

Abstract
The identification of complement activity in serum and immunohistochemical samples represents a core element of nephropathology. On the basis of this observation, different experimental models and molecular studies have shown the role of this cascade in glomerular disease etiology, but the absence of inhibiting drugs have limited its importance. Since 2006, the availability of target-therapies re-defined this ancient pathway, and its blockage, as the new challenging frontier in renal disease treatment. In the graft, the complement cascade is able to initiate and propagate the damage in ischemia-reperfusion injury, C3 glomerulopathy, acute and chronic rejection, atypical hemolytic uremic syndrome and, probably, in many other conditions. The importance of complement-focused research is revealed by the evidence that eculizumab, the first complement-targeting drug, is now considered a valid option in atypical hemolytic uremic syndrome treatment but it is also under investigation in all the aforementioned conditions. In this review we evaluate the importance of complement cascade in renal transplantation diseases, focusing on available treatments, and we propose a speculative identification of areas where complement inhibition may be a promising strategy.
Keywords: Complement, Eculizumab, Kidney transplantation
Core tip: Complement cascade is involved in different types of renal disease, from glomerulonephritides to pre-eclampsia, and the availability of new drugs, able to inhibit different steps of the cascade, re-defined this ancient pathway, and its blockage both in native and transplanted kidneys, as a new challenging frontier in renal disease treatment. In this review we evaluate the importance of complement cascade in renal transplantation diseases, focusing on available treatments, and we propose a speculative identification of areas where complement inhibition may be a promising strategy.
INTRODUCTION
Complement was initially identified by Ehrlich and Morgenroth in the 19th century as a factor that “complemented” the bactericidal action of immune cells[1]. Muller-Eberhard in 1960 described the cascade and hypothized its involvement in some different diseases[2,3]. Despite the overt demonstration of a complement activation in serum and immunohistochemical samples, the exact role of this cascade remained unclear for a long time.
Recently, the increasing evidence of a key role of complement cascade in many renal diseases, from glomerulonephritides to pre-eclampsia[4-9], and the availability of new drugs able to inhibit different steps of the cascade, re-defined this ancient pathway, and its blockage, as the new challenging frontier in renal disease treatment.
In this review we evaluate the importance of complement cascade in renal transplantation diseases, with an initial general description and a subsequent in-depth evaluation. A separate section is dedicated to treatment, analyzing areas where complement inhibition may represent a promising strategy.
Description of complement cascade
The complement cascade represents a direct defense against bacterial infection and a “scavenger” for immune complexes and apoptotic cells[10].
As summarized in Figure 1, three pathways are involved in complement activation: (1) Classical pathway: C1q binds IgG and IgM, apoptotic cells, or acute-phase proteins promoting cascade progression; (2) Lectin pathways: two trigger proteins (mannose-binding lectin-MBL- and ficolins) activate complement after the adhesion to bacterial surface; and (3) Alternative pathway: in this important and well studied pathway, the first step is represented by the constant and spontaneous hydrolysis of C3.
Figure 1.
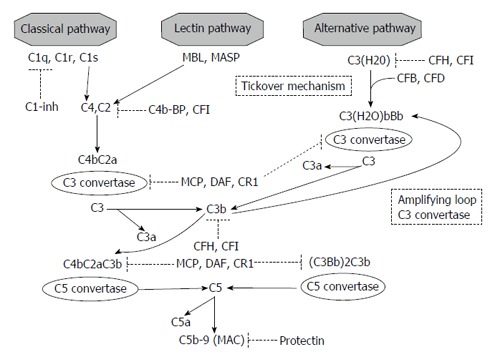
Complement cascade and regulatory proteins. MBL: Mannose binding protein; MASP: MBL-associated serine proteases; CFB: Complement factor B; CFD: Complement factor D; CFH: Complement factor H; CFI: Complement factor I; C1-inh: C1 inhibitor; C4b-BP: C4 binding protein; MCP: Membrane cofactor protein; DAF: Decay accelerating factor; MAC: Membrane attack complex.
In the classical pathway, the conformational change induced by C1q binding causes C1r and C1s activation. These proteases are both able to generate C4b from C4. After C2 cleavage and generation of C4b2a (a C3 covertase), the last step (C3 cleavage), generates the C5 convertase C4b2a3b[11,12].
In the lectin pathways, MBL interaction with one of its two associated serine proteases (MASPs-1 or MASP-2), promotes C3 cleavage[13,14].
Although the activation in previously reported pathways is due to a direct stimulation derived from binding on different epitopes, the alternative pathway is constitutively active, a condition called “tickover mechanism”. In this manner, if necessary in response to an appropriate stimulus, the host enables a rapid and strong activation of the cascade.
As first step, the modification induced by factors B and D on hydrolyzed C3 creates C3(H2O)Bb, a C3 covertase. C3bBb is the result, after C3 cleavage and C3b generation, of another interaction with factors B and D (“amplification loop”). C3bBb is also able to generate C3b and to enhance alternative pathway. The interaction of C3b with C3bBb is the final causative factor for alternative pathway C5 convertase formation (C3bBb3b)[13].
C5a, one of the product of C5 cleavage, is able to combine with final components of the cascade (C6, C7, C8, C9): this final step generates, in all three pathways, the membrane attack complex (MAC)[11,12].
Regulatory mechanism
The cascade regulation has a key role, because an uncontrolled activation, secondary to defects or mutations in regulatory proteins, is referred to be the triggering cause of some complement-mediated diseases (i.e., atypical hemolytic syndrome).
The alternative pathway is the more controlled cascade, as predictable by the evidence of its continuous and spontaneous activation depending on C3 hydrolysis.
The regulatory proteins are divided in two types: fluid-phase and membrane-bound molecules (Figure 1): (1) The fluid-phase regulators are serum proteins. C1 inhibitor is able to arrest classical activation by blocking C1q dissociation, and, in high concentration, can inhibit both the lectin-binding and alternative pathways[11]. Factors H and I act a pivotal role cleaving C3b and consequently regulating the alternative pathway. Factor I is also able to hydrolyze C4b in presence of C4 binding protein; and (2) The second type of regulatory proteins, not pathway specific, are membrane-bound regulators. In most cases, these molecule are anchored to the cell membrane through a glycophosphatidylinositol component. Factor I inhibits complement in association with two of them, membrane cofactor protein (MCP) and CR1; CD35 and complement receptor of immunoglobulin also accelerate C3 and C5 convertase degradation. CD55 enhances C3 convertase spontaneous degradation. Protectin (CD59) directly inhibits MAC formation[11,14].
Effects of complement cascade activation
Three different effectors are activated by complement cascade: (1) Anaphylatoxins (C3a and C5a): stimulators of different inflammatory pathways (tumor necrosis factor-α, upregulation of histamine, cytokine, chemokine, and eicosanoid production)[15]; (2) opsonins (C3b, iC3b, and C3d): these molecules are responsible for the “scavenger” ability of complement by direct binding to the surface of target cells and immune complexes facilitating their elimination[16]; and (3) MAC: C5b-9 generation on cell membrane leads to opsonization, and finally to osmotic lysis of pathogens and damaged cells[17].
COMPLEMENT-MEDIATED DISEASES IN KIDNEY TRANSPLANTATION
In this section, we summarized renal transplant diseases where, at the present time, complement activation shows its involvement. In every paragraph we enunciate the effect of complement inhibition in the mitigation of the damage, if present; a separate section is dedicated to available treatments and future perspectives.
Ischemia-reperfusion injury
An important limitation in long-term outcomes of cadaveric transplants is the inevitable ischemia-reperfusion injury. In the past decade, the use of preservation fluids and machine-reperfusion instruments have expanded the availability of organs reducing delayed graft function, but did not abrogate this kind of damage. The ischemia-reperfusion injury remains, for the same age and risk factors, the corner-stone which differentiate living from deceased donors.
As known, the reperfusion of the graft activates some different processes, such as release of reactive oxygen species, alteration of endothelial permeability and gene transcription. All these conditions ended on an inflammatory infiltrate and kidney injury[18,19].
The complement cascade is activated in the ischemia-reperfusion mediated damage. The alternative pathway seems to explain the major role, as suggested by the absence of antibody deposition in kidney biopsy after reperfusion in animal models[20]. As a further confirmation, C3 and CFB deficient mice are resistant to ischemia-reperfusion injury, despite C4 knockout mice suffered from this damage[21]. C3 expression after reperfusion, probably related to methylation of C3 gene promoter as observed after cold ischemia[22], is associated with a diminished graft survival[23].
Recent data have shown that a kidney from a mice with C3 deficiency is not affected from ischemia reperfusion injury after transplantation in a normal mice, and per contrast, the inverted transplant combination is associated with damage, suggesting the idea that the C3 production in the kidney, and not circulating C3, can be considered[24].
In a swine model, the presence of MASP and MBL deposits after ischemia-reperfusion suggests also an activation of lectine pathway[25] and a more complex overall picture.
In humans, increased serum C5a and C5aR expression in kidney biopsy are both found in brain-dead donors[26], and also a transient MAC-release is demonstrated after reperfusion[27].
The blockage of complement cascade shows a protective effect: small interfering RNA (siRNA) against C5a receptor, C5a antagonists and C1q inhibitors display a limitation of ischemia-reperfusion injury in different models[21,25].
Atypical hemolytic uremic syndrome
Microangiopathic hemolysis, thrombocytopenia and renal damage characterize atypical hemolytic uremic syndrome (aHUS)[28], a condition with poor prognosis both on native and transplanted kidneys. aHUS has also an important recurrence rate in the graft (from 60% to 100%)[29], and this evidence paves the way to the application of preventive strategies to mitigate/abrogate this damage.
In the past decade, the identification of alternative pathway role in aHUS pathogenesis[30] has completely changed the treatment and management of aHUS. It is now well-known that an overactivation of alternative pathway explains almost all cases of aHUS[29,31].
Patients with mutations on C3 or on regulatory proteins complement factor H (CFH), complement factor I (CFI), complement factor B (CFB) experienced high recurrence rates[31]. Mutations on MCP or on diacylglycerol kinase-ε, on the contrary, exposed patients to a very low rate of recurrence, because the graft expressed normal proteins[31,32].
Patients who had autoantibodies against regulatory protein (i.e., anti-CFH antibodies) are in midstream: surprisingly cases of recurrent aHUS are observed even when the immunosuppressive regimen have successfully reduced the antibody titer[33].
The role of eculizumab in aHUS, both on treatment and prevention of recurrences, is discussed separately. Here we anticipate that all previously adopted strategies (plasma infusion, abrogation or minimization of calcineurine inhibitors (CNI)-for its potential inductive effect in aHUS) failed in treatment and prevention of aHUS[29]; also mammalian target of rapamycin inhibitors, proposed as alternative to CNI, are recognized as an independently associated risk factors for aHUS[34].
C3 glomerulopathies
One of the most important innovation in nephropathology is the introduction of C3 glomerulopathy definition. This entity is intended as a condition in which the immunohistochemical samples show C3 glomerular accumulation with scanty immunoglobulin deposition[35], including deposit dense disease and membranoproliferative type I and type III in which the C3 deposition is isolated or predominant[36].
This pathological reclassification implies a new clinical insight: C3 glomerulopathy is a complement-mediated condition where alternative pathway activation plays, as in aHUS, a pivotal role. Genetic and serological tests revealed CFH, CFI, MCP, C3 and factor B mutations in a low but significant percentage of patients, and positive C3 nephritic factor (an immunoglobulin able to stabilize C3bBb reducing its inactivation)[37]. In most cases, with or without positive genetic/serological tests, elevated levels of soluble C5b-9 are demonstrable, surrounding evidence of a smoldering complement activation[36,37].
The recurrence rate of C3 glomerulopathy on renal transplantation could be approximately estimated on about 60%, as derived from two small case series of Servais et al[37] and Little et al[38] and confirmed in the recent paper of Zand et al[39].
The treatment, or the prevention, of the recurrence is far from being defined. Anti-cellular immunosuppression with rituximab, cyclofosfamide or mycophenolate mofetil with or without plasma therapy are ineffective both in native and transplanted kidneys[40,41]; as observed in aHUS, a post-treatment negativization of C3 nephritic factor is not sufficient to prevent a recurrence in the graft[41]. As for aHUS, the role of eculizumab in C3 glomerulopathies treatment is discussed in a separate section. We anticipate that no clinical trial have already evaluate the potential role of anti-C5 drugs in disease prevention, but some papers suggest promising effects of Eculizumab in recurrence treatment[41,42].
Other glomerulonephritides
The role of complement activation is demonstrated in some different glomerulonephritides.
In animal model of membranous glomerulonephritis, for example, nephrotic-range proteinuria was abrogated after complement deprivation[43]. Recent studies in humans showed a direct correlation between podocyte injury and complement activation[44]. Recently, antibodies against the phospholipase A2 receptors (PLA2R) were correlated with idiopathic membranous nephritis[45]: curiously, the anti-PLA2R antibodies are mainly an IgG4, which is the subtype with a low ability in classical pathway activation.
Complement cascade is also directly involved in systemic lupus erythematous (SLE). Patients with genetic deficiency in classical pathway proteins may develop SLE[46] and complement products excretion (C5a and MAC) increased during disease “flares”[47].
Recently, an complement involvement has been demonstrated in other antibody-mediated nephritis, i.e., ANCA-vasculitis[48], antiphospholipid antibody syndrome[49] and crioglobulinemic disease[50]. As a consequence, complement inhibition is a key point of interest in these conditions.
Renal allograft rejection
Complement exerts an effect in the immune activity, and C5a-C5aR interaction plays the critical role. It has been shown that natural regulatory T cells express both C5aR and C3aR, and a blockage of these receptors in mice induces a tolerance profile in autoimmune condition and organ transplants[51]. It was also reported that anaphylatoxins can stimulate T-cell proliferation and activation[52]. Moreover, an indirect confirmation of the alternative pathway causative role is suggested by the evidence that a murine model with C4 deficiency transplanted with a kidney graft derived from a donor with the same defect experienced a T-cell mediated rejection[53].
The cascade involvement in acute antibody mediated rejection (ABMR) represents an interesting issue, both for therapeutic and diagnostic point-of-views.
Anti-Human Leukocyte Antigen antibodies, especially against donor specific molecules (anti-DSA), are identified as the pivotal cause[54] of ABMR, but, at the same time, a wide spectrum of lesions in presence of positive DSA tests has been reported, ranging from nearly normal histological samples to acute and rapidly destructive rejections[55]. So, the differentiation between “bad” or “not so bad” antibodies acquired a growing interest.
Moreover, some data show that the “bad” activity of antibodies can be also involved in previously considered “chronic” lesions (i.e., transplant glomerulopathy)[56,57] and the application of acute rejection therapeutic approaches in these conditions seems to be able to abrogate or at least mitigate the progression to chronic allograft disfunction[58,59].
We know that antibodies can activate complement cascade both with classical and lectin pathway. This activation leads to C4 cleavage and C4d deposition on peritubular capillaries, which is considered a “footprint” of both acute and chronic ABMR[60].
Recently, patients with C1q-binding antibodies demonstrates a poorer outcome than patients with no-fixing antibodies at 1 and 5 years, with a significant difference in acute rejection rates and C4d positive staining[61]. This evidence definitely paves the way to evaluate the efficacy of complement blockage drugs to prevent or treat antibody-related diseases.
The reduction in C4d deposition with a better long term outcomes in patients with an increased expression of CD55 provides a further confirmation[62].
ANTI-COMPLEMENT STRATEGIES
Eculizumab
Eculizumab (Soliris®, Alexion Pharma) is an anti-C5 fully humanized monoclonal antibody able to abrogate terminal complement activation[63]. Eculizumab completely changed the prognosis and the clinical course of paroxysmal nocturnal hemoglobinuria, a complement-dependent disease[63,64]. Efficacy, well-toleration, absence of major side-effects and the potential usefulness in all condition in which complement is involved increased the interest about this drug worldwide.
An important precaution is to adopt a combined anti-meningococcal vaccination and antibiotic treatment, because MAC-inhibition is associated with a “splenectomy-like” effect (and a consequent high risk of infection) and already available vaccines are unable to cover all meningococcal subgroups, as demonstrated by the development of meningococcal infection in a patient vaccinated before eculizumab treatment[65].
At the present time 50 studies are evaluating the effect of this drug in different subsets: more in detail 10 of these refers to transplant area, 15 to nephrologic diseases.
Regarding to C3 glomerulopathy, McCaughan et al[41] successfully treated a patient with a recurrence of deposit dense disease which had no previous response to rituximab and plasmapheresis despite a normalization in C3 nephritic factor titer; Bomback et al[42] in the first trial, demonstrated a diffuse but not consistent response: best outcomes are obtained in subjects with high levels of serum C5b-9, suggesting the idea that patients with a “burst” of complement are the best candidate to this therapy. In renal transplantation, eculizumab has been successfully adopted in desentitization protocols[66] and, as a rescue treatment, in some cases of ABMR[67,68].
Regulatory agencies in United States and Europe (Food and Drug Administration and European Medicine Agency) approved eculizumab in aHUS treatment on native kidneys after struggle results of some experiences[69,70]. Also in renal transplantation eculizumab is effective in aHUS treatment[71] and recurrence prevention[72,73].
Future perspectives
Some different strategies of complement cascade blockage are part of ongoing trials. One research hypothesis is to silence the expression of C5a receptor. Mice treated with an infusion of siRNA have a prolonged graft survival, with reduced inflammatory response on kidney biopsies[74].
In a murine model of crescentic glomerulonephritis CCX168, a small molecule able to inhibit C5a receptor, abrogates extracapillary proliferation[48]. Inhibition of antibody-mediated complement activation can be also provided by stimulating the natural inhibitory activity, or interfering with other parts of classical pathway.
Berinert® (CSL Bhering) is a concentrate of C1q esterase approved for the treatment of ereditary angioedema[75]: a prospective and randomized trial in sensitized patients [NCT01134510] is investigating its effect in reducing antibody-mediated rejection rate.
Some authors proposed Mirococept (APT070), a derived form of complement receptor 1 able to attenuate myocardial and intestinal ischemia-reperfusion damage in rats[76], as a promising option for the reduction of delayed graft function.
Other molecules (a C3 cyclic peptide inhibitor - POT-4, Alcon-, an anti-C5 antibody-LFG316, Novartis-, and a C5 aptamer-ARC1905, Ophthotech) are under investigation in age-related macular degeneration.
CONCLUSION
In the past few years we acquired an enormous amount of evidences that undoubtedly confirmed the central role of complement cascade in different pathologies and an inhibitor of complement activation, Eculizumab, shows is safety and usefulness, also in renal transplantation. However, the issue is far from settled.
At the present time, the next step is, not any more, if the use of the drug is a successful strategy, but when we have to consider this therapy and, nevertheless, how long. For example, in aHUS, eculizumab prevents the recurrence when used in induction regimen but, as previously reported on native kidneys, the risk of relapses in cases of suspension or prolongation of the treatment over the defined period is intolerable[63]. Moreover, the treatment of the relapse with a readoption of eculizumab fails in some cases[69]. If the drug is started only at the time of the recurrence, the outcomes were strictly time-dependent, with poorer results in patient treated afterwards[62]. Similar data are emerging in the treatment of other conditions, as C3 glomerulopathy[36]. In theory, a suspension is a safe option only in those conditions where complement activation is a transient phenomenon, as in ischemia-reperfusion damage. In antibody-mediated damage the question is still debating.
As known, a limit of the lifelong approach is the cost-per-patient, so the adoption of instruments able to individuate the subset of patients where a minimization/interruption of the drug could be safe (i.e., functional or serological tests of complement activation) is one of the future challenges.
In conclusion, the re-discovery of the key role of complement activation and the availability of new drugs with a direct ability in complement inhibition are modifying our treatment protocols and offer a new potential amelioration in graft and patient survival rates. Long term outcomes and randomized clinical trials for the new indications are desirable, in view of a better therapy individualization.
Footnotes
P- Reviewer: Esposito P, Garcia-Roca R, Martins LSS S- Editor: Song XX L- Editor: A E- Editor: Liu SQ
References
- 1.Ehrlich P, Morgenroth J. Zur Theorie der Lysenwirkung. Berlin Klin Wchsr. 1899;36:6. [Google Scholar]
- 2.Morse JH, Muller-eberhard HJ, Kunkel HG. Anti-nuclear factors and serum complement in systemic lupus erythematosus. Bull N Y Acad Med. 1962;38:641–651. [PMC free article] [PubMed] [Google Scholar]
- 3.Polley MJ, Müller-Eberhard HJ. Chemistry and mechanism of action of complement. Prog Hematol. 1966;5:2–25. [PubMed] [Google Scholar]
- 4.Seelen MA, Trouw LA, Daha MR. Diagnostic and prognostic significance of anti-C1q antibodies in systemic lupus erythematosus. Curr Opin Nephrol Hypertens. 2003;12:619–624. doi: 10.1097/00041552-200311000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Ma H, Sandor DG, Beck LH. The role of complement in membranous nephropathy. Semin Nephrol. 2013;33:531–542. doi: 10.1016/j.semnephrol.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sethi S, Nester CM, Smith RJ. Membranoproliferative glomerulonephritis and C3 glomerulopathy: resolving the confusion. Kidney Int. 2012;81:434–441. doi: 10.1038/ki.2011.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai KN. Pathogenesis of IgA nephropathy. Nat Rev Nephrol. 2012;8:275–283. doi: 10.1038/nrneph.2012.58. [DOI] [PubMed] [Google Scholar]
- 8.Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–1279. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denny KJ, Woodruff TM, Taylor SM, Callaway LK. Complement in pregnancy: a delicate balance. Am J Reprod Immunol. 2013;69:3–11. doi: 10.1111/aji.12000. [DOI] [PubMed] [Google Scholar]
- 10.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 11.Ehrnthaller C, Ignatius A, Gebhard F, Huber-Lang M. New insights of an old defense system: structure, function, and clinical relevance of the complement system. Mol Med. 2011;17:317–329. doi: 10.2119/molmed.2010.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171:715–727. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith RJ, Harris CL, Pickering MC. Dense deposit disease. Mol Immunol. 2011;48:1604–1610. doi: 10.1016/j.molimm.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger SP, Daha MR. Complement in glomerular injury. Semin Immunopathol. 2007;29:375–384. doi: 10.1007/s00281-007-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu ZM, Zhu SM, Qin XJ, Cheng ZD, Liu MY, Zhang HM, Liu DX. Silencing of C5a receptor gene with siRNA for protection from Gram-negative bacterial lipopolysaccharide-induced vascular permeability. Mol Immunol. 2010;47:1325–1333. doi: 10.1016/j.molimm.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 16.He JQ, Wiesmann C, van Lookeren Campagne M. A role of macrophage complement receptor CRIg in immune clearance and inflammation. Mol Immunol. 2008;45:4041–4047. doi: 10.1016/j.molimm.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25:1256–1264. doi: 10.1038/nbt1344. [DOI] [PubMed] [Google Scholar]
- 18.Kosieradzki M, Kuczynska J, Piwowarska J, Wegrowicz-Rebandel I, Kwiatkowski A, Lisik W, Michalak G, Danielewicz R, Paczek L, Rowinski WA. Prognostic significance of free radicals: mediated injury occurring in the kidney donor. Transplantation. 2003;75:1221–1227. doi: 10.1097/01.TP.0000065282.46425.87. [DOI] [PubMed] [Google Scholar]
- 19.Thurman JM, Lucia MS, Ljubanovic D, Holers VM. Acute tubular necrosis is characterized by activation of the alternative pathway of complement. Kidney Int. 2005;67:524–530. doi: 10.1111/j.1523-1755.2005.67109.x. [DOI] [PubMed] [Google Scholar]
- 20.Park P, Haas M, Cunningham PN, Bao L, Alexander JJ, Quigg RJ. Injury in renal ischemia-reperfusion is independent from immunoglobulins and T lymphocytes. Am J Physiol Renal Physiol. 2002;282:F352–F357. doi: 10.1152/ajprenal.00160.2001. [DOI] [PubMed] [Google Scholar]
- 21.Quigg RJ. Complement and the kidney. J Immunol. 2003;171:3319–3324. doi: 10.4049/jimmunol.171.7.3319. [DOI] [PubMed] [Google Scholar]
- 22.Parker MD, Chambers PA, Lodge JP, Pratt JR. Ischemia- reperfusion injury and its influence on the epigenetic modification of the donor kidney genome. Transplantation. 2008;86:1818–1823. doi: 10.1097/TP.0b013e31818fe8f9. [DOI] [PubMed] [Google Scholar]
- 23.Damman J, Nijboer WN, Schuurs TA, Leuvenink HG, Morariu AM, Tullius SG, van Goor H, Ploeg RJ, Seelen MA. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol Dial Transplant. 2011;26:2345–2354. doi: 10.1093/ndt/gfq717. [DOI] [PubMed] [Google Scholar]
- 24.Vieyra MB, Heeger PS. Novel aspects of complement in kidney injury. Kidney Int. 2010;77:495–499. doi: 10.1038/ki.2009.491. [DOI] [PubMed] [Google Scholar]
- 25.Castellano G, Melchiorre R, Loverre A, Ditonno P, Montinaro V, Rossini M, Divella C, Battaglia M, Lucarelli G, Annunziata G, et al. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol. 2010;176:1648–1659. doi: 10.2353/ajpath.2010.090276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Werkhoven MB, Damman J, van Dijk MC, Daha MR, de Jong IJ, Leliveld A, Krikke C, Leuvenink HG, van Goor H, van Son WJ, et al. Complement mediated renal inflammation induced by donor brain death: role of renal C5a-C5aR interaction. Am J Transplant. 2013;13:875–882. doi: 10.1111/ajt.12130. [DOI] [PubMed] [Google Scholar]
- 27.de Vries DK, van der Pol P, van Anken GE, van Gijlswijk DJ, Damman J, Lindeman JH, Reinders ME, Schaapherder AF, Kooten Cv. Acute but transient release of terminal complement complex after reperfusion in clinical kidney transplantation. Transplantation. 2013;95:816–820. doi: 10.1097/TP.0b013e31827e31c9. [DOI] [PubMed] [Google Scholar]
- 28.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–1687. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 29.Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, Chatelet V, Mousson C, Mourad G, Bridoux F, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant. 2013;13:663–675. doi: 10.1111/ajt.12077. [DOI] [PubMed] [Google Scholar]
- 30.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8:622–633. doi: 10.1038/nrneph.2012.195. [DOI] [PubMed] [Google Scholar]
- 31.Noris M, Remuzzi G. Thrombotic microangiopathy after kidney transplantation. Am J Transplant. 2010;10:1517–1523. doi: 10.1111/j.1600-6143.2010.03156.x. [DOI] [PubMed] [Google Scholar]
- 32.Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, Fakhouri F, Taque S, Nobili F, Martinez F, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45:531–536. doi: 10.1038/ng.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dragon-Durey MA, Sethi SK, Bagga A, Blanc C, Blouin J, Ranchin B, André JL, Takagi N, Cheong HI, Hari P, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21:2180–2187. doi: 10.1681/ASN.2010030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sartelet H, Toupance O, Lorenzato M, Fadel F, Noel LH, Lagonotte E, Birembaut P, Chanard J, Rieu P. Sirolimus-induced thrombotic microangiopathy is associated with decreased expression of vascular endothelial growth factor in kidneys. Am J Transplant. 2005;5:2441–2447. doi: 10.1111/j.1600-6143.2005.01047.x. [DOI] [PubMed] [Google Scholar]
- 35.Fakhouri F, Frémeaux-Bacchi V, Noël LH, Cook HT, Pickering MC. C3 glomerulopathy: a new classification. Nat Rev Nephrol. 2010;6:494–499. doi: 10.1038/nrneph.2010.85. [DOI] [PubMed] [Google Scholar]
- 36.Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 38.Little MA, Dupont P, Campbell E, Dorman A, Walshe JJ. Severity of primary MPGN, rather than MPGN type, determines renal survival and post-transplantation recurrence risk. Kidney Int. 2006;69:504–511. doi: 10.1038/sj.ki.5000084. [DOI] [PubMed] [Google Scholar]
- 39.Zand L, Lorenz EC, Cosio FG, Fervenza FC, Nasr SH, Gandhi MJ, Smith RJ, Sethi S. Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J Am Soc Nephrol. 2014;25:1110–1117. doi: 10.1681/ASN.2013070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nester CM, Smith RJ. Treatment options for C3 glomerulopathy. Curr Opin Nephrol Hypertens. 2013;22:231–237. doi: 10.1097/MNH.0b013e32835da24c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCaughan JA, O’Rourke DM, Courtney AE. Recurrent dense deposit disease after renal transplantation: an emerging role for complementary therapies. Am J Transplant. 2012;12:1046–1051. doi: 10.1111/j.1600-6143.2011.03923.x. [DOI] [PubMed] [Google Scholar]
- 42.Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, Stokes MB, Markowitz GS, D’Agati VD, Canetta PA, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol. 2012;7:748–756. doi: 10.2215/CJN.12901211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Camussi G, Noble B, Van Liew J, Brentjens JR, Andres G. Pathogenesis of passive Heymann glomerulonephritis: chlorpromazine inhibits antibody-mediated redistribution of cell surface antigens and prevents development of the disease. J Immunol. 1986;136:2127–2135. [PubMed] [Google Scholar]
- 44.Nangaku M, Shankland SJ, Couser WG. Cellular response to injury in membranous nephropathy. J Am Soc Nephrol. 2005;16:1195–1204. doi: 10.1681/ASN.2004121098. [DOI] [PubMed] [Google Scholar]
- 45.Beck LH, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11–21. doi: 10.1056/NEJMoa0810457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pickering MC, Walport MJ. Links between complement abnormalities and systemic lupus erythematosus. Rheumatology (Oxford) 2000;39:133–141. doi: 10.1093/rheumatology/39.2.133. [DOI] [PubMed] [Google Scholar]
- 47.Kusunoki Y, Akutsu Y, Itami N, Tochimaru H, Nagata Y, Takekoshi Y, Sagawa A, Kataoka Y, Nagasawa S. Urinary excretion of terminal complement complexes in glomerular disease. Nephron. 1991;59:27–32. doi: 10.1159/000186513. [DOI] [PubMed] [Google Scholar]
- 48.Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, Seitz LC, Penfold ME, Gan L, Hu P, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol. 2014;25:225–231. doi: 10.1681/ASN.2013020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lonze BE, Zachary AA, Magro CM, Desai NM, Orandi BJ, Dagher NN, Singer AL, Carter-Monroe N, Nazarian SM, Segev DL, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459–465. doi: 10.1111/ajt.12540. [DOI] [PubMed] [Google Scholar]
- 50.Gupta N, Wang ES. Long-term response of refractory primary cold agglutinin disease to eculizumab therapy. Ann Hematol. 2014;93:343–344. doi: 10.1007/s00277-013-1800-7. [DOI] [PubMed] [Google Scholar]
- 51.Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med. 2013;210:257–268. doi: 10.1084/jem.20121525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, Medof ME. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin T, Zhou W, Farrar CA, Hargreaves RE, Sheerin NS, Sacks SH. Deficiency of C4 from donor or recipient mouse fails to prevent renal allograft rejection. Am J Pathol. 2006;168:1241–1248. doi: 10.2353/ajpath.2006.050360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Terasaki PI, Ozawa M. Predicting kidney graft failure by HLA antibodies: a prospective trial. Am J Transplant. 2004;4:438–443. doi: 10.1111/j.1600-6143.2004.00360.x. [DOI] [PubMed] [Google Scholar]
- 55.Colvin RB. Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. J Am Soc Nephrol. 2007;18:1046–1056. doi: 10.1681/ASN.2007010073. [DOI] [PubMed] [Google Scholar]
- 56.Einecke G, Sis B, Reeve J, Mengel M, Campbell PM, Hidalgo LG, Kaplan B, Halloran PF. Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant. 2009;9:2520–2531. doi: 10.1111/j.1600-6143.2009.02799.x. [DOI] [PubMed] [Google Scholar]
- 57.Sis B, Mengel M, Haas M, Colvin RB, Halloran PF, Racusen LC, Solez K, Baldwin WM, Bracamonte ER, Broecker V, et al. Banff ‘09 meeting report: antibody mediated graft deterioration and implementation of Banff working groups. Am J Transplant. 2010;10:464–471. doi: 10.1111/j.1600-6143.2009.02987.x. [DOI] [PubMed] [Google Scholar]
- 58.Billing H, Rieger S, Ovens J, Süsal C, Melk A, Waldherr R, Opelz G, Tönshoff B. Successful treatment of chronic antibody-mediated rejection with IVIG and rituximab in pediatric renal transplant recipients. Transplantation. 2008;86:1214–1221. doi: 10.1097/TP.0b013e3181880b35. [DOI] [PubMed] [Google Scholar]
- 59.Rostaing L, Guilbeau-Frugier C, Fort M, Mekhlati L, Kamar N. Treatment of symptomatic transplant glomerulopathy with rituximab. Transpl Int. 2009;22:906–913. doi: 10.1111/j.1432-2277.2009.00896.x. [DOI] [PubMed] [Google Scholar]
- 60.Feucht HE, Schneeberger H, Hillebrand G, Burkhardt K, Weiss M, Riethmüller G, Land W, Albert E. Capillary deposition of C4d complement fragment and early renal graft loss. Kidney Int. 1993;43:1333–1338. doi: 10.1038/ki.1993.187. [DOI] [PubMed] [Google Scholar]
- 61.Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong van Huyen JP, Mooney N, Suberbielle C, Frémeaux-Bacchi V, Méjean A, Desgrandchamps F, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med. 2013;369:1215–1226. doi: 10.1056/NEJMoa1302506. [DOI] [PubMed] [Google Scholar]
- 62.Brodsky SV, Nadasdy GM, Pelletier R, Satoskar A, Birmingham DJ, Hadley GA, Obeidat K, Nadasdy T. Expression of the decay-accelerating factor (CD55) in renal transplants--a possible prediction marker of allograft survival. Transplantation. 2009;88:457–464. doi: 10.1097/TP.0b013e3181b0517d. [DOI] [PubMed] [Google Scholar]
- 63.Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, Röth A, Szer J, Elebute MO, Nakamura R, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355:1233–1243. doi: 10.1056/NEJMoa061648. [DOI] [PubMed] [Google Scholar]
- 64.McKeage K. Eculizumab: a review of its use in paroxysmal nocturnal haemoglobinuria. Drugs. 2011;71:2327–2345. doi: 10.2165/11208300-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 65.Struijk GH, Bouts AH, Rijkers GT, Kuin EA, ten Berge IJ, Bemelman FJ. Meningococcal sepsis complicating eculizumab treatment despite prior vaccination. Am J Transplant. 2013;13:819–820. doi: 10.1111/ajt.12032. [DOI] [PubMed] [Google Scholar]
- 66.Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, Cosio FG, Gandhi MJ, Kremers W, Gloor JM. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. 2011;11:2405–2413. doi: 10.1111/j.1600-6143.2011.03757.x. [DOI] [PubMed] [Google Scholar]
- 67.Kocak B, Arpali E, Demiralp E, Yelken B, Karatas C, Gorcin S, Gorgulu N, Uzunalan M, Turkmen A, Kalayoglu M. Eculizumab for salvage treatment of refractory antibody-mediated rejection in kidney transplant patients: case reports. Transplant Proc. 2013;45:1022–1025. doi: 10.1016/j.transproceed.2013.02.062. [DOI] [PubMed] [Google Scholar]
- 68.Ghirardo G, Benetti E, Poli F, Vidal E, Della Vella M, Cozzi E, Murer L. Plasmapheresis-resistant acute humoral rejection successfully treated with anti-C5 antibody. Pediatr Transplant. 2014;18:E1–E5. doi: 10.1111/petr.12187. [DOI] [PubMed] [Google Scholar]
- 69.Nester CM, Brophy PD. Eculizumab in the treatment of atypical haemolytic uraemic syndrome and other complement-mediated renal diseases. Curr Opin Pediatr. 2013;25:225–231. doi: 10.1097/MOP.0b013e32835df4a3. [DOI] [PubMed] [Google Scholar]
- 70.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–2181. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 71.Zuber J, Le Quintrec M, Krid S, Bertoye C, Gueutin V, Lahoche A, Heyne N, Ardissino G, Chatelet V, Noël LH, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012;12:3337–3354. doi: 10.1111/j.1600-6143.2012.04252.x. [DOI] [PubMed] [Google Scholar]
- 72.Weitz M, Amon O, Bassler D, Koenigsrainer A, Nadalin S. Prophylactic eculizumab prior to kidney transplantation for atypical hemolytic uremic syndrome. Pediatr Nephrol. 2011;26:1325–1329. doi: 10.1007/s00467-011-1879-9. [DOI] [PubMed] [Google Scholar]
- 73.Nester C, Stewart Z, Myers D, Jetton J, Nair R, Reed A, Thomas C, Smith R, Brophy P. Pre-emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2011;6:1488–1494. doi: 10.2215/CJN.10181110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng X, Zhang X, Feng B, Sun H, Suzuki M, Ichim T, Kubo N, Wong A, Min LR, Budohn ME, et al. Gene silencing of complement C5a receptor using siRNA for preventing ischemia/reperfusion injury. Am J Pathol. 2008;173:973–980. doi: 10.2353/ajpath.2008.080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Craig TJ, Bewtra AK, Bahna SL, Hurewitz D, Schneider LC, Levy RJ, Moy JN, Offenberger J, Jacobson KW, Yang WH, et al. C1 esterase inhibitor concentrate in 1085 Hereditary Angioedema attacks--final results of the I.M.P.A.C.T.2 study. Allergy. 2011;66:1604–1611. doi: 10.1111/j.1398-9995.2011.02702.x. [DOI] [PubMed] [Google Scholar]
- 76.Souza DG, Esser D, Bradford R, Vieira AT, Teixeira MM. APT070 (Mirococept), a membrane-localised complement inhibitor, inhibits inflammatory responses that follow intestinal ischaemia and reperfusion injury. Br J Pharmacol. 2005;145:1027–1034. doi: 10.1038/sj.bjp.0706286. [DOI] [PMC free article] [PubMed] [Google Scholar]