

Highlights
-
•
Quercetin-3-glucoside can increase LDLR expression by hepatocytes.
-
•
Quercetin-3-glucoside can reduce PCSK9 secretion by hepatocytes.
-
•
Quercetin-3-glucoside can down regulate sortilin expression.
-
•
Quercetin-3-glucoside can increase LDL uptake by hepatocytes.
-
•
Quercetin-3-glucoside is a potential anti-cholesterolemic agent.
Abbreviations: ABCA1, ATP-binding cassette transporter A1; ApoB, apolipoprotein B; HMGCoAR, 3-hydroxy-3-methylglutaryl-CoA reductase; LDL-C, low-density lipoprotein-cholesterol; LDLR, low-density lipoprotein receptor; PCSK9, proprotein convertase subtilisin/kexin 9; Q3G, quercetin-3-β-d-glucoside; SREBP-2, sterol regulatory element-binding protein 2; TGN, trans Golgi network
Keywords: Quercetin, Proprotein convertase, Low-density lipoprotein receptor, Cholesterol, Sortilin, Hepatocytes
Abstract
Low-density lipoprotein receptor (LDLR) mediates hepatic clearance of plasma cholesterol; proprotein convertase subtilisin/kexin 9 (PCSK9) opposes this clearance by promoting LDLR degradation. The plant flavonoid quercetin-3-β-d-glucoside (Q3G) has been shown to reduce hypercholesterolemia in experimental animals. Here, we examined how it affects LDLR and PCSK9 expression as well as LDL uptake by human Huh7 hepatocytes. At low micromolar concentrations, Q3G increased LDLR expression, reduced PCSK9 secretion, and stimulated LDL uptake. It also diminished intracellular sortilin, a sorting receptor known to facilitate PCSK9 secretion. Thus, as an LDLR inducer and a PCSK9 anti-secretagogue, Q3G may represent an effective anti-cholesterolemic agent.
1. Introduction
Elevated circulating low-density lipoprotein-cholesterol (LDL-C) is a risk factor for cardiovascular disease [1]. Clearance of plasma LDL-C is primarily mediated by hepatic LDL receptors (LDLR) [2]. This clearance is opposed by proprotein convertase subtilisin/kexin type 9 (PCSK9) [3], a member of the proprotein convertase family of endoproteinases known to activate precursor proteins in the secretory pathway [4,5]. In human hepatocytes, PCSK9 is biosynthesized in the endoplasmic reticulum (ER) as a 72-kDa proPCSK9. For this zymogen to exit the ER, it must undergo autocatalytic cleavage after the prodomain, generating a 14-kDa propeptide and a glycosylated 63-kDa mature enzyme. The two moieties form a stable, non-covalent, and enzymatically inactive complex which is secreted into circulation [6,7]. This complex binds to LDLR at the surface of hepatocytes, gets endocytosed with it and, preventing its return to the cell surface, reroutes it into lysosomes where it is degraded [8]. PCSK9-mediated LDLR degradation reduces LDL-C clearance. These observations provided a mechanism for the hypercholesterolemia associated with certain PCSK9 genetic mutations [9–11].
Hypercholesterolemia is commonly treated with statins, a class of drugs that inhibit cholesterol biosynthesis by inactivating 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CoAR), the rate-limiting enzyme in this process [12]. Because they reduce intracellular cholesterol, statins induce feedback up-regulation of nuclear sterol regulatory element-binding protein 2 (SREBP-2), the transcription factor that drives cholesterol biosynthesis. This factor also activates transcription of the LDLR and PCSK9 genes [13]. The coordinated up-regulation of these two functionally opposing genes limits the therapeutic efficacy of statins. Consistently, induced or genetic deficiency of PCSK9 expression or activity has been shown to increase this efficacy in animals and man [14–16]. Furthermore, results from clinical trials indicate that PCSK9 inhibitors represent a novel class of statin-potentiating agents [17–19].
Many plant parts contain compounds with anti-cholesterolemic properties [20]. Quercetin appears to be one such phytochemical [21]. Found in a broad range of fruits and vegetables, predominantly as quercetin-3-O-β-d-glucoside (Q3G) [22] (Fig. 1A), this bioflavonoid has been shown to reduce diet-induced hyperlipidemia and atherosclerosis in rabbits [23,24] and to attenuate metabolic syndrome in obese Zucker rats [25]. In this study, we examined how Q3G affects LDLR and PCSK9 expression in human Huh7 hepatocytes.
Fig. 1.
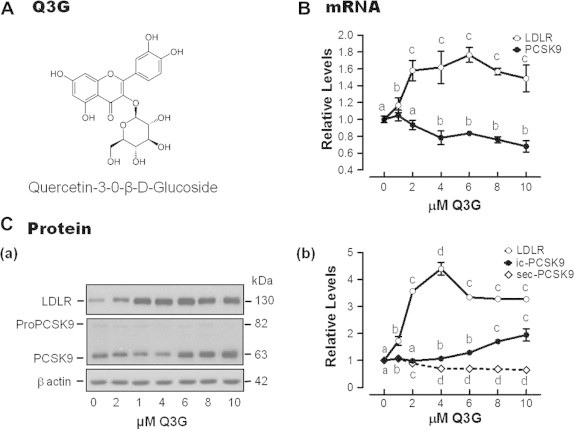
(A) Structure of Q3G. (B–C) Q3G effects on the levels of LDLR and PCSK9 expression Cells were incubated for 24 h in medium containing the indicated concentrations of Q3G. Levels of mRNA were determined by qRT-PCR (B); those of proteins by sq-immunoblotting in cell extracts or ELISA in spent media (C). Values are means of triplicate experiments ± and standard errors of means (SEM). They are expressed relative to untreated cells. Significant difference (P < 0.05) is represented in the graph by different letters above symbols of means ± SEM. PCSK9 prefixes ic- and sec- stand for intracellular and secreted, respectively. Note that the LDLR band shown in B, a represents the glycosylated mature form of the receptor. A co-regulated immature form can be observed below on overexposed X-ray films (not shown).
2. Materials and methods
2.1. Materials
Huh7 human liver cells were obtained from the Japanese Collection of Research Bioresources. The rabbit anti-human PCSK9 antibody used for immunoblotting was obtained from Dr. Nabil G. Seidah (Clinical Research Institute of Montreal). The rabbit anti-human PCSK9 antibody for immunoprecipitation was produced in house. The following antibodies were from commercial sources: anti-human LDLR (R&D Systems), anti-β-actin (Sigma), anti-SREBP-2 (Santa Cruz), anti-HMG-CoAR (Millipore), and anti-sortilin (Abcam), Horseradish peroxidase (HRP)-conjugated anti-rabbit or mouse immunoglobulins (Ig) (GE HealthCare) or anti-goat Ig (Santa Cruz). The chemiluminescence revelation kit was from PerkinElmer; the PCSK9 ELISA kit from Circulex; the RNeasy extraction kit from Qiagen; the Superscript II RNase H– Reverse Transcriptase, bodipy-LDL, non-conjugated LDL and Alexa Fluor 488™ were from Invitrogen; lipoprotein-depleted serum (LPDS), Q3G and simvastatin from Sigma; the FastStart TaqMan ProbeMaster-Rox master mix, primer pairs, and Universal Probe Library (UPL) fluorescent probes, and the Protease Inhibitor Cocktail (PIC) from Roche; the Amplify fluor solution from Amersham Biosciences, the Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) from Vector Laboratories.
2.2. Cell culture and lysis
Unless otherwise specified, all Huh7 cell incubations were carried out at 37 °C in a humidified 5% CO2–95% air atmosphere. At passage, cells were seeded at sub-confluence (∼106 cells/3.5-cm dish) in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) for maintenance or LPDS for experiments, and 50 μg/ml gentamycin; they were incubated overnight and then treated or not with Q3G, or simvastatin, or both, at defined concentrations, and for defined lengths of time; media were collected, centrifuged at 200g for 5 min to sediment suspended cells, and collected supernatants were supplemented 0.02 volumes of a 50× stock PIC; cell monolayers were rinsed with ice-cold phosphate-buffered saline (PBS), overlaid with 0.5 ml of the RIPA lysis buffer (50 mM Tris–HCl, pH 8, 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate and 0.1% SDS) supplemented with 1× PIC, and placed on an ice bath for 20 min; lysates were centrifuged at 14,000g and 4 °C for 20 min, and supernatants collected. Spent media and cell lysates were stored at −20 °C until analysis.
2.3. Metabolic labeling
Huh7 cells were seeded onto 12-well plates at 8 × 105 cells/well in 1.5 ml/well of complete medium and incubated overnight; after a rinse with Dulbecco’s PBS (PBS-D), they were overlaid with 1.5 ml of DMEM/10% LPDS without or with 5 μM Q3G, and re-incubated for 24 h; Met/Cys-depleted serum-free DMEM (SFM, 1.5 ml) was substituted, and incubation resumed for 20 min to use up endogenous Met and Cys; this medium was removed, fresh SFM (0.75 ml/well) containing 300 μCi/ml 35S-Met/Cys was added in its place, and incubation was carried on for another 20 min to label de novo biosynthesized proteins (pulse-labeling); the radioactive medium was replaced with DMEM/0.5% LPDS containing 10 mM non-radioactive Met/Cys and, incubation reinstated for 0, 15, 30, 60, 90 and 120 min (chase); conditioned media were collected and supplemented with 0.33 volumes of 3× RIPA lysis buffer containing 30 mM Met/Cys; cells were lysed in 0.5 ml of a 3-fold dilution of this same buffer.
2.4. Indirect immunofluorescence flow cytometry
Cells were seeded in 6-well plates, at 106 cells/well in 3 ml of complete medium and incubated overnight; LPDS medium containing or not 5 μM Q3G was substituted and incubation resumed for 24 h; after two rinses with PBS-D, cell monolayers were overlaid with 2 ml of Versene and incubated for 20 min; detached cells were suspended and their concentration determined using an hemocytometer; they were sedimented by a 5-min centrifugation and suspended in 0.5 ml of DMEM containing 5% bovine serum albumin (DMEM-5% BSA); 2 × 106 cells were transferred into an Eppendorf tube and rinsed 3 times as above; using ice-cold solutions and, keeping them at 4 °C to the end of the protocol, they were suspended in 0.2 ml of DMEM-5% BSA containing 10 μg/ml of goat antibody against human LDLR and mixed by rotation for 1 h; after 3 rinses with DMEM-5% BSA, they were suspending in 0.2 ml of the same buffer supplemented with Alexa Fluor-488-conjugated donkey antibody against goat immunoglobulins, and mixed for another 1 h; after 3 other rinses with DMEM-5% BSA, they were suspended in PBS-D and analyzed in a Benson–Dickenson XL 488 Laser flow cytometer. Cell autofluorescence and non-specific fluorescence were assessed using cells not treated with the secondary and the primary antibody, respectively.
2.5. Confocal microscopy
Huh7 cells were seeded in 4-well chamber slides at 2.4 × 105 cells/well and grown for 24 h; LPDS medium containing or not 5 μM Q3G was substituted and incubation resumed for 24 h; kept at room temperature to the end of the protocol, cells were fixed with 1% paraformaldehyde in PBS for 15 min; they were treated for indirect immunofluorescence as for flow cytometry, except that PBS was substituted for DMEM in the various solutions; they were mounted on the Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI); confocal images were captured with an Olympus 1X70 inverted microscope equipped with a BioRad MRC-1024 confocal laser-scanning unit at 488 nm and 522 nm excitation and emission wavelengths, respectively, using an Olympus UApo 40_ (NA 1.15) water immersion lens. Sections were taken every 50 μm (Z-series) through the thickness of the cells.
2.6. LDL uptake assay
Huh7 cells were seeded in 96-well black-bottom plates at 4 × 104 cells/well in 0.1 ml complete medium and allowed to attach by overnight incubation; they were then rinsed with PBS-D, overlaid with 0.1 ml of DMEM/10% LPDS and incubated for 24 h; after a PBS-D rinse, they were overlaid with 0.1 ml DMEM/0.5% LPDS containing or not 5 μM Q3G and incubated for 24 h. To assay for LDL uptake, cells were rinsed, first with pre-warmed (37 °C) PBS-D, then with pre-warmed DMEM/0.5% LPDS; they were overlaid with 75 μl of the latter medium containing 20 μg/ml bodipy-LDL, and then incubated at 37 °C for 15 min or 30 min to allow LDLR-mediated endocytosis of the fluorescent lipoprotein; the process was stopped by substituting ice-cold DMEM/0.5% LPDS; after 3 rinses with 0.2 ml of ice-cold PBS-D, the cells were fixed with 0.1 ml of isopropanol for 20 min, in the dark, with gentle shaking; intracellular fluorescence was measured in a SpectraMax Gemini XS fluorescence plate reader (Molecular Devices) at the excitation and emission wavelengths of 485 and 535 nm, respectively. Non-specific fluorescence was measured by incubating cells in medium containing bodipy-LDL (20 μg/ml) and a 12.5× excess of non-fluorescent LDL (250 μg/ml).
2.7. RT-qPCR
Total RNA was extracted using the Qiagen RNeasy extraction kit. It was reverse-transcribed into cDNA using oligo-dT primers and the Superscript II RNase H– Reverse Transcriptase. The levels of specific cDNAs were quantified by PCR-based fluorogenic Taqman assays, using FastStart TaqMan ProbeMaster-Rox master mix, primer pairs and the appropriate fluorescent UPL probes (Table 1), in a Mx3005P thermocycler (Stratagene). Standard curves were established using varying amounts of purified and quantified cDNA amplicons of each mRNA. The level of mRNA for the TATA-binding protein (TBP) was used for normalization.
Table 1.
UPL primers and probes.a
| Gene | Exon number: Primer sequence |
Amplicon |
||
|---|---|---|---|---|
| Forward | Reverse | Size (bp) | Probe # | |
| HMGCOAR | Exon 14: gggatgaacatgatttcaaagg | Exon 15: ttttcctcttccctctatccaa | 147 | 62 |
| LDLR | Exon 17: aggacggctacagctaccc | Exon18: ctccaggcagatgttcacg | 73 | 88 |
| PCSK9 | Exon 2: catgtcttccatggccttct | Exon3: tagtcgacatggggcaactt | 89 | 62 |
| SORTILIN | Exon 9: gcagcaaatgatgacatggt | Exon 10: gaggtaaagattgtgccaaacc | 74 | 80 |
| SREBP2 | Exon 2: gcctttgatataccagaatgcag | Exon 3: cctgctgctgaatggtgac | 107 | 88 |
| TBP | Exon 2: gaacatcatggatcagaacaaca | Exon 3: atagggattccgggagtcat | 87 | 87 |
The probes were designed using an online algorithm at the Roche Universal Probe Library Assay Design Center (www.roche-applied-science.com/sis/rtpcr/upl/adc.jsp).
2.8. Immunoblotting
Cell lysates were fractionated by SDS–PAGE and electrophoretically transferred onto a polyvinylidene fluoride membrane. The membrane was incubated with a goat anti-human LDLR, rabbit anti-PCSK9, rabbit anti-SREBP-2 polyclonal antibody, rabbit anti HMG-CoAR, or rabbit anti-sortilin at 1:1000, 1:1500, 1:200, 1:2000, or 1:2000 dilutions, respectively, and then with a HRP-conjugated heterospecific secondary antibody against the primary Igs at a 1:5000 and 1:10,000 dilution for goat and rabbit or mouse antibody, respectively. It was probed for HRP reaction using the Western Lightning Chemiluminescence Reagent Plus a chemiluminescence-based revelation kit. The signal was captured on X-ray film and immunoreactive bands analyzed by densitometry on a Syngene’s ChemiGenius2XE Bio Imaging System within the dynamic range of the instrument. The membrane was stripped and reprobed with the anti-β-actin monoclonal primary antibody at 1:20,000 dilution and HRP-conjugated rabbit anti-mouse IgG secondary antibody at a 1:10,000 dilution. The densitometric values of β-actin bands were using for normalization of experimental samples.
2.9. Immunoprecipitation
Radioactive conditioned media or cell lysates (0.1 ml) were supplemented with 0.2 μl of normal rabbit serum and 15 μl of a 50% (w/v) suspension of Protein A-agarose. After a 1-h incubation at 4 °C with rotational mixing, the samples were centrifuged at 3000g for 5 min at 4 °C. Supernatants were supplemented 2 μl of rabbit anti-PCSK9, and incubated as above. The resin with bound immune complexes was then sedimented by centrifugation as above, rinsed three times with RIPA buffer, twice with a buffer containing 1 M NaCl, 10 mM Tris–HCl and 1 mM EDTA, pH 8, and twice with PBS containing 1 mM EDTA. Pellets were suspended each in 25 μl of 2x Tricine-DTT sample buffer (100 mM Tris–HCl, pH 6.8, 24% glycerol, 8% SDS, 5% β-mercaptoethanol, 50 mM DTT, and 0.02% bromophenol blue), boiled for 5 min, and sedimented as above. Supernatant was subjected to electrophoresis through Tris-Tricine polyacrylamide gels (8% or 12%). Gels were fixed for 30 min in a 50% methanol–10% acetic acid solution, treated for 30 min with Amplify fluor solution, dried under vacuum and exposed to phosphorimaging screen overnight. Specific radioactive protein bands were visualized and quantified on a Typhoon Phosphorimager (Molecular Dynamics).
2.10. ELISA
PCSK9 levels in conditioned media were measured using a human PCSK9-specific sandwich ELISA kit, as specified by the manufacturer.
2.11. Statistical analysis
All experiments were repeated at least twice and in triplicates. Values are expressed as means ± standards errors of means (SEM). Significance of differences among several experimental conditions was evaluated by ANOVA, between two conditions by Student’s t test, using the GraphPad Prism 5 software.
3. Results
3.1. Q3G increased LDLR expression and reduced PCSK9 secretion
Huh7 cells were incubated for 24 h in medium containing 0–10 μM Q3G. The level of LDLR and PCSK9 mRNA was measured by qRT-PCR. The treatment increased LDLR mRNA levels in a concentration-dependent manner, by a maximum 60% at 2 μM (P < 0.01); in contrast, starting at 4 μM, it reduced PCSK9 mRNA levels by 20–30% (P < 0.05) (Fig. 1B).
The levels of corresponding proteins in cell extracts and cell media were evaluated by sq-immunoblotting and ELISA, respectively. The treatment remarkably increased total LDLR levels starting at 2 μM (by 300–400%; P < 0.005); interestingly, starting at 4 μM, it gradually increased intracellular PCSK9 levels (by 20–90%; P < 0.05), while reducing its levels in spent media (by 30–35%; P < 0.05) (Fig. 1C).
Time course immunoblotting analysis of extracts of cells treated with 2 μM Q3G for 0.5–48 h revealed that cellular accumulation of PCSK9 and LDLR begun after a 3-h and a 6-h lag, respectively; it reached a maximum after 27 h for both (not shown).
3.2. Q3G increases nuclear SREBP-2 and delays PCSK9 secretion
The increase in LDLR mRNA content could be attributed to up-regulation of its gene transcription by SREBP-2 [26], a nuclear factor generated through two successive cleavages of its ER membrane-bound precursor form by the Golgi proteases S1P and S2P [27]. The effect of Q3G on SREBP-2 expression was therefore examined. The flavonoid had no effect on the level of SREBP-2 mRNA (Fig. 2A, a), but, starting at 4 μM, it increased that of HMGCoAR (Fig. 2A, b; P < 0.05) and decreased that ATP-binding cassette transporter A1 (ABCA1) (Fig. 2A, c; P < 0.01), two proteins implicated in cholesterol homeostasis and whose gene transcription is regulated by SREBP-2, positively for the former [26] and negatively for the latter [28].
Fig. 2.
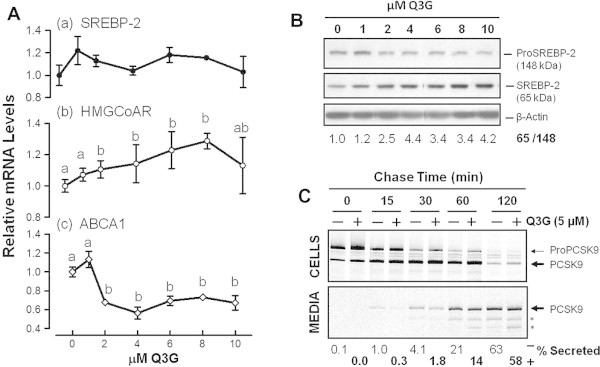
Q3G stimulates nuclear SREBP-2 production and delays PCSK9 secretion. Cells were incubated for 24 h in medium containing the indicated concentrations of Q3G. (A) qRT-PCR for mRNA levels of SREBP-2, HMGCoAR and ABCA1. Values are means of triplicate experiments ± SEM. They are expressed relative to untreated cells. Significant difference (P < 0.05) is represented in the graph by different letters above symbols of means ± SEM. (B) Sq-immunoblotting for cellular SREBP-2-related proteins. Density ratios of the 65-kDa SREBP over the 148-kDa precursor SREBP after normalization for β-actin were derived from two separate experiments. (C) Cells were pre-incubated for 24 h in medium 5 μM Q3G. After metabolic labeling with radioactive amino acids, labeled proteins were chased in Q3G-free non-radioactive medium, for varying lengths of time. PCSK9-related proteins were immunoprecipitated, fractionated by SDS–PAGE, and quantified by phosphorimaging. Upper panel: PCSK9-related proteins in cell lysates. Lower panel: PCSK9-related proteins in spent media. The percents of secreted PCSK9 were based on densitometric values of intracellular and extracellular bands corresponding to proPCSK9 and PCSK9. Asterisks (∗) indicate possible products of further PCSK9 processing.
Increased SREBP-2 activity, as suggested by the regulation of HMGCoAR and ABCA1 mRNA levels, could have resulted from greater post-translational conversion of the inactive 148-kDa ER precursor of SREBP-2 to its active 65-kDa nuclear form. Indeed, immunoblotting analysis of these proteins showed that, starting at 2 μM, Q3G stimulated this conversion 2–4-fold (Fig. 2B).
Q3G treatment apparently reduced PCSK9 secretion causing its intracellular accumulation. Since PCSK9 can be secreted only after endoproteolytic cleavage of its precursor [6,7], the possibility of impaired proPCSK9 maturation was examined after radioactively pulse labeling it during biosynthesis. As shown in Fig. 2C, chase of untreated and treated cells revealed a gradual intracellular conversion of proPCSK9 to mature PCSK9 (upper panel), associated with a gradual appearance of the latter in spent media (lower panel). No obvious difference in the rate of intracellular precursor processing was observed. However, when cumulative levels of secreted PCSK9 were expressed as percentages of total PCSK9 proteins (precursor PCSK9 and mature PCSK9 in cells and media), they were found to be consistently lower in spent media of Q3G-treated cells, relative to those of untreated cells, most noticeably at early chase time points (lower panel), suggesting delayed secretion.
It has been suggested that PCSK9 transport in the secretory pathway is facilitated by active packaging into SEC24A-coated vesicles at the ER-cis Golgi interface and by the sorting receptor sortilin in the trans-Golgi network (TGN), since knockdown or knockout of expression of either protein is associated with reduced PCSK9 secretion and its overexpression with stimulation of this secretion [29,30]. We therefore examined whether down regulation of PCSK9 secretion in Q3G-treated cells could be associated with diminished expression of these proteins. Treatment of Huh7 cells with 5 mM Q3G for 24 h reduced the intracellular level of sortilin by about 50% (P < 0.001) (Fig. 3A), and that of its mRNA by about 40% (P < 0.01) (Fig. 3B). It induced no change in Sec24A content (not shown). These observations suggested that PCSK9 retention in Q3G-treated Huh7 cells might be partly due to induced sortilin deficiency.
Fig. 3.
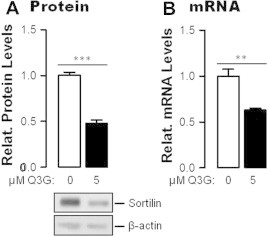
Q3G inhibit sortilin expression. Cells were pre-treated or not with 5 μM Q5G for 24 h. Their content in sortilin protein was evaluated by sq-immunoblotting (A); that of sortilin mRNA by qRT-PCR (B). Values are expressed relative to untreated cells. They represent means of triplicate experiments ± SEM. ∗∗∗P < 0.001; ∗∗P < 0.01 by Student t test.
3.3. Q3G increased cell surface expression of LDLR and accelerated LDL uptake
To be functionally relevant, Q3G-induced LDLR must reach the cell surface where it could mediate LDL uptake. We stained cells at 4 °C for surface LDLR by indirect immunofluorescence and assessed its level by flow cytometry. Pretreatment with 5 μM Q3G increased cell surface LDLR 1.7-fold (Fig. 4A; P < 0.001), suggesting that it rendered the cells more capable of taking up more exogenous LDL. This prediction was verified using a fluorescent LDL uptake assay. Compared to untreated cells, cells pretreated with 5 μM Q3G accumulated 4-fold and 2.5-fold more bodipy-LDL after 15 min and 30 min, respectively (Fig. 4B, P < 0.005).
Fig. 4.
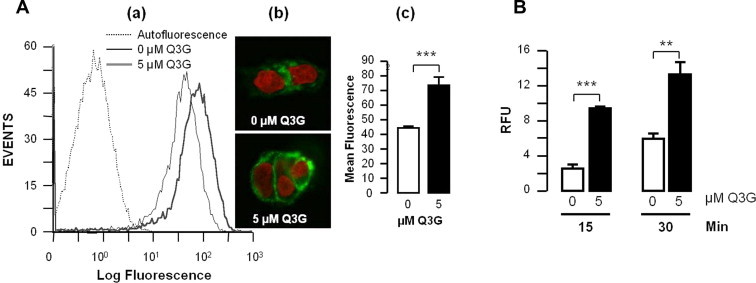
Q3G increases cell surface LDLR and LDL uptake. Cells were pre-treated or not with 5 μM Q5G; (A) they were then stained for LDLR indirect immunofluorescence and analyzed by (a) flow cytometry, or (b) confocal microscopy, (c) means of fluorescence ± SEM were compared. The experiment was conducted in triplicates. (B) Cells were incubated bodipy-LDL for up to 30 min; uptake of the lipoprotein was measured by fluorescence spectrometry. Values represents means of 6 replicates ± SEM. ∗∗∗P < 0.005; ∗∗P < 0.01 by Student t test.
3.4. Q3G potentiates statin-induced LDLR increase but not PCSK9 secretion
Statins increase LDLR expression as well as PCSK9 secretion [13,31,32]. Q3G, as this study indicated, stimulate LDLR expression while reducing PCSK9 expression and secretion. An experiment was conducted to determine whether, at an optimum concentration of Q3G (5 μM), simvastatin at 0.2 and 1 μM could further up regulate LDLR expression; and, inversely, whether the bioflavonoid can reduce statin-stimulated PCSK9 secretion. As shown in Fig. 5. In the absence of Q3G (open bars), simvastatin treatment, in a concentration-dependent manner, increased the levels of cellular LDLR (Fig. 5A.a), cellular PCSK9 (Fig. 5A.b), and secreted PCSK9 (Fig. 5B). Co-treatment with 5 μM Q3G (black bars), increased cellular LDLR to the level induced by the bioflavonoid alone (Fig. 5A.a); it further increased the amount of cellular PCSK9 (Fig. 5A.b), while maintaining or slightly reducing its level in spent media (Fig. 5B). Thus, while both simvastatin and Q3G stimulated LDLR expression; Q3G could additionally reduce PCSK9 secretion.
Fig. 5.
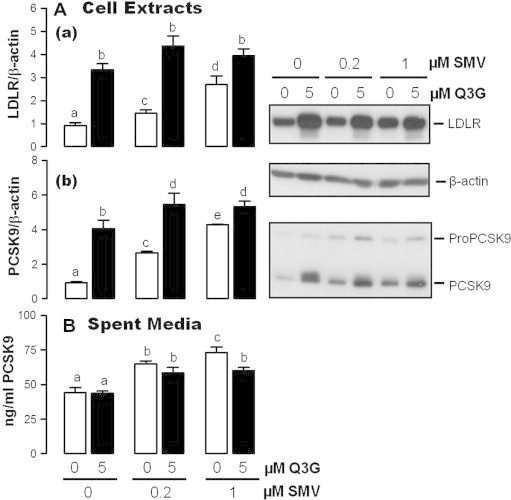
Q3G reduces statin-induced PCSK9 secretion. Cells were incubated for 24 h in culture medium containing 5% FBS and simvastatin (SMV: 0, 0.2, or 1 μM), without or with 5 μM Q3G. The levels of LDLR and PCSK9 in cell extracts were evaluated by sq-immunoblotting; that of PCSK9 in spent media by ELISA. Significant difference (P < 0.05) is represented in the graph by different letters above symbols of means ± SEM.
4. Discussion
Data presented in this report indicate that, at low micromolar concentrations, Q3G accelerates hepatocyte uptake of exogenous LDL by increasing expression and cell surface localization of LDLR. The increased expression of the receptor could be partly explained by transcriptional activation of its gene following stimulated conversion of the ER precursor form of SREBP-2 to its nuclear active form. This conversion is known to be promoted by a deficit of free cholesterol in the ER [27]. It is possible that, like red grape juice polyphenols [33], Q3G causes such a deficit by delaying the transport of endocytosed LDL and its breakdown components from endosomes, through lysosomes, into the cytoplasm.
The PCSK9 promoter also carries a sterol-regulatory element responsive to activation by nuclear SREBP-2 [13]. But its sustained activation by this factor strongly depends on the presence of co-activators including of hepatocyte nuclear factor 1α (HNF-1α) [34,35] and histone nuclear factor P (HINFP) [36]. Reduced expression of either factor has been shown to repress PCSK9 gene transcription. We found that Q3G does not alter HNF1α expression in Huh7 cells (not shown). Its effect of HINFP expression remains to be verified.
Why and where PCSK9 was retained within Q3G-treated cells is unclear. The retention was not caused by impaired autoproteolytic processing of its precursor, a prerequisite for its exit from the ER [7] as a cargo in COPII-coated vesicles. The presence of SEC24A in the COPII complex is necessary for the efficiency of this exit [29]. Our analysis indicated that SEC24A expression is not affected by Q3G. In the TGN, PCSK9 packaging into secretory vesicles is facilitated by the protein sorting receptor sortilin [30]. Here we report that treatment of Huh7 hepatocytes Q3G significantly inhibits sortilin expression at the mRNA and protein levels. This inhibition could partly explain the intracellular retention of PCSK9. Interestingly, quercetin is also known to reduced secretion of apolipoprotein B (ApoB) [37], the major protein component of LDL particles. ApoB, like PCSK9, depends on sortilin for efficient secretion [38,30]. If Q3G can inhibit sortilin expression in vivo, it could reduce the negative impact of PCSK9 secretion on the half-life of hepatic LDLR and thus promote plasma LDL-C clearance. An LDL-C-reducing effect of sortilin deficiency has been reported in mice [39,30]. However, other mouse studies have observed the opposite, i.e. lower plasma LDL-C was caused by sortilin overexpression [40,41]. These discrepancies may be due to differences of induced modifications in the experimental mice which may have differentially affected the ability of sortilin to target its ligands towards either secretion (ApoB, PCSK9) or intracellular degradation in lysosomes (LDL, VLDL) [42].
Interestingly, we did not observe any alteration of PCSK9 secretion or LDLR expression in Huh7 cells exposed to quercetin aglycone at single-digit micromolar concentrations (data not shown). LDLR up regulation has reportedly been achieved in HepG2 hepatocytes treated with this unmodified flavonoid at 75 μM [43]. The greater efficiency of Q3G could be explained by its more active receptor-mediated uptake by cells [44,45]. It should be noted however that, in vivo, orally administered Q3G is converted to quercetin through deglycosylation by hydrolases found in brush border membrane cells of the small intestine. In enterocytes, quercetin is largely metabolized to glucuronidated and/or methylated forms which are released into the bloodstream and taken up by multiple tissues, including the liver [46,47]. Quercetin uptake and metabolism by hepatocytes ex vivo have been studied [48]. A similar study for Q3G remains to be conducted.
In conclusion, as an LDLR inducer, a sortilin inhibitor and a PCSK9 anti-secretagogue, Q3G could make an effective anti-cholesterol agent.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
The authors thank Mrs. Elaine Coderre for her assistance with confocal microscopy, and Mrs. Denise Joanisse for her assistance with bibliographical searches. Ms. Sonia Simoes was a trainee from the Institut Universitaire de Technologie Créteil/Vitry, Paris Est, France. The work was supported by grants from the Canadian Institute of Heath Research, the Edith and Richard Strauss Foundation, and La Foundation J.-Louis Lévesque. MM conceived the project, designed it, and wrote the paper; FS and SS acquired the data; MM, FS, SS, JM and MC analyzed and interpreted the data, and revised the paper.
Contributor Information
Majambu Mbikay, Email: mmbikay@ohri.ca.
Francine Sirois, Email: Francine.sirois@ircm.qc.ca.
Sonia Simoes, Email: simoes.sonia@hotmail.fr.
Janice Mayne, Email: jmayn2@uottawa.ca.
Michel Chrétien, Email: mchretien@ohri.ca.
References
- 1.Bittner V. Non-high-density lipoprotein cholesterol and cardiovascular disease. Curr. Opin. Lipidol. 2003;14(4):367–371. doi: 10.1097/00041433-200308000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Brown M.S., Goldstein J.L. Lipoprotein receptors in the liver. Control signals for plasma cholesterol traffic. J. Clin. Invest. 1983;72(3):743–747. doi: 10.1172/JCI111044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mbikay M., Mayne J., Chretien M. Proprotein convertases subtilisin/kexin type 9, an enzyme turned escort protein: hepatic and extra hepatic functions. J. Diabetes. 2013;5(4):391–405. doi: 10.1111/1753-0407.12064. [DOI] [PubMed] [Google Scholar]
- 4.Seidah N.G. The proprotein convertases, 20 years later. Methods Mol. Biol. 2011;768:23–57. doi: 10.1007/978-1-61779-204-5_3. [DOI] [PubMed] [Google Scholar]
- 5.Chretien M. My road to Damascus: how I converted to the prohormone theory and the proprotein convertases. Biochem. Cell Biol. 2012;90(6):750–768. doi: 10.1139/o2012-031. [DOI] [PubMed] [Google Scholar]
- 6.Seidah N.G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S.B., Stifani S., Basak A., Prat A., Chretien M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 2003;100(3):928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayne J., Dewpura T., Raymond A., Bernier L., Cousins M., Ooi T.C., Davignon J., Seidah N.G., Mbikay M., Chretien M. Novel loss-of-function PCSK9 variant is associated with low plasma LDL cholesterol in a French–Canadian family and with impaired processing and secretion in cell culture. Clin. Chem. 2011;57(10):1415–1423. doi: 10.1373/clinchem.2011.165191. [DOI] [PubMed] [Google Scholar]
- 8.Horton J.D., Cohen J.C., Hobbs H.H. PCSK9: a convertase that coordinates LDL catabolism. J. Lipid Res. 2009;50(Suppl.):S172–S177. doi: 10.1194/jlr.R800091-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abifadel M., Varret M., Rabes J.P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003;34(2):154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 10.Naoumova R.P., Tosi I., Patel D., Neuwirth C., Horswell S.D., Marais A.D., van Heyningen C., Soutar A.K. Severe hypercholesterolemia in four British families with the D374Y mutation in the PCSK9 gene: long-term follow-up and treatment response. Arterioscler. Thromb. Vasc. Biol. 2005;25(12):2654–2660. doi: 10.1161/01.ATV.0000190668.94752.ab. [DOI] [PubMed] [Google Scholar]
- 11.Abifadel M., Guerin M., Benjannet S., Rabes J.P., Le Goff W., Julia Z., Hamelin J., Carreau V., Varret M., Bruckert E. Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis. 2012;223(2):394–400. doi: 10.1016/j.atherosclerosis.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 12.Delahoy P.J., Magliano D.J., Webb K., Grobler M., Liew D. The relationship between reduction in low-density lipoprotein cholesterol by statins and reduction in risk of cardiovascular outcomes: an updated meta-analysis. Clin. Ther. 2009;31(2):236–244. doi: 10.1016/j.clinthera.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 13.Dubuc G., Chamberland A., Wassef H., Davignon J., Seidah N.G., Bernier L., Prat A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2004;24(8):1454–1459. doi: 10.1161/01.ATV.0000134621.14315.43. [DOI] [PubMed] [Google Scholar]
- 14.Rashid S., Curtis D.E., Garuti R., Anderson N.N., Bashmakov Y., Ho Y.K., Hammer R.E., Moon Y.A., Horton J.D. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc. Natl. Acad. Sci. U.S.A. 2005;102(15):5374–5379. doi: 10.1073/pnas.0501652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berge K.E., Ose L., Leren T.P. Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler. Thromb. Vasc. Biol. 2006;26(5):1094–1100. doi: 10.1161/01.ATV.0000204337.81286.1c. [DOI] [PubMed] [Google Scholar]
- 16.Pisciotta L., Sallo R., Rabacchi C., Wunsch A., Calandra S., Bertolini S. Leucine 10 allelic variant in signal peptide of PCSK9 increases the LDL cholesterol-lowering effect of statins in patients with familial hypercholesterolaemia. Nutr. Metab. Cardiovasc. Dis. 2012;22(10):831–835. doi: 10.1016/j.numecd.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Chrétien M., Seidah N.G., Basak A., Mbikay M. Proprotein convertases as therapeutic targets. Expert Opin. Ther. Targets. 2008;12(10):1289–1300. doi: 10.1517/14728222.12.10.1289. [DOI] [PubMed] [Google Scholar]
- 18.Hooper A.J., Burnett J.R. Anti-PCSK9 therapies for the treatment of hypercholesterolemia. Expert Opin. Biol. Ther. 2013;13(3):429–435. doi: 10.1517/14712598.2012.748743. [DOI] [PubMed] [Google Scholar]
- 19.Seidah N.G. Proprotein convertase subtilisin kexin 9 (PCSK9) inhibitors in the treatment of hypercholesterolemia and other pathologies. Curr. Pharm. Des. 2013;19(17):3161–3172. doi: 10.2174/13816128113199990313. [DOI] [PubMed] [Google Scholar]
- 20.Mukherjee P.K. Plant products with hypocholesterolemic potentials. Adv. Food Nutr. Res. 2003;47:277–338. doi: 10.1016/s1043-4526(03)47005-8. [DOI] [PubMed] [Google Scholar]
- 21.Romano B., Pagano E., Montanaro V., Fortunato A.L., Milic N., Borrelli F. Novel insights into the pharmacology of flavonoids. Phytother. Res. 2013;27(11):1588–1596. doi: 10.1002/ptr.5023. [DOI] [PubMed] [Google Scholar]
- 22.Kelly G.S. Quercetin Monograph. Altern. Med. Rev. 2011;16(2):172–194. [PubMed] [Google Scholar]
- 23.Juzwiak S., Wojcicki J., Mokrzycki K., Marchlewicz M., Bialecka M., Wenda-Rozewicka L., Gawronska-Szklarz B., Drozdzik M. Effect of quercetin on experimental hyperlipidemia and atherosclerosis in rabbits. Pharmacol. Rep. 2005;57(5):604–609. [PubMed] [Google Scholar]
- 24.Kamada C., da Silva E.L., Ohnishi-Kameyama M., Moon J.H., Terao J. Attenuation of lipid peroxidation and hyperlipidemia by quercetin glucoside in the aorta of high cholesterol-fed rabbit. Free Radic. Res. 2005;39(2):185–194. doi: 10.1080/10715760400019638. [DOI] [PubMed] [Google Scholar]
- 25.Rivera L., Moron R., Sanchez M., Zarzuelo A., Galisteo M. Quercetin ameliorates metabolic syndrome and improves the inflammatory status in obese Zucker rats. Obesity. 2008;16(9):2081–2087. doi: 10.1038/oby.2008.315. [DOI] [PubMed] [Google Scholar]
- 26.Hua X., Yokoyama C., Wu J., Briggs M.R., Brown M.S., Goldstein J.L., Wang X. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl. Acad. Sci. U.S.A. 1993;90(24):11603–11607. doi: 10.1073/pnas.90.24.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown M.S., Goldstein J.L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 28.Zeng L., Liao H., Liu Y., Lee T.S., Zhu M., Wang X., Stemerman M.B., Zhu Y., Shyy J.Y. Sterol-responsive element-binding protein (SREBP) 2 down-regulates ATP-binding cassette transporter A1 in vascular endothelial cells: a novel role of SREBP in regulating cholesterol metabolism. J. Biol. Chem. 2004;279(47):48801–48807. doi: 10.1074/jbc.M407817200. [DOI] [PubMed] [Google Scholar]
- 29.Chen X.W., Wang H., Bajaj K., Zhang P., Meng Z.X., Ma D., Bai Y., Liu H.H., Adams E., Baines A. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. eLife. 2013;2:e00444. doi: 10.7554/eLife.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gustafsen C., Kjolby M., Nyegaard M., Mattheisen M., Lundhede J., Buttenschon H., Mors O., Bentzon J.F., Madsen P., Nykjaer A. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab. 2014;19(2):310–318. doi: 10.1016/j.cmet.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Gouni-Berthold I., Berthold H.K., Gylling H., Hallikainen M., Giannakidou E., Stier S., Ko Y., Patel D., Soutar A.K., Seedorf U. Effects of ezetimibe and/or simvastatin on LDL receptor protein expression and on LDL receptor and HMG-CoA reductase gene expression: a randomized trial in healthy men. Atherosclerosis. 2008;198(1):198–207. doi: 10.1016/j.atherosclerosis.2007.09.034. [DOI] [PubMed] [Google Scholar]
- 32.Mayne J., Dewpura T., Raymond A., Cousins M., Chaplin A., Lahey K.A., Lahaye S.A., Mbikay M., Ooi T.C., Chretien M. Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. 2008;7:22. doi: 10.1186/1476-511X-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davalos A., Fernandez-Hernando C., Cerrato F., Martinez-Botas J., Gomez-Coronado D., Gomez-Cordoves C., Lasuncion M.A. Red grape juice polyphenols alter cholesterol homeostasis and increase LDL-receptor activity in human cells in vitro. J. Nutr. 2006;136(7):1766–1773. doi: 10.1093/jn/136.7.1766. [DOI] [PubMed] [Google Scholar]
- 34.Li H., Dong B., Park S.W., Lee H.S., Chen W., Liu J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J. Biol. Chem. 2009;284(42):28885–28895. doi: 10.1074/jbc.M109.052407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong B., Wu M., Li H., Kraemer F.B., Adeli K., Seidah N.G., Park S.W., Liu J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J. Lipid Res. 2010;51(6):1486–1495. doi: 10.1194/jlr.M003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H., Liu J. The novel function of HINFP as a co-activator in sterol-regulated transcription of PCSK9 in HepG2 cells. Biochem. J. 2012;443(3):757–768. doi: 10.1042/BJ20111645. [DOI] [PubMed] [Google Scholar]
- 37.Casaschi A., Wang Q., Dang K., Richards A., Theriault A. Intestinal apolipoprotein B secretion is inhibited by the flavonoid quercetin: potential role of microsomal triglyceride transfer protein and diacylglycerol acyltransferase. Lipids. 2002;37(7):647–652. doi: 10.1007/s11745-002-0945-8. [DOI] [PubMed] [Google Scholar]
- 38.Strong A., Ding Q., Edmondson A.C., Millar J.S., Sachs K.V., Li X., Kumaravel A., Wang M.Y., Ai D., Guo L. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J. Clin. Invest. 2012;122(8):2807–2816. doi: 10.1172/JCI63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kjolby M., Andersen O.M., Breiderhoff T., Fjorback A.W., Pedersen K.M., Madsen P., Jansen P., Heeren J., Willnow T.E., Nykjaer A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010;12(3):213–223. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 40.Linsel-Nitschke P., Heeren J., Aherrahrou Z., Bruse P., Gieger C., Illig T., Prokisch H., Heim K., Doering A., Peters A. Genetic variation at chromosome 1p13.3 affects sortilin mRNA expression, cellular LDL-uptake and serum LDL levels which translates to the risk of coronary artery disease. Atherosclerosis. 2010;208(1):183–189. doi: 10.1016/j.atherosclerosis.2009.06.034. [DOI] [PubMed] [Google Scholar]
- 41.Musunuru K., Strong A., Frank-Kamenetsky M., Lee N.E., Ahfeldt T., Sachs K.V., Li X., Li H., Kuperwasser N., Ruda V.M. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466(7307):714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thaker A.M., Frishman W.H. Sortilin: the mechanistic link between genes, cholesterol, and coronary artery disease. Cardiol. Rev. 2014;22(2):91–96. doi: 10.1097/CRD.0000000000000008. [DOI] [PubMed] [Google Scholar]
- 43.Moon J., Lee S.M., Do H.J., Cho Y., Chung J.H., Shin M.J. Quercetin up-regulates LDL receptor expression in HepG2 cells. Phytother. Res. 2012;26(11):1688–1694. doi: 10.1002/ptr.4646. [DOI] [PubMed] [Google Scholar]
- 44.Cermak R., Landgraf S., Wolffram S. The bioavailability of quercetin in pigs depends on the glycoside moiety and on dietary factors. J. Nutr. 2003;133(9):2802–2807. doi: 10.1093/jn/133.9.2802. [DOI] [PubMed] [Google Scholar]
- 45.Reinboth M., Wolffram S., Abraham G., Ungemach F.R., Cermak R. Oral bioavailability of quercetin from different quercetin glycosides in dogs. Br. J. Nutr. 2010;104(2):198–203. doi: 10.1017/S000711451000053X. [DOI] [PubMed] [Google Scholar]
- 46.Paulke A., Eckert G.P., Schubert-Zsilavecz M., Wurglics M. Isoquercitrin provides better bioavailability than quercetin: comparison of quercetin metabolites in body tissue and brain sections after six days administration of isoquercitrin and quercitrin. Pharmazie. 2012;67:991–996. [PubMed] [Google Scholar]
- 47.Valentova K., Vrba J., Bancirova M., Ulrichova J., Kren V. Isoquercitrin: pharmacology, toxicology, and metabolism. Food Chem. Toxicol. 2014;68:267–282. doi: 10.1016/j.fct.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 48.Omar K., Grant M.H., Henderson C., Watson D.G. The complex degradation and metabolism of quercetin in rat hepatocyte incubations. Xenobiotica. 2014:1–9. doi: 10.3109/00498254.2014.932032. [DOI] [PubMed] [Google Scholar]