduces hepatic TAG but not DAG content. A, expression of Mogat1 and Mogat2 in the indicated tissues of mice fed LF or HTF-C diet and treated with control or Mogat1 ASOs. *, p < 0.05 versus LF controls. **, p < 0.05 versus LF and HTF-C diet controls. AU, arbitrary units. B, liver weight and liver weight/body weight (BW) ratio. *, p < 0.05 versus LF controls. C, liver TAG content. Averages ± S.E. (error bars) for each group are shown. *, p < 0.05 versus LF controls. **, p < 0.05 versus both HTF-C and LF. D, liver free cholesterol content. *, p < 0.05 versus LF controls.
Mogat1 ASO Administration Failed to Reduce Hepatic DAG Content
Interestingly, hepatic content of the product of the reaction catalyzed by MGAT1, DAG, which was increased in the livers of HTF-C control mice compared with LF control mice, tended to be further increased by Mogat1 ASO (Fig. 4A). Indeed, all species of DAG that were increased in HTF-C mice were unchanged by Mogat1 inhibition (Fig. 4B). These included DAG composed of saturated, monounsaturated, and polyunsaturated fatty acids. These findings are consistent with our recent work demonstrating that inhibition of Mogat1 in mice fed a 60% fat diet did not reduce, but actually increased, hepatic DAG content (13). We also found that neither HTF-C diet nor Mogat1 inhibition had an effect on total liver free fatty acid content (Fig. 4C). However, HTF-C diet significantly reduced the hepatic content of 14:0, 16:0, and 22:6 free fatty acids in liver (Fig. 7D), possibly reflecting a paucity of these fatty acid species in the diet. Neither HTF-C feeding nor Mogat1 ASO treatment significantly affected plasma TAG, free fatty acids, or cholesterol concentrations (Table 2).
FIGURE 4.

Mogat1 inhibition does not affect liver DAG or free fatty acid content. A, total liver DAG content. Averages ± S.E. (error bars) for each group are shown. Data shown are the ion intensity of the specified DAG molecular species normalized to the ion intensity of the internal standard 20:0–20:0 DAG/mg of tissue. *, p < 0.05 versus LF. B, liver DAG species by fatty acid chain length and saturation index. C, liver FFA content; D, FFA species. Averages ± S.E. for each group are shown. *, p < 0.05 versus LF.
FIGURE 7.

Gene expression markers for liver injury, inflammation, and NASH. Hepatic expression of inflammatory and cytokine markers (A) and fibrogenic and hepatic stellate cell activation markers (B) in livers of mice fed an LF or HTF-C diet and treated with control or Mogat1 ASOs. *, p < 0.05 versus LF; **, p < 0.05 versus both HTF-C and LF. Error bars, S.E. AU, arbitrary units.
TABLE 2.
Plasma parameters
| Plasma parameter | LF control | HTF-C control | HTF-C Mogat1 |
|---|---|---|---|
| TAG (mg/dl) | 109 ± 14 | 89 ± 10 | 91 ± 4 |
| Cholesterol (mg/dl) | 153 ± 9 | 174 ± 14 | 189 ± 20 |
| FFA (mmol/liter) | 0.6 ± 0.1 | 0.65 ± 0.1 | 0.5 ± 0.06 |
Increased Fatty Acid Oxidation and Reduced Lipogenesis following Mogat1 Knockdown
To evaluate the biochemical mechanism leading to reduced hepatic TAG content, we assessed rates of fatty acid oxidation, de novo lipogenesis, and TAG synthesis in hepatocytes of mice treated with LF or HTF-C diet with or without Mogat1 knockdown. Rates of palmitate oxidation were significantly increased (Fig. 5A), whereas rates of fatty acid synthesis were significantly reduced (Fig. 5B), by Mogat1 ASO treatment. Rates of TAG synthesis were increased in both HTF-C diet-fed groups compared with LF controls, and Mogat1 ASO significantly reduced TAG synthetic rates in HTF-C hepatocytes (Fig. 5C). Mogat1 ASO treatment was also associated with an increase in TG turnover in isolated hepatocytes (Fig. 5C).
FIGURE 5.

Mogat1 ASO increases fatty acid oxidation and suppresses lipogenesis. A, rates of hepatocyte palmitate oxidation. Averages ± S.E. (error bars) for each group are shown. *, p < 0.05 versus LF and HTF-C controls. B, rates of de novo lipogenesis. *, p < 0.05 versus LF controls. **, p < 0.05 versus LF and HTF-C controls. C, rates of triglyceride synthesis and turnover. *, p < 0.05 versus LF controls. **, p < 0.05 versus LF and HTF-C controls. D, expression of genes encoding fatty acid oxidation and lipogenic enzymes. *, p < 0.05 versus controls. AU, arbitrary units.
To determine whether these metabolic changes were associated with altered expression of genes involved in these processes, we measured the expression of genes encoding key enzymes in fatty acid oxidation, de novo lipogenesis, and TAG synthesis. We detected no significant effect of Mogat1 ASO on the hepatic expression of fatty acid oxidation (Cpt1a, Acadvl, and Acadm), lipogenic (Acaca, Fasn), or TAG synthetic enzymes (Lpin1, Lpin2, and Dgat1) that was congruent with the metabolic effects observed in biochemical assays (Fig. 5D). The sole exception to this statement was that the expression of Dgat2, which acylates DAG to form TAG, was down-regulated by Mogat1 ASO. This change correlates with decreased TAG synthesis rates and increased turnover.
Mogat1 ASO Fails to Prevent Liver Injury in Mice on HTF-C Diet
Inspection of histological liver sections from these mice did not grossly suggest any marked improvement in hepatic steatosis, injury, or leukocyte infiltration following Mogat1 inhibition, although HTF-C clearly caused the accumulation of lipid droplets and accumulation of non-parenchymal cells (Fig. 6A). We examined this in a more detailed analysis by having a histopathologist, blinded to treatment group, evaluate the samples and generate a NAS. Mogat1 inhibition did not improve any histological measures of steatohepatitis, including steatosis, hepatocyte ballooning, inflammation, or fibrosis grades in HTF-C-fed mice (Fig. 6B). Overall NAS, which was increased by HTF-C compared with LF in control ASO mice, was also not improved (Fig. 6B). Plasma AST and ALT levels were increased with HTF-C diet feeding and were not affected by Mogat1 ASO treatment (Fig. 6C).
FIGURE 6.

Hepatic Mogat1 inhibition does not result in histological improvement of steatohepatitis. A, H&E- and trichrome-stained liver sections from mice fed an LF or HTF-C diet and treated with control or Mogat1 ASOs. B, graphs depict results of NAS by a histopathologist blinded to treatment group. Data points represent individual mice, and averages (open boxes) for each group are also shown. Steatosis grading was done on low to medium power evaluation of parenchymal involvement by steatosis (0 for <5%, 1 for 5–33%, 2 for 33–66%, and 3 for more than 66%). Inflammation was graded by overall assessment of all inflammatory foci on a ×200 field (0 for no foci, 1 for <2, 2 for 2–4 foci, and 3 for >4 foci). Ballooning score was 0 for no ballooned cells per field, 1 for few, and 2 for many. Fibrosis was graded 0 for no fibrosis, 1 for perisinusoidal or periportal/portal, 2 for perisinusoidal and periportal/portal, 3 for bridging fibrosis, and 4 for cirrhosis. NAS is the sum of steatosis, inflammation, and ballooning scores. C, plasma ALT and AST concentrations are shown. U/L, units/liter. *, p < 0.05 versus LF. Error bars, S.E.
Mogat1 Inhibition Does Not Suppress Hepatic Expression of Gene Markers of Injury, Leukocyte Infiltration, or Stellate Cell Activation
Multiple cytokine and inflammatory markers that are commonly induced in NASH were up-regulated with the HTF-C diet. These included pro-inflammatory cytokines (Tnfa, Il1a, and Il1b), chemokines (Ccl2, Ccl7, Cxcl1, and Cxcl10), and macrophage cell surface markers (Cd68 and Cd11b). There was no change in these markers with Mogat1 ASO treatment, with the exception of Ccl2 and Cxcl10, which were further induced by Mogat1 ASO treatment (Fig. 7A). The expression of Ccl5 and Cxcl9 was also significantly increased by Mogat1 ASO treatment. Although the majority of mice in the HTF-C control group failed to develop fibrosis histologically, the HTF-C diet induced expression of several genes involved in liver fibrogenesis and hepatic stellate cell activation, including Col1a, Col3a, Timp1, Timp3, Mmp2, Fn1, Tgfb, Thbs, and Asma (Fig. 7B). Mogat1 inhibition in the liver did not alter the expression of these genes except that the HTF-C-induced increase in Timp1 expression was significantly exacerbated in the Mogat1 ASO group (Fig. 7B). Collectively, these data suggest that inhibition of Mogat1 in mice fed the HTF-C diet improves glucose tolerance and reduces hepatic TAG content but does not suppress markers of liver inflammation or injury.
DISCUSSION
The accumulation of excessive or toxic species of lipids is believed to lead to hepatocyte injury and death, driving the development of NASH. The expression of the MGAT family of enzymes has recently been shown to be highly activated in the liver of several obese mouse models as well as in obese human subjects (10, 12). Two recent studies have shown that inhibition of Mogat1 in the liver improves hepatic and systemic metabolic indices (12, 13). However, from those studies, it was not clear whether liver inflammation and injury were also improved. Interestingly, although Mogat1 inhibition reduced hepatic TAG content, enhanced fatty acid oxidation, reduced de novo lipogenesis, and markedly improved glucose tolerance in mice fed HTF-C diet, there was no beneficial effect on hepatic content or other lipids linked to liver injury, NASH indices, or gene expression markers for inflammation and fibrosis. Thus, improvements in glucose tolerance were surprisingly dissociated from corrections in DAG, free fatty acid, and cholesterol content as well as hepatic inflammatory end points, which have previously been associated with hepatic metabolic abnormalities. These findings also suggest that improvements in metabolic parametes may be separable from the mechanisms that regulate the development of liver injury.
It is widely believed that reducing intrahepatic lipid content will have protective effects on the development of NASH. There are several examples of knock-out mice lacking key proteins involved in lipid synthesis (27) or storage (28–31) being protected from diet-induced hepatic steatosis. However, most of these studies showing beneficial effects detect a global reduction in intrahepatic lipid species, whereas we see a selective effect on TAG content without affecting other quantified lipids. Moreover, some work has suggested that inhibition of TAG synthesis is not necessarily beneficial toward these end points. For example, injections of ASOs against Dgat2, which catalyzes the step downstream of Mogat1, in mice on a methionine/choline-deficient diet were associated with worse liver fibrosis and apoptosis, despite a reduction in liver TAG content (32). In our study, decreased TAG content in the livers of mice that received Mogat1 ASO on the HTF-C diet was not associated with improvement in inflammation, injury, or markers of fibrosis. TAG is probably not a pathogenic mediator in the development of NASH, and there may actually be a protective role of storing FFA in TAG. Hepatic free fatty acids and intermediates in the TAG synthesis pathway have been implicated in the pathology of NASH (33, 34). Impeding incorporation of FFA into TAG, the preferred and probably well buffered lipid storage form, could cause accumulation of other potentially toxic lipids, leading to hepatocyte injury or apoptosis. In this study, we found no increase in free fatty acid content of the liver with this diet. We also found dissociation between DAG and TAG accumulation, such that DAG levels remained high despite a reduction in hepatic TAG. This DAG may be produced by the other, probably predominant, triglyceride synthesis pathway in an enzymatic step catalyzed by lipin family phosphatidate phosphohydrolases. We have shown previously that the accumulation of DAG in livers of mice with diminished MGAT activity is associated with deactivation of DGAT activity (13). Mogat1 ASO also reduced Dgat2 expression in this model, and we found that TAG turnover was increased, possibly due to reduced re-esterification of lipolytic products by DGAT and MGAT. Congruent with this, Mogat1 knockdown increased rates of palmitate oxidation independent of changes in the expression of oxidative enzymes, suggesting that more of the intrahepatic fat was directed toward an oxidative fate. Finally, rates of de novo lipogenesis were reduced by Mogat knockdown. Whether these metabolic effects are a cause or consequence of the metabolic improvements is not clear, and the specific lipid or classes of lipids that mediate liver injury in this and other models of NASH remain to be clarified.
Typical rodent high fat diets employed to cause obesity and produce hepatic steatosis are usually not associated with development of severe liver inflammation and fibrosis in mice unless they are fed for extremely long durations. In the past few years, attempts have been made to induce liver injury by using a number of permutations of the high fat diet formula (35) and/or by superimposing other insults with the dietary regimen (36–41). Studies utilizing a combination of high fat diets containing high levels of trans fatty acids, often in combination with increased dietary fructose or the addition of cholesterol have enjoyed success at provoking liver injury (15, 42, 43). In the present study, we employed one of these previously used diets (15) and found that hepatocyte ballooning and other histologic indices of NASH were increased by feeding this diet for 16 weeks. Furthermore, several gene expression markers for leukocyte infiltration, inflammation, and stellate cell activation were elevated. However, histologic scoring of liver sections and hydroxyproline assays (data not shown) failed to detect fibrosis in the majority of the mice in this study. Also, compared with LF control diet mice, HTF-C diet-fed mice were not more obese. It is possible that the purified LF diet, which contains supplemented glucose, is very palatable, causing the control mice to overeat because the body weight of the LF control mice was quite high for mice of the C57BL/6 strain at this age. Nonetheless, HTF-C diet causes glucose intolerance, hepatic steatosis, and liver inflammation and injury, despite the lack of effect on body weight.
Although not a perfect model of human NASH, there are a number of aspects to this diet that make it attractive for testing hypotheses regarding the utility of novel therapeutics for NASH. For example, our previous work with inhibition of Mogat1 in mice on a 60% fat diet also showed marked improvements in hepatic insulin signaling and systemic glycemia (13). However, we were unable to assess the effects on liver injury because that diet provokes relatively little inflammation or injury. A methionine/choline-deficient diet is associated with weight loss and increased insulin sensitivity (44, 45) and is not a faithful model for NASH in the setting of overnutrition. Our previous work, with the present study, suggests that inhibiting MGAT activity may be an effective insulin-sensitizing therapy but that this may not reduce clinical measures of NASH. The present data illustrate the need for careful assessment of potential adverse outcomes of MGAT inhibition in what might seem to be a promising therapeutic target for metabolic syndrome.
In conclusion, the current study demonstrated that inhibiting hepatic Mogat1 in a mouse model of NASH reduces weight gain, hepatic TAG content, and glucose tolerance but does not improve inflammation, injury, or markers of NASH. It may be worthwhile to re-examine these findings in other mouse models of NASH and by using other methods to inhibit MGAT activity. Additional work on inhibiting other members of the MGAT family as well as the development of mice with liver-specific knockout of these genes could help shed more light on the role of the MGAT pathway in metabolic abnormalities, simple hepatic steatosis, and NASH.
Acknowledgments
We thank George Schweitzer and Connie Gan for assistance with mouse injections and feeding.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK078187 (to B. N. F.), K01 DK087821 (to A. M. H.), and HL074214 and HL111906 (to D. A. F.) and the core services of the Digestive Diseases Research Core Center (Grant P30 DK52574), Diabetes Research Center (Grant P30 DK020579), and the Nutrition Obesity Research Center (Grant P30 DK56341) at Washington University School of Medicine.
- NAFLD
- nonalcoholic fatty liver disease
- NASH
- steatohepatitis
- DAG
- diacylglycerol
- TAG
- triacylglycerol
- MGAT
- monoacylglycerol acyltransferase
- ASO
- antisense oligonucleotide
- DIO
- diet-induced obese
- NAS
- NAFLD activity score
- AST
- aspartate aminotransferase
- ALT
- alanine aminotransferase
- FFA
- free fatty acid
- LF
- low fat
- HTF-C diet
- diet containing high levels of trans fatty acids, fructose, and cholesterol.
REFERENCES
- 1. Vernon G., Baranova A., Younossi Z. M. (2011) Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 34, 274–285 [DOI] [PubMed] [Google Scholar]
- 2. Fan J. G., Cao H. X. (2013) Role of diet and nutritional management in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 28, 81–87 [DOI] [PubMed] [Google Scholar]
- 3. Sookoian S., Pirola C. J. (2011) Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 53, 1883–1894 [DOI] [PubMed] [Google Scholar]
- 4. Zhu L., Baker S. S., Gill C., Liu W., Alkhouri R., Baker R. D., Gill S. R. (2013) Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57, 601–609 [DOI] [PubMed] [Google Scholar]
- 5. Yue Y. G., Chen Y. Q., Zhang Y., Wang H., Qian Y. W., Arnold J. S., Calley J. N., Li S. D., Perry W. L., 3rd, Zhang H. Y., Konrad R. J., Cao G. (2011) The acyl coenzyme A:monoacylglycerol acyltransferase 3 (MGAT3) gene is a pseudogene in mice but encodes a functional enzyme in rats. Lipids 46, 513–520 [DOI] [PubMed] [Google Scholar]
- 6. Cao J., Cheng L., Shi Y. (2007) Catalytic properties of MGAT3, a putative triacylgycerol synthase. J. Lipid Res. 48, 583–591 [DOI] [PubMed] [Google Scholar]
- 7. Shi Y., Cheng D. (2009) Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am. J. Physiol. Endocrinol. Metab. 297, E10–E18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xia T., Mostafa N., Bhat B. G., Florant G. L., Coleman R. A. (1993) Selective retention of essential fatty acids: the role of hepatic monoacylglycerol acyltransferase. Am. J. Physiol. 265, R414–R419 [DOI] [PubMed] [Google Scholar]
- 9. Bhat B. G., Wang P., Coleman R. A. (1994) Hepatic monoacylglycerol acyltransferase is regulated by sn-1,2-diacylglycerol and by specific lipids in Triton X-100/phospholipid-mixed micelles. J. Biol. Chem. 269, 13172–13178 [PubMed] [Google Scholar]
- 10. Hall A. M., Kou K., Chen Z., Pietka T. A., Kumar M., Korenblat K. M., Lee K., Ahn K., Fabbrini E., Klein S., Goodwin B., Finck B. N. (2012) Evidence for regulated monoacylglycerol acyltransferase expression and activity in human liver. J. Lipid Res. 53, 990–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cortés V. A., Curtis D. E., Sukumaran S., Shao X., Parameswara V., Rashid S., Smith A. R., Ren J., Esser V., Hammer R. E., Agarwal A. K., Horton J. D., Garg A. (2009) Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab. 9, 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee Y. J., Ko E. H., Kim J. E., Kim E., Lee H., Choi H., Yu J. H., Kim H. J., Seong J. K., Kim K. S., Kim J. W. (2012) Nuclear receptor PPARγ-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc. Natl. Acad. Sci. U.S.A. 109, 13656–13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hall A. M., Soufi N., Chambers K. T., Chen Z., Schweitzer G. G., McCommis K. S., Erion D. M., Graham M. J., Su X., Finck B. N. (2014) Abrogating monoacylglycerol acyltransferase activity in liver improves glucose tolerance and hepatic insulin signaling in obese mice. Diabetes 63, 2284–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clapper J. R., Hendricks M. D., Gu G., Wittmer C., Dolman C. S., Herich J., Athanacio J., Villescaz C., Ghosh S. S., Heilig J. S., Lowe C., Roth J. D. (2013) Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G483–G495 [DOI] [PubMed] [Google Scholar]
- 15. Trevaskis J. L., Griffin P. S., Wittmer C., Neuschwander-Tetri B. A., Brunt E. M., Dolman C. S., Erickson M. R., Napora J., Parkes D. G., Roth J. D. (2012) Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G762–G772 [DOI] [PubMed] [Google Scholar]
- 16. Chen Z., Gropler M. C., Norris J., Lawrence J. C., Jr., Harris T. E., Finck B. N. (2008) Alterations in hepatic metabolism in fld mice reveal a role for lipin 1 in regulating VLDL-triacylglyceride secretion. Arterioscler. Thromb. Vasc. Biol. 28, 1738–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burgess S. C., Leone T. C., Wende A. R., Croce M. A., Chen Z., Sherry A. D., Malloy C. R., Finck B. N. (2006) Diminished hepatic gluconeogenesis via defects in tricarboxylic acid cycle flux in peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α)-deficient mice. J. Biol. Chem. 281, 19000–19008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin X., Schonfeld G., Yue P., Chen Z. (2002) Hepatic fatty acid synthesis is suppressed in mice with fatty livers due to targeted apolipoprotein B38.9 mutation. Arterioscler. Thromb. Vasc. Biol. 22, 476–482 [DOI] [PubMed] [Google Scholar]
- 19. Chen Z., Norris J. Y., Finck B. N. (2010) Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) stimulates VLDL assembly through activation of cell death-inducing DFFA-like effector B (CideB). J. Biol. Chem. 285, 25996–26004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim S. Y., Volsky D. J. (2005) PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics 6, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshino J., Mills K. F., Yoon M. J., Imai S. (2011) Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14, 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hjelkrem M., Stauch C., Shaw J., Harrison S. A. (2011) Validation of the non-alcoholic fatty liver disease activity score. Aliment. Pharmacol. Ther. 34, 214–218 [DOI] [PubMed] [Google Scholar]
- 23. Kleiner D. E., Brunt E. M., Van Natta M., Behling C., Contos M. J., Cummings O. W., Ferrell L. D., Liu Y. C., Torbenson M. S., Unalp-Arida A., Yeh M., McCullough A. J., Sanyal A. J., and Nonalcoholic Steatohepatitis Clinical Research Network (2005) Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 [DOI] [PubMed] [Google Scholar]
- 24. Bowden J. A., Albert C. J., Barnaby O. S., Ford D. A. (2011) Analysis of cholesteryl esters and diacylglycerols using lithiated adducts and electrospray ionization-tandem mass spectrometry. Anal. Biochem. 417, 202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brown R. J., Shao F., Baldán A., Albert C. J., Ford D. A. (2013) Cholesterol efflux analyses using stable isotopes and mass spectrometry. Anal. Biochem. 433, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang Y., Kuwano T., Lagor W. R., Albert C. J., Brenton S., Rader D. J., Ford D. A., Brown R. J. (2014) Lipidomic analyses of female mice lacking hepatic lipase and endothelial lipase indicate selective modulation of plasma lipid species. Lipids 49, 505–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hammond L. E., Gallagher P. A., Wang S., Hiller S., Kluckman K. D., Posey-Marcos E. L., Maeda N., Coleman R. A. (2002) Mitochondrial glycerol-3-phosphate acyltransferase-deficient mice have reduced weight and liver triacylglycerol content and altered glycerolipid fatty acid composition. Mol. Cell. Biol. 22, 8204–8214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chang B. H., Li L., Paul A., Taniguchi S., Nannegari V., Heird W. C., Chan L. (2006) Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol. Cell. Biol. 26, 1063–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McManaman J. L., Bales E. S., Orlicky D. J., Jackman M., MacLean P. S., Cain S., Crunk A. E., Mansur A., Graham C. E., Bowman T. A., Greenberg A. S. (2013) Perilipin-2-null mice are protected against diet-induced obesity, adipose inflammation, and fatty liver disease. J. Lipid Res. 54, 1346–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou L., Xu L., Ye J., Li D., Wang W., Li X., Wu L., Wang H., Guan F., Li P. (2012) Cidea promotes hepatic steatosis by sensing dietary fatty acids. Hepatology 56, 95–107 [DOI] [PubMed] [Google Scholar]
- 31. Li J. Z., Ye J., Xue B., Qi J., Zhang J., Zhou Z., Li Q., Wen Z., Li P. (2007) Cideb regulates diet-induced obesity, liver steatosis, and insulin sensitivity by controlling lipogenesis and fatty acid oxidation. Diabetes 56, 2523–2532 [DOI] [PubMed] [Google Scholar]
- 32. Yamaguchi K., Yang L., McCall S., Huang J., Yu X. X., Pandey S. K., Bhanot S., Monia B. P., Li Y. X., Diehl A. M. (2007) Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45, 1366–1374 [DOI] [PubMed] [Google Scholar]
- 33. Anderson N., Borlak J. (2008) Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol. Rev. 60, 311–357 [DOI] [PubMed] [Google Scholar]
- 34. Cheung O., Sanyal A. J. (2008) Abnormalities of lipid metabolism in nonalcoholic fatty liver disease. Semin. Liver Dis. 28, 351–359 [DOI] [PubMed] [Google Scholar]
- 35. Nagarajan P., Mahesh Kumar M. J., Venkatesan R., Majundar S. S., Juyal R. C. (2012) Genetically modified mouse models for the study of nonalcoholic fatty liver disease. World J. Gastroenterol. 18, 1141–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kubota N., Kado S., Kano M., Masuoka N., Nagata Y., Kobayashi T., Miyazaki K., Ishikawa F. (2013) A high-fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non-alcoholic steatohepatitis in mice. Clin. Exp. Pharmacol. Physiol. 40, 422–430 [DOI] [PubMed] [Google Scholar]
- 37. Kita Y., Takamura T., Misu H., Ota T., Kurita S., Takeshita Y., Uno M., Matsuzawa-Nagata N., Kato K., Ando H., Fujimura A., Hayashi K., Kimura T., Ni Y., Otoda T., Miyamoto K., Zen Y., Nakanuma Y., Kaneko S. (2012) Metformin prevents and reverses inflammation in a non-diabetic mouse model of nonalcoholic steatohepatitis. PloS One 7, e43056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fukunishi S., Nishio H., Fukuda A., Takeshita A., Hanafusa T., Higuchi K., Suzuki K. (2009) Development of fibrosis in nonalcoholic steatosis through combination of a synthetic diet rich in disaccharide and low-dose lipopolysaccharides in the livers of Zucker (fa/fa) rats. J. Clin. Biochem. Nutr. 45, 322–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang S. Q., Lin H. Z., Lane M. D., Clemens M., Diehl A. M. (1997) Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. U. S. A. 94, 2557–2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sato W., Horie Y., Kataoka E., Ohshima S., Dohmen T., Iizuka M., Sasaki J., Sasaki T., Hamada K., Kishimoto H., Suzuki A., Watanabe S. (2006) Hepatic gene expression in hepatocyte-specific Pten deficient mice showing steatohepatitis without ethanol challenge. Hepatol. Res. 34, 256–265 [DOI] [PubMed] [Google Scholar]
- 41. Svegliati-Baroni G., Candelaresi C., Saccomanno S., Ferretti G., Bachetti T., Marzioni M., De Minicis S., Nobili L., Salzano R., Omenetti A., Pacetti D., Sigmund S., Benedetti A., Casini A. (2006) A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-α and n-3 polyunsaturated fatty acid treatment on liver injury. Am. J. Pathol. 169, 846–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kohli R., Kirby M., Xanthakos S. A., Softic S., Feldstein A. E., Saxena V., Tang P. H., Miles L., Miles M. V., Balistreri W. F., Woods S. C., Seeley R. J. (2010) High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 52, 934–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matsuzawa N., Takamura T., Kurita S., Misu H., Ota T., Ando H., Yokoyama M., Honda M., Zen Y., Nakanuma Y., Miyamoto K., Kaneko S. (2007) Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 46, 1392–1403 [DOI] [PubMed] [Google Scholar]
- 44. Rinella M. E., Green R. M. (2004) The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 40, 47–51 [DOI] [PubMed] [Google Scholar]
- 45. Rizki G., Arnaboldi L., Gabrielli B., Yan J., Lee G. S., Ng R. K., Turner S. M., Badger T. M., Pitas R. E., Maher J. J. (2006) Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J. Lipid Res. 47, 2280–2290 [DOI] [PubMed] [Google Scholar]