

Highlights
-
•
The structure of C-terminal domain of stomatin operon partner protein PH1510 was determined.
-
•
C-terminal domain of PH1510 (1510-C) forms a five-stranded β-barrel known as an OB-fold.
-
•
1510-C could assemble into multimers based on a dimer as a basic unit.
-
•
1510-C functions as a scaffold protein to form a multimeric assembly with stomatin.
Abbreviations: 1510-C, residues 371–441 of PH1510; 1510-N, residues 16–236 of PH1510; 471-C, residues 72–143 of PH0471; OB-fold, oligosaccharide/oligonucleotide-binding fold; r.m.s.d., root-mean-square deviation; STOPP, stomatin operon partner protein
Keywords: Stomatin operon partner protein, OB-fold, SPFH, Pyrococcus horikoshii, Crystal structure
Abstract
Stomatin, prohibitin, flotillin, and HflK/C (SPFH) domain proteins are found in the lipid raft microdomains of various cellular membranes. Stomatin/STOPP (stomatin operon partner protein) gene pairs are present in both archaeal and bacterial species, and their protein products may be involved in the quality control of membrane proteins. In the present study, the crystal structure of the C-terminal soluble domain of STOPP PH1510 (1510-C) from the hyperthermophilic archaeon Pyrococcus horikoshii was determined at 2.4 Å resolution. The structure of 1510-C had a compact five-stranded β-barrel fold known as an oligosaccharide/oligonucleotide-binding fold (OB-fold). According to crystal packing, 1510-C could assemble into multimers based on a dimer as a basic unit. 1510-C also formed a large cylinder-like structure composed of 24 subunits or a large triangular prism-like structure composed of 12 subunits. These results indicate that 1510-C functions as a scaffold protein to form the multimeric assembly of STOPP and stomatin.
1. Introduction
Stomatin, prohibitin, flotillin, and HflK/C (SPFH) domain proteins are found in the lipid raft microdomains of various cellular membranes [1,2]. More than 1,300 predicted members of annotated SPFH domain proteins have been identified to date [3]. Of these, human stomatin is a major component of vesicles produced by red cells [4]. In a form of hemolytic anemia known as hereditary stomatocytosis, the stomatin protein is deficient in the erythrocyte membrane due to mis-trafficking [5,6]. Stomatin has been shown to interact with and modulate the activity of GLUT-1 [7–9] and acid-sensing ion channels (ASICs) [10,11]. Stomatin is organized into high order homo-oligomeric complexes of approximately 300 kDa, comprising 9- to 12-mers [12], and is localized in detergent-resistant membrane domains [13,14]. Stomatin-like proteins have been found in most species of eukaryotes, bacteria, and archaea [1].
We previously determined the first crystal structure of the core domain of stomatin PH1511 from the hyperthermophilic archaeon Pyrococcus horikoshii. In this structure, the SPFH domain was found to form a stable trimer, while three C-terminal α-helical domains extended from the apexes of the triangle [15]. Another stomatin, PH0470, also has the SPFH domain structure [16]. The crystal structures of the mouse stomatin-domain were shown to assemble into a banana-shaped dimer [17]. An autosomal-recessive disorder, nephritic syndrome type 2, has been specifically associated with mutations in NPHS2, which encodes podocin, and belongs to the stomatin protein family. Podocin accumulates in dimeric or oligomeric forms in lipid raft microdomains at the podocyte slit diaphragm [18]. The disease phenotype has been attributed to the altered heterodimerization and mislocalization of podocin, and the pococin homodimer model has been calculated based on the crystal structure of stomatin from P. horikoshii [15,18].
Stomatin and STOPP (stomatin operon partner protein) genes form an operon in more than 350 archaeal and bacterial genomes [16,19]. STOPP is also known as nfeD (nodulation formation efficiency D). Simple sequence analyses [20] revealed that this family was comprised of long (∼460 aa, long-STOPP) and short (∼145 aa, short-STOPP) forms. Long-STOPP contains an N-terminal serine protease domain and C-terminal STOPP-specific domain [21]. Two sets of STOPP/stomatin gene pairs, PH1510 (long-STOPP)/PH1511 (stomatin) and PH0471 (short-STOPP)/PH0470 (stomatin), have been identified in P. horikoshii. The N-terminal region of long-STOPP PH1510 (residues 16–236, 1510-N) is a serine protease with a catalytic Ser-Lys dyad (Ser97 and Lys138) and specifically cleaves the C-terminal hydrophobic region of the stomatin PH1511 [22]. When 1510-N cleaves a substrate, two degraded products are produced via acyl-enzyme intermediates [23]. The crystal structure of 1510-N [24,25] is known to be similar to those of bacterial ClpP protease [26] and signal peptide peptidase [27]. Surface plasmon resonance (SPR) analysis revealed that two proteins, long-STOPP PH1510 and stomatin PH0470, interacted with each other [24]. No direct interaction between long-STOPP PH1510 and stomatin PH1511 was observed by SPR; however, these two proteins may interact because 1510-N is known to specifically cleave the C-terminal hydrophobic region of PH1511. Links between proteases and other SPFH proteins have also been observed in Escherichia coli. The E. coli SPFH proteins, HflK and HflC, were previously shown to form a complex with FtsH, a protease involved in protein quality control, and regulate FtsH activity [28]. Another E. coli SPFH protein, YbbK, which was renamed QmcA, forms an oligomer and interacts with FtsH [29].
The C-terminal region of long-STOPP PH1510 (residues 371–441, 1510-C) was found to be homologous to the soluble core domain of short-STOPP PH0471 (residues 72–143, 471-C) with 33% sequence identity [21]. The solution structure of 471-C was determined to be a compact five β-strand barrel [30], which was structurally similar to the soluble OB-fold domain of YuaF from Bacillus subtilis (sYuaF) [31]. We recently performed cross-linking and blue native polyacrylamide gel electrophoresis (BN-PAGE) experiments on 1510-C. In these studies, the 1510-C domain formed 12- to 24-mer oligomers [21]. However, the multimeric structure of STOPP has not yet been examined. To understand the multimeric properties and possible molecular function of 1510-C in more detail, we herein described the crystal structure of 1510-C, which indicated the formation of a multimeric assembly.
2. Results and discussion
2.1. Overall structure
The crystal structure of 1510-C, the C-terminal soluble domain of STOPP PH1510, was determined at 2.4 Å resolution by the molecular replacement method (Table 1). The structure contained one 1510-C molecule in an asymmetric unit. The refined model contained residues 373–436 of the PH1510 and 21 solvent molecules. Clear and continuous electron densities were observed for all these residues. Based on the stereochemistry of the protein model evaluated with RAMPAGE [32], no residues were located in the outlier region (Table 1).
Table 1.
Data collection and refinement statistics.
| Data collection | |
| Space group | P6222 |
| Cell dimensions (Å) | a = b = 75.85, c = 43.63 |
| Wavelength (Å) | 0.9800 |
| Resolution range (Å) | 19.57–2.40 (2.53–2.40)a |
| No. of observed reflections | 31,740 |
| No. of unique reflections | 3,165 (443) |
| Rmerge (I)b | 0.077 (0.404) |
| Completeness | 0.997 (1.000) |
| Average I/σ | 25.9 (6.9) |
| Wilson B-factor (Å2) | 27.6 |
| Refinement | |
| Resolution range (Å) | 18.96–2.40 |
| No. of reflections used | 2,865 |
| Rc/Rfreed | 0.221/0.258 |
| No. of non-hydrogen atoms | |
| Protein | 507 |
| Solvent | 21 |
| Average B factors (Å2) | |
| Protein | 31.3 |
| Solvent | 35.7 |
| r.m.s. deviations from ideality | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.316 |
| Ramachandran plote(%) | |
| Favored region | 95.2 |
| Allowed region | 4.8 |
| Outlier region | 0.0 |
Values in parentheses are for the highest-resolution shell.
Rmerge (I) = ΣhklΣj|Ij (hkl) − 〈I (hkl)〉|/ΣhklΣjIj (hkl), where Ij (hkl) is the intensity of an individual reflection and 〈I (hkl)〉 is the mean intensity of that reflection.
R = Σhkl||Fobs| − |Fcalc||/Σhkl|Fobs|, where |Fobs| and |Fcalc| are the observed and calculated structure factor amplitudes, respectively.
Rfree is calculated for 10% of the reflections randomly excluded from the refinement.
Values were calculated with RAMPAGE [32].
1510-C was previously shown to be organized into a compact five-stranded β-barrel fold with a 1–2–3–5–4–1 topology, which is known as an OB-fold domain [33]. Strands β3 and β5 were oriented in a parallel manner, while the other strands were oriented in an antiparallel manner (Fig. 1). These structural features have also been observed in other related proteins, 471-C and sYuaF [30,31]. Ser407, Val421, Leu429, and Val431 are perfectly conserved residues among several archaea and bacteria (Supplementary Fig. 1) [21], and are located inside the β-barrel. As previously described [21], 1510-C does not contain the conserved surface residues necessary for binding to nucleic acids.
Fig. 1.
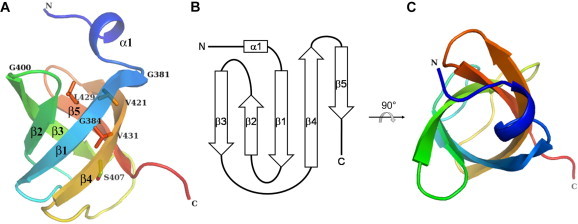
Overall structure of 1510-C. 1510-C is shown as a ribbon model using a rainbow of colors, from purple at the N-terminus to red at the C-terminus. The figures are viewed from the front (A) and top (C). N and C denote the N- and C-termini, respectively. Each secondary structure element is labeled. Perfectly conserved residues among several strains are labeled, and those, except for Gly, are shown in stick models. (B) Topology diagram of 1510-C, where an α-helix is represented as a rectangle and β-strands as arrows. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.2. Comparison of 1510-C with 471-C and sYuaF
Using PDBeFold [34], each one of the solution structures of 471-C [30] and sYuaF [31] was individually superposed onto the crystal structure of 1510-C (Fig. 2). 471-C had a root-mean-square deviation (r.m.s.d.) of 1.69 Å for 57 Cα atoms, while sYuaF had an r.m.s.d. of 1.32 Å for 57 Cα atoms (Supplementary Fig. 2). The five β strands were superposed well, whereas the N-terminus and loop region between the two β strands of 1510-C differed from those of 471-C and sYuaF. The location of Leu390 between β1 and β2 largely differed between 1510-C and 471-C (Supplementary Fig. 2A). The loop between β1 and β2 of 1510-C was closer to the neighboring loop between β3 and β4 than those of 471-C. Due to intra-molecular interactions, residues in the loop between β1 and β2 had low B-factors (Supplementary Fig. 3). Pro392 was also located between β1 and β2, and its location was different between 1510-C and sYuaF (Supplementary Fig. 2B). This residue is involved in an inter-molecular interaction as described later. Gly400 was located between β2 and β3, and Phe409 was located between β3 and β4 (Supplementary Fig. 2). Gly400 and adjacent Arg399 were exposed to the solvent, and, thus, had relatively high B-factors (Supplementary Fig. 3). Phe409 is an insertion residue, and the corresponding residue is absent in 471-C (Supplementary Fig. 1). This residue is partly involved in inter-molecular interactions as described later.
Fig. 2.
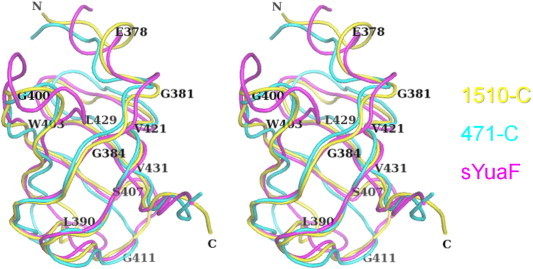
Superposition of 1510-C, 471-C, and sYuaF in a stereo view. Each one of the solution structures of 471-C (PDB: 2EXD) [30] and sYuaF (PDB: 2K14) [31] was individually superposed onto the crystal structure of 1510-C. 1510-C (yellow) and superposed 471-C (cyan) and sYuaF (magenta) are shown as tube models, and several residues of 1510-C were labeled. The view is almost the same as that in Fig. 1A. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.3. Interaction with symmetry-related molecules
The structure contained one 1510-C molecule in an asymmetric unit. However, according to our previous BN-PAGE results, 1510-C (monomeric Mw = 9.2 kDa) showed a broad band of 110–220 kDa, indicating that 1510-C forms 12- to 24-mer oligomers [21]. We previously predicted a hexameric ring model of 1510-C based on the structure of subcomplex I of proteasomal-activating nucleotidase, the monomer of which contained an OB-fold domain [21,35]. However, the multimeric structure of STOPP has not been reported until now.
The interface areas between 1510-C and its symmetry-related molecules were calculated based on the crystal structure of 1510-C (Supplementary Table 1). Five pairs of 1510-C and its symmetry-related molecules showing large interface areas are shown in Fig. 3, and all these pairs were related by crystallographic twofold axes. The largest interface area of 1510-C (interface 1) was 698 Å2, which was 16.3% of the total surface area (4,280 Å2). Perfectly conserved residues Gly381, Val421, and Val431 were included in interface 1. Interface 1 was mainly hydrophobic (Fig. 3): Ile380, Phe409, Val422, and Val432 were involved in hydrophobic interactions (Fig. 4A). Phe409 (84 Å2) and Arg435 (125 Å2) were major contributors to the interface area of 698 Å2. The aromatic side chain of Phe409 was in a van der Waals interaction with the side chain of Glu377. A total of eight water molecules were located at the interface, and were involved in forming a hydrogen-bonding network. The side chains of the charged residues, Lys376, Lys408, Asp423, Glu434, and Arg435, and the main chain atoms of Glu377, Ile380, Gly381, Val422, and Arg435 formed hydrogen bonds via water molecules.
Fig. 3.

Interactions between 1510-C and its symmetry-related molecules. One 1510-C molecule is shown as a surface model, and other symmetry-related molecules in the crystal are shown as ribbon models. Negatively charged residues (Asp and Glu) are colored red and positively charged residues (Lys, Arg, and His) blue in the surface model. Each molecule forming interface 1, 2, 3, 4, and 5 shown in Supplementary Table 1 is colored cyan, magenta, orange, light pink, and green, respectively, and labeled. The molecule in green is located behind the molecule shown as a transparent surface model. The figures are viewed from the front (A) and side (B). One 1510-C molecule shown as a surface model of (A) is viewed from almost the same direction as Fig. 1A. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.

Detailed interactions between 1510-C and its symmetry-related molecules (stereo view). Interactions of interface 1 (A), interface 2 (B), and interface 3 (C) are shown. The views of (A) and (B) are almost the same as that in Fig. 3A, and the view of (C) is almost the same as that in Fig. 3B. The crystallographic twofold axes are shown as black lines with ellipses. One molecule is colored yellow, and the symmetry-related molecules forming interfaces 1, 2, and 3 are shown in the same colors as those in Fig. 3. Residues involved in the interaction are shown as stick models. Water molecules are shown as spheres colored red (one molecule), cyan (symmetry-related molecule forming interface 1), magenta (interface 2), and orange (interface 3). Hydrogen bonds are shown as dashed lines. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The second largest interface area of 1510-C was 337 Å2 (Supplementary Table 1), and the interface mainly consisted of charged residues (Fig. 3), such as Glu414, Lys415, Glu417, Arg433, and Lys436 (Fig. 4B). Two water molecules were hydrogen-bonded to the main-chain atoms of Lys415 and Lys436, and bridged the two molecules. However, no hydrogen bonds were observed other than these water molecules.
The interface area of interface 3 was 316 Å2 (Supplementary Table 1), and the side chain of almost conserved residue, Trp403, was in a stacking interaction with the side chain of Arg373 of symmetry-related molecule-forming interface 3 (Fig. 4C). Arg373 was stabilized by hydrogen bonds to the Glu401 Oε1 and Leu402 O atoms. The side chains of Leu427 that faced each other were in van der Waals interactions.
The interface area of interface 4 was 233 Å2 (Supplementary Table 1), and the interface mainly consisted of hydrophobic residues, Val387, Met395, and Leu402 (Supplementary Fig. 4A). The interface area of interface 5 was 181 Å2 (Supplementary Table 1). Pro392 and Phe409 were involved in hydrophobic interactions (Supplementary Fig. 4B). A sodium ion and water molecules were located at the interface, and the water molecule was hydrogen-bonded to the main-chain O atoms of Ser407 and Asn410. These solvent-mediated hydrogen bonds contributed to stabilizing interface 5.
2.4. A cylinder-like multimeric structure along the crystallographic sixfold helical axis
Given that dimer-forming interface 1 is considered to be a basic unit in the formation of a multimer, 1510-C formed a cylinder-like multimeric structure along the crystallographic sixfold helical axis (Fig. 5 and Supplementary Fig. 5). In Fig. 5A, a dimer (shown in orange and surrounded by a rectangle with broad lines) and another dimer related by interface 4 (a rectangle with dashed lines) were located in a straight line. As the crystal belonged to space group P6222, the interaction related by interface 2 (a rectangle with thin lines) produced a sixfold helical axis. The helical axis corresponded to the crystallographic c axis, which also corresponded to the long axis of rod-shaped crystals, indicating that the proteins were easy to be arranged along with the helical axis. The N-termini of all dimers were located outside the cylinder, while the C-termini of all dimers were located inside the cylinder (Fig. 5B). The distance between the Cα atoms of the N-terminal residue Arg373 of the two molecules located oppositely around the center of the cylinder was 79.4 Å. Furthermore, the distance between the Cα atoms of the C-terminal residue Lys436 of the two molecules located oppositely around the center of the cylinder was 40.7 Å. These values indicated that the outer diameter of the cylinder was approximately 80 Å, while the inner diameter of the cylinder was approximately 40 Å (Fig. 5B). 1510-C formed a 24 mer multimer as a basic unit of the cylinder, and the multimeric unit grew to a long cylinder along the sixfold helical axis. To the best of our knowledge, such a cylinder-like multimeric structure has not been reported in other OB-fold proteins until now.
Fig. 5.

A sixfold helical structure of 1510-C in a stereo view. 1510-C forms a cylinder-like multimeric structure along the crystallographic sixfold helical axis in the crystal belonging to space group P6222. Each basic dimer forming interface 1 shown in Supplementary Table 1 is in the same color, and 14 dimers are shown in different colors. Each molecule is shown as a ribbon and transparent surface model. The N- and C-termini of one dimer are labeled as N and C, respectively. A rectangle with broad lines, thin lines, and dashed lines represent the dimers forming interface 1, interface 2, and interface 4, respectively. The figures are viewed from the front (A) and top (B). The N-termini of all dimers are located outside the cylinder. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.5. A triangular prism-like multimeric structure along the crystallographic threefold helical axis
Another multimer was present given that the dimer-forming interface 1 was a basic unit. 1510-C formed a triangular prism-like multimeric structure along the crystallographic threefold helical axis (Fig. 6 and Supplementary Fig. 5). As shown in Fig. 6A, a dimer (shown in orange and surrounded by a rectangle with black lines) and another dimer related by interface 4 (a rectangle with black dashed lines) were located in a straight line. As the crystal belonged to space group P6222, the interactions related by interface 5 (a rectangle with red dashed lines) and interface 3 (a rectangle with red lines) produced a threefold helical axis. The helical axis corresponded to the c axis, similar to the cylinder-like multimer shown in Fig. 5. The N-termini of all dimers were located around the interface of the two molecules as well as outside the triangular prism. The C-termini of all dimers were located outside the triangular prism (Fig. 6B). The distance between the Cα atoms of the C-terminal residue Lys436 of the two adjacent molecules was 49.6 Å. This value indicated that one side of the triangle was approximately 50 Å (Fig. 6B). No vacant space existed inside the triangular prism. 1510-C formed a 12-mer multimer to form two turns of the triangular prism. To the best of our knowledge, such a triangular prism-like multimeric structure has not yet been reported in other OB-fold proteins until now.
Fig. 6.

A threefold helical structure of 1510-C in a stereo view. 1510-C forms a triangular prism-like multimeric structure along the crystallographic threefold helical axis in the crystal belonging to space group P6222. Each basic dimer forming interface 1 shown in Supplementary Table 1 is in the same color, and 6 dimers are shown in different colors. The N- and C-termini of one dimer are labeled as N and C, respectively. A rectangle with black lines, red lines, black dashed lines, and red dashed lines represent the dimers forming interface 1, interface 3, interface 4, and interface 5, respectively. The figures are viewed from the front (A) and top (B). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.6. A hypothetical model of PH1510
According to the calculation using the SOSUI server [36], long-STOPP PH1510 (441 residues) had five membrane-spanning regions. PH1510 possessed the N-terminal serine protease domain (residues 16–236, 1510-N) and C-terminal OB-fold domain (residues 371–441, 1510-C), as described (Fig. 7A). The crystal structure of 1510-N [25] indicated that the 1510-N dimer bound to one substrate peptide of stomatin PH1511. The 1510-N dimer also exhibited protease activity based on activity staining using casein-copolymerized PAGE [22]. These findings indicated that the dimer of 1510-N may be a functional unit. Therefore, the 1510-C dimer may also be considered as a functional unit. Dimer-forming interface 1 is the most probable candidate for this unit due to having the largest interface area as well as several perfectly conserved residues such as Gly381, Val421, and Val431. The intact PH1510 has not been successfully prepared due to the presence of the membrane-spanning regions [22], and the structure of PH1510 has not been determined. Then, hypothetical models of PH1510 were manually constructed (Fig. 7B and C) based on the two types of multimeric structures of 1510-C described above and the crystal structure of 1510-N [25]. Although the arrangements of the four membrane-spanning regions between 1510-N and 1510-C remain unclear, the 1510-N dimer was placed by considering that these molecules did not clash with each other, and the C-terminus of 1510-N was located in as close proximity to the N-terminus of 1510-C as possible. In both multimeric structures of 1510-C, 1510-N can bind to the outer surface of the 1510-C multimer. In Fig. 7B, the distance between the Cα atoms of the Pro137 of two 1510-N molecules located oppositely around the center of the cylinder was 171.5 Å, indicating that the approximate diameter of the cylinder-like multimeric model was 170 Å. In Fig. 7C, the distance between the Cα atoms of the Lys178 of two 1510-N molecules located oppositely around the center of the triangular prism was 136.7 Å, indicating that the approximate diameter of the triangular prism-like multimeric model was 140 Å. According to these models, 1510-C may be a scaffold domain that forms the multimeric assembly of STOPP.
Fig. 7.

Hypothetical models of PH1510. (A) The domain arrangements and positions of PH1510. Putative membrane-spanning regions are shown as filled triangles with residue numbers. (B and C) Hypothetical models of PH1510 based on the structures of the 1510-C dimer and 1510-N dimer in a stereo view. The structure of the 1510-N dimer (residues 16–236 of PH1510) [25] is manually placed into the cylinder-like multimeric structure (B) or triangular prism-like multimeric structure (C) of 1510-C. The dashed ellipses show one unit of the 1510-N dimer and 1510-C dimer (One monomer is colored orange, while the other is colored magenta). In the other units, the 1510-N dimer and 1510-C dimer are shown as the same colors. N and C denote the N- and C-termini of both 1510-N and 1510-C. The views of (B) and (C) are almost the same as that in Figs. 5 and 6B, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
471-C, the C-terminal OB-fold domain of short-STOPP, also formed 12- to 24-mer oligomers in our previous studies [21]. It remains unclear whether 471-C similarly forms either of the two multimeric structures of 1510-C. However, 471-C may form dimer-forming interface 1 by inter-molecular interactions with conserved residues. Stomatin has also been shown to form high order homo-oligomeric complexes comprising 9- to 12-mers [12], and the SPFH domain of stomatin forms a stable trimer according to the crystal structure of the core domain of stomatin PH1511 from P. horikoshii [15]. The multimeric properties of the C-terminal domain of STOPP are also important, and may be involved in the formation of the multimeric complexes of stomatin and STOPP.
In conclusion, the structure of 1510-C provides an insight into the multimeric assembly of 1510-C by crystal packing. By assuming that the 1510-C dimer is a basic unit, 1510-C forms a large cylinder-like structure composed of 24 subunits, or a large triangular prism-like structure composed of 12 subunits. In our previous study involving cross-linking and BN-PAGE experiments on 1510-C, the 1510-C domain formed 12- to 24-mer oligomers [21]. The multimeric assembly formed by the crystal packing appears to conform to the cross-linking and BN-PAGE results. 1510-C may function as a scaffold protein to form a multimeric assembly of STOPP and stomatin.
3. Materials and methods
3.1. Protein preparation and crystallization
1510-C (residues 371–441 of PH1510) was mostly prepared as described previously [21,22]. In the final stage, the protein was purified with a HiTrap SP cation-exchange column in an AKTAprime plus system (GE Healthcare) using a buffer containing 20 mM MES–NaOH (pH 6.0) and then eluted with a linear gradient from 0 to 1 M NaCl. Fractions containing the target protein were concentrated with a Vivaspin-20 centrifugal concentrator (Sartorius) with a cut-off molecular mass of 5 kDa, and a buffer containing 50 mM Tris–HCl (pH 7.5) and 50 mM NaCl was added to the Vivaspin-20 concentrator. The concentration and addition of the buffer were repeated twice to change the buffer completely.
Crystallization drops were prepared by mixing equal volumes (0.5 μl) of the protein and reservoir solutions. The protein solution contained 5.7 mg/mL 1510-C in a buffer containing 50 mM Tris–HCl (pH 7.5) and 50 mM NaCl. The reservoir solution was the Crystal Screen II (Hampton Research) #31 condition that contained 20% (v/v) Jeffamine M-600 and 0.1 M HEPES–NaOH (pH 7.5). Crystals were grown at 20 °C by the sitting-drop vapor diffusion method. Rod-shaped crystals grew to an approximate size of 0.20 × 0.02 × 0.02 mm.
3.2. Data collection and structure determination
A crystal was transferred into a solution of 20% (v/v) glycerol in the reservoir solution, and flash-frozen at 95 K. X-ray diffraction data were collected at beamline BL17A of the Photon Factory in KEK (Tsukuba, Japan) with a Quantum 315r CCD detector, and were then integrated and scaled with XDS [37] and SCALA [38].
The structure was determined by the molecular replacement method with the program PHENIX [39]. The core region of the solution structure of the C-terminal region of short-STOPP PH0471 (residues 85–143) was used as an initial model (PDB: 2EXD) [30]. After auto MR and auto model building steps with PHENIX, R and Rfree values became 0.28 and 0.34, respectively, although the sequence identity between 1510-C and 471-C was relatively low, 33% [21]. The model was subjected to several cycles of maximum-likelihood refinement with REFMAC5 [40] in the CCP4 suite [41], followed by manual model fitting with COOT [42]. Data collection and refinement statistics are summarized in Table 1.
Interface areas were calculated with Protein interfaces, surfaces and assemblies service PISA at the European Bioinformatics Institute (<http://www.ebi.ac.uk/pdbe/prot_int/pistart.html>), authored by E. Krissinel and K. Henrick. The least-squares fit between two structures were performed with PDBeFold [34]. All molecular figures were produced with PyMOL (<http://www.pymol.org/>).
The atomic coordinates and structure factors have been deposited in the Protein Data Bank Japan (PDBj) with the accession code PDB: 3WWV.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported in part by a Grant-in-Aid for Young Scientists (B) Grant Number 21770122 to H.Y. from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Professor H. Hashimoto (University of Shizuoka, Japan) for providing us with technical and valuable advice. We thank the Photon Factory staff for their assistance with data collection.
Appendix A. Supplementary data
This document file contains Table S1 and Figs. S1–S5.
References
- 1.Tavernarakis N., Driscoll M., Kyrpides N.C. The SPFH domain: implicated in regulating targeted protein turnover in stomatins and other membrane-associated proteins. Trends Biochem. Sci. 1999;24:425–427. doi: 10.1016/s0968-0004(99)01467-x. [DOI] [PubMed] [Google Scholar]
- 2.Browman D.T., Hoegg M.B., Robbins S.M. The SPFH domain-containing proteins: more than lipid raft markers. Trends Cell Biol. 2007;17:394–402. doi: 10.1016/j.tcb.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Lapatsina L., Brand J., Poole K., Daumke O., Lewin G.R. Stomatin-domain proteins. Eur. J. Cell Biol. 2012;91:240–245. doi: 10.1016/j.ejcb.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 4.Salzer U., Zhu R., Luten M., Isobe H., Pastushenko V., Perkmann T., Hinterdorfer P., Bosman G.J. Vesicles generated during storage of red cells are rich in the lipid raft marker stomatin. Transfusion. 2008;48:451–462. doi: 10.1111/j.1537-2995.2007.01549.x. [DOI] [PubMed] [Google Scholar]
- 5.Stewart G.W., Argent A.C., Dash B.C.J. Stomatin: a putative cation transport regulator in the red cell membrane. Biochim. Biophys. Acta. 1993;1225:15–25. doi: 10.1016/0925-4439(93)90116-i. [DOI] [PubMed] [Google Scholar]
- 6.Fricke B., Parsons S.F., Knöpfle G., von Düring M., Stewart G.W. Stomatin is mis-trafficked in the erythrocytes of overhydrated hereditary stomatocytosis, and is absent from normal primitive yolk sac-derived erythrocytes. Br. J. Haematol. 2005;131:265–277. doi: 10.1111/j.1365-2141.2005.05742.x. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J.Z., Hayashi H., Ebina Y., Prohaska R., Ismail-Beigi F. Association of stomatin (band 7.2b) with Glut1 glucose transporter. Arch. Biochem. Biophys. 1999;372:173–178. doi: 10.1006/abbi.1999.1489. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J.Z., Abbud W., Prohaska R., Ismail-Beigi F. Overexpression of stomatin depresses GLUT-1 glucose transporter activity. Am. J. Physiol. Cell Physiol. 2001;280:C1277–C1283. doi: 10.1152/ajpcell.2001.280.5.C1277. [DOI] [PubMed] [Google Scholar]
- 9.Rungaldier S., Oberwagner W., Salzer U., Csaszar E., Prohaska R. Stomatin interacts with GLUT1/SLC2A1, band 3/SLC4A1, and aquaporin-1 in human erythrocyte membrane domains. Biochim. Biophys. Acta. 2013;1828:956–966. doi: 10.1016/j.bbamem.2012.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Price M.P., Thompson R.J., Eshcol J.O., Wemmie J.A., Benson C.J. Stomatin modulates gating of acid-sensing ion channels. J. Biol. Chem. 2004;279:53886–53891. doi: 10.1074/jbc.M407708200. [DOI] [PubMed] [Google Scholar]
- 11.Wetzel C., Hu J., Riethmacher D., Benckendorff A., Harder L., Eilers A., Moshourab R., Kozlenkov A., Labuz D., Caspani O. A stomatin-domain protein essential for touch sensation in the mouse. Nature. 2007;445:206–209. doi: 10.1038/nature05394. [DOI] [PubMed] [Google Scholar]
- 12.Snyers L., Umlauf E., Prohaska R. Oligomeric nature of the integral membrane protein stomatin. J. Biol. Chem. 1998;273:17221–17226. doi: 10.1074/jbc.273.27.17221. [DOI] [PubMed] [Google Scholar]
- 13.Umlauf E., Csaszar E., Moertelmaier M., Schuetz G.J., Parton R.G., Prohaska R. Association of stomatin with lipid bodies. J. Biol. Chem. 2004;279:23699–23709. doi: 10.1074/jbc.M310546200. [DOI] [PubMed] [Google Scholar]
- 14.Umlauf E., Mairhofer M., Prohaska R. Characterization of the stomatin domain involved in homo-oligomerization and lipid raft association. J. Biol. Chem. 2006;281:23349–23356. doi: 10.1074/jbc.M513720200. [DOI] [PubMed] [Google Scholar]
- 15.Yokoyama H., Fujii S., Matsui I. Crystal structure of a core domain of stomatin from Pyrococcus horikoshii illustrates a novel trimeric and coiled-coil fold. J. Mol. Biol. 2008;376:868–878. doi: 10.1016/j.jmb.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 16.Kuwahara Y., Unzai S., Nagata T., Hiroaki Y., Yokoyama H., Matsui I., Ikegami T., Fujiyoshi Y., Hiroaki H. Unusual thermal disassembly of the SPFH domain oligomer from Pyrococcus horikoshii. Biophys. J. 2009;97:2034–2043. doi: 10.1016/j.bpj.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brand J., Smith E.S., Schwefel D., Lapatsina L., Poole K., Omerbašić D., Kozlenkov A., Behlke J., Lewin G.R., Daumke O. A stomatin dimer modulates the activity of acid-sensing ion channels. EMBO J. 2012;31:3635–3646. doi: 10.1038/emboj.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tory K., Menyhárd D.K., Woerner S., Nevo F., Gribouval O., Kerti A., Stráner P., Arrondel C., Cong E.H., Tulassay T. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat. Genet. 2014;46:299–304. doi: 10.1038/ng.2898. [DOI] [PubMed] [Google Scholar]
- 19.Green J.B., Fricke B., Chetty M.C., von Düring M., Preston G.F., Stewart G.W. Eukaryotic and prokaryotic stomatins: the proteolytic link. Blood Cells Mol. Dis. 2004;32:411–422. doi: 10.1016/j.bcmd.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 20.Green J.B., Lower R.P.J., Young J.P.W. The NfeD protein family and its conserved gene neighbours throughout prokaryotes: functional implications for stomatin-like proteins. J. Mol. Evol. 2009;69:657–667. doi: 10.1007/s00239-009-9304-8. [DOI] [PubMed] [Google Scholar]
- 21.Yokoyama H., Matsui E., Hiramoto K., Forterre P., Matsui I. Clustering of OB-fold domains of the partner protease complexed with trimeric stomatin from Thermococcales. Biochimie. 2013;95:1494–1501. doi: 10.1016/j.biochi.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Yokoyama H., Matsui I. A novel thermostable membrane protease forming an operon with a stomatin homolog from the hyperthermophilic archaebacterium Pyrococcus horikoshii. J. Biol. Chem. 2005;280:6588–6594. doi: 10.1074/jbc.M411748200. [DOI] [PubMed] [Google Scholar]
- 23.Yokoyama H., Kobayashi D., Takizawa N., Fujii S., Matsui I. Structural and biochemical analysis of a thermostable membrane-bound stomatin-specific protease. J. Synchrotron. Rad. 2013;20:933–937. doi: 10.1107/S0909049513021328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yokoyama H., Matsui E., Akiba T., Harata K., Matsui I. Molecular structure of a novel membrane protease specific for a stomatin homolog from the hyperthermophilic archaeon Pyrococcus horikoshii. J. Mol. Biol. 2006;358:1152–1164. doi: 10.1016/j.jmb.2006.02.052. [DOI] [PubMed] [Google Scholar]
- 25.Yokoyama H., Takizawa N., Kobayashi D., Matsui I., Fujii S. Crystal structure of a membrane stomatin-specific protease in complex with a substrate peptide. Biochemistry. 2012;51:3872–3880. doi: 10.1021/bi300098k. [DOI] [PubMed] [Google Scholar]
- 26.Kim D.Y., Kim K.K. The structural basis for the activation and peptide recognition of bacterial ClpP. J. Mol. Biol. 2008;379:760–771. doi: 10.1016/j.jmb.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 27.Kim A.C., Oliver D.C., Paetzel M. Crystal structure of a bacterial signal peptide peptidase. J. Mol. Biol. 2008;376:352–366. doi: 10.1016/j.jmb.2007.11.080. [DOI] [PubMed] [Google Scholar]
- 28.Kihara A., Akiyama Y., Ito K. A protease complex in the Escherichia coli plasma membrane: HflKC (HflA) forms a complex with FtsH (HflB), regulating its proteolytic activity against SecY. EMBO J. 1996;15:6122–6131. [PMC free article] [PubMed] [Google Scholar]
- 29.Chiba S., Ito K., Akiyama Y. The Escherichia coli plasma membrane contains two PHB (prohibitin homology) domain protein complexes of opposite orientations. Mol. Microbiol. 2006;60:448–457. doi: 10.1111/j.1365-2958.2006.05104.x. [DOI] [PubMed] [Google Scholar]
- 30.Kuwahara Y., Ohno A., Morii T., Yokoyama H., Matsui I., Tochio H., Shirakawa M., Hiroaki H. The solution structure of the C-terminal domain of NfeD reveals a novel membrane-anchored OB-fold. Protein Sci. 2008;17:1915–1924. doi: 10.1110/ps.034736.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker C.A., Hinderhofer M., Witte D.J., Boos W., Möller H.M. Solution structure of the soluble domain of the NfeD protein YuaF from Bacillus subtilis. J. Biomol. NMR. 2008;42:69–76. doi: 10.1007/s10858-008-9261-3. [DOI] [PubMed] [Google Scholar]
- 32.Lovell S.C., Davis I.W., Arendall W.B., III, de Bakker P.I.W., Word J.M., Prisant M.G., Richardson J.S., Richardson D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Struct. Funct. Genet. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 33.Theobald D.L., Mitton-Fry R.M., Wuttke D.S. Nucleic acid recognition by OB-fold proteins. Annu. Rev. Biophys. Biomol. Struct. 2003;32:115–133. doi: 10.1146/annurev.biophys.32.110601.142506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krissinel E., Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 35.Zhang F., Hu M., Tian G., Zhang P., Finley D., Jeffrey P.D., Shi Y. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol. Cell. 2009;34:473–484. doi: 10.1016/j.molcel.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirokawa T., Boon-Chieng S., Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 37.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 39.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murshudov G.N., Skubák P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This document file contains Table S1 and Figs. S1–S5.