

Highlights
-
•
Heat shock (HS) preconditioning protects against ER stress-induced apoptosis upstream of BAX activation.
-
•
HS preconditioning elevates the expression of HSPA1, a key pro-survival marker.
-
•
The primary mechanism by which HS preconditioning renders cells resistant to ER stress is via downregulation of BIM.
-
•
HSPA1 does not play a role in the regulation of BIM protein levels.
Abbreviations: ER, endoplasmic reticulum; HS, heat shock; TG, thapsigargin; TM, tunicamycin; UPR, unfolded protein response
Keywords: Apoptosis, BCL-2 family, BIM, Endoplasmic reticulum stress, Heat shock, Unfolded protein response
Abstract
A mild heat shock (HS) preconditioning and acquisition of thermotolerance protects cells against a variety of cytotoxic agents that otherwise induce apoptosis. Here we tested whether there is a molecular link between HS preconditioning and endoplasmic reticulum (ER) stress-induced apoptosis. ER stress results from a loss of ER lumen homeostasis, culminating in an accumulation of unfolded/misfolded proteins in the ER and activation of unfolded protein response (UPR). Unresolved, ER stress leads to activation of BH3-only proteins, mitochondrial membrane permeabilization, caspase activation and apoptotic cell death. HS preconditioning (1 h at 42 °C) induced a rapid increase in HSPA1 (HSP70) levels which remained elevated for at least 48 h post-HS. HS preconditioning significantly reduced BAX, caspase activation and apoptosis in cell cultures treated with the ER stress-inducing agents thapsigargin (TG) and tunicamycin (TM). HS-mediated protection was found to be due to regulation of the BH3-only protein BIM. Further, overexpression of HSPA1 could not mimic the effect of HS on BIM expression, suggesting that other HS factors may play a role in inhibiting ER stress-induced apoptosis by regulating BIM.
1. Introduction
Endoplasmic reticulum (ER) stress occurs as a consequence of accumulation of unfolded and misfolded proteins in the ER lumen [1]. As a result, the cells evoke the unfolded protein response (UPR) [1,2]. Three ER transmembrane proteins, PRK (RNA-dependent protein kinase)-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1α (IRE1α) monitor the “health” of the ER [3,4]. Upon accumulation of unfolded proteins in the ER lumen, BiP/GRP78 (HSPA5) dissociates from PERK, ATF6 and IRE1α permitting their activation and initiation of the UPR signaling pathways. The outcome of the cell’s response to stress depends on the severity and duration of the stress. Initially, cells mount a pro-survival response, however, if the effects of the stressor cannot be resolved, cell death ensues [3,4]. ER stress-induced cell death is primarily mediated via the intrinsic apoptotic pathway leading to BAX/BAK-mediated mitochondrial outer membrane permeabilization (MOMP), dissipation of mitochondrial inner-membrane potential (ΔΨm) and release of pro-apoptotic factors, including cytochrome c [5,6]. This leads to activation of caspase-9 and caspase-3. Members of the BCL-2 family of proteins are the key regulators of MOMP and therefore, represent critical regulators of cell fate [7]. BCL-2 homology domain 3 (BH3)-only proteins, e.g. BIM and NOXA, are stressor specific modulators of apoptosis. BIM has been shown to be central to ER stress-induced apoptosis in a number of cell lines, and RNAi-mediated knockdown of BIM results in significant protection against ER stress-induced cell death [8–11].
Heat shock proteins (HSPs) are a set of evolutionary conserved protein chaperones that can modulate the intrinsic apoptotic pathway at multiple levels [12–14]. Several members of HSP family are stress inducible and their expression increases after exposure of cells to environmental and physiological stresses such as non-lethal elevations in temperature or oxidative stress [15]. The transient increase in the expression of HSPs following heat shock (HS) has been shown to prevent cell death-induced by a variety of toxic insults [16–18].
Here we investigated whether HS preconditioning can block ER stress-induced apoptosis. PC12 and HeLa cell lines were heat shocked by incubation for 1 h at 42 °C. This treatment resulted in a rapid induction of HSPA1 (Hsp70) protein and its levels remained elevated for up to 48 h. HS preconditioned cells had significantly reduced BAX activation, caspase processing and apoptosis compared to control cells following treatment with ER stress inducing agents. The inhibition of apoptosis was due to reduced levels of the BH3-only protein BIM, as HS did not confer any further protection in BIM shRNA-expressing cells. HS-mediated suppression of BIM is therefore critical for cell survival upon ER stress. Our data suggests that there are HS-induced factors, apart from HSPA1, that alter BIM protein levels and protect against ER stress-induced apoptosis.
2. Results and discussion
2.1. Heat shock preconditioning protects against TG-induced cytotoxicity
In order to determine the dose response of HeLa and PC12 cells to TG, an MTT assay was used to measure cell viability in cultures treated with TG concentrations ranging from 0.1 to 2 μM for 24 h and 48 h. Doses which caused an approximately 25% decrease in cell viability after 24 h of treatment were used in subsequent experiments (Fig. 1A and B). Similar studies were carried out with TM (data not shown) and a concentration of 1.5 μM or 2 μM of TM was used in HeLa and PC12 cells respectively throughout this study.
Fig. 1.
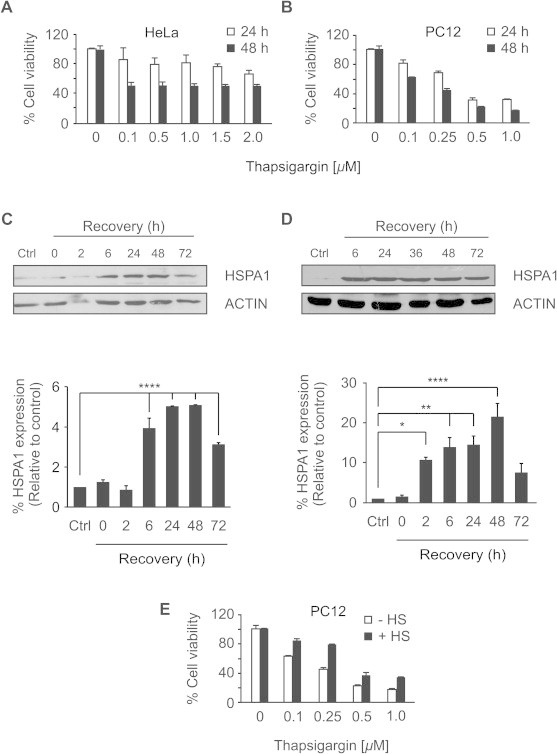
Heat shock preconditioning reduces thapsigargin-induced cytotoxicity in HeLa and PC12 cells. (A) HeLa cells and (B) PC12 cells were treated with indicated concentrations of TG for 24 h and 48 h followed by MTT assessment of cell viability. (C) HeLa cells and (D) PC12 cells were heat shocked in HEPES buffered media for 1 h at 42 °C and after recovery at 37 °C for indicated periods of time the induction of HSPA1 was determined by Western blotting (upper panel) using specific antibody against HSPA1. ACTIN was used as a loading control. Densitometric analysis (lower panel) of HSPA1 induction was normalized to ACTIN and expressed relative to untreated cells. Values shown are the mean ± SEM of three independent determinations. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗∗P < 0.001 (E) PC12 cells were heat shocked or left untreated, and after a 6 h recovery period they were treated with increasing doses of TG for 48 h and an MTT assay was performed after that time. Average and error bars represent mean ± SD from two independent experiments.
To investigate whether there is a molecular link between the HS response and ER stress-induced apoptosis, we set up a HS preconditioning experiment. It is well established that HSPs expression can be induced by exposing cells to a sub-lethal HS followed by a recovery period [16,19–21]. Cells were exposed to a non-lethal thermal stress by placing cells in a water bath at 42 °C for 1 h. Cells were allowed to recover for 6–72 h and expression of the HSPs (HSPA1) was assessed as a marker of activation of the HS response by Western blotting (Fig. 1C and D). The maximal level of HSPA1 was induced by 6 h of recovery and remained elevated for 48 h in HeLa and PC12 cells (Fig. 1C and D). The 6 h time point was selected as the optimal recovery time for subsequent HS preconditioning experiments as HSPs were significantly increased in both cell lines at this time point.
We have reported previously that these conditions can render cells resistant to toxic insults [16,20]. We hypothesized that thermal preconditioning confers a survival advantage to cells under ER stress conditions. Indeed, heat shock of PC12 cells resulted in 34% reduction in cytotoxicity induced by 0.25 μM TG, as assessed by MTT assay (Fig. 1E).
2.2. Heat shock preconditioning attenuates ER stress-induced caspase activation and apoptosis
To examine the effect of HS preconditioning on TG-induced apoptosis we carried out sub-G1 analysis of control versus heat shocked cells treated with TG. Indeed, the rate of ER stress-induced apoptosis was reduced by 10% in both HS preconditioned HeLa and PC12 cells (Fig. 2A and B). Equally, HS preconditioning reduced caspase-3 cleavage in HeLa cells treated with either TG or TM (Fig. 2C). Similar results were obtained in PC12 cells (Fig. 2D). The reduction in caspase cleavage reflected a 40% reduction in DEVDase activity in heat shocked HeLa cells relative to control cells 48 h after treatment with 1.5 μM TG (Fig. 2E). In PC12 cells thermal preconditioning led to a 50% reduction in caspase activity at 24 h relative to control cells (Fig. 2F). This suggests that induction of HSPs is an adaptive feature that can act to help cells prevent ER stress-induced apoptosis.
Fig. 2.
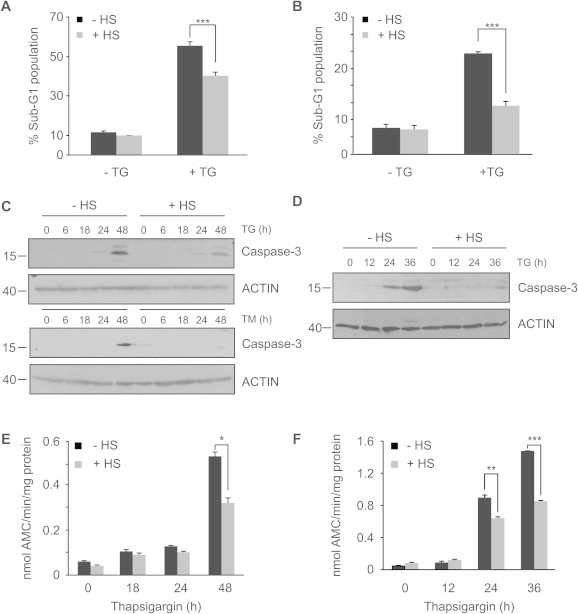
Thermal preconditioning reduces ER stress-induced apoptosis. (A) HeLa cells and (B) PC12 cells were heat shocked or left untreated, and after a 6 h recovery period they were treated with 1.5 μM or 0.25 μM TG respectively for 48 h. Cells were fixed in 70% ethanol and Sub-G1 analysis was determined by PI staining. Values are the mean ± SEM of three separate determinations. ∗∗∗P < 0.001 compared to TG alone (C) Pro-caspase-3 cleavage was assessed in whole cell lysate from HeLa and (D) PC12 cells by Western blotting using anti-cleaved caspase-3 antibody. ACTIN was used as a loading control. Data are representative of three independent repeats. (E) DEVD-MCA cleavage activity was measured in whole cell extracts in HeLa and (F) PC12 cells. Values shown are the mean ± SEM of three independent repeats. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 compared to TG alone.
2.3. Heat shock preconditioning augments a pro-survival UPR response
We examined expression levels of UPR markers in the preconditioned and control cells in order to determine whether HS preconditioning also affected UPR signaling upstream of apoptotic events. Expression of phospho-eIF2α and CHOP were comparable between the HS and control cells (Fig. 3A, B and D). However, the expression of key pro-survival UPR markers Grp78 and XBP1s was elevated upon HS (Fig. 3C and E) and is in line with our previous publication [25]. This suggests that HS preconditioned cells are better equipped to deal with ER stress due to a more robust activation of a pro-survival UPR response. As such, thermal preconditioning primes cells, enabling them to mount a much stronger pro-survival response to a subsequent ER stress insult.
Fig. 3.
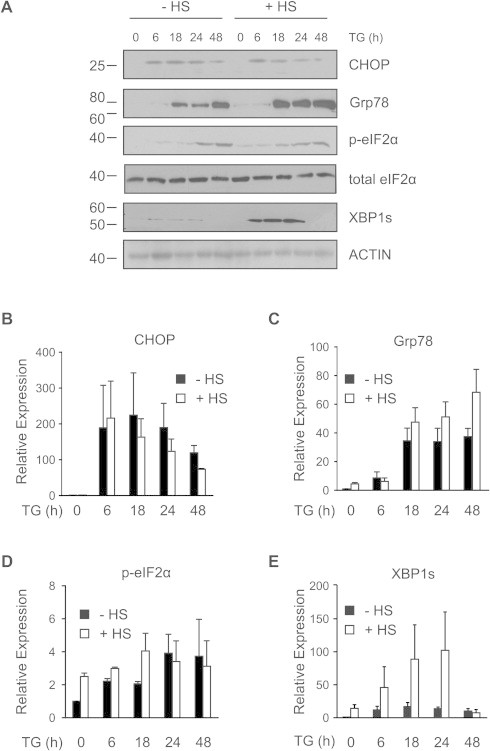
Heat shock preconditioning augments an adaptive UPR response. (A) HeLa cells were heat shocked or left untreated, and after a 6 h recovery, they were treated with 1.5 μM TG for indicated periods of time. Western blotting analysis has been performed using antibody against CHOP, Grp78, phospho-eIF2α (p-eIF2α), total eIF2α and XBP1s. ACTIN was used as a loading control. (B–E) Densitometric analysis of (B) CHOP, (C) Grp78, (D) p-eIF2α and (E) XBP1s band intensity expression relative to control. Average and error bars represent mean ± SEM from three independent experiments.
2.4. Heat shock preconditioning attenuates apoptosis upstream of mitochondria
HSPs can positively modulate cell fate by regulating the apoptotic pathway. We have previously demonstrated that HS preconditioning inhibits HS-induced apoptosis at the level of the mitochondria and prior to BAX activation [22]. Here we investigated how HS preconditioning regulates apoptotic events during ER stress. Apoptosis induced by ER stress occurs via the mitochondrial (intrinsic) pathway; therefore we investigated this pathway in thermally conditioned cells and non-conditioned control cells. To this end we assessed BAX activation by flow cytometry. TG treatment caused a 45% increase in the number of cells containing active BAX, while only 25% of HS preconditioned cells expressed active BAX following TG treatment (Fig. 4A). This data demonstrates that HS preconditioning regulates BAX activation and affords protection upstream of mitochondria.
Fig. 4.
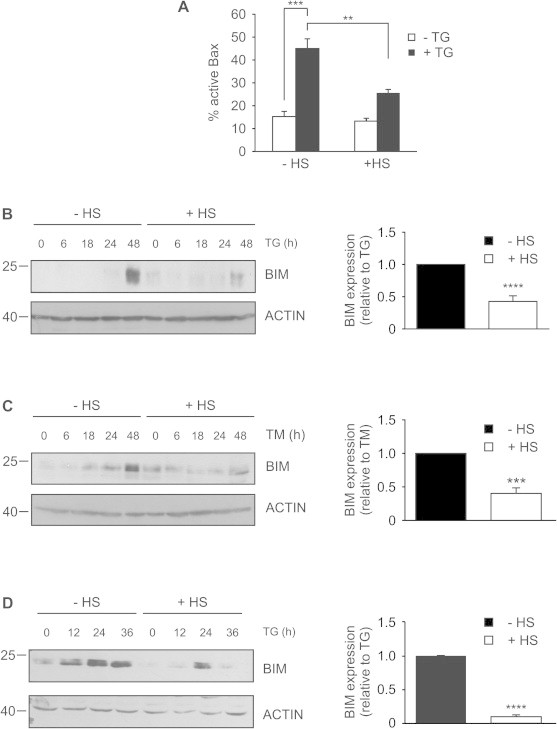
Thermal preconditioning regulates BIM levels and affects mitochondrial apoptosis. (A–C) HeLa cells were heat shocked or left untreated, after a 6 h recovery period they were either treated with 1.5 μM TG or 1.5 μM TM for up to 48 h. (A) Flow cytometry-based measurement of the active conformation of BAX was performed using anti-active BAX. Values are the mean ± SEM of three independent repeats. ∗∗P < 0.01 compared to TG alone, ∗∗∗P < 0.001 compared with control. Western blotting was used to visualize BIM protein levels (left panel) in (B) TG-treated and (C) TM-treated cells. ACTIN was used as loading control. Densitometry analysis (right panel) for BIM expression after 48 h of TG treatment was normalized to ACTIN and expressed as fold change from TG alone. Values are the mean ± SEM of three independent experiments. ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001 compared to TG alone. (D) PC12 cells were heat shocked or left untreated; after a 6 h recovery period they were treated with 0.25 μM TG for 0–36 h. Samples were prepared for SDS–PAGE gels and probed using anti-BIM (left panel). ACTIN was used as a loading control. Densitometry analysis (right panel) for BIM expression after 36 h of TG treatment was normalized to ACTIN and expressed as fold change from TG alone. Values are the mean ± SEM of three independent experiments. ∗∗∗∗P < 0.0001 compared to TG alone.
2.5. Heat shock preconditioning protects against ER stress-induced apoptosis by regulation of BIM
BAX activation is regulated by members of BCL-2 protein family. We have previously demonstrated that an increase in expression of the pro-apoptotic BH3-only protein BIM is crucial for ER stress-induced apoptosis [7]. BIM has also been shown to directly interact with BAX leading to its activation [23,24] Therefore, we examined if the expression of BIM was modulated by HS preconditioning. TG treatment caused an increase in expression of the pro-apoptotic protein BIM at 48 h in HeLa cells (Fig. 4B). However, the expression of BIM was significantly reduced in HS preconditioned cells (Fig. 4B). HS preconditioned cells also had reduced TM-induced expression of BIM (Fig. 4C). This demonstrates that activation of the heat shock response inhibited ER stress-induced BIM expression. Similar results were obtained in PC12 cells indicating that regulation of BIM by the heat shock response is a general, and not a cell type specific response (Fig. 4D).
To further delineate the involvement of BIM expression in the protection afforded by HS preconditioning we took control cells (PLKO) and BIM shRNA-expressing PC12 cells and subjected them to HS or control conditions. We reasoned that if there are other factors, that contribute in parallel with BIM to the protective effects of HS preconditioning, we would be still be able to see a HS-mediated reduction in the level of ER stress-induced apoptosis even in BIM shRNA-expressing cells. We determined the efficiency of knockdown achieved by the BIM shRNA by carrying out densitometric analysis of control cells (PLKO empty vector) versus BIM shRNA expressing cells in both untreated and TG-treated conditions (Fig. 5C). It was found that 48 h of TG treatment caused a 35-fold increase in BIM expression in PLKO expressing cells relative to untreated conditions (P value < 0.0001) while in BIM shRNA cells induction of BIM by TG was not significant. Upon treatment with TG, HS PLKO cells had lower BIM expression, lower caspase-9 and -3 cleavage and a 36% reduction in apoptosis relative to control cells (Fig. 5A and B). BIM shRNA-expressing PC12 cells were protected against apoptosis to the same extent as their HS-treated PLKO vector expressing counterparts following TG treatment. We could not detect caspase-9 and -3 processing in these cells (Fig. 5B). HS preconditioning of BIM shRNA cells failed to further enhance the level of protection afforded by BIM knockdown (Fig. 5A). These data suggest that BIM is essential for ER stress-induced apoptosis and that the primary mechanism by which HS preconditioning renders cells resistant to ER stress is via regulation of BIM.
Fig. 5.
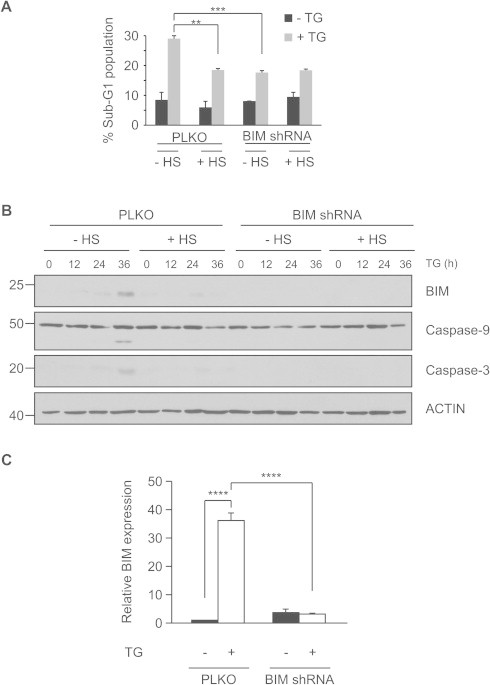
Heat shock preconditioning renders cells resistant to ER stress via regulation of BIM. (A) PLKO and BIM shRNA expressing PC12 cells were untreated or heat shocked followed by a 6 h recovery after which cells were treated for 48 h with 0.25 μM TG. Cell death was assessed by quantifying the sub-G1 population by PI staining and flow cytometry. Values are the mean ± SEM of three independent repeats. ∗∗P < 0.01 compared to TG-only treated cells, ∗∗∗P < 0.001 compared to PLKO cells. (B) Western blot analysis of BIM in control and heat shocked PLKO and BIM shRNA expressing PC12 cells treated with TG for indicated periods of time. ACTIN was used as a loading control. Data are representative of at least three independent repeats. (C) Densitometry analysis for BIM band intensity after 36 h of TG treatment was performed, normalized to ACTIN and expressed as fold change from untreated control. Values are the mean ± SEM of three independent repeats. ∗∗∗∗P < 0.0001 compared to control or to TG-treated PLKO expressing cells.
HSPA1 is highly stress inducible and we have previously shown that overexpression of HSPA1 alone can protect from ER stress-induced apoptosis. By interacting with IRE1α and enhancing its ability to splice XBP1, HSPA1 increases the expression of XBP1s target genes [25]. Since we observed increased expression of XBP1s in HS preconditioned cells, we looked at BIM protein levels in HSPA1 overexpressing PC12 cells. Interestingly no reduction in BIM protein levels was observed in HSPA1-overexpressing cells following TM treatment (Fig. 6C). In response to TG treatment HSPA1 cells were found to have higher levels of BIM protein despite being protected from ER stress-induced apoptosis (Fig. 6B). Such a paradoxical observation has previously been reported for expression of BIM in ER stress resistant cells [26]. It has been reported that the pro-apoptotic ability of BIM can be inhibited via phosphorylation on Ser87. Taking that into account, HSPA1 might protect against ER stress-induced apoptosis by post-translational modifications of BIM.
Fig. 6.
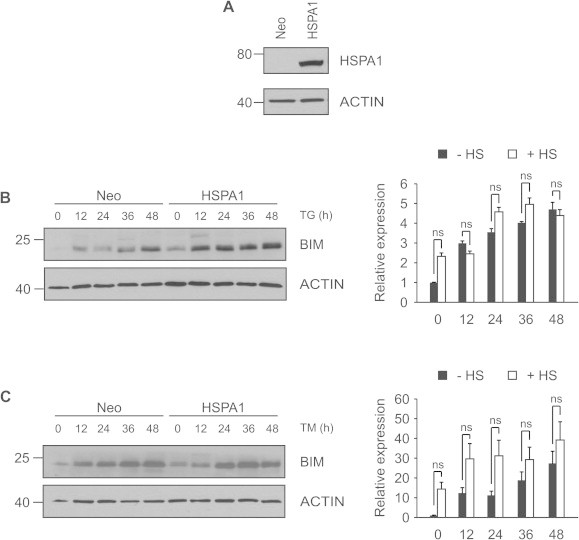
HSPA1 does not regulate BIM levels following ER stress. (A) Protein extracts from Neo and HSPA1 overexpressing PC12 cells were analyzed by Western blotting using a specific anti-HSPA1 antibody. ACTIN was used as a loading control. Neo and HSPA1 overexpressing PC12 cells were treated with (B) 0.25 μM TG or (C) 2 μM TM for 0–48 h and protein extracts were assessed for levels of BIM. ACTIN was used as a loading control. Images are representative of three independent experiments.
Our data point to downregulation of BIM by a component of the HSR, however this effect is not mediated by HSPA1. Numerous potential mechanisms, possibly parallel, may be involved in the regulation of BIM following HS. BIM expression can be regulated transcriptionally and post-transcriptionally in response to ER stress. CHOP has been reported to regulate BIM at transcriptional level following ER stress, however, CHOP knockdown does not affect BIM expression or cell viability in PC12 cells treated with TG [27]. We have also already shown that microRNAs belonging to miR-106b-25 cluster contribute to ER stress-induced up-regulation of BIM [27]. BIM is also subject to multiple levels of post-translational regulation. In particular alteration in the phosphorylation of BIM can affect its activity, interaction with BCL-2 family proteins, stability and cellular localization [10,28–30]. Indeed we have observed an electrophoretic mobility shift of BIM protein, suggestive of its increased phosphorylation upon ER stress in both HeLa and PC12 cells. HS preconditioning was associated with decline of both upper and lower bands corresponding to BIM, which likely suggests that HS preconditioning might inhibit BIM synthesis (reduction in lower band) and phosphorylation/degradation (reduction in upper band). The protective effect of HS preconditioning may be acting via any of these mechanisms.
3. Conclusions
The UPR is a biphasic signaling program activated in response to ER stress. However, the switching mechanisms leading to execution of the cell death program remain elusive. Here we demonstrated that HS preconditioning, but not HSPA1 expression, regulates BIM expression to protect cells from ER stress-induced apoptosis. HS regulates apoptosis at the level of UPR and upstream from mitochondrial changes associated with BAX activation. We propose that mechanisms which augment expression and/or activity of HSPs represent a potential therapeutic target in conditions where excessive ER stress-induced apoptosis is maladaptive. HSPs are potent anti-apoptotic modulators and previously have been shown to protect from numerous apoptotic stimuli. However, the precise molecular mechanisms have not been well described. We show for the first time that HS preconditioning protects from ER stress-induced apoptosis by regulation of the pro-apoptotic protein BIM.
HSPs, in particular HSPA1 have been linked to interference with the intrinsic apoptotic pathway via inhibition of cytochrome c release and subsequent caspase activation [31]. We also observed reduced caspase activation and activity in HS preconditioned cells. In addition we saw significant down regulation of BIM in those cells. Regulation of the BCL-2 family by HSPs has previously been reported with the small HSP HSPB1 involved in the regulation of BAD [32] and HSPA1 involved in the stabilization of the pro-survival protein, MCL1 [33]. We have previously shown that HSPA1 could protect from ER stress. However to date, no reports have linked HSPs to the regulation of BIM as the protective mechanism against apoptosis. Given the crucial role of BIM in ER stress-induced apoptosis we were interested in identifying if HSPA1 overexpression could reduce ER stress-induced BIM expression. Interestingly, no reduction in BIM was observed in cells overexpressing HSPA1 following treatment with TG or TM. Collectively these data point to molecular link between HSR and ER stress-induced apoptosis, which involves down regulation of BIM.
4. Experimental procedures
4.1. Cell culture and treatments
Rat pheochromocytoma cell line, PC12 (ECACC), and cervical cancer cell line, HeLa (ATCC), were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma, D6429). The media for PC12 cells were supplemented with 10% heat inactivated horse serum, 5% fetal bovine serum (FBS), while the media for HeLa cells were supplemented with 10% FBS. In addition 1% penicillin/streptomycin was also added. All cells were cultured at 37 °C, 5% CO2 in humidified incubator. HeLa cells and PC12 cells were seeded at 6 × 103 and 6 × 104 cells/cm2 density respectively 24 h prior to treatments. PC12 cells were treated with 0.25 μM thapsigargin (TG) or 2 μM tunicamycin (TM) and HeLa cells were treated with 1.5 μM TG or 1.5 μM TM.
4.2. Cell viability assay
The mitochondrial metabolic function of cells was assayed by monitoring the conversion of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazonium bromide) (Sigma) to purple formazan crystals in viable cells. HeLa and PC12 cells were plated into 96-well plates at 6 × 103 and 6 × 104 cells/cm2, respectively. After carrying out an experiment, cells were incubated with 0.5 mg/ml of MTT for 3 h at 37 °C. To stop the reaction and solubilize the formazan crystals 1 volume of 20% SDS in 50% dimethyl formamide was added and the absorbance was measured at 550 nm by a Wallac 1420 plate-reader with a reference wavelength of 650 nm. Cell viability was expressed as percent of the control cells.
4.3. Heat shock preconditioning
To subject the cells to heat shock, the culture flasks were sealed by wrapping parafilm around the lids and immersed in a water bath at 42 °C for 1 h in HEPES buffered media. Culture media was replaced and cells were allowed to recover at 37 °C for 6 h.
4.4. Western blotting
Cells were harvested, washed once in PBS and lysed in RIPA buffer (50 mM Tris–HCl, pH 8.8, containing 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40) plus a cocktail of protease and phosphatase inhibitors (100 mM PMSF, 1 mg/ml pepstatin, 10 mM leupeptin, 2.3 mg/ml aprotinin, 250 mM ALLN, 10 mM NaF, 1 mM Na3VO4). Protein concentration was determined by bicinchoninic acid (BCA) assay. Lysates were denatured by adding 1/4 volume 5× Laemmli’s buffer and incubating for 5 min at 96 °C. Proteins were separated on SDS–polyacrylamide gels. The proteins were transferred onto a nitrocellulose membrane and blocked with 5% milk in PBS containing 0.05% Tween. The membrane was incubated in a 1:1000 dilution of primary antibody in 5% milk in PBS containing 0.05% Tween and probed with specific antibodies for BIM (Stressgen, AAP-330), cleaved caspase-3 (Cell Signaling Technologies, #9644), caspase-9 (Cell Signaling Technologies, #9508 rat, #9502 human), ACTIN (Sigma, A2066), HSPA1 (Stressgen, SPA811), eIF2α (Cell Signaling Technologies, #9722), phopsho-eIF2α (Cell Signaling Technologies, #9721), CHOP (Santa Cruz, sc793), GRP78 (Stressgen, SPA826), XBP1s (Biolegend, 619501). They were further incubated in a 1:5000 dilution of appropriate horseradish peroxidase-conjugated secondary antibody (Jackson) in 5% milk in PBS containing 0.05% Tween for 90 min. Signals were detected using Pierce ECL western blotting substrate [34].
4.5. Detection of caspase activity
The activity of group II caspases, DEVDases, was determined fluorometrically with Ac-Asp-Glu-Val-Asp-a-(4-methyl-coumaryl-7-amide) (DEVD-MCA), obtained from the Peptide Institute, Osaka, Japan. Briefly, cell lysates and substrate (DEVD-MCA) were combined in reaction buffer (100 M N-2-hydroxyethyl-piperazine-NO-2-ethanesulphonic acid (HEPES), pH 7.5, 10% sucrose, 0.1% 3[(3cholamidopropyl)-dimethylammonio]-1-propanesulphonate, 5 mM dithiothreitol, 10% Nonidet P40, and 50 μM DEVD-MCA). Substrate cleavage leading to the release of free AMC was monitored at 37 °C using a Wallac Victor multilabel counter (excitation 355 nm, emission 460 nm). Fluorescent units were converted to micromoles of AMC released using a standard curve generated with free AMC and subsequently related to protein concentration.
4.6. Sub-G1 DNA content analysis
Following treatment cells were harvested by trypsinization and washed in PBS. Cells were fixed by adding 500 μl of ice-cold 70% ethanol drop-wise, while gently vortexing. Cells were placed on ice for a minimum of 1 h for efficient fixation. Once fixed, cells were stained with 20 μg/ml propidium Iodide (PI) for 30 min in the dark on ice. Cells were then analyzed using a FACS Canto flow cytometer (BD Biosciences) with a total of 10,000 events acquired per sample. Analysis was carried out with Cyflogic software.
4.7. Detection of BAX activation
Cells were trypsinized and then fixed in 2% formaldehyde for 10 min at room temperature. Washed cells were resuspended in PBS. Anti-BAX antibody (BD Biosciences clone 6A7, 1 μg) was added to 100 μl of permeabilization buffer (0.1% saponin, 0.5% BSA in PBS), in which cells were incubated for 1 h at 4 °C. Mouse IgG isotype control (Biolegend) served as an autofluorescence control. Samples were washed and incubated with a 1:200 dilution of FITC-conjugated anti-mouse antibodies in PBS for 1 h at 4 °C. Samples were washed and resuspended in 300 μl PBS and analyzed by flow cytometry. Histograms were overlaid using FCS express.
4.8. Generation of stable knockdown of Bim using shRNA
PC12 cells with reduced levels of Bim were generated using a method previously described [35] by targeting Bim mRNA with shRNA using the lentiviral expression vector PLKO and puromycin selection (3 μg/ml for 7 days). Constructs were generated by Broad Institute (Boston, MA, USA) based on different criteria for shRNA design (http://www.broad.mit.edu/genome_bio/trc/rnai.html).
4.9. Generation of HSPA1 overexpressing PC12 cells
The plasmid expressing full-length human HSPA1 (GenBank: NM_005346) under the CMV promoter was a kind gift from Dr. Tomomi Gotoh, Kumamoto University, Japan. Cells were transiently transfected with pcDNA3.1 control plasmid (Neo) or pcDNA3.1-HSPA1. Transient transfections were carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Cells were then cultured in presence of 800 ng of G418, enabling selection of plasmid expressing PC12 cells.
4.10. Densitometric analysis
Quantitative analysis of Western blotting results was carried out using densitometric analysis with ImageJ software. Expression was normalized to the loading control (ACTIN) and expressed relative to untreated sample (otherwise indicated).
4.11. Statistical analysis
Experiments were repeated independently at least 3 times. Error bars represent means ± SEM of replicates. Significance was determined using Two-way ANOVA followed by Tukey’s post hoc analysis, with P-value < 0.05 being considered significant and annotated by ∗.
Author contribution
D.K., K.M. and A.S. planned experiments and analyzed data; D.K. and K.M. performed experiments and acquired the data, A.S., D.K. and K.M. wrote the paper, AS secured the funding.
Acknowledgments
We would like to thank all members of our lab and Apoptosis Research Centre for their help and input in this study. D.K. was funded by a fellowship from the College of Science of NUI Galway. This work was supported by the research grants from Science foundation Ireland (Grant number 09/RFP/BIC2371 or 09/RFP/BMT2153) and Belgian grant – Interuniversity Attraction Poles, IAP 7/32.
References
- 1.S.M. Healy, T. Verfaillie, R. Jager, P. Agostinis, A. Samali, Biology of the Endoplasmic Reticulum, in: P. Agostinis, S. Afshin (Eds.), Springer, Netherlands, 2012, pp. 3–22.
- 2.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 3.Gorman A.M., Healy S.J.M., Jager R., Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012;134:306–316. doi: 10.1016/j.pharmthera.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Szegezdi E., Logue S.E., Gorman A.M., Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta S., Cuffe L., Szegezdi E. Mechanisms of ER stress-mediated mitochondrial membrane permeabilization. Int. J. Cell Biol. 2010;2010:170215. doi: 10.1155/2010/170215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei M.C., Zong W., Cheng E.H., Lindsten T., Panoutsakopoulou V., Ross A.J., Roth K.A., MacGregor G.R., Thompson C.B., Korsmeyer S.J. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szegezdi E., MacDonald D., Catriona C., Triona, Gupta S., Samali A. Bcl-2 family on guard at the ER. Am. J. Physiol. Cell Physiol. 2009;296:C941–C953. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- 8.Wali J.A., Rondas D., McKenzie M.D., Zhao Y., Elkerbout L., Fynch S., Gurzov E.N., Akira S., Mathieu C., Kay T.W.H., Overbergh L., Strasser A., Thomas H.E. The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 2014;5:e1124. doi: 10.1038/cddis.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szegezdi E., Reed Herbert K., Kavanagh E.T., Samali A., Gorman A.M. Nerve growth factor blocks thapsigargin-induced apoptosis at the level of the mitochondrion viaregulation of Bim. J. Cell Mol. Med. 2008;12:2482–2496. doi: 10.1111/j.1582-4934.2008.00268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puthalakath H., O’Reilly L.A., Gunn P., Lee L., Kelly P.N., Huntington N.D., Hughes P.D., Michalak E.M., McKimm-Breschkin J., Motoyama N., Gotoh T., Akira S., Bouillet P., Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L., Lopez H., George N.M., Liu X., Pang X., Luo X. Selective involvement of BH3-only proteins and differential targets of Noxa in diverse apoptotic pathways. Cell Death Differ. 2011;18:864–873. doi: 10.1038/cdd.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samali A., Orrenius S. Heat shock proteins: regulators of stress response and apoptosis. Cell Stress Chaperones. 1998;3:228–236. doi: 10.1379/1466-1268(1998)003<0228:hspros>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy D., Jäger R., Mosser D.D., Samali A. Regulation of apoptosis by heat shock proteins. IUBMB Life. 2014;66(5):327–338. doi: 10.1002/iub.1274. [DOI] [PubMed] [Google Scholar]
- 14.Lanneau D., Brunet M., Frisan E., Solary E., Fontenay M., Garrido C. Heat shock proteins: essential proteins for apoptosis regulation. J. Cell Mol. Med. 2008;12:743–761. doi: 10.1111/j.1582-4934.2008.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morimoto R.I. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 16.Quigney D.J., Gorman A.M., Samali A. Heat shock protects PC12 cells against MPP+ toxicity. Brain Res. 2003;993:133–139. doi: 10.1016/j.brainres.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Cheng L., Smith D.J., Anderson R.L., Nagley P. Modulation of cellular Hsp72 levels in undifferentiated and neuron-like SH-sy5y cells determines resistance to staurosporine-induced apoptosis. PLoS ONE. 2011;6:e24473. doi: 10.1371/journal.pone.0024473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerner E.W., Schneuder M.J. Induced thermal resistance in HeLa cells. Nature. 1975;256:500–502. doi: 10.1038/256500a0. [DOI] [PubMed] [Google Scholar]
- 19.Ito H., Shimojo T., Fujisaki H., Tamamori M., Ishiyama S., Adachi S., Abe S., Marumo F., Hiroe M. Thermal preconditioning protects rat cardiac muscle cells from doxorubicin-induced apoptosis. Life Sci. 1999;64:755–761. doi: 10.1016/s0024-3205(98)00617-1. [DOI] [PubMed] [Google Scholar]
- 20.Samali A., Cotter T.G. Heat shock proteins increase resistance to apoptosis. Exp. Cell Res. 1996;223:163–170. doi: 10.1006/excr.1996.0070. [DOI] [PubMed] [Google Scholar]
- 21.Samali A., Holmberg C., Sistonen L., Orrenius S. Thermotolerance and cell death are distinct cellular responses to stress: dependence on heat shock proteins. FEBS Lett. 1999;461:306–310. doi: 10.1016/s0014-5793(99)01486-6. [DOI] [PubMed] [Google Scholar]
- 22.Samali A., Robertson J.D., Peterson E., Manero F., Van Zeijl L., Paul C., Cotgreave I.A., Arrigo A., Orrenius S. Hsp27 protects mitochondria of thermotolerant cells against apoptotic stimuli. Cell Stress Chaperones. 2001;6:49–58. doi: 10.1379/1466-1268(2001)006<0049:hpmotc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vela L., Gonzalo O., Naval J., Marzo I. Direct interaction of Bax and Bak proteins with Bcl-2 homology domain 3 (BH3)-only proteins in living cells revealed by fluorescence complementation. J. Biol. Chem. 2013;288:4935–4946. doi: 10.1074/jbc.M112.422204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harada H., Quearry B., Ruiz-Vela A., Korsmeyer S.J. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15313–15317. doi: 10.1073/pnas.0406837101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta S., Deepti A., Deegan S., Lisbona F., Hetz C., Samali A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1α-XBP1 signaling through a physical interaction. PLoS Biol. 2010;8:e1000410. doi: 10.1371/journal.pbio.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cawley K., Logue S.E., Gorman A.M., Zeng Q., Patterson J., Gupta S., Samali A. Disruption of microRNA biogenesis confers resistance to ER stress-induced cell death upstream of the mitochondrion. PLoS ONE. 2013;8:e73870. doi: 10.1371/journal.pone.0073870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta S., Read D.E., Deepti A., Cawley K., Gupta A., Oommen D., Verfaillie T., Matus S., Smith M.A., Mott J.L., Agostinis P., Hetz C., Samali A. Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell Death Dis. 2012;3:e333. doi: 10.1038/cddis.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lei K., Davis R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi X., Wildey G.M., Howe P.H. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J. Biol. Chem. 2006;281:813–823. doi: 10.1074/jbc.M505546200. [DOI] [PubMed] [Google Scholar]
- 30.Puthalakath H., Huang D.C.S., O’Reilly L.A., King S.M., Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell. 1999;3:287–296. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- 31.Concannon C.G., Orrenius S., Samali A. Hsp27 inhibits cytochrome c-mediated caspase activation by sequestering both pro-caspase-3 and cytochrome c. Gene Expr. 2001;9:195–201. doi: 10.3727/000000001783992605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zoubeidi A., Zardan A., Wiedmann R.M., Locke J., Beraldi E., Fazli L., Gleave M.E. Hsp27 promotes insulin-like growth factor-I survival signaling in prostate cancer via p90Rsk-dependent phosphorylation and inactivation of BAD. Cancer Res. 2010;70:2307–2317. doi: 10.1158/0008-5472.CAN-09-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stankiewicz A.R., Livingstone A.M., Mohseni N., Mosser D.D. Regulation of heat-induced apoptosis by Mcl-1 degradation and its inhibition by Hsp70. Cell Death Differ. 2009;16:638–647. doi: 10.1038/cdd.2008.189. [DOI] [PubMed] [Google Scholar]
- 34.MacPhee D.J. Methodological considerations for improving Western blot analysis. J. Pharmacol. Toxicol. Methods. 2010;61:171–177. doi: 10.1016/j.vascn.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 35.Hetz C., Thielen P., Fisher J., Pasinelli P., Brown R.H., Korsmeyer S., Glimcher L. The proapoptotic BCL-2 family member BIM mediates motoneuron loss in a model of amyotrophic lateral sclerosis. Cell Death Differ. 2007;14:1386–1389. doi: 10.1038/sj.cdd.4402166. [DOI] [PubMed] [Google Scholar]