

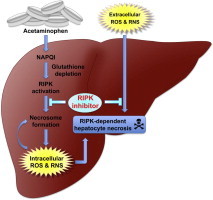
Graphical abstract
Abbreviations: ABTS, 2,2′-azino-bis (3-ethylbenzothiazoline)-6-sulfonic acid; ALF, acute liver failure; ALT, alanine aminotransferase; APAP, acetaminophen; AST, aspartate aminotransferase; bFGF, basic fibroblast growth factor; CM-H2DCFDA, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester; CXCL1, chemokine (C-X-C motif) ligand 1; CYP2E1, cytochrome P450 2E1; DMSO, dimethyl sulfoxide; Drp1, dynamin-related protein 1; FBS, fetal bovine serum; GSH, glutathione; LDH, lactate dehydrogenase; NAPQI, N-acetyl-p-benzoquinone; Nec-1, necrostatin-1; NO, nitric oxide; PGAM5, phosphoglycerate mutase family member 5; PI, propidium iodide; RIPK, receptor-interacting protein kinase; ROS, reactive oxygen species; SNAP, S-nitroso-N-acetyl-dl-penicillamine; WST-8, 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium; λPP, lambda protein phosphatase
Keywords: Hepatocytes, Acetaminophen, Acute liver failure, RIPK-dependent necrosis, Necroptosis, Reactive oxygen species
Highlights
-
•
RIPK-dependent necrosis is involved in acetaminophen (APAP)-induced hepatotoxicity.
-
•
Necrostatin-1 (Nec-1) protects mice against APAP-induced acute liver damage.
-
•
Nec-1 suppresses APAP-induced ROS generation in hepatocytes.
-
•
Nec-1 promotes resistance to oxidative stress in hepatocytes.
Abstract
Excessive acetaminophen (APAP) use is one of the most common causes of acute liver failure. Various types of cell death in the damaged liver are linked to APAP-induced hepatotoxicity, and, of these, necrotic cell death of hepatocytes has been shown to be involved in disease pathogenesis. Until recently, necrosis was commonly considered to be a random and unregulated form of cell death; however, recent studies have identified a previously unknown form of programmed necrosis called receptor-interacting protein kinase (RIPK)-dependent necrosis (or necroptosis), which is controlled by the kinases RIPK1 and RIPK3. Although RIPK-dependent necrosis has been implicated in a variety of disease states, including atherosclerosis, myocardial organ damage, stroke, ischemia–reperfusion injury, pancreatitis, and inflammatory bowel disease. However its involvement in APAP-induced hepatocyte necrosis remains elusive. Here, we showed that RIPK1 phosphorylation, which is a hallmark of RIPK-dependent necrosis, was induced by APAP, and the expression pattern of RIPK1 and RIPK3 in the liver overlapped with that of CYP2E1, whose activity around the central vein area has been demonstrated to be critical for the development of APAP-induced hepatic injury. Moreover, a RIPK1 inhibitor ameliorated APAP-induced hepatotoxicity in an animal model, which was underscored by significant suppression of the release of hepatic enzymes and cytokine expression levels. RIPK1 inhibition decreased reactive oxygen species levels produced in APAP-injured hepatocytes, whereas CYP2E1 expression and the depletion rate of total glutathione were unaffected. Of note, RIPK1 inhibition also conferred resistance to oxidative stress in hepatocytes. These data collectively demonstrated a RIPK-dependent necrotic mechanism operates in the APAP-injured liver and inhibition of this pathway may be beneficial for APAP-induced fulminant hepatic failure.
1. Introduction
Acetaminophen, or N-acetyl-para-amino-phenol (APAP), is the most widely used analgesic and antipyretic [1]. The use of APAP is safe at therapeutic doses, but high doses can lead to acute liver failure (ALF). In 1998, 28% of all ALF cases in the United States were attributed to APAP overdose, which increased to 51% in 2003 [2]. Furthermore, the percentage was much higher in the United Kingdom, where 57% of ALF cases were attributed to APAP use from 1999 to 2008 [3]. The toxicity of APAP is ascribed to N-acetyl-p-benzoquinone imine (NAPQI), a highly reactive metabolite of APAP, which reacts with glutathione (GSH) and leads to a profound depletion of hepatocellular GSH [4], resulting in mitochondrial permeability transition and necrotic cell death [5]. Of note, although necrosis is the major contributing mechanism of APAP-induced hepatic injury [6], various types of cell death resulting from several complicated mechanisms are assumed to play a role in this process [7].
Necrosis has been considered as an accidental and non-regulated cell death process; however, recent studies have shed light on a new concept of regulated necrosis called receptor-interacting protein kinase (RIPK)-dependent necrosis (or necroptosis). The most prominent characteristics of this type of cell death are as follows: (i) RIPK1 kinase activation, which can be assessed by monitoring RIPK1 phosphorylation, and (ii) cell-death, which can be suppressed by several RIPK1 inhibitors, including necrostatin-1 (Nec-1) [8]. Necroptosis results from RIPK1 and RIPK3 kinase activity in the form of a necrosome, which is regulated by ubiquitination and phosphorylation of RIPK1 and RIPK3 [9]. RIPK1–RIPK3 necrosome formation, which is induced by several factors, including tumor necrosis factor alpha (TNF), leads to the overproduction of reactive oxygen species (ROS) and the induction of mitochondrial dysfunction mediated by mitochondrial complex I [10]. Furthermore, the mitochondrial phosphatase PGAM5 and the mitochondrial fission factor Drp1, which cause mitochondrial fragmentation and may up-regulate ROS generation, are intimately involved in RIPK-dependent necrosis [11]. Nec-1 allosterically blocks RIPK1 kinase activity and inhibits RIPK-dependent necrosis, though the activation of RIPK1-mediated NF-κB, mitogen-activated protein kinase p38 and JNK1 remain [12,13]. Nec-1 blocks the formation of the RIPK1–RIPK3 complex, indicating that kinase activity of RIPK1 is required for necrosome formation [13]. The cytoprotective effects of Nec-1 has been shown in several experimental settings, in ischemic brain injury [14], myocardial ischemia–reperfusion [15], as well as radiation-induced cell death in anaplastic thyroid and adrenocortical cancers [16].
Here, we studied the molecular mechanisms involved in APAP-induced ALF and found that RIPK-dependent necrosis is involved in APAP-induced hepatocyte death, suggesting that hepatotoxicity is, at least partly, due to druggable cellular events. Indeed, we also provide evidence that Nec-1 successfully protects against APAP-induced acute hepatotoxicity through the acquisition of resistance to oxidative stress as well as by suppressing ROS production in hepatocytes.
2. Materials and methods
2.1. Animals
Male C57BL/6 mice, 8–10 weeks old, were purchased from CLEA Japan (Tokyo, Japan). To confirm the time-dependent development of hepatotoxicity and the involvement of RIPK1 activation in APAP-induced hepatotoxicity, overnight-fasted mice received a single intraperitoneal injection of 800 mg/kg APAP (Sigma–Aldrich, St. Louis, MO, USA) and were sacrificed 0, 1, 3, and 6 h after administration. To investigate the outcome of RIPK1 inhibition, fasted mice received an intravenous injection of 12.5 mg/kg Nec-1 (Calbiochem, San Diego, CA, USA), which was dissolved in dimethyl sulfoxide (DMSO) diluted in a warm saline solution. Control mice were intravenously injected with the same volume of DMSO in a warm saline solution, because DMSO reduces APAP-induced liver damage [17]. All mice received an intraperitoneal injection of 800 mg/kg acetaminophen dissolved in warm saline 15 min after pretreatment with Nec-1 or DMSO, and were sacrificed 6 h after administration. Blood was collected from the vena cava under general anesthesia, and serum was separated to measure aspartate aminotransferase (AST), alanine aminotransferase (ALT), and lactate dehydrogenase (LDH). Portions of the liver tissue specimens were frozen immediately in liquid nitrogen for further use, and the remaining portions were fixed in 10% neutral buffered formalin for microscopic analysis. The Animal Research Committee of Kyoto University approved the animal protocol, and all experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals promulgated by the National Institutes of Health.
2.2. Immunohistochemical analysis
Formalin-fixed, paraffin-embedded sections were cut into a thickness of 4 μm and mounted on Matsunami adhesive silane-coated glass slides (Matsunami Glass, Osaka, Japan). After deparaffinization and rehydration, the slides were autoclaved in 10 mM citrate buffer for 20 min to retrieve the antigens. Then, endogenous peroxidase was quenched with 0.3% hydrogen peroxide (H2O2) in methanol at room temperature for 10 min. After blocking, the sections were incubated at 4 °C overnight with the following primary diluted antibodies: anti-CYP2E1 (dilution, 1:500; Abcam, Cambridge, UK), anti-RIPK1 (dilution, 1:200; Santa Cruz Biotechnology, Dallas, TX, USA), and anti-RIPK3 (dilution, 1:300; Imgenex, San Diego, CA, USA). Subsequently, the sections were incubated with peroxidase-labeled polymer conjugated secondary antibody (Dako Japan, Tokyo, Japan) for 30 min at room temperature. Immunoreactivity was detected with a diaminobenzidine substrate kit (Dako Japan), and the sections were counterstained with hematoxylin. ImageJ imaging analysis software (National Institutes of Health, Bethesda, MD, USA) was used to quantitate the percentage of necrotic area. Field images at ×100 magnification were selected at random from different individuals. The percentage of necrosis was determined by measuring the total dimension of the field and comparing it with the dimension of the necrotic area.
2.3. Measurement of inflammatory and regenerative cytokines
The frozen mouse liver tissues were homogenized using the FastPrep-24 tissue homogenizer (MP Biomedicals Japan, Tokyo, Japan) with 2.0 mm zirconia beads for 1 min while cooling. Bio-Plex multiplex system (Bio-Rad Laboratories, Hercules, CA, USA) was used in conjunction with the Bio-Plex 200 (Bio-Rad Laboratories) to measure inflammatory mediators, according to the manufacturer’s directions. Data were analyzed using Bio-Plex Manager 6.1 software (Bio-Rad Laboratories).
2.4. Isolation and culture of primary mouse hepatocytes
Primary hepatocytes were isolated from C57BL/6 mice as described previously [18] and cultured on a collagen-coated plastic dish in Williams’ medium E (Life Technologies, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified atmosphere of 5% CO2. The medium was replaced with serum-free Williams’ medium E, including the vehicle (0.05% DMSO) or each of the tested Nec-1 concentrations 3 h after plating. An hour later, the cells were treated with 10 mM APAP (Sigma–Aldrich) and incubated for an additional 18 h. The APAP incubation period was relatively short after isolation, because cultured hepatocytes have less CYP2E1 and are less sensitive to APAP with time [19].
2.5. Cell death assay
Cell death was assayed by measuring LDH in the supernatant using the Cytotoxicity Detection Kit Plus (Roche Diagnostics GmbH, Mannheim, Germany). Then, the cells were incubated for 60–120 min with 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST-8; Dojindo Laboratories, Kumamoto, Japan) reagent supplemented in culture media prior to absorbance readings at 450 nm with an iMark Microplate Absorbance Reader (Bio-Rad Laboratories). Thereafter, the cells were stained with Hoechst 33342 (Dojindo Laboratories) and propidium iodide (PI; Dojindo Laboratories). PI-positive cells were considered as dead cells.
2.6. Total GSH measurements
GSH levels in cultured hepatocytes treated with or without APAP were determined using the Total Glutathione Quantification Kit (Dojindo Laboratories), according to the manufacturer’s instructions. Each GSH value was expressed as a ratio of the control value.
2.7. ROS measurements
ROS content was quantified using the fluorescent dye 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Life Technologies) as described previously [20]. Hepatocytes were isolated from C57BL/6 mice and cultured in 96-well black plates with transparent bottoms in Williams’ medium E containing 10% FBS, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C for 3 h. The cells were then incubated in serum-free media, including 0.05% DMSO and several Nec-1 concentrations for 1 h. Then, 10 mM APAP diluted in serum-free media was added, and the incubation was continued for an additional 6 h. The hepatocytes were subsequently loaded with CM-H2DCFDA (10 μM) diluted in phenol red and serum-free media for 30 min at 37 °C. The cells were then rinsed twice with Williams’ medium E without phenol red. CM-H2DCFDA fluorescence was detected at excitation and emission wavelengths of 490 nm and 520 nm, respectively. ROS formation was measured over a time period of 60 min using a FlexStation 3 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Hepatic ROS generation was detected microscopically using the MitoSOX Red Mitochondrial Superoxide Indicator (Life Technologies), which selectively targets superoxide production in mitochondria, according to the manufacturer’s instructions.
2.8. Protein blotting
Frozen liver tissue or cultured hepatocytes were homogenized in RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) containing benzylsulfonyl fluoride (Tokyo Chemical Industry, Tokyo, Japan), Halt protease inhibitor cocktail (Pierce Biotechnology, Rockford, IL, USA), and PhosSTOP phosphatase inhibitor cocktail (Roche Diagnostics GmbH) or in a buffer containing 20 mM Tris–HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and Halt protease inhibitor cocktail. Aliquots of hepatocyte extracts were fractionated by electrophoresis on a 7.5% or 10% sodium dodecyl sulfate (SDS) polyacrylamide gel (Bio-Rad Laboratories). In some cases, phosphate-affinity gel electrophoresis was performed using gels containing 8.5% acrylamide, 50 μM MnCl2, and 25 μM acrylamide-pendant Phos-tag ligand (NARD Institute, Ltd., Amagasaki, Japan) [21], and an equal aliquot was incubated for 30 min at 30 °C with or without 400 U λPP (New England Biolabs Inc., Ipswich, MA, USA) prior to electrophoresis. The proteins were transferred onto polyvinylidene fluoride membranes (Bio-Rad Laboratories), blocked with Blocking One blocking solution (Nacalai Tesque, Ltd., Kyoto, Japan), and incubated at 4 °C overnight with anti-CYP2E1 (dilution, 1:1000), anti-RIP1 (dilution, 1:1000; Cell Signaling Technology), and anti-β-actin (dilution, 1:1000; Sigma–Aldrich). The membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibodies. Chemiluminescence was detected using Clarity Western ECL substrate (Bio-Rad Laboratories), the membranes were subjected to direct densitometric analysis, and images were captured using a charge-coupled device camera system (LAS-4000 mini; Fujifilm, Tokyo, Japan). Band intensity was quantified using ImageJ software and normalized with respect to β-actin levels as an internal control.
2.9. Cell death induction by an exogenous ROS inducer
To evaluate the effects of ROS on RIPK1-inhibited hepatocytes, the medium was replaced with serum-free Williams’ medium E, including the vehicle (0.05% DMSO), or each of the tested Nec-1 concentrations. After 1 h, H2O2 (30% v/v; Wako Pure Chemical Industries, Osaka, Japan) or the nitric oxide (NO) donor S-nitroso-N-acetyl-dl-penicillamine (SNAP; Sigma–Aldrich), which was diluted to a final concentration of 250 μM and 2 mM in serum-free Williams’ medium E, was added, and the cells were incubated for 6 h. Thereafter, cell death assays were performed.
2.10. 2,2′-Azino-bis (3-ethylbenzothiazoline)-6-sulfonic acid (ABTS) free radical decolorization assay
This spectrophotometric decolorization assay, which is widely used for the assessment of antioxidant activity, measures the loss of color when an antioxidant is added to the blue–green chromophore ABTS radical cation (ABTS∗+). The radical scavenging capacity of a sample was previously determined [22] with some modifications. In brief, ABTS (Wako Pure Chemical Industries) was dissolved in UltraPure Distilled Water (Life Technologies) to a concentration of 7 mM. The ABTS radical cation (ABTS∗+) was produced by reacting ABTS stock solution at a final concentration of 2.45 mM potassium persulfate and allowing the mixture to stand in the dark at room temperature for 12–16 h before use. Because ABTS and potassium persulfate react stoichiometrically at a ratio of 1:0.5, this will result in incomplete oxidation of the ABTS. Oxidation of the ABTS commenced immediately, but the absorbance was not maximal and stable until more than 6 h had elapsed. The radical was stable in this form for more than two days when stored in the dark at room temperature. The ABTS∗+ solution was diluted with UltraPure Distilled Water or ethanol to an absorbance of 0.70–0.80 at 734 nm. After the addition of 100 μL diluted ABTS∗+ solution to 20 μL of necrostatin-1, Inactive control (Calbiochem), which is an inactive analogue of Nec-1, or l(+)-Ascorbic Acid (Wako Pure Chemical Industries), absorbance (734 nm) was measured at exactly 4 min after initial mixing using a FlexStation 3 microplate reader (Molecular Devices).
2.11. Concanavalin A-induced hepatitis
Concanavalin A (Sigma–Aldrich) was dissolved in pyrogen-free normal saline solution at a concentration of 2.0 mg/mL and injected intravenously at a dose of 20 mg/kg body weight to induce hepatitis, as previously described [23].
2.12. Quantitative real-time reverse transcription polymerase chain reaction (qPCR)
Total RNA of mouse liver was isolated and transcribed into cDNA using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) and Superscript III reverse transcriptase (Life Technologies) according to the manufacturer’s instructions. qPCR was performed using THUNDERBIRD SYBR qPCR Mix (Toyobo, Osaka, Japan) on a LightCycler 480 (Roche Diagnostics GmbH). The normalization of relative expression was calculated by the comparative Ct (2−ΔΔCt) method with 18s gene expression. The primer sequences used were as follows: TNF, forward 5′-AGGGTCTGGGCCATAGAACT-3′, reverse 5′-CCACCACGCTCTTCTGTCTAC-3′; Heme Oxygenase-1, forward 5′-CACAGATGGCGTCACTTCGTC-3′, reverse 5′-GTGAGGACCCACTGGAGGAG-3′; 18s, forward 5′-AGTCCCTGCCCTTTGTACACA-3′, reverse 5′-CGATCCGAGGGCCTCACTA-3′.
2.13. Statistical analyses
Results are presented as means ± standard errors of the mean. The two groups were compared using the unpaired Student’s t test and analysis of variance, where appropriate. Multiple groups were compared by one-way analysis of variance using the Tukey’s multiple comparison test or the Kruskal–Wallis test followed by Dunn’s multiple comparisons. A probability (P) value <0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism version 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
3. Results
3.1. APAP-induced hepatotoxicity is associated with RIPK1 activation
Initially, we confirmed the time-dependent development of APAP-induced liver damage in C57BL/6 mice with a single dose of APAP (800 mg/kg). Serum AST, ALT, and LDH levels were increased to 3–6 h after APAP administration (Fig. 1A). Next, we evaluated RIPK1 expression and activation in mouse liver to examine the possibility that RIPK-dependent necrosis is involved in APAP-induced hepatotoxicity. RIPK1 protein expression was observed even in the absence of APAP administration, where APAP administration led to the induction of slow-migrating bands at 1–3 h (Fig. 1B). The upper RIPK1 bands disappeared following treatment of the cell lysates with λPP (Fig. 1B and C), indicating that those bands represented phosphorylated-RIPK1, which has been characterized as a hall mark of RIPK1 activation [24]. These data imply the possibility that RIPK1 may play a role in APAP-induced hepatocyte injury in mice and that RIPK-dependent necrosis is involved in this model. To further assess the involvement of RIPK-dependent necrosis, the localization of RIPK1 and RIPK3 was immunohistochemically investigated (Fig. 1D). RIPK3 was strongly expressed in hepatocytes around the central vein area and co-localized with CYP2E1, which is a key enzyme in the conversion of APAP to highly reactive NAPQI, whereas RIPK1 was diffusely expressed in hepatocytes throughout the entire liver. Of note, APAP-induced cell death mainly proceeds around the central vein [25], where CYP2E1, RIPK1, and RIPK3 expression occur. Interestingly, Dot-like RIPK1 expression was strongly induced in and around the necrotic area after APAP administration (Fig. 1D). These results also suggested that APAP-induced cell death in mice involved RIPK-dependent necrosis.
Fig. 1.
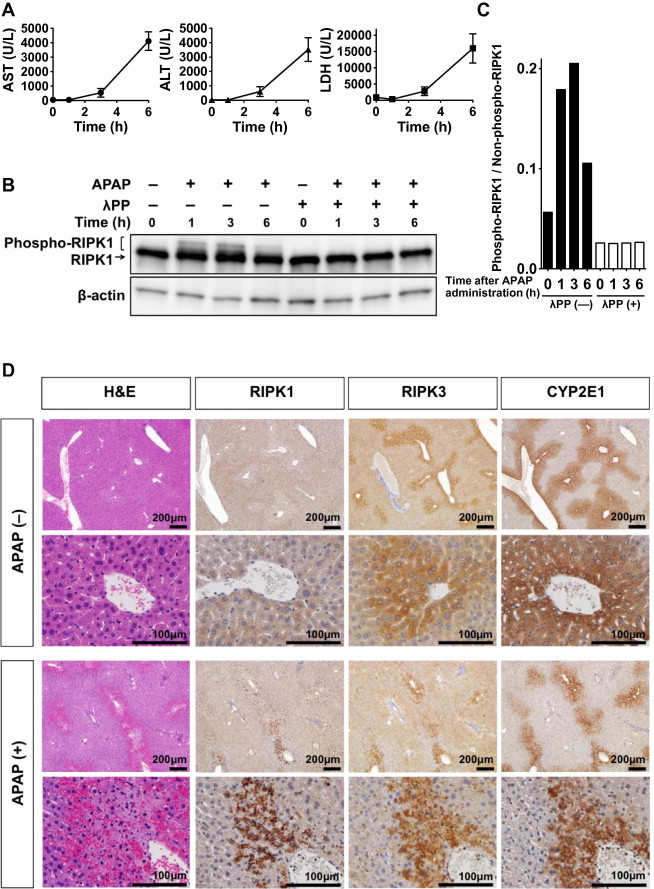
RIPK1 activation during acetaminophen (APAP)-induced liver injury and localization of RIPK1 and RIPK3 in the liver. (A) Time course for the development of APAP toxicity (n = 3 animals per time point). (B) Protein blotting of RIPK1 and phospho-RIPK1. (C) Densitometric analysis of blotted proteins of SDS–PAGE (B). (D) Localization of RIPK1, RIPK3, and CYP2E1 in mouse liver with or without APAP administration. Liver sections stained with hematoxylin and eosin (H and E) are also shown.
3.2. Nec-1 protects against APAP-induced hepatic injury in vivo
To clearly demonstrate the involvement of RIPK1-dependent necrosis in APAP-induced hepatotoxicity, we utilized a murine model of APAP-induced ALF. Hepatic injury was induced by intraperitoneal injection of APAP. Mice that were administered DMSO as a vehicle showed severe hepatic injury, as assessed histologically by hematoxylin and eosin staining (Fig. 2A). We observed increased numbers of swollen hepatocytes around the central vein (Fig. 2A), and severe hemorrhage was visible in the perinecrotic area (Fig. 2A). However, mice treated with Nec-1 showed mild injury, and the necrotic area was localized only to the pericentral vein area (Fig. 2A and B). These results were confirmed by serum transaminase and LDH levels, i.e., Nec-1 showed a significant protective effect against APAP-induced hepatic injury (Fig. 2C). Without administration of APAP, no significant difference was observed between hepatocytes treated either with the vehicle alone or 50 μM Nec-1 alone (data not shown). Nec-1 significantly decreased the production of the inflammatory and regenerative cytokines IL-1β, IL-10, CXCL1, and basic FGF as compared with the vehicle (Fig. 2D). Although, no significant difference was detected in IL-6 concentrations, a tendency towards decreased expression was observed (Fig. 2D). TNF induction, which is a virulence determinant of concanavalin A- and LPS/d-galactosamine-induced hepatitis [23,26–28], was not observed in our experimental ALF settings, i.e., the liver samples were harvested within 6 h after APAP administration (Fig. 3A). However, heme oxygenase-1, which is known to be induced by APAP, was observed even in the early phase of liver injury [29] (Fig. 3B), which is in agreement with previous reports that showed APAP-induction of TNF only occurred in the later phase and not in the acute phase [30,31].
Fig. 2.
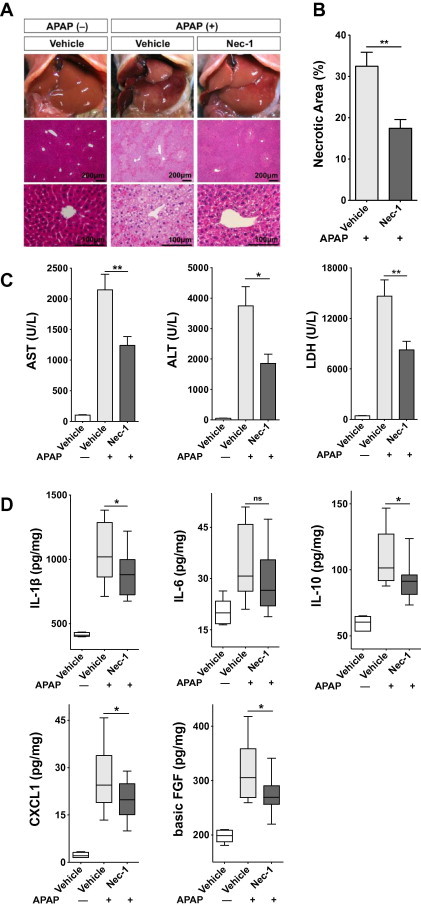
Specific inhibition of RIPK1 kinase by Nec-1 protects mice against acetaminophen (APAP)-induced hepatic injury. (A) Macroscopical and histological examination of Nec-1 protection against APAP. Representative hematoxylin and eosin-stained liver sections are shown. (B) The percent necrotic area for vehicle-treated (n = 9) or Nec-1-treated (n = 9) mice was determined by random evaluation of each hematoxylin and eosin-stained sections. (C) Serum AST, ALT, and LDH levels (n = 21–22). (D) Hepatic IL-1β, IL-6, IL-10, CXCL1, and basic FGF expression was measured by bead-based immunoassays (n = 12). ∗P < 0.05, ∗∗P < 0.01.
Fig. 3.
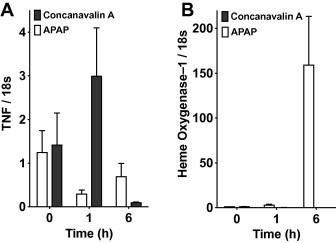
Differential regulation of TNF vs. heme oxygenase-1 mRNA expression in APAP- and concanavalin A-induced acute hepatic injury; mRNA expression of TNF (A) and heme oxygenase-1 (B) in liver tissues. Mice were administered with APAP (800 mg/kg, intraperitoneal injection, n = 3 per time point) or concanavalin A (20 mg/kg, intravenous injection, n = 4 per time point) to induce hepatic injury. Liver samples were harvested before or 1 h and 6 h after administration. Total RNA was prepared at each indicated time point and subjected to quantitative RT-PCR. Error bars show standard error of the mean.
Altogether, these results clearly demonstrated that RIPK1-specific agent Nec-1 has a protective effect against APAP-induced hepatotoxicity and that APAP-induced cell death is due, at least partly, to RIPK-dependent necrosis.
3.3. Nec-1 inhibits ROS production and suppresses mitochondrial dysfunction in APAP-damaged hepatocytes
To investigate the mechanism of how RIPK1 inhibition protects hepatocytes from APAP-induced regulated necrosis, we isolated primary hepatocytes and performed cell-based in vitro assays. In agreement with our in vivo findings, RIPK1-phosphorylation also increased in APAP-treated hepatocytes in a time-dependent manner (Figs. 1B and 4A). Nec-1 protected primary hepatocytes from APAP-induced cytotoxicity in a dose-dependent manner, as shown by the reduction of LDH in the supernatant (Fig. 4B) and by increased cell viability (Fig. 4C). The protective effect of Nec-1 against APAP-induced cell death was also observed microscopically, as assessed by staining with the membrane-impermeant fluorescent molecule PI to evaluate cell death and the membrane-permeant fluorescent dye Hoechst 33342 to stain cell nuclei (Fig. 4D).
Fig. 4.
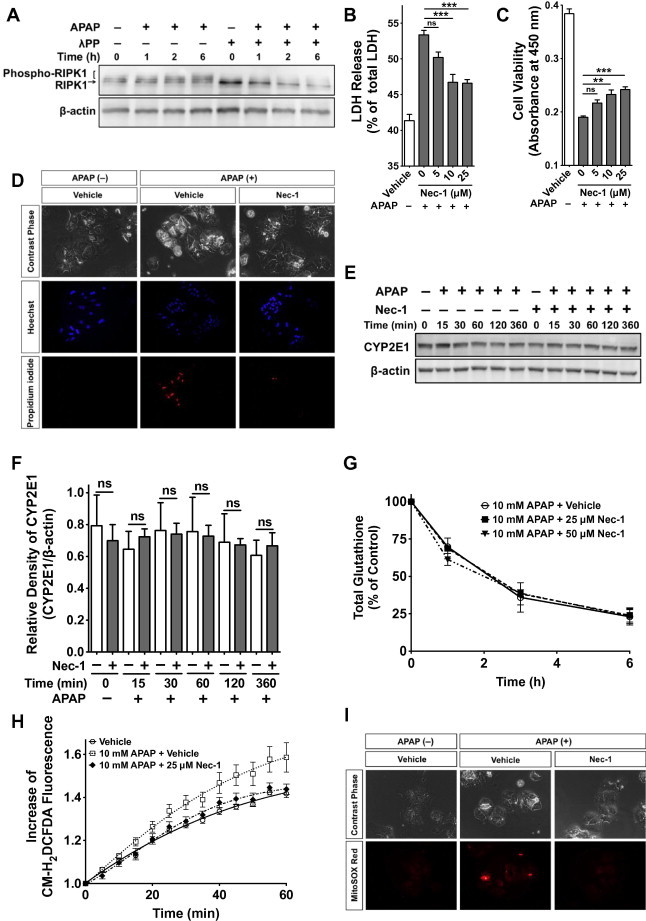
Nec-1 inhibits ROS production and suppresses mitochondrial dysfunction in acetaminophen (APAP)-damaged hepatocytes without affecting CYP2E1 and cellular GSH levels. (A) APAP-induced activation of RIPK1. SDS–PAGE of RIPK1 and phospho-RIPK1 in cultured mouse hepatocytes with or without λPP is shown. (B) Effect of Nec-1 on APAP-induced LDH release (18 h after APAP treatment, n = 6). (C) Effect of Nec-1 on APAP-induced hepatocyte death (18 h after APAP treatment, n = 6) (D) Microscopical analysis of hepatocytes with or without Nec-1 (6 h after APAP treatment). (E) SDS–PAGE of CYP2E1 expression (F) Densitometric analysis of (E) (n = 3). (G) APAP-induced depletion of cellular GSH levels in primary hepatocytes with 25 μM and 50 μM Nec-1 or the vehicle. Data are expressed as percent of non-APAP-treated controls (n = 3). (H) The redox-sensitive dye CM-H2DCFDA (10 μM) was loaded for 30 min. Fluorescent signals were quantified continuously for 60 min (n = 8). (I) Cells were loaded with MitoSOX (2.5 μM) for 6 h and live images were captured. Pictures are representative of three different experiments. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Next, we compared CYP2E1 expression levels to determine whether the Nec-1 protective effect against hepatic injury resulted from altered APAP metabolism. Therefore, we could not detect any significant differences in CYP2E1 expression levels between Nec-1- and vehicle-treated hepatocytes during APAP-induced hepatocyte death (0–6 h post APAP-treatment) (Fig. 4E and F). In addition, we examined the total GSH to assess the detoxification status of the cells, because highly reactive NAPQI is detoxified primarily by GSH conjugation. Nevertheless, total GSH levels in APAP-injured hepatocytes decreased with time regardless of Nec-1 treatment (Fig. 4G).
After GSH depletion, it is considered that NAPQI becomes hepatotoxic by binding to cellular macromolecules, which leads to cellular organ damage including mitochondrial dysfunction through ROS generation [32]. Therefore, we examined the effect of Nec-1 on intracellular ROS induction by APAP and found that Nec-1 suppressed the cellular ROS levels in APAP-treated hepatocytes to levels similar to those in APAP-untreated hepatocytes (Fig. 4H). Similarly, Nec-1 suppressed the mitochondrial ROS induction in APAP-treated hepatocytes, as determined using MitoSOX Red, whereas mitochondrial ROS were strongly induced in APAP-damaged hepatocytes treated with the vehicle alone (Fig. 4I).
These findings raise the possibility that Nec-1 protects against APAP-induced hepatocyte injury by suppressing the intracellular burden of ROS formation, including mitochondrial superoxide production, without affecting CYP2E1 expression or depleting GSH.
3.4. RIPK1 inhibition confers hepatocytes resistance to oxidative stress
Extensive mitochondrial GSH depletion is associated with a significant increase in H2O2 released from stressed mitochondria [33]. Peroxynitrite (ONOO−), which is formed by the reaction of nitric oxide (NO) with superoxide, is detoxified by GSH during APAP-induced hepatotoxicity [34]. These oxidizing agents produced in living cells and/or leaked from dead cells not only damage organs intracellularly, but may also affect neighboring cells extracellularly [35,36]. Therefore, we attempted to test whether RIPK1 inhibition influenced hepatocyte response to exogenous oxidative stress. We treated mouse hepatocytes with H2O2 or SNAP, which serves as a NO donor. Low SNAP concentrations provide protection from cell death in various cell types, including hepatocytes [37–39]; however, at higher concentrations, SNAP is highly toxic [39–42]. Thus, we used SNAP in a concentration range that was toxic to hepatocytes (data not shown) and found a dose-dependent protective effect of Nec-1 against exogenous oxidative stress in cell viability and LDH-release assays (Fig. 5A, B, D and E). A microscopic analysis confirmed that RIPK1 inhibition by Nec-1 protected the primary hepatocytes from exogenous oxidizing agents (Fig. 5C and F). However, there was no antioxidant activity in Nec-1 compared with l(+)-Ascorbic Acid, which is an antioxidant well-known as Vitamin C, as determined by an ABTS free-radical decolorization assay (Fig. 5G and H). This suggests that Nec-1 does not act as a scavenger for H2O2 or SNAP. These results collectively indicate that RIPK1 inhibition not only suppresses ROS production in hepatocytes but also ameliorates hepatocyte damage caused by extracellular ROS (Fig. 6); although the exact mechanism remains to be addressed.
Fig. 5.
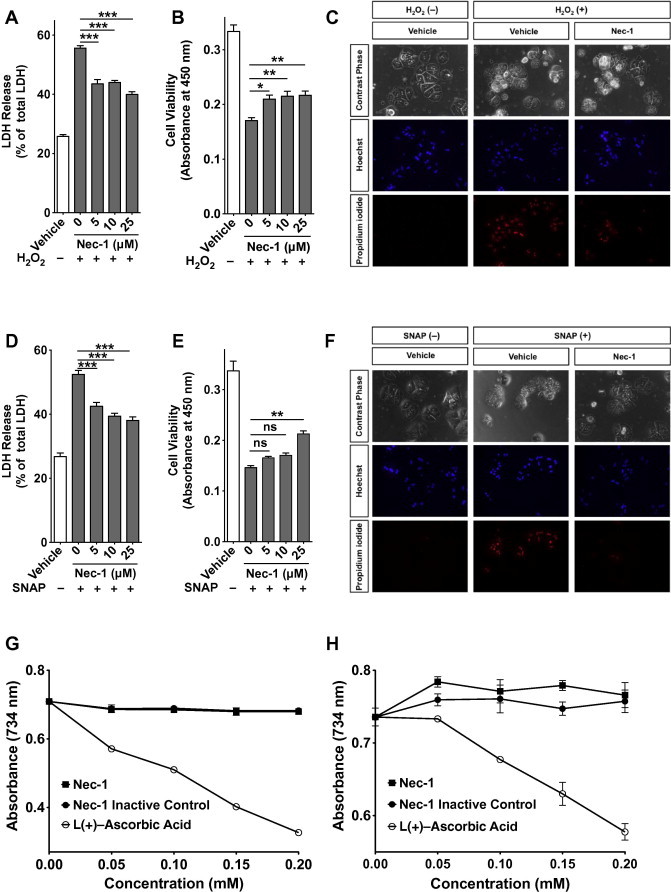
RIPK1 inhibition by Nec-1 confers hepatocytes resistance to oxidative stress, isolated hepatocytes were cultured with different Nec-1 concentrations or the vehicle alone for 1 h and then treated with 250 μM H2O2 or 2 mM SNAP. (A and D) LDH was measured 6 h after H2O2 (A) or SNAP (D) administration (n = 6). (B and E) Cell viability was measured 6 h after H2O2 (B) or SNAP (E) administration using WST-8 (n = 6). (C and F) The cells were stained with Hoechst 33342 and propidium iodide 6 h after adding H2O2 (C) or SNAP (F), and then examined by fluorescent microscopy. (G and H) The antioxidant effect of Nec-1 was evaluated using the ABTS free-radical decolorization assay. An Nec-1 inactive control and l(+)-ascorbic acid were used as controls. Samples were dissolved in either distilled water (G) or ethanol (H) (n = 6). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Fig. 6.
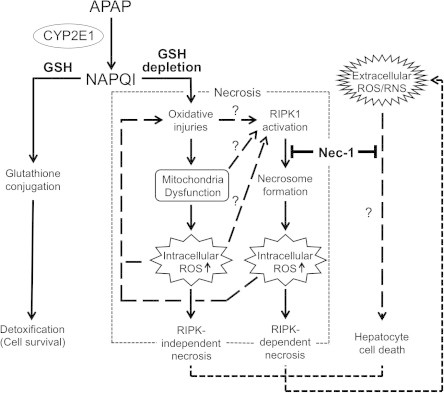
Schematic illustration of the model for the protective effect(s) of Nec-1 against acetaminophen (APAP)-induced hepatocyte necrosis. APAP is converted to the highly reactive metabolite NAPQI by CYP2E1 and other cytochrome P450 enzymes (CYPs) in the liver. NAPQI is inactivated by conjugation to GSH and detoxified. However, excess NAPQI leads to GSH depletion and unconjugated NAPQI binds to cellular proteins, causing further formation of ROS. Resultant oxidative injuries, including mitochondria dysfunction, result in massive hepatic necrosis. Although the mechanism of RIPK1 activation remains unclear, Nec-1 inhibits necrosome formation and intracellular ROS production, which eventually prevent RIPK-dependent necrosis. ROS and reactive nitrogen species (RNS) produced by living cells and/or leaked from dead cells (extracellular ROS/RNS) also provoke hepatocyte cell death. By unknown mechanisms, Nec-1 also inhibits this cell death pathway.
4. Discussion
Our results demonstrate that RIPK-dependent necrosis is involved in the pathological manifestation of APAP-induced acute liver damage. Indeed, RIPK1 inhibition by Nec-1 showed a protective effect against APAP-induced hepatocyte injury both in vivo and in vitro. The in vitro studies revealed that RIPK1 inhibition reduces APAP-induced ROS production, which is one of the main causes of APAP-induced hepatocyte damage (Fig. 5) [43], whereas CYP2E1 expression and APAP-induced GSH exhaustion were unaffected (Fig. 4E–G). A pathological examination of the liver before and after APAP injury demonstrated co-localization of RIPK1 with RIPK3 and CYP2E1 in hepatocytes around the central vein area (Fig. 1D), which is the most vulnerable area for APAP-induced hepatocyte injury [25], also supporting the notion that RIPK-dependent mechanisms operate in APAP liver injury. Furthermore, although Nec-1 has no antioxidant activity (Fig. 4G and H) [14], we found that Nec-1-treated hepatocytes acquired resistance to oxidative stress. This resistance mechanism remains to be elucidated; however, the resistance would be beneficial to further protect against APAP-induced liver damage, because extracellular ROS increase in response to initial hepatocyte cell death [35,36], and those ROS together with inflammatory cytokines lead to secondary hepatocyte damage [44] (Figs. 5 and 6).
APAP-induced cell death is predominantly necrotic [6]. Our results clearly demonstrated that APAP-induced necrosis includes, if not all, regulated forms of necrosis that are a druggable target of RIPK1 inhibitors (Fig. 6). Nec-1 has a short half-life span [45], but demonstrated efficacy against APAP-induced ALF. Thus, the development of inhibitors with better pharmacokinetic properties may pave the way for therapeutic intervention of human APAP-induced ALF, for which efficient treatment is truly required.
Author contributions
K. Takemoto, K.I., E.H. and M.A. designed the study. K. Takemoto, K.I., M.T., and N.N. performed experiments and analyzed data. S.O., K. Toriguchi, K. Tanabe, H.T., S. Seo, K. Taura, K.M., S. Saji and S.U. provided technical and analytical assistance. K. Takemoto, E.H., N.T. and M.A. wrote and edited the manuscript.
Conflicts of interest
M.A. receives a grant from Innovation Center for Immunoregulation and Therapeutics (AK Project). However, the study was designed, conducted, analyzed, and reported independently of the funding. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We are grateful to Dr. Yuji Takemoto and Dr. Takeshi Watanabe for their continuous support and fruitful discussion. This work was supported in part by a grant from the Japanese Ministry of Health, Labour and Welfare (K.I., N.T., S.U., and M.A.); a Grant-in-Aid for Young Scientists (M.A.) and a Grant-in-Aid for Challenging Exploratory Research (M.A.) from Japan Society for the Promotion of Science; and a grant from the Kanae Foundation for the Promotion of Medical Science (M.A.).
Contributor Information
Etsuro Hatano, Email: etsu@kuhp.kyoto-u.ac.jp.
Masataka Asagiri, Email: masa-asagiri@umin.org.
References
- 1.Kaufman D.W., Kelly J.P., Rosenberg L., Anderson T.E., Mitchell A.A. Recent patterns of medication use in the ambulatory adult population of the United States: the Slone survey. JAMA. 2002;287:337–344. doi: 10.1001/jama.287.3.337. [DOI] [PubMed] [Google Scholar]
- 2.Larson A.M., Polson J., Fontana R.J., Davern T.J., Lalani E., Hynan L.S., Reisch J.S., Schiodt F.V., Ostapowicz G., Shakil A.O., Lee W.M., Grp A.L.F.S. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 3.Bernal W., Auzinger G., Dhawan A., Wendon J. Acute liver failure. Lancet. 2010;376:190–201. doi: 10.1016/S0140-6736(10)60274-7. [DOI] [PubMed] [Google Scholar]
- 4.Dahlin D.C., Miwa G.T., Lu A.Y., Nelson S.D. N-Acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. U.S.A. 1984;81:1327–1331. doi: 10.1073/pnas.81.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kon K., Kim J.S., Jaeschke H., Lemasters J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- 6.Gujral J.S., Knight T.R., Farhood A., Bajt M.L., Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol. Sci. 2002;67:322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- 7.El-Hassan H., Anwar K., Macanas-Pirard P., Crabtree M., Chow S.C., Johnson V.L., Lee P.C., Hinton R.H., Price S.C., Kass G.E. Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: roles of cytochrome c, Bax, Bid, and caspases. Toxicol. Appl. Pharmacol. 2003;191:118–129. doi: 10.1016/s0041-008x(03)00240-0. [DOI] [PubMed] [Google Scholar]
- 8.Galluzzi L., Vitale I., Abrams J.M., Alnemri E.S., Baehrecke E.H., Blagosklonny M.V., Dawson T.M., Dawson V.L., El-Deiry W.S., Fulda S., Gottlieb E., Green D.R., Hengartner M.O., Kepp O., Knight R.A., Kumar S., Lipton S.A., Lu X., Madeo F., Malorni W., Mehlen P., Nuñez G., Peter M.E., Piacentini M., Rubinsztein D.C., Shi Y., Simon H.U., Vandenabeele P., White E., Yuan J., Zhivotovsky B., Melino G., Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M., Chan F.K. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vandenabeele P., Declercq W., Van Herreweghe F., Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 2010;3 doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z., Jiang H., Chen S., Du F., Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 12.Degterev A., Hitomi J., Germscheid M., Ch’en I.L., Korkina O., Teng X., Abbott D., Cuny G.D., Yuan C., Wagner G., Hedrick S.M., Gerber S.A., Lugovskoy A., Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 14.Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G.D., Mitchison T.J., Moskowitz M.A., Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 15.Oerlemans M.I., Liu J., Arslan F., den Ouden K., van Middelaar B.J., Doevendans P.A., Sluijter J.P. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia–reperfusion in vivo. Basic Res. Cardiol. 2012;107:270. doi: 10.1007/s00395-012-0270-8. [DOI] [PubMed] [Google Scholar]
- 16.Nehs M.A., Lin C.I., Kozono D.E., Whang E.E., Cho N.L., Zhu K., Moalem J., Moore F.D., Ruan D.T. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery. 2011;150:1032–1039. doi: 10.1016/j.surg.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Park Y., Smith R.D., Combs A.B., Kehrer J.P. Prevention of acetaminophen-induced hepatotoxicity by dimethyl sulfoxide. Toxicology. 1988;52:165–175. doi: 10.1016/0300-483x(88)90202-8. [DOI] [PubMed] [Google Scholar]
- 18.Tamaki N., Hatano E., Taura K., Tada M., Kodama Y., Nitta T., Iwaisako K., Seo S., Nakajima A., Ikai I., Uemoto S. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G498–G505. doi: 10.1152/ajpgi.00482.2007. [DOI] [PubMed] [Google Scholar]
- 19.Wu D.F., Clejan L., Potter B., Cederbaum A.I. Rapid decrease of cytochrome P-450IIE1 in primary hepatocyte culture and its maintenance by added 4-methylpyrazole. Hepatology. 1990;12:1379–1389. doi: 10.1002/hep.1840120620. [DOI] [PubMed] [Google Scholar]
- 20.Paik Y.H., Iwaisako K., Seki E., Inokuchi S., Schnabl B., Osterreicher C.H., Kisseleva T., Brenner D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology. 2011;53:1730–1741. doi: 10.1002/hep.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosako H., Yamaguchi N., Aranami C., Ushiyama M., Kose S., Imamoto N., Taniguchi H., Nishida E., Hattori S. Phosphoproteomics reveals new ERK MAP kinase targets and links ERK to nucleoporin-mediated nuclear transport. Nat. Struct. Mol. Biol. 2009;16:1026–1035. doi: 10.1038/nsmb.1656. [DOI] [PubMed] [Google Scholar]
- 22.Re R., Pellegrini N., Proteggente A., Pannala A., Yang M., Rice-Evans C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med. 1999;26:1231–1237. doi: 10.1016/s0891-5849(98)00315-3. [DOI] [PubMed] [Google Scholar]
- 23.Gantner F., Leist M., Lohse A.W., Germann P.G., Tiegs G. Concanavalin a-induced T-cell-mediated hepatic-injury in mice – the role of tumor-necrosis-factor. Hepatology. 1995;21:190–198. doi: 10.1016/0270-9139(95)90428-x. [DOI] [PubMed] [Google Scholar]
- 24.Declercq W., Vanden Berghe T., Vandenabeele P. IP kinases at the crossroads of cell death and survival. Cell. 2009;138:229–232. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 25.Roberts D.W., Bucci T.J., Benson R.W., Warbritton A.R., McRae T.A., Pumford N.R., Hinson J.A. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. Am. J. Pathol. 1991;138:359–371. [PMC free article] [PubMed] [Google Scholar]
- 26.Maeda S., Chang L.F., Li Z.W., Luo J.L., Leffert H., Karin M. IKK beta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNF alpha. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 27.Pasparakis M., Alexopoulou L., Episkopou V., Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lehmann V., Freudenberg M.A., Galanos C. Lethal toxicity of lipopolysaccharide and tumor-necrosis-factor in normal and d-galactosamine-treated mice. J. Exp. Med. 1987;165:657–663. doi: 10.1084/jem.165.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mobasher M.A., Gonzalez-Rodriguez A., Santamaria B., Ramos S., Martin M.A., Goya L., Rada P., Letzig L., James L.P., Cuadrado A., Martin-Perez J., Simpson K.J., Muntane J., Valverde A.M. Protein tyrosine phosphatase 1B modulates GSK3 beta/Nrf2 and IGFIR signaling pathways in acetaminophen-induced hepatotoxicity. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardner C.R., Laskin J.D., Dambach D.M., Sacco M., Durham S.K., Bruno M.K., Cohen S.D., Gordon M.K., Gerecke D.R., Zhou P., Laskin D.L. Reduced hepatotoxicity of acetaminophen in mice lacking inducible nitric oxide synthase: potential role of tumor necrosis factor-alpha and interleukin-10. Toxicol. Appl. Pharmacol. 2002;184:27–36. [PubMed] [Google Scholar]
- 31.Blazka M.E., Wilmer J.L., Holladay S.D., Wilson R.E., Luster M.I. Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 1995;133:43–52. doi: 10.1006/taap.1995.1125. [DOI] [PubMed] [Google Scholar]
- 32.Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J. Pharmacol. Exp. Ther. 1990;255:935–941. [PubMed] [Google Scholar]
- 33.Han D., Canali R., Rettori D., Kaplowitz N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol. Pharmacol. 2003;64:1136–1144. doi: 10.1124/mol.64.5.1136. [DOI] [PubMed] [Google Scholar]
- 34.Sies H., Sharov V.S., Klotz L.O., Briviba K. Glutathione peroxidase protects against peroxynitrite-mediated oxidations. A new function for selenoproteins as peroxynitrite reductase. J. Biol. Chem. 1997;272:27812–27817. doi: 10.1074/jbc.272.44.27812. [DOI] [PubMed] [Google Scholar]
- 35.Laskin J.D., Heck D.E., Gardner C.R., Laskin D.L. Prooxidant and antioxidant functions of nitric oxide in liver toxicity. Antioxid. Redox Signal. 2001;3:261–271. doi: 10.1089/152308601300185214. [DOI] [PubMed] [Google Scholar]
- 36.Dragomir A.C., Laskin J.D., Laskin D.L. Macrophage activation by factors released from acetaminophen-injured hepatocytes: potential role of HMGB1. Toxicol. Appl. Pharmacol. 2011;253:170–177. doi: 10.1016/j.taap.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farinelli S.E., Park D.S., Greene L.A. Nitric oxide delays the death of trophic factor-deprived PC12 cells and sympathetic neurons by a cGMP-mediated mechanism. J. Neurosci. 1996;16:2325–2334. doi: 10.1523/JNEUROSCI.16-07-02325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatano E., Bennett B.L., Manning A.M., Qian T., Lemasters J.J., Brenner D.A. NF-kappaB stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis. Gastroenterology. 2001;120:1251–1262. doi: 10.1053/gast.2001.23239. [DOI] [PubMed] [Google Scholar]
- 39.Shen Y.H., Wang X.L., Wilcken D.E.L. Nitric oxide induces and inhibits apoptosis through different pathways. FEBS Lett. 1998;433:125–131. doi: 10.1016/s0014-5793(98)00844-8. [DOI] [PubMed] [Google Scholar]
- 40.Bal-Price A., Brown G.C. Nitric-oxide-induced necrosis and apoptosis in PC12 cells mediated by mitochondria. J. Neurochem. 2000;75:1455–1464. doi: 10.1046/j.1471-4159.2000.0751455.x. [DOI] [PubMed] [Google Scholar]
- 41.Figueroa S., Oset-Gasque M.J., Arce C., Martinez-Honduvilla C.J., Gonzalez M.P. Mitochondrial involvement in nitric oxide-induced cellular death in cortical neurons in culture. J. Neurosci. Res. 2006;83:441–449. doi: 10.1002/jnr.20739. [DOI] [PubMed] [Google Scholar]
- 42.Uchiyama T., Otani H., Okada T., Ninomiya H., Kido M., Imamura H., Nogi S., Kobayashi Y. Nitric oxide induces caspase-dependent apoptosis and necrosis in neonatal rat cardiomyocytes. J. Mol. Cell. Cardiol. 2002;34:1049–1061. doi: 10.1006/jmcc.2002.2045. [DOI] [PubMed] [Google Scholar]
- 43.Jaeschke H., McGill M.R., Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012;44:88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Degterev A., Maki J.L., Yuan J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013;20:366. doi: 10.1038/cdd.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]