

Graphical abstract
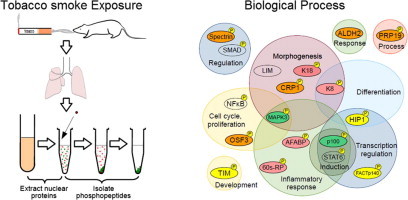
Abbreviations: COPD, chronic obstructive pulmonary disorder; FACTp140, FACT complex subunit SPT16; K18, keratin type 1 cytoskeletal 18; AFABP, adipocyte fatty acid-binding protein; OSF3, peroxiredoxin-1; SPTBN1, spectrin β chain brain 1; STAT, signal transducer and activator of transcription; IL, interleukin; K8, keratin type 2 cytoskeletal 8; PRP19, pre-mRNA-processing factor 19; CRP1, cysteine and glycine-rich protein 1; Jak2, tyrosine-protein kinase JAK2; pSTAT3-Tyr705, phosphorylated STAT3; TIM, mitochondrial import inner membrane translocase subunit Tim9; HIP1, Huntingtin-interacting protein 1; 60s-RP, 60s ribosomal protein L10E; TNF, tumor necrosis factor; ALDH2, aldehyde dehydrogenase, mitochondrial; PRP19, pre-mRNA-processing factor 19; ROS, reactive oxygen species; TNFR2, tumor necrosis factor receptor 2; JNK, c-Jun NH2-terminal kinase; ERK(1/2), extracellular signal regulated kinase 1/2; NF-κB, nuclear factor-kappa B; PKC-α, protein kinase C-α; p100, serine protease P100; MAPK3, mitogen-activated protein kinase 3; TGF-β, Transforming growth factor-β; TRAP1, heat shock protein 75 kDa; LIM, LIM/homeobox protein
Keywords: Tobacco smoke exposure, Nuclear phosphoprotein, Phosphoproteomic analysis, Signaling pathways
Highlights
-
•
We analyzed nuclear phosphoprotein expression activated by tobacco smoke exposure.
-
•
253 phosphoproteins were identified in 1-day and 7-day exposure groups.
-
•
Of these, 33 were significantly differentially expressed in control and exposed groups.
-
•
Identified proteins were related to inflammation, response to stress and nicotine.
-
•
OSF3 and spectrin β chain were identified as candidate tobacco smoke markers.
Abstract
Smoking is a risk factor for lung diseases, including chronic obstructive pulmonary disease and lung cancer. However, the molecular mechanisms mediating the progression of these diseases remain unclear. Therefore, we sought to identify signaling pathways activated by tobacco-smoke exposure, by analyzing nuclear phosphoprotein expression using phosphoproteomic analysis of lung tissue from mice exposed to tobacco smoke. Sixteen mice were exposed to tobacco smoke for 1 or 7 days, and the expression of phosphorylated peptides was analyzed by mass spectrometry. A total of 253 phosphoproteins were identified, including FACT complex subunit SPT16 in the 1-day exposure group, keratin type 1 cytoskeletal 18 (K18), and adipocyte fatty acid-binding protein, in the 7-day exposure group, and peroxiredoxin-1 (OSF3) and spectrin β chain brain 1 (SPTBN1), in both groups. Semi-quantitative analysis of the identified phosphoproteins revealed that 33 proteins were significantly differentially expressed between the control and exposed groups. The identified phosphoproteins were classified according to their biological functions. We found that the identified proteins were related to inflammation, regeneration, repair, proliferation, differentiation, morphogenesis, and response to stress and nicotine. In conclusion, we identified proteins, including OSF3 and SPTBN1, as candidate tobacco smoke-exposure markers; our results provide insights into the mechanisms of tobacco smoke-induced diseases.
1. Introduction
Smoking is a risk factor for various lung diseases, including chronic obstructive pulmonary disease (COPD) [1–4] and lung cancer [4–7]. Many studies have attempted to clarify the molecular mechanisms responsible for the induction and progression of these diseases [8]; however, these mechanisms have not yet been fully elucidated. Moreover, while many studies have demonstrated that accumulation of repeated tobacco-related cell injury is involved in the pathogenesis of these diseases [9,10], few reports have investigated the effect of acute smoke exposure [11,12]. In animal models, short-term exposure to tobacco smoke has been shown to cause activation of various pathways and processes [13], such as immunity [14,15], response to oxidative stress [16,17], somatic mutations, gene expression [18–23], and epigenetic mechanisms [16,24]; it is thought that these pathways may also be activated in response to short-term tobacco smoke exposure in humans.
In order to elucidate the molecular mechanisms associated with tobacco smoke exposure, approaches focusing on the induction of toxic changes during disease induction and progression, such as cell proliferation [25], chronic inflammation [26,27], and inhibition of apoptosis [28], have been considered previously. Moreover, biomarkers induced in lung cancer [29], COPD [30], and diseases caused by exposure to environmental toxic substances have been investigated actively in recent years, because such factors were expected to provide effective tools for the early diagnosis, prevention, and cure of these diseases. cDNA microarrays have also been applied to identify markers related to tobacco smoke [31,32]. However, information on mRNA expression alone is not enough to elucidate the underlying regulatory mechanisms. For example, when tobacco smoke causes inflammation of the lung, the signal transducer and activator of transcription 3 (STAT3) is phosphorylated by interleukin (IL)-6 and translocates into the nucleus, where it regulates transcription. Despite its usefulness, cDNA microarray analysis cannot be used to analyze such events.
However, functional changes in proteins are often regulated by post-translational modifications, such as phosphorylation. Therefore, proteomic methods, which are able to detect protein expression and post-translational modifications that are crucial to biological events, can be particularly useful for the detection of functional molecules. Proteomic approaches facilitate identification of markers for early diagnosis of diseases. In contrast to other research methods that identify individual genes, proteins, or pathways, proteomic methods provide a more systematic perspective, which can enrich our understanding of pulmonary diseases related to tobacco smoke exposure. Proteomic approaches can be used to identify and track cell signaling pathways; this can play an important role in estimating biological changes that occur in human diseases. However, changes in specific signaling pathway components in response to toxic substances have not yet been clarified. Therefore, it is important to establish new methods for detecting changes in regulatory systems, proteins making up these systems, and post-translational modifications, such as phosphorylation, of such proteins in the tissues of animals and humans.
Therefore, in the current study, we sought to use proteomic methods to clarify the nuclear phosphoproteins involved in signaling pathways induced by short-term tobacco smoke exposure, representing the acute phase response, in the mouse, by using a nose-only, flow-past inhalation exposure chamber system to best mimic human exposure [33,34]. We established a novel strategy for eluting and purifying nuclear phosphoproteins, and analyzed these proteins by mass spectrometry. Our analysis provided insights into the proteins activated in the acute phase response to tobacco smoke exposure and may lead to identification of novel tobacco smoke exposure-related biomarkers.
2. Materials and methods
2.1. Animals and tobacco smoke exposure conditions
Mice were treated according to the Japanese National Animal Welfare Regulations, and the study protocol was approved by the Animal Care and Use Committee of Kumamoto University. A total of 32 male ICR mice weighing 45–50 g each (age: 10 weeks) were used. Mice were obtained from Japan SLC, Inc. (Shizuoka, Japan). They were housed in a room illuminated for 12 h (lights on 07:00–19:00) and kept at 22 ± 2 °C during the experiment. Food (CE2, CLEA Japan, Inc., Tokyo, Japan) and water were freely available. Mice were divided into 4 groups: 1-day exposure, 1-day control, 7-day exposure, and 7-day control (n = 8 mice per group).
To expose the mice to tobacco smoke, a nose-only, flow-past inhalation exposure chamber system was used. The system consists of 3 parts: a tobacco smoke generator (SCIREQ Inc., Montreal, Canada), exposure chamber (CR Equipment S.A., Gland, Switzerland), and an air filtering system and compressor (NES-1000; SHINTECHNO, Fukuoka, Japan) (Fig. 1A). Compared to conventional systems, this nose-only, flow-past system suppresses the effects of secondhand smoke (Fig. 1B). The exposure experiment was conducted in an iteration process; tobacco smoke was puffed in the mouse’s muzzle and the mouse inspired the smoke, and then exhaled the smoke. The exhaled smoke (and the rest of the smoke) was removed by the vacuum system. Mice were exposed to tobacco smoke in a chamber for 30 min once a day, for 1 or 7 days. Twenty-four University Kentucky 3R4F cigarettes for animal tobacco exposure experiments were burned per day and each cigarette was puffed 10 times. Control mice were held in mouse holders and were exposed to room air for the same duration as mice in the tobacco smoke exposure groups.
Fig. 1.
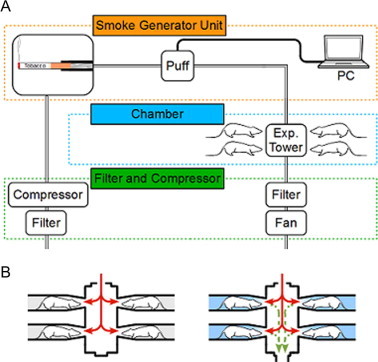
Panel A: schematic of the nose-only, flow-past inhalation exposure chamber system. Panel B: schematic of the exposure system. This system removes the exhaled smoke and suppresses the effect of secondhand smoke (right).
2.2. Sample preparation
For morphological studies, 4 mice in each group were sacrificed by intraperitoneal injection of pentobarbital, and the lung tissues were isolated and fixed with 4% phosphate-buffered paraformaldehyde. Tissues were embedded in paraffin and sections were stained with hematoxylin and eosin (HE) stain.
For proteomic studies, 4 mice in each group were sacrificed within 1 h after the last exposure. The right lung was used for protein extraction, and the left lung of mice in the 7-day control and tobacco smoke exposure group was used for immunostaining. After perfusion with phosphate-buffered saline (PBS) containing protease and phosphatase inhibitors, lung tissue was isolated. The left lung was kept frozen at −80 °C; thin sections were prepared and subjected to immunofluorescence staining. To enrich for nuclear proteins, the lung tissue from the right lung was homogenized on ice in elution buffer from a nuclear protein dissociation kit (Thermo Scientific Pierce, Rockford, IL, USA). The homogenate was vortexed for 15 s, followed by a 10-min incubation on ice. After adding cytosolic elution buffer, the homogenate was vortexed for 5 s, followed by a 1-min incubation on ice. The homogenate was then centrifuged at 15,000 rpm for 5 min, and the supernatant was removed as the cytosolic protein extract. The pellet was vortexed in nuclear dissociation buffer for 15 s every 10 min, for 40 min in total. The extract was again centrifuged at 15,000 rpm for 10 min, and the supernatant, including nuclear proteins, was collected. Nuclear proteins were digested with trypsin prior to peptide elution. To selectively isolate phosphopeptides, polymer-based metal ion affinity capture (PolyMAC; Tymora Analytical Operations, LLC, West Lafayette, IN, USA) was used. The nuclear peptide extracts were allowed to react with TiO2-coated magnetic capture beads to isolate phosphopeptides [35]. After agitation, of the beads, nuclear phosphopeptides were eluted. The samples were resolved in Trifluoroacetic acid (TFA) (0.1% TFA/2% methyl cyanide (MeCN)/DW) and analyzed by electrospray ionization (ESI)-TRAP.
2.3. Western blotting
For confirmation of protein phosphorylation, the cytosolic protein extracts and nuclear eluates of the 7-day control and exposure groups were used for Western blotting. The respective cytosolic protein extracts and nuclear eluates from 4 mice in each group were mixed, electrophoresed on polyacrylamide gels, and transferred to polyvinylidene fluoride membrane (GE Healthcare UK Ltd., Little Chalfont, England); the membrane was then blocked with Tris-buffered saline (pH 7.4) containing 0.1% Tween 20 (Wako, Osaka, Japan) and 5% skim milk. The membrane was then incubated with a primary rabbit antibody against phosphorylated STAT3 (Tyr705; Cell Signaling Technology, Inc., Boston, MA, USA) and a primary mouse antibody against β-actin (Sigma–Aldrich, St. Louis, MO, USA) for 12 h and washed with Tween-TBS. Next, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (GE Healthcare) against mouse or rabbit IgG for 1 h, and washed with Tween-TBS. Immunoreactivities to antibodies were visualized using an enhanced chemiluminescence system (PerkinElmer, Inc., Winter Street Waltham, MA, USA).
2.4. Mass spectrometry
The samples were analyzed by mass spectrometry (MS). The μ-HPLC/NSI-MS/MS system was comprised of an Advance Nano UHPLC (Michrom Bioresources Inc., Auburn, CA, USA) for high-performance liquid chromatography (HPLC), HTS-PAL auto sampler (CTC Analysis, Zwingen, Switzerland), and mass spectrometer (amaZonTM ETD, Bruker, Billerica, MA, USA). The samples were loaded onto a reverse-phase column (Zaplous column C18, 3 μm 0.1 mm ID × 150 mm; AMR Inc., Tokyo, Japan) for separation, and nuclear phosphoproteins were identified as described below.
2.5. Data analysis
Investigation of all MS/MS data was performed using the MASCOT search engine (Matrix Science, Ltd., London, UK) against the Swiss-Prot database. The data obtained from 4-protein digests were examined against the other mammalian subsets of the sequences. The MS/MS data search was performed using the Mus musculus (mouse) subsets of sequences. We searched the database in view of fixed modifications on cysteine residues (carbamidomethyl, +57 Da), variable modifications on methionine residues (oxidation, +16 Da), serine/threonine residues (phosphorylation), and tyrosine residues (phosphorylation). The peptide and fragment mass tolerances were set to ±1.2 Da and ±0.6 Da, respectively.
2.6. Immunostaining
Among the candidate nuclear proteins identified by MS, we focused on OSF3 and spectrin β chain, and performed immunofluorescence staining for these molecules in order to confirm the expression pattern and localization. Control and tobacco exposure lung tissues were embedded with OCT compound. Frozen sections of 10-μm thickness were prepared. After activation, lung tissue sections were blocked with 1% bovine serum albumin for 30 min, and were washed 3 times with PBS before being incubated with primary antibodies, diluted at 1:1000, for 2 h. After washing with PBS 3 times, sections were incubated with secondary antibodies, diluted at 1:100, for 1 h. Immunoreactive signals were detected using Alexa488 fluorescent dye from the tyramide signal amplification kit (T20922; Life Technologies, Carlsbad, CA, USA). DAPI was diluted with the mounting agent, and this DAPI-containing mounting agent used to seal the lung tissue. Immunoreactive fluorescent signals were observed using an epifluorescence microscope system (Olympus, Tokyo, Japan). An anti-OSF3 antibody (AB41906; Abcam, Cambridge, UK) and an anti-SPTBN1 antibody (19722-1-AP, Protein Tech Group. Inc., Chicago, IL, USA) were used as primary antibodies in this study.
3. Results
3.1. Translocation of phosphorylated STAT3 to the nucleus after tobacco smoke exposure
Previous studies have shown that exposure to tobacco smoke activates inflammatory signaling pathways; for example, in response to tobacco smoke, cytoplasmic STAT3 is phosphorylated by tyrosine-protein kinase2 (JAK2) and translocates into the nucleus, where it functions as a transcription factor [36]. Therefore, we used detection of phosphorylated STAT3 (pSTAT3-Tyr705) to indicate that nuclear proteins were properly purified and that the tobacco smoke exposure was effective. As expected, Western blotting analysis using an anti-pSTAT3-Tyr705 antibody demonstrated an increase in pSTAT3-Tyr705 in the nuclear fraction of mouse lung homogenates after exposure to tobacco smoke (Fig. 2).
Fig. 2.
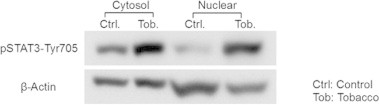
Changes in phosphorylation of STAT3 at tyrosine 705 (pSTAT3-Tyr705) as assessed by Western blotting. The panel shows the immunoreactive bands of pSTAT3-Tyr705. Lane 1: cytosolic fraction of control mice, Lane 2: cytosolic fraction of tobacco smoke exposed mice, Lane 3: nuclear fraction of control mice, Lane 4: nuclear fraction of tobacco smoke exposed mice. β-Actin was used as a loading control.
3.2. Histological changes induced by tobacco smoke exposure
In order to verify that the tissues were modified after exposure to tobacco smoke, we observed HE-stained lung tissues, including the trachea, terminal tracheal, and alveoli, of the tobacco smoke exposed mice. These results showed hyperplasia of the tracheal tract epithelium (Fig. 3A), prominent Clara cells (Fig. 3B), and accumulation of inflammatory cells in the alveoli (Fig. 3C).
Fig. 3.
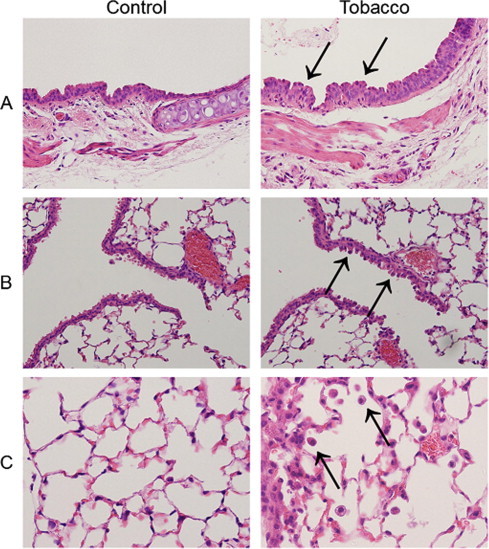
Micrographs of lung tissues obtained from control mice and mice exposed to tobacco smoke for 7 days. Panel A shows the tracheal epithelium of a control mouse (left) and a mouse exposed to tobacco smoke for 7 days (right; magnification 400×). Arrows indicate hyperplasia of the epithelium. Panel B shows terminal bronchiole. Arrows indicate prominent Clara cells. Panel C shows the alveoli. Arrows indicate macrophages.
3.3. Biological functional analysis of identified nuclear phosphoproteins
Purified nuclear phosphoproteins were analyzed by MS (Figs. 4 and 5). Nuclear phosphopeptides that were expressed specifically in each group, as well as across groups, were identified (Fig. 6). A total of 253 phosphoproteins were identified in this experiment (data not shown). Among them, 21 proteins were commonly expressed in the 1-day and 7-day exposure groups. Additionally, 70 proteins were identified as 1-day exposure-specific proteins, while 49 proteins were identified as 7-day exposure-specific proteins (Fig. 7).
Fig. 4.
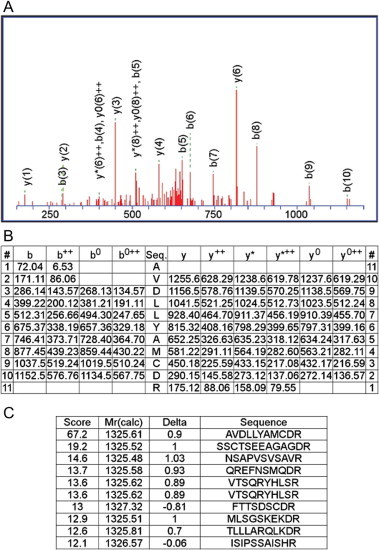
MS/MS data of the AP-2 complex subunit alpha-2 peptide as a representative phosphoprotein detected in this study. Panel A shows one of the MS/MS spectra used to identify the m/z 1199 ion-trap peak as a fragment of the AP-2 complex subunit alpha-2 peptide AVDLLYAMCDR. Spectra were obtained by LC–MS/MS. b++, y++: divalent ion, b0, y0: –H2O, y∗: –NH3.
Fig. 5.
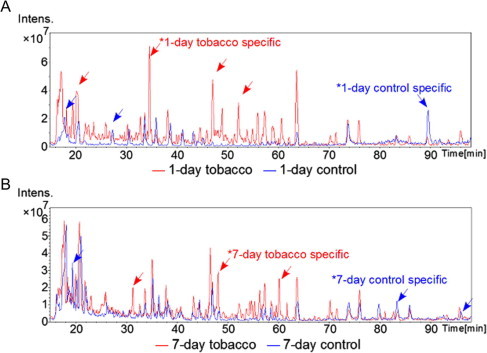
Spectrum of the detected peptides (A: 1-day control group in blue and 1-day exposed group in red; and B: 7-day control group in blue and 7-day exposed group in red). Blue arrows indicate control-specific peaks and red arrows indicate tobacco exposure-specific peaks. The vertical axis indicates intensity, and the horizontal axis indicates retention time, which has a linear relationship with the molecular mass of the protein. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 6.
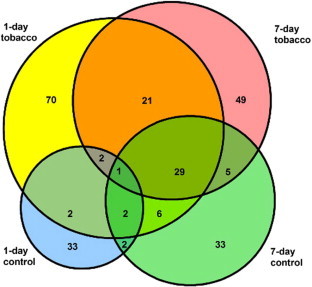
Venn diagram showing protein profile overlaps between each group. The area of each circle is proportional to the number of identified proteins.
Fig. 7.
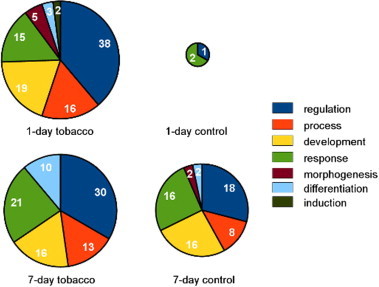
Pie charts representing the biological functional classification of identified proteins in each group. Identified proteins were classified using ProteoIQ software. The area of each circle is proportional to the number of proteins identified in each group.
The identified phosphoproteins were analyzed semi-quantitatively in silico by ProteoIQ software, and a heat map of the proteins was constructed to show the differential expression of proteins in each group. A total of 33 proteins showed significantly different expression between control and exposure groups. Ten nuclear phosphoproteins were specifically expressed in the 1-day exposure group, 8 in the 7-day exposure group, and 13 were specifically expressed in both the tobacco smoke exposed groups. Additionally, expression of 2 phosphoproteins was detected specifically in the control groups (Fig. 8).
Fig. 8.
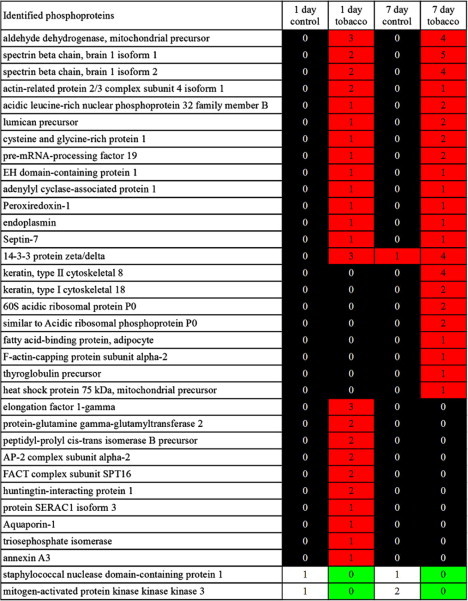
Heat map of phosphoproteins exhibiting differential expression after exposure to tobacco smoke. The table shows the result of semi-quantitative analysis using ProteoIQ software. “Identified phosphoproteins” represents the list of protein names detected by MS. The number in each cell indicates the number of peptides detected by MS.
The identified phosphoproteins were classified according to biological function using ProteoIQ software. This revealed that the differentially expressed proteins were related to inflammation, regeneration, repair, proliferation, differentiation, morphogenesis, and response to stress and nicotine. After 1 day of tobacco smoke exposure, the number of detected proteins that were related to induction and morphogenesis increased significantly. Additionally, the expression of the mitochondrial import inner membrane translocase subunit Tim9 (TIM), FACT complex subunit SPT16 (FACTp140), Huntingtin-interacting protein 1 (HIP1), proteins that function in development, transcription, and cell differentiation, were induced by 1 day of exposure to tobacco smoke.
In the 7-day exposure group, the number of detected proteins related to differentiation and response was increased. Among them were AFABP, keratin type 2 cytoskeletal 8 (K8), and 60s ribosomal protein L10E (60s-RP), which respond to inflammatory factors, such as IL-4, IL-6, or tumor necrosis factor (TNF), and induce cell differentiation and morphogenesis. OSF3 and septin-7, which were detected in both of the exposure groups, play crucial roles in the cell cycle, proliferation, and mitosis. Expression of aldehyde dehydrogenase, mitochondrial (ALDH2), was also detected in both exposure groups.
3.4. Nuclear localization of OSF3 and SPTBN1
We examined the immunostaining for OSF3 and SPTBN1 in the lung tissue. OSF3 and SPTBN1 were highly expressed in the exposure groups compared to the control groups. These results corresponded to the result of MS analysis, which indicated that these proteins were expressed specifically in the exposure groups. Moreover, co-immunostaining with DAPI, a nuclear stain, indicated that OSF3 and SPTBN1 were localized in the nucleus. This demonstrates that nuclear proteins were appropriately purified using our method (Fig. 9).
Fig. 9.
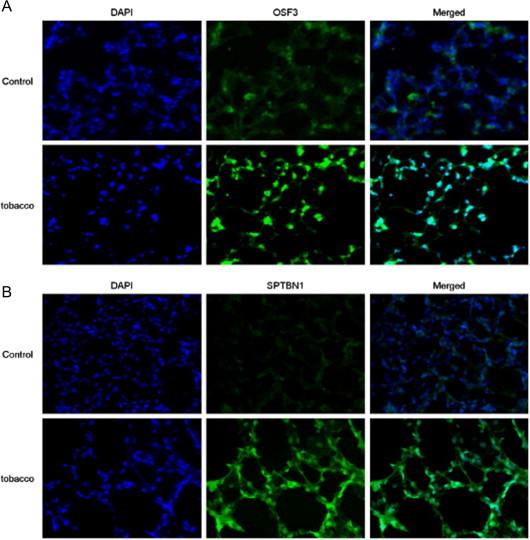
Micrographs of the immunofluorescently stained lung tissues obtained from control and mice exposed to tobacco smoke for 7 days. Panel A shows micrographs of DAPI staining (left), OSF3 staining (center), and the merged image (right). Panel B shows micrographs of DAPI staining (left), SPTBN1 staining (center), and the merged image (right; magnification 400×).
4. Discussion
Using our novel proteomic analysis approach, we found that ALDH2, SPTBN1, OSF3, and pre-RNA processing factor 19 (PRP19) showed increased expression in the tobacco smoke exposure groups. This is the first study to report these proteins as tobacco smoke exposure-related markers. ALDH2 has previously been shown to be expressed at high levels in the liver, heart, kidneys, and muscles of mice, and at low levels in most other tissues, in response to nicotine [37]. We did observe increased expression of ALDH2 in response to tobacco smoke exposure in this study; ALDH2 expression could be induced to protect cells from cell death related to the generation of reactive oxygen species (ROS), since this protein has been reported to attenuate hyperoxia-induced cell death [38]. Spectrin β chain has been shown to mediate TGF-β signaling [39], and in a study using heterozygous knockout mice [40], TGF-β has been shown to function in sustaining alveolar cell membranes. This implies that the expression of SPTBN1 may have been increased in order to repair and regenerate impaired lung tissue. OSF3 is induced by ROS to function as an antioxidant protein [41,42] and protects cells from oxidant-mediated damage to DNA [43,44]. Thus, OSF3 may have been upregulated in order to protect lung cells from being damaged by oxidants generated in response to tobacco smoke. PRP19 has been shown to function in the repair of DNA double-stranded breaks [45], suggesting that its enhanced expression may be related to the repair of DNA that has been damaged by toxic substances in tobacco smoke, particularly ROS. Expression of heat shock protein 75 kDa (TRAP1) was increased in the 7-day exposure group, and TRAP1 has been shown to function in moderating inner cell accumulation of ROS, protecting cells from damage by ROS and inhibiting apoptosis [46]. Thus, the tobacco smoke exposure-related molecules detected in the present study may have been induced to protect against cell injury.
Our proteomic analysis also revealed an increase in the expression of annexin in both 1- and 7-day exposure groups and an increase in the expression of K8 and K18 in the 7-day exposure group. Previous studies have reported the upregulation of annexin and K8 in lung tissue of rats exposed that had been exposed to tobacco smoke [47]. Annexin has been demonstrated to play a role in mediating the anti-inflammatory effects of glucocorticoids [48], which suggests that annexin could function in suppressing inflammation induced by tobacco smoke exposure. K8 and K18 have been shown to be phosphorylated under conditions of heat stress [49] and cell cycle arrest at G2/M transition [50]. Phosphorylation of K18, in particular, modulates the S and G2/M phases of cell cycle in relation to filament reorganization [51]. Phosphorylation of K18 during mitosis is reported to regulate its binding to 14-3-3 protein, one of the proteins found to exhibit a high expression level in the exposure groups in this study. Therefore, upregulation of phosphorylated K8 and K18 may have resulted from activation of a signaling pathway involved in cell cycle arrest and cell injury repair in response to tobacco smoke exposure. Additionally, K8 and K18 have been reported to bind to the cytoplasmic domain of TNF receptor 2 (TNFR2), which moderates the c-Jun NH2-terminal kinase (JNK) signaling pathway and inactivates nuclear factor-kappa B (NF-κB) [52]. Therefore, K8 and K18 may have been upregulated in order to moderate JNK signaling and to suppress inflammation and apoptosis. In contrast, another study in rats that had been exposed to tobacco smoke for 5 days reported that the intensity of staining for JNK in epithelial cells in lung parenchyma and for protein kinase C-α (PKC-α) in macrophages were increased, suggesting that PKC-α is activated by exposure to tobacco smoke and leads to the activation of NF-κB through the activation of JNK [53]. In addition, another study has reported increased expression of JNK and ERK2 following prolonged (over 1 month) exposure to tobacco smoke [54]. Upregulation of ERK1/2 has also been reported in the rat lung following a 5-day exposure to tobacco smoke [55]. Furthermore, AFABP, which we found to be increased in the 7-day exposure group, activates the NF-κB signaling pathway in macrophages and causes inflammation [56]. Considering the above changes in phosphoprotein expression, the following events occur in response to tobacco smoke: (1) during the acute phase, the expression of proteins that cause inflammation, such as AFABP in alveolar macrophages, was increased; (2) as inflammation was exacerbated, another signaling pathway could be activated to moderate inflammation, and annexin and other proteins related to the moderation of inflammatory pathways were increased.
The expression of HIP1 and FACTp140 was increased specifically in the 1-day exposure group. Over-expression of HIP1 can induce cellular apoptosis [57], which suggests that some lung cells damaged by tobacco smoke underwent cell death accompanying increased HIP1 expression. FACTp140 is known to play a crucial role in transcription and DNA replication [58]. The observed increase in FACTp140 expression likely indicated that this protein was induced in order to enhance cell proliferation and to allow the repair of lung tissue damaged by tobacco smoke exposure.
Compared with the control groups, the expression of serine protease P100 (p100) was down-regulated in the 1- and 7-day exposure groups. P100 has been reported to interact with STAT6 and regulate IL-4 signaling [59]. IL-4 is known to cause asthma and allergy in the lungs [60]. In addition, MAPK3, which functions in the activation of the JNK signaling pathway, exhibited decreased expression in tobacco-exposed groups in the present study [61]. Therefore, our results demonstrated that many complex and contradictory phenomena function simultaneously during short-term exposure to tobacco smoke. Some proteins could protect cells from being damaged by toxic tobacco smoke substances, particularly ROS, and from cell death induced by tobacco smoke exposure. At the same time, severely damaged cells undergo apoptosis. During the repairing phase, cell proliferation is induced in order to heal damaged tissue, or due to hyperplasia. Therefore, down-regulation of these molecules suggested that many complex signaling pathways were involved in the cellular response to tobacco smoke exposure.
In conclusion, here, in order to investigate the effect of tobacco smoke exposure in the acute phase, we analyzed nuclear phosphoproteins extracted from murine lung tissue after exposed of mice to tobacco smoke for 1 or 7 days. We found that most of the proteins detected by the present proteomic study are associated with signaling pathways related to inflammation, repair, regeneration, proliferation, differentiation, morphogenesis, and response to stress and nicotine, as indicated in Fig. 10. These molecules are potential markers of short-term tobacco smoke exposure.
Fig. 10.
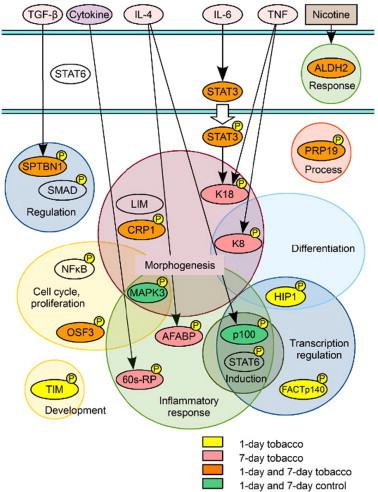
Proposed signaling pathways and associated proteins identified as being activated by tobacco smoke exposure in this study.
Author contributions
Kanako Niimori-Kita and Kiyoshi Ogino contributed to this work equally.
Specific contributions of each author
K.N.-K. and T.I. designed experiments. K.N.-K., K.O. and T.I. wrote the manuscript. K.N.-K., K.O., S.K., D.K., N.K., F.N. and T.I. performed the experiments. N.N. provided assistance in experimental assessment and in manuscript preparation. K.O., S.K., N.K., N.F., M.M., T.S., K.H. and T.I. performed the tobacco exposure experiments. The nuclear phosphoprotein analysis was developed by K.N.-K., with support from T.I. and S.M., F.U. and N.N. assisted in the proteomic analyses. The authors thank Ms. Takako Maeda for technical assistance.
Conflicts of interest
The authors have no competing financial interest.
Acknowledgements
This work was supported in part by a grant from the Smoking Research Foundation, by Grants-in-Aid for Young Scientists (B), and by Shibasaburo Program from Kumamoto University. The funding source had no role in study design and concept; data collection, analysis, and interpretation, or in manuscript preparation and submission.
Contributor Information
Kanako Niimori-Kita, Email: nimokana7@gmail.com.
Takaaki Ito, Email: takaito@kumamoto-u.ac.jp.
References
- 1.Hylkema M.N., Sterk P.J., de Boer W., Postma D. Tobacco use in relation to COPD and asthma. Eur Respir J. 2007;29:438–445. doi: 10.1183/09031936.00124506. [DOI] [PubMed] [Google Scholar]
- 2.Taylor J.D. COPD and the response of the lung to tobacco smoke exposure. Pulm Pharmacol Ther. 2010;23:376–383. doi: 10.1016/j.pupt.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Ward S.A., Casaburi R. 21st century perspective on chronic obstructive pulmonary disease. Respiration. 2011;68:557–561. doi: 10.1159/000063541. [DOI] [PubMed] [Google Scholar]
- 4.Powell H., Iyen-Omofoman B., Baldwin D., Hubbard R., Tata L. Chronic obstructive pulmonary disease and risk of lung cancer: the importance of smoking and timing of diagnosis. J Thorac Oncol. 2013;8:6–11. doi: 10.1097/JTO.0b013e318274a7dc. [DOI] [PubMed] [Google Scholar]
- 5.Wingo P.A. Annual report to the nation on the status of cancer, 1973–1996, with a special section on lung cancer and tobacco smoking. J Natl Cancer Inst. 1999;91:675–690. doi: 10.1093/jnci/91.8.675. [DOI] [PubMed] [Google Scholar]
- 6.Hackshaw A., Law M., Wald N. The accumulated evidence on lung cancer and environmental tobacco smoke. BMJ. 1997;315:980–988. doi: 10.1136/bmj.315.7114.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thun M.J., Lally C., Flannery J.T., Calle E.E., Flanders W.D., Heath C.W., Jr. Cigarette smoking and changes in the histopathology of lung cancer. J Natl Cancer Inst. 1997;89:1580–1586. doi: 10.1093/jnci/89.21.1580. [DOI] [PubMed] [Google Scholar]
- 8.Adcock I.M., Caramori G., Barnes P.J. Chronic obstructive pulmonary disease and lung cancer: new molecular insights. Respiration. 2011;81:265–284. doi: 10.1159/000324601. [DOI] [PubMed] [Google Scholar]
- 9.Stinn W. Lung inflammatory effects, tumorigenesis, and emphysema development in a long-term inhalation study with cigarette mainstream smoke in mice. Toxicol Sci. 2013;131:596–611. doi: 10.1093/toxsci/kfs312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Churg A., Cosio M., Wright J.L. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol. 2008;294:612–631. doi: 10.1152/ajplung.00390.2007. [DOI] [PubMed] [Google Scholar]
- 11.van der Vaart H., Postma D., Timens W., Hacken N. Acute effects of cigarette smoke on inflammation and oxidative stress: a review. Thorax. 2004;59:713–721. doi: 10.1136/thx.2003.012468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Churg A. Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am J Respir Cell Mol Biol. 2002;27:36874. doi: 10.1165/rcmb.4791. [DOI] [PubMed] [Google Scholar]
- 13.Carter C.A., Misra M., Pelch S. Proteomic analysis of lung lysates from short-term exposure to Fischer 344 rats to cigarette smoke. J Proteome Res. 2011;10:3720–3731. doi: 10.1021/pr200345y. [DOI] [PubMed] [Google Scholar]
- 14.Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 15.Chung K., Adcoc T. Multifaced mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31:1334–1356. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- 16.Yao H., Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol. 2011;254:72–85. doi: 10.1016/j.taap.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barreiro E. Cigarette smoke-induced oxidative stress in skeletal muscles of mice. Respir Physiol Neurobiol. 2012;182:9–17. doi: 10.1016/j.resp.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Hecht S.S. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 19.Hecht S.S. Lung carcinogenesis by tobacco smoke. Int J Cancer. 2012;131:2724–2732. doi: 10.1002/ijc.27816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osada H., Takahashi T. Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene. 2002;21:7421–7434. doi: 10.1038/sj.onc.1205802. [DOI] [PubMed] [Google Scholar]
- 21.Witschi H., Espiritu I., Dance S.T., Miller M.S. A mouse lung tumor model of tobacco smoke carcinogenesis. Toxicol Sci. 2002;68:322–330. doi: 10.1093/toxsci/68.2.322. [DOI] [PubMed] [Google Scholar]
- 22.Lòpez-Boado Y.S., Li J.U., Clayton C.L., Wright J.L., Churg A. Modification of the rat airway explant transcriptome by cigarette smoke. Inhal Toxicol. 2010;22:234–244. doi: 10.3109/08958370903191437. [DOI] [PubMed] [Google Scholar]
- 23.Brody J.S. Transcriptome alterations induced by cigarette smoke. Int J Cancer. 2012;131:2754–2762. doi: 10.1002/ijc.27829. [DOI] [PubMed] [Google Scholar]
- 24.Marshall H. Genetic and epigenetic factors in development of lung cancer. Lancet Oncol. 2012;13:1188. doi: 10.1016/s1470-2045(12)70523-5. [DOI] [PubMed] [Google Scholar]
- 25.Catassi A., Servent D., Paleari L., Cesario A., Russo P. Multiple roles of nicotine on cell proliferation and inhibition of apoptosis: implications on lung carcinogenesis. Mutat Res. 2008;659:221–231. doi: 10.1016/j.mrrev.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Moerloose K.B., Pauwels R.A., Joos G.F. Short-term cigarette smoke exposure enhances allergic airway inflammation in mice. Am J Respir Crit Care Med. 2005;172:168–172. doi: 10.1164/rccm.200409-1174OC. [DOI] [PubMed] [Google Scholar]
- 27.Arnson Y., Shoenfeld Y., Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun. 2010;34:258–265. doi: 10.1016/j.jaut.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Wright S.C., Zhong J., Zhng H., Lariuce J.W. Nicotine inhibition of apoptosis suggests a role in tumor promotion. FASEB J. 1993;7:1045–1051. [PubMed] [Google Scholar]
- 29.Brambilla C., Fievet F., Jeanmart M., de Fraipont F., Lantuejoul S., Frappat V. Early detection of lung cancer: role of biomarkers. Eur Respir J. 2003;2:36–44. doi: 10.1183/09031936.02.00062002. [DOI] [PubMed] [Google Scholar]
- 30.Borrill Z.L., Roy K., Singh D. Exhaled breath condensate biomarkers in COPD. Eur Respir J. 2008;32:472–486. doi: 10.1183/09031936.00116107. [DOI] [PubMed] [Google Scholar]
- 31.Gebel S., Gerstmayer B., Bosio A., Haussmann H.-J., van Miert E., Müller T. Gene expression profiling in respiratory tissues from rats exposed to mainstream cigarette smoke. Carcinogenesis. 2004;25:169–178. doi: 10.1093/carcin/bgg193. [DOI] [PubMed] [Google Scholar]
- 32.de Flora S. Smoke-induced microRNA and related proteome alterations. Modulation by chemo-preventive agents. Int J Cancer. 2012;131:2763–2773. doi: 10.1002/ijc.27814. [DOI] [PubMed] [Google Scholar]
- 33.Stevenson C.S., Birrell M.A. Moving towards a new generation of animal models for asthma and COPD with improved clinical relevance. Pharmacol Ther. 2011;130:93–105. doi: 10.1016/j.pharmthera.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Nemmar A. Evaluation of the pulmonary effects of short-term nose-only cigarettes smoke exposure in mice. Exp Biol Med. 2012;237:1449–1456. doi: 10.1258/ebm.2012.012103. [DOI] [PubMed] [Google Scholar]
- 35.Xue L. Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc Natl Acad Sci USA. 2012;109:5615–5620. doi: 10.1073/pnas.1119418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halappanavar S., Russell M., Stampfli M.R., Williams A., Yauk C.L. Induction of the interleukin 6/signal transducer and activator of transcription pathway in the lungs of mice sub-chronically exposed to mainstream tobacco smoke. BMC Med Genomics. 2009;2:56. doi: 10.1186/1755-8794-2-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart M.J., Dipple K.M., Estonius M., Nakshatri H., Everett L.M., Crabb D.W. Binding and activation of the human aldehyde dehydrogenase 2 promoter by hepatocyte nuclear factor 4. Biochim Biophys Acta. 1998;1399:181–186. doi: 10.1016/s0167-4781(98)00115-8. [DOI] [PubMed] [Google Scholar]
- 38.Xu D., Guthrie J.R., Mabry S., Sack T.M., Truog W.E. Mitochondrial aldehyde dehydrogenase attenuates hyperoxia-induced cell death through activation of ERK/MAPK and PI3K–Akt pathways in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;291:966–975. doi: 10.1152/ajplung.00045.2006. [DOI] [PubMed] [Google Scholar]
- 39.Tang Y., Katuri V., Dillner A., Mishra B., Deng C.-X., Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- 40.Gao C. Sensitivity of heterozygous α 1,6-fucosyltransferase knock-out mice to cigarette smoke-induced emphysema. J Biol Chem. 2012;287:16699–16708. doi: 10.1074/jbc.M111.315333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hofmann B., Hecht H.-J., Flohé L. Peroxiredoxins. Biol Chem. 2002;383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 42.Wood Z.A., Schröder E., Harris J.R., Poole L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2008;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 43.Toyokuni S. Reactive oxygen species-induced molecular damage and its application in pathology. Pathol Int. 1999;49:91–102. doi: 10.1046/j.1440-1827.1999.00829.x. [DOI] [PubMed] [Google Scholar]
- 44.Neumann C.A., Krause D.S., Carman C.V., Das S., Dubey D.P., Abraham J.L. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 45.Mahajan K.N., Mitchell B.S. Role of human Pso4 in mammalian DNA repair and association with terminal deoxynucleotidyl transferase. Proc Natl Acad Sci USA. 2003;1001:10746–10751. doi: 10.1073/pnas.1631060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hua G., Zhang Q., Fan Z. Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme M-mediated apoptosis. J Biol Chem. 2007;282:20553–20560. doi: 10.1074/jbc.M703196200. [DOI] [PubMed] [Google Scholar]
- 47.Zhang S., Xu N., Nie J., Dong L., Li J., Tong J. Proteomic alteration in lung tissue of rats exposed to cigarette smoke. Toxicol Lett. 2008;178:191–196. doi: 10.1016/j.toxlet.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 48.Yang Y. Modulation of inflammation and response to dexamethasone by Annexin 1 in antigen-induced arthritis. Arthritis Rheum. 2004;50:976–984. doi: 10.1002/art.20201. [DOI] [PubMed] [Google Scholar]
- 49.Liao J., Lowthert L.A., Omary M.B. Heat stress or rotavirus infection of human epithelial cells generates a distinct hyperphosphorylated form of keratin 8. Exp Cell Res. 1995;219:348–357. doi: 10.1006/excr.1995.1238. [DOI] [PubMed] [Google Scholar]
- 50.Chou C.-F., Omary M.B. Mitotic arrest-associated enhancement of 0-linked glycosylation and phosphorylation of human keratins 8 and 18. J Biol Chem. 1993;268:4465–4472. [PubMed] [Google Scholar]
- 51.Liao J., Lowthert L.A., Ku N.-O., Fernandez R., Omary M.B. Dynamics of human keratin 18 phosphorylation: polarized distribution of phosphorylated keratins in simple epithelial tissues. J Cell Biol. 1995;131:1291–1301. doi: 10.1083/jcb.131.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caulin C., Ware C.F., Magin T.M., Oshima R.G. Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J Cell Biol. 2000;149:17–22. doi: 10.1083/jcb.149.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter C.A., Mirsa M. Effects of short-term cigarette smoke exposure of Fischer 344 rats and on selected lung proteins. Toxicol Pathol. 2010;38:402–415. doi: 10.1177/0192623310364028. [DOI] [PubMed] [Google Scholar]
- 54.Kuo W.-H., Chen J.-H., Lin H.-H., Chen B.-C., Hsu J.D., Wang C.J. Induction of apoptosis in the lung tissue from rats exposed to cigarette smoke involves p38/JNK MAPK pathway. Chem Biol Interact. 2005;155:31–42. doi: 10.1016/j.cbi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 55.Carter C.A., Mirsa M., Maronpot R.R. Tracheal morphologic and protein alterations following short-term cigarette mainstream smoke exposure to rats. J Toxicol Pathol. 2012;25:201–207. doi: 10.1293/tox.25.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makowski L., Brittingham K.C., Reynolds J.M., Suttles J., Hotamisligil G.S. The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of AP2 impacts peroxisome proliferator-activated receptor and IkappaB kinase activities. J Biol Chem. 2004;280:12888–12895. doi: 10.1074/jbc.M413788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hackam A.S., Yassa A.S., Singaraja R., Metzler M., Gutekunst C.A., Gan L. Huntingtin interacting protein 1 induces apoptosis via a novel caspase-dependent death effector domain. J Biol Chem. 2000;275:41299–41308. doi: 10.1074/jbc.M008408200. [DOI] [PubMed] [Google Scholar]
- 58.Morrison A.J., Shen X. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat Rev Mol Cell Biol. 2009;10:373–384. doi: 10.1038/nrm2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang J. Identification of p100 as a coactivator for STAT6 that bridges STAT6 with RNA polymerase II. EMBO J. 2002;21:4950–4958. doi: 10.1093/emboj/cdf463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X., Liu X., Fang J., Lu Y., He J., Yao X. Coactivator p100 protein enhances STAT6-dependent transcriptional activation but has no effect on STAT1-mediated gene transcription. Anat Rec (Hoboken) 2010;293:1010–1016. doi: 10.1002/ar.21143. [DOI] [PubMed] [Google Scholar]
- 61.Ichijo H. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]