

Key Points
Deletion of Mysm1 impairs development of steady-state DC, but no other myeloid lineages; monocyte, macrophage, and granulocyte.
Mysm1 governs DC differentiation from CMP by regulating Flt3 expression via modulating histone modifications and mediating Pu.1 recruitment.
Abstract
The mechanisms controlling the development of dendritic cells (DCs) remain incompletely understood. Using an Mysm1 knockout (Mysm1−/−) mouse model, we identified the histone H2A deubiquitinase Mysm1, as a critical regulator in DC differentiation. Mysm1−/− mice showed a global reduction of DCs in lymphoid organs, whereas development of granulocytes and macrophages were not severely affected. Hematopoietic progenitors and DC precursors were significantly decreased in Mysm1−/− mice and defective in Fms-like tyrosine kinase-3(Flt3) ligand–induced, but not in granulocyte macrophage–colony-stimulating factor (GM-CSF)-induced DC differentiation in vitro. Molecular studies demonstrated that the developmental defect of DCs from common myeloid progenitor (CMP) in Mysm1−/− mice is associated with decreased Flt3 expression and that Mysm1 derepresses transcription of the Flt3 gene by directing histone modifications at the Flt3 promoter region. Two molecular mechanisms were found to be responsible for the selective role of Mysm1 in lineage determination of DCs from CMPs: the selective expression of Mysm1 in a subset of CMPs and the different requirement of Mysm1 for PU.1 recruitment to the Flt3 locus vs GM-CSF-α and macrophage–colony-stimulating factor receptor loci. In conclusion, this study reveals an essential role of Mysm1 in epigenetic regulation of Flt3 transcription and DC development, and it provides a novel mechanism for lineage determination from CMP.
Introduction
Dendritic cell (DC) differentiation is tightly regulated by the coordination of signal transduction pathways and transcriptional networks. Fms-like tyrosine kinase-3 (Flt3), macrophage colony-stimulating factor (M-CSF) receptor, and granulocyte macrophage–colony-stimulating factor (GM-CSF)-α receptor play major roles in DC development.1 GM-CSF can derive DCs from murine bone marrow (BM) or human monocytes in vitro2-4 and plays essential roles in generating tumor necrosis factor-α–producing DCs and inducible nitric oxide synthase–producing DCs in infection.5 However, deletion of the GM-CSF receptor gene in mice does not result in a severe reduction of DCs,6 which implies a redundant role of GM-CSF in DC development. In contrast, Flt3 ligand (Flt3L) is an indispensable cytokine in steady-state DC development; Flt3L induces both conventional DC (cDC) and plasmacytoid DCs (pDCs) development from murine BM, whereas GM-CSF generates only cDCs in vitro.5,7,8 Flt3 and Flt3L knockout (KO) studies showed a dramatic reduction in the number of DCs in mice.9-11 Furthermore, BM progenitor cells gain DC-differentiating potential along with the Flt3 expression in DC progenitors and precursors.12,13 Therefore, Flt3L–Flt3 is a dominant cytokine stimuli and signaling network for steady state DC development.
Epigenetic factors play pivotal roles in controlling lineage-specific gene expression in hematopoiesis. A histone deubiquitinase, Mysm1, has been reported in regulating gene expression by interacting with histone acetylase p300/CBP-associated factor (PCAF) and promoting H1 dissociation from nucleosomes that is often linked with gene activation.14 Recently, we reported the role of Mysm1 in B-cell development and natural killer cell maturation by derepressing expression of transcription.15,16 Furthermore, Nijnik et al17 demonstrated the role of Mysm1 in maintaining BM hematopoietic stem cell (HSC) function and development of lymphoid and erythroid lineages. However, the cellular and physiological functions of Mysm1 are still largely unknown.
In this study, we demonstrated the essential role of histone H2A deubiquitinase Mysm1 in steady state DC development. We showed that Mysm1 regulates the expression of Flt3 by altering histone modifications at its encoding locus and recruiting the transcription factor PU.1 to the promoter region, which contributes to DC lineage specification from common myeloid progenitor (CMP).
Methods
Generation of Mysm1−/− mice
Mysm1−/− mice (Mysm1-KO first-floxed mice) were generated through a KO-first strategy as previously described.15 All animal breeding and experiments were approved and performed in accordance with the University of Southern California Institutional Animal Care and Use Committee.
Flow cytometry (FACS) and cell sorting
Sample preparation, cytometric analysis, and cell sorting were performed as described previously.18,19 For each staining, at least 100 000 events were collected for analysis. For sorting of the Lin- progenitor population, cells from BM were first depleted of mature hematopoietic cells using a lineage cell depletion kit (Miltenyi Biotec), then sorted by a fluorescence-activated cell sorter (FACS) using the anti-mouse lineage cocktail. More details are included in the supplemental Methods on the Blood Web site.
Chromatin immunoprecipitation
Chromatin was immunoprecipitated according to the manufacturer’s instructions (#9002; Cell Signaling). Briefly, sorted cells were crosslinked with 1% formaldehyde and blocked with glycine. Cells were then washed and digested by micrococcal nuclease. The nuclear pellet was suspended in chromatin immunoprecipitation (ChIP) buffer and sheared by sonication. The sheared chromatin was then incubated with various antibodies, uH2A (05-678), H3K27me3 (ab-6002), PU.1 (T-21, sc-352), Mysm1, or control anti-IgG (Cell Signaling), bound to protein A/G plus agarose beads. The immunoprecipitated chromatins were then eluted with ChIP elution buffer. The DNA fragments were released by treatment of ribonuclease A and then proteinase K at 65°C for 2 hours. The released DNA fragments were purified with columns and amplified by site-specific primers by quantitative polymerase chain reaction (qPCR). For sequential 2-step ChIP experiments,20 crosslinked chromatin was immunoprecipitated with anti-Mysm1 or control IgG (#9002; Cell Signaling) and was then eluted in a solution of 30 mM dithiothreitol, 500 mM NaCl, and 0.1% sodium dodecyl sulfate. Eluted chromatin was reimmunoprecipitated with anti-Pu.1 or control IgG. The binding capability was expressed as a ratio of immunoprecipitated DNA to that in the input sample. The ratio of IgG-precipitated DNA to that in the input sample was set as 1. Primer sequences are available upon request.
Lentivirus and retrovirus production and transduction
Recombinant lentiviral vectors were generated as described.21 293T cells were cotransfected with package plasmids VSVg, Rev, Gag/Pol, and a recombinant lentiviral vector expressing Mysm1-enhanced green fluorescent protein (eGFP) (LV-Mysm1) or lentiviral vector expressing eGFP (LV-GFP) for recombinant lentiviral production. Retroviruses were produced as previously described.15 Phoenix cells were transfected with recombinant plasmid pMIG-Flt3-IRES-GFP (pMIG-Flt3)22 or pMIG-GFP. The pMIG-Flt3 was a gift from Dr Stephen L. Nutt (The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia). For transduction, hematopoietic cells seeded in the RetroNectin (TaKaRa)-pretreated culture dishes were added with lentiviral or retroviral supernatants and centrifuged at 3000 rpm for 90 minutes and were then incubated at 37°C in the presence of polybrene (4 μg/mL) for an additional 48 hours.
BM-derived DC culture
Murine BM-derived DCs were prepared as previously described.19 Briefly, murine BM was flushed from the hind limbs and depleted of red blood cells with red blood cell lysis buffer (#555899; BD Bioscience). After extensive washing with RPMI-1640, cells were cultured with RPMI-1640 supplemented with 20 ng/mL of GM-CSF and IL-4 (PeproTech) or 100 ng/mL of Flt3L (eBioscience). For GM-CSF culture, the culture media were changed every other day until day 7. For Flt3L culture, the cells were cultured for 8 days without disruption.
Statistics
Groups of 3 to 8 mice were used for statistical analysis. P values were calculated using the Student t test.
Results
Mysm1 is required for cDC and pDC development
Our previous studies showed that Mysm1−/− mice had overall decreased cellularity in the BM, spleen, and thymus, which resulted from the impaired HSC maintenance and self-renewal.15 To further define development of myeloid cell lineages in Mysm1−/− mice, several hematopoietic organs including the BM and spleen were harvested from 4- to 6-week-old Mysm1−/− or wild-type (WT) littermates, and the development of myeloid populations including DCs, macrophages, and granulocytes were analyzed by FACS (Figure 1A-H). Both cDC (CD11chi MHCII+) and pDC (CD11cmid PDCA1+) (Figure 1A) were significantly decreased in frequency as well as in absolute number in the spleens of Mysm1−/− mice (Figure 1B). The proportions of splenic cDC subsets, CD8+ and CD8− cDCs, were not significantly changed (Figure 1A-B). Since pDCs fully develop in BM and migrate to lymphoid organs, we analyzed the development of pDCs in BM (Figure 1C-D). The results showed a significant reduction in the absolute number as well as in the frequency of pDCs in Mysm1−/− BM. However, granulocytes (CD11b+Ly6g+Ly6clow), monocytes (CD11b+ Ly6gneg-lowLy6chigh), and macrophages (CD11b+Gr1−F4/80+) had no significant alteration in their frequencies in Mysm1−/− spleen (Figure 1E-F) and BM (Figure 1G-H), although their absolute numbers dramatically decreased due to reduction of cellularity (Figure 1F-H, lower panel). The results suggest that Mysm1 may more profoundly be required by DC lineage differentiation compared with the differentiation of other myeloid lineages such as macrophages and granulocytes. Because DCs diverge their differentiation pathway from macrophages/granulocytes at the stage of CMP, the results also imply that Mysm1 may be specifically required for DC lineage commitment from CMP.
Figure 1.
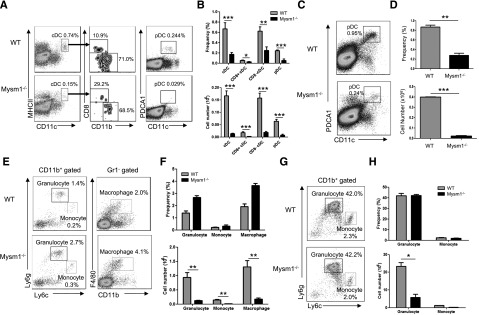
Mysm1 is required for DC development. (A-H) Spleens and BM from 4- to 6-week-old WT or Mysm1−/− mice were analyzed by FACS for the indicated cell populations (n = 4). (A) FACS analyses of splenic cDCs (CD11chighMHCII+) and the subsets: CD8+ cDCs (CD11chighMHCII+CD8+CD11b−), CD8− cDCs (CD11chighMHCII+CD8−CD11b+), and splenic pDCs (CD11cintPDCA1+). (B) Frequency and cell numbers of splenic cDCs and pDCs from WT and Mysm1−/− mice. (C) FACS analyses of BM pDCs (CD11c+PDCA1+). (D) Frequency and cell numbers of pDCs from BM of WT and Mysm1−/− mice. (E) FACS analyses of splenic granulocytes (CD11b+Ly6g+Ly6clow), monocytes (CD11b+ Ly6gneg-lowLy6chigh), and macrophages (CD11b+Gr1−F4/80+). (F) Frequency and cell numbers of each cell type. (G) FACS analyses of BM-derived granulocytes and monocytes. (H) Frequency and cell numbers of each cell type. The data were shown form 1 of 3 independent experiments. *P < .05; **P < .01; ***P < .001.
Intrinsic role of Mysm1 in DC development
To test whether the defect in DC development in Mysm1−/− mice is a cell intrinsic effect, BM cells from CD45.2+ Mysm1−/− or WT littermates were transplanted into lethally irradiated CD45.1+ recipient mice at 1:1 ratio with CD45.1+ WT competitor cells. Three weeks later, spleen, BM, and peripheral blood were harvested and analyzed by FACS for reconstitution of DCs (CD11c+) (Figure 2A). The results showed that Mysm1−/− BM (CD45.2+) failed to develop DCs, which implies an intrinsic role of Mysm1 in the differentiation of BM progenitors into DCs.
Figure 2.

Intrinsic role of Mysm1 in DC development. (A) CD45.2+ (2 × 106) WT or Mysm1−/− BM cells were transplanted into lethally irradiated CD45.1+ recipient mice at 1:1 ratios with CD45.1+ WT competitor cells (2 × 106). CD45.2+ cell populations in recipients were determined by FACS at 3 weeks posttransplantation in BM, peripheral blood (PBL), and spleen (SP). The representative FACS data from 2 mice were shown. The experiments were repeated more than 3 times with similar results. (B) BM cells were harvested from the femurs and tibiae of Mysm1−/− and WT littermates and 2 × 106 cells/mL and were then plated in a 6-well plate. For Flt3L-induced DC differentiation, BM cells were cultured for 8 days without disruption in DC culture media supplemented with 100 ng/mL Flt3L. Cells were harvested and analyzed via FACS for pDCs (CD11c+B220+) and cDCs (CD11c+B220-−). The experiments were repeated more than 3 times with similar results. (C) For GM-CSF-induced DC culture, BM cells were cultured for 5 days in DC culture media supplemented with 20 ng/mL GM-CSF and 20 ng/mL IL-4. Cells were harvested and analyzed via FACS for cDC (CD11c+CD11b+). (D) BM cells from Mysm1−/− mice were transduced with either lentivirus-expressing mouse Mysm1 (LV-Mysm1) or control lentivirus (LV-GFP) for 8 hours. Cells were washed and cultured in Flt3L (100 ng/mL) supplemented media for 8 days and CD11c+ DC differentiation was analyzed by FACS. All the experiments were repeated more than 3 times with similar results. SSC, side scatter.
Flt3L is an essential cytokine for steady-state DC differentiation9 (including cDC and pDC), whereas GM-CSF generates only cDCs in vitro.8,23 To further examine the intrinsic role of Mysm1 in DC differentiation, BM cells from Mysm1−/− and WT littermates were cultured in Flt3L- or GM-CSF/IL-4-supplemented media and DC differentiation was analyzed by FACS. Strikingly, Mysm1−/− BM failed to generate cDC (CD11c+B220−) or pDC (CD11c+B220+), instead, it differentiated into an undefined population with a granulocyte-like phenotype (side scatter/forward scatterhigh) upon Flt3L stimulation (Figure 2B). In contrast, Mysm1−/− BM was able to differentiate into DCs upon GM-CSF/IL4 stimulation (Figure 2C). To confirm that the failure of Mysm1−/− BM in Flt3L-induced DC culture results from an absence of Mysm1, we reconstituted Mysm1 expression in Mysm1−/− BM cells using a recombinant lentiviral vector that coexpresses Mysm1 and GFP markers (LV-Mysm1), and cultured the cells in Flt3L-supplemented media (Figure 2D). Mysm1−/− BM transduced with LV-Mysm1 were able to differentiate into CD11c+ DCs in Flt3L culture, indicating that the defect in DC differentiation of Mysm1−/− BM can be rescued by forced expression of Mysm1.
Mysm1 is selectively required for Flt3L-induced DC development from early hematopoietic precursors
To determine whether the defect in DC development in Mysm1−/− mice is due to a decrease of hematopoietic progenitors or DC precursors, hematopoietic progenitors in the BM of Mysm1−/− and WT littermates were analyzed by FACS (Figure 3). The HSC (Lin−Sca1+cKit+), CLPs (Lin−IL7R+Sca1intcKitint), CMP (Lin−IL7R−Sca1−cKit+CD34+CD16/32int), and GMPs (Lin−IL7R−Sca1−cKit+CD34+CD16/32hi) were markedly decreased in frequency and absolute number in Mysm1−/− mice (Figure 3A-B). Similarly, DC-committed precursor populations, Lin−CD115+ (macrophage and DC progenitor (MDP)-enriched population10) and common DC progenitor (CDP) (Lin−Flt3+CD115+cKitint), were significantly decreased in Mysm1−/− BM. Also, the migratory DC precursors, pre-cDC (Lin−CD11c+MHCII+Flt3+ SIRPαneg-low),24,25 showed a marked reduction in Mysm1−/− spleen (Figure 3C-D,F). These results indicate that Mysm1 contributes to the development of hematopoietic progenitors and DC precursors.
Figure 3.
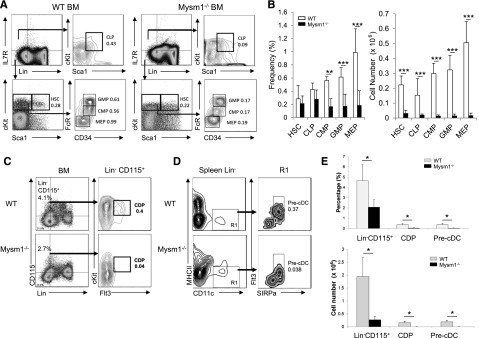
Decrease in DC progenitors. BM cells and splenocytes were analyzed via FACS for the indicated DC progenitor populations. (A) Lineage markers, Sca1, cKit, CD34, and CD16/32 were used to analyze HSC, common lymphoid progenitor (CLP), megakaryocyte and erythrocyte progenitor (MEP), granulocyte macrophage progenitor (GMP), and CMP populations. (B) Frequency and cell numbers of the hematopoietic precursor subpopulations in whole BM compartment. (C) BM from WT or Mysm1−/− were analyzed for Lin−CD115+ and CDP (Lin−CD115+c-KitintFlt3+). (D) Splenocytes were analyzed for pre-cDC (CD11c+MHCII−signal-regulatory protein alpha [SIRPα]+ Flt3+). (E) Frequency and cell numbers of Lin−CD115+, CDPs, and pre-cDCs in BM or spleen. Data presented are mean values (± standard error of the mean) (n = 4). The experiments were repeated more than 3 times with similar results. *P < .05; **P < .01; ***P < .001.
Next, we tested whether there is a differentiation defect in residual Mysm1−/− DC precursors. Lin−, Lin-sca1+cKit+ (LSK), CMP, and Lin−CD115+ populations from WT or Mysm1−/− BM were sorted by FACS and cultured with feeder cells from CD45.1+ syngeneic mice in Flt3L-supplemented media (Figure 4A) or GM-CSF/IL-4-supplemented media (Figure 4B). We found that deletion of Mysm1 abolished DC generation from all of the precursors tested in Flt3L culture. In contrast, Mysm1−/− DC precursors generated DC upon GM-CSF/IL4 stimulation, albeit with less efficiency compared with WT DC precursors (Figure 4C), which was likely due to impaired stem cell maintenance and self-renewal. These data indicate that Mysm1 is required for Flt3-induced DC development from early stage of hematopoiesis.
Figure 4.
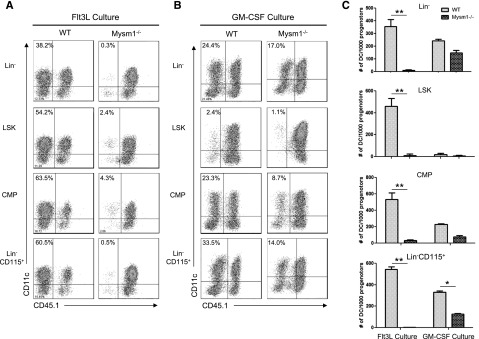
Mysm1−/− DC progenitors fail to differentiate into DC with Flt3L in vitro stimulation. DC progenitor cells Lin−LSKs (Lin−Sca1+cKit+), Lin−CD115+, and CMPs (Lin−IL7R−Sca1−c-Kit+CD34+CD16/32int) were isolated from WT or Mysm1−/− BM by FACS sorter and 10 000 progenitor cells were cultured with 1.5 × 105 CD45.1 BM feeder cells in the presence of Flt3L (100 ng/mL) (A) or GM-CSF (20 ng/mL) and IL-4 (20 ng/mL) (B) in a 96-well plate for 8 days. Cells were harvested and analyzed by FACS for DC differentiation (CD11c+ CD45.1−). (C) The number of DCs (CD11c+ CD45.1−) generated from 1000 progenitors of each culture was calculated and presented as an average from 2 to 3 wells. The experiments were repeated 3 times with similar results. *P < .05; **P < .001.
Mysm1 upregulates expression of Flt3 through direct binding at the promoter locus
Expression of Flt3 is essential for steady-state DC development.24 Flt3 is used as a surface marker to identify DC-committed progenitors26 including CDP, MDP, and pre-cDC given their high expression of Flt3. Our results revealed that Mysm1−/− mice had approximately 10-fold decrease in the frequency of CDP (Lin−CD115+cKit+Flt3+) in BM and pre-cDC (Lin−SIRPa+Flt3+) in the spleen (Figure 3C-D) due to loss of Flt3 expression. Our in vitro experiments explored that Mysm1−/− BM failed to differentiate into DCs upon Flt3L stimulation as described above. The observations promoted us to examine Flt3 expression in Mysm1−/− hematopoietic progenitors and DC precursors. The FACS results showed that expression of Flt3 on Mysm1−/− Lin− and CMP was almost completely abrogated (Figure 5A). To test if the reduction occurred at the transcription level, the amount of Flt3 mRNA was measured by qPCR and the results showed a significant downregulation of Flt3 transcription in Mysm1−/− Lin− and CMP (Figure 5B). Collectively, the results suggest that Mysm1 is required for Flt3 expression.
Figure 5.
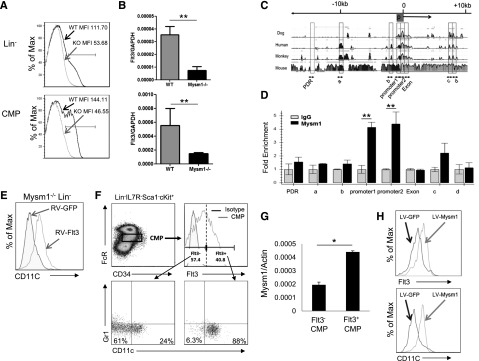
Mysm1 drives CMP toward DC lineage specification by upregulating Flt3 expression. Expression of Flt3 in Lin− BM cells and CMP population was analyzed by (A) FACS and (B) qPCR; the messenger RNA (mRNA) expression was normalized to glyceraldehyde 3-phosphate dehydrogenase messenger RNA amount. (C) Sequence alignment of dog, human, monkey, and mouse Flt3 genes; transcription start site (0); and promoter site (P). (D) ChIP assay analyzed recruitment of Mysm1 to Flt3 gene locus in WT Lin− BM cells. The prepared chromatin was immunoprecipitated with either an anti-Mysm1 antibody or control IgG, and pulled-down DNA was eluted and subjected to qPCR using the primers designed from conserved region (boxed) among species. (E) Lin− cells were sorted from Mysm1−/− BM and transduced with retrovirus-expressing GFP (RV-GFP) or Flt3 (RV-Flt3) for 12 hours. Cells were cultured in DC culture media supplemented with 100 ng/mL of Flt3L (at a density of 1 × 105 cells/100 uL) for 8 days. FACS was used to analyze the differentiation of CD11c+ DCs. (F-H) Mysm1 drives CMPs to DC lineage specification. CMPs from WT BM was sorted and divided into 2 populations by Flt3 expression: Flt3− CMPs and Flt3+ CMPs. The sorted cells were cultured in Flt3L-supplemented media for 8 days and surface markers CD11c and Gr1 was used to analyze DC differentiation by FACS (F), and Mysm1 levels in these 2 populations were measured by qPCR (G). Flt3− CMP population was infected with lentivirus expressing either GFP or Mysm1, and was cultured in Flt3L-supplemented media for 8 days. Surface expression of CD11c and Flt3 was analyzed by FACS (H). *P < .05; **P < .01.
Despite the critical role of Flt3 and its signaling pathway in hematopoiesis, the regulating mechanism of Flt3 expression is poorly understood. To examine whether Mysm1 directly regulates expression of Flt3, we surveyed the Flt3 gene for Mysm1 binding sites by ChIP assay (Figure 5C-D) in WT Lin− populations. The genomic sequences of Flt3 from dog, human, monkey, and mouse were aligned, and primers were designed from highly conserved regions including the promoter (promoter 1 and promoter 2) and the first exon (Exon) (Figure 5C). Mysm1-chromatin complex was immunoprecipitated by anti-Mysm1 followed by qPCR analysis with site-specific primers. We found that the signal was highly enriched at the Flt3 promoter site (Figure 5D), indicating a direct Mysm1 binding at the Flt3 promoter.
Mysm1 induces DC lineage specification from CMP by activating Flt3 expression
To ascertain a direct correlation between reduced levels of Flt3 and DC developmental defects in Mysm1−/−, we restored Flt3 in Mysm1−/− Lin− cells by transducing with a retrovirus expressing Flt3 or GFP control and culturing in Flt3L media (Figure 5E). FACS analysis of CD11c showed that forced expression of Flt3 restored DC development in Mysm1−/− Lin− cells, which indicates that a reduction of Flt3 contributed to the DC developmental defect in Mysm1−/− mice.
We have shown that development of macrophage and granulocyte was not as severely affected by deleting Mysm1 as DC development (Figure 1), although they all differentiate from CMP. The CMP is a heterogeneous population in which Flt3 expression level varies. A previous report showed that Flt3+ CMPs give rise to DC progenitors including MDPs and CDPs, whereas Flt3− CMPs possess GMP potential that differentiates into macrophages and granulocytes.13 As shown in Figure 5, Mysm1 regulates Flt3 expression via a direct association with its promoter region. Therefore, we hypothesized that Mysm1 may induce DC lineage specification from CMPs by upregulating Flt3 expression. To test this hypothesis, we first compared the DC differentiation potential from Flt3− and Flt3+ CMPs. Flt3− and Flt3+ CMPs were sorted and cultured in Flt3L-supplemented media. The cells were then stained with CD11c and Gr1 antibodies to exclude granulocyte populations and analyzed by FACS analysis. As expected, differentiation of DCs was significantly decreased from Flt3− CMPs as compared with Flt3+ CMPs, in which the majority of cells gave rise to DCs (88%) (Figure 5F). We observed that 24% of Flt3− CMP differentiated into CD11c+ DCs, suggesting that a portion of Flt3− CMP may have acquired DC differentiation capacity by unknown mechanisms under our culture condition. Given a close correlation between Mysm1 and Flt3 expression, we compared the mRNA level of Mysm1 in Flt3− and Flt3+ CMPs by qPCR and found that Mysm1 expression was higher in the Flt3+ CMP population (Figure 5G). To further investigate the role of Mysm1 in CMP lineage differentiation and Flt3 expression, we transduced WT Flt3− CMPs with a lentiviral vector expressing GFP (LV-GFP) or Mysm1 (LV-Mysm1) and cultured the cells in Flt3L-supplemented media. Forced expression of Mysm1 not only restored Flt3 expression in Flt3− CMPs, but also induced DC differentiation (Figure 5H). The results demonstrate that Mysm1 plays an important role in DC lineage specification from CMPs by positively regulating Flt3 expression.
Mysm1 regulates modifications of histone, RNA polymerase activity at the Flt3 promoter region
Given that Mysm1 has histone deubiquitinase activity, we hypothesized that the histone ubiquitination status may be altered at the Flt3 promoter site in Mysm1−/−. We immunoprecipitated the uH2A-chromatin complex from WT or Mysm1−/− BM with an anti-uH2A antibody, and enrichments in promoter sites, the first exon, and promoter desert region were compared (Figure 6A). A significant increase of uH2A was found at both the promoter and promoter desert regions of Flt3 in Mysm1−/− BM. Since the ubiquitination status on histones affects other histone modifications,27,28 we tested the status of other histone modifications at the Flt3 regulatory locus by ChIP assay. Figure 6B shows a significant increase of repressive trimethylated histone H3 at lysine 27 (H3K27me3) at the Flt3 promoter sites.
Figure 6.
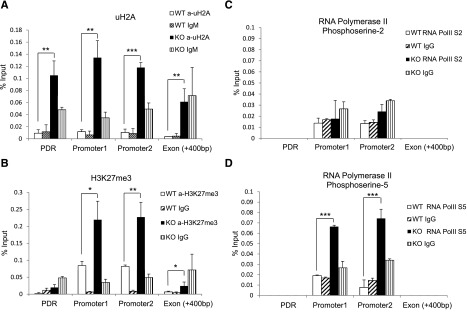
Altered histone modification and RNA polymerase activity at Flt3 locus in Mysm1−/− BM. ChIP assay analyzed BM Lin− cells from WT or Mysm1−/− mice via (A) anti-uH2A, (B) anti-H3K27me3, (C) anti-phosphoserine-2 RNA polymerase, or (D) anti-phosphoserine-5 RNA polymerase II. The precipitated DNA was analyzed by qPCR with primers amplifying Flt3 promoter regions (promoter 1 and 2), promoter deprived region (control primer), and Flt3 exon 1 region. *P < .05; **P < .01; ***P < .001. PDR, promoter-deprived region.
Previous reports demonstrated that histones act as a restraint for poised RNA polymerase II on gene promoters and a block for transcriptional elongation.20,29 RNA polymerase II assembled at the promoter is phosphorylated at serine 5 (S5) of the carboxy-terminal domain repeat through transcription factor IIH. As elongation proceeds, the level of phosphorylation of the carboxy-terminal domain at serine 5 decreases and phosphorylation at serine-2 (S2) increases.30 To examine activity of RNA polymerase II at the Flt3 promoter, we pulled down chromatin by antibodies that recognize RNA polymerase II with either the S2 or S5 and measured enrichment of Flt3 promoter DNA by qPCR. The level of S2 RNA polymerase II remained similar in WT and Mysm1−/− (Figure 6C), but we observed an accumulation of S5 RNA polymerase II at the Mysm1−/− Flt3 promoter site (Figure 6D). This result suggests that an increase of uH2A may cause RNA polymerase to halt at the promoter, which, in turn, blocks it from proceeding to elongation. Collectively, these data suggest that Mysm1 derepresses Flt3 transcription by downregulating the level of uH2A and H3K27me3, thereby promoting transcription elongation at the promoter.
Mysm1 is required for the recruitment of PU.1 to the Flt3 locus, but not to the GM-CSF-α receptor and M-CSF receptor loci
PU.1 has been proposed as a crucial regulator of DC development.31−34 A recent study reported that PU.1 activates expression of Flt3 by directly associating with the Flt3 promoter region.22 We tested the functionality of Mysm1 on the activating transcription of Flt3 by luciferase reporter assay. The results showed that Mysm1 enhances Flt3 promoter activity (−800 to +44) and PU.1 synergized activity of Mysm1 (supplemental Figure 1). Next, we tested whether deletion of Mysm1 affects PU.1 recruitment to the Flt3 promoter by comparing PU.1 occupancy at the Flt3 locus in WT and Mysm1−/− BM via ChIP assay. Figure 7A shows that recruitment of PU.1 was significantly decreased at the Flt3 promoter in Mysm1−/−. Because both Mysm1 and PU.1 directly bind to the Flt3 promoter region, we examined if there is an interaction between Mysm1 and PU.1 at the Flt3 promoter by sequential 2-step ChIP assay. For the first ChIP, crosslinked chromatin from WT Lin− cells was immunoprecipitated with an anti-Mysm1 antibody or a control IgG. The precipitated chromatin was eluted, reimmunoprecipitated with anti-Pu.1 or control IgG (second ChIP) and the precipitated DNA was amplified by primers encompassing the Flt3 promoter by qPCR. The results showed that anti-Pu.1 antibody specifically precipitated the DNA fragment spanning from −0.8 Kb to 0 of the Flt3 promoter that was precipitated by anti-Mysm1 antibody, implying that Mysm1 and PU.1 occupy the same Flt3 locus (Figure 7B). Coimmunoprecipitation assay supported a direct interaction between Mysm1 and PU.1 (Figure 7C).
Figure 7.
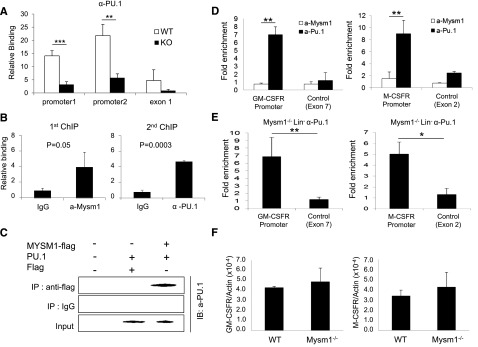
Mysm1 mediates localization of PU.1 to the Flt3 locus, but not to the GM-CSF-α receptor and M-CSF receptor loci. (A) ChIP analyses of PU.1 recruitment in WT or Mysm1−/− BM Lin− cells. The precipitated DNA was analyzed by qPCR via 2 sets of primers amplifying the Flt3 promoter region (promoter 1 and promoter 2) or the first exon (exon1) and normalized with IgG control. (B) Sequential 2-step ChIP analyses of Mysm1 and PU.1 recruitment to the Flt3 promoter in WT BM Lin− progenitors. Chromatin was immunoprecipitated with anti-Mysm1 or IgG for the first ChIP. The pulled-down chromatin was eluted and reimmunoprecipitated with anti-Pu.1 or IgG for the second ChIP. The released DNA from the first and second ChIP were subjected to qPCR analysis using primers encompassing Flt3 promoter. Relative binding was defined by determining the ratio of immunoprecipitated DNA to that in the input sample. The ratios of the control IgG-immunoprecipitated DNA to that in the input samples were set as 1.0. (C) Coimmunoprecipitation assay of Mysm1 and PU1. pCMV-Pu.1 plasmid was cotransfected with either pCMV-Flag-Mysm1 or pCMV-Flag expression plasmid into HEK293T cells and cell lysates were incubated with anti-Flag antibody and immunoprecipitated proteins were analyzed by anti-PU.1 antibody via western blot analysis. (D) ChIP analysis of Mysm1 and PU.1 recruitment to GM-CSF-α receptor or M-CSF receptor gene loci in WT Lin− cells. The signal was normalized to IgG control. (E) ChIP analysis of PU.1 recruitment to the promoter loci of GM-CSF-α receptor and M-CSF receptor. Lin− cells were isolated from Mysm1−/− BM and chromatin was immunoprecipitated with anti-PU.1 or IgG. The pulled-down DNA was eluted and amplified by qPCR via primers spanning the promoter or exon regions of GM-CSF-α receptor and M−CSF receptor. The signal was normalized to IgG control. (F) The qPCR analyzing messenger RNA of GM-CSF-α receptor (GM-CSFR) and M-CSF receptor (M-CSFR) in WT and Mysm12/2 Lin2 cells. *P < .05; **P < .01; ***P < .001.
PU.1 is a central regulator of many hematopoietic cell lineages and is essential for final differentiation of myeloid cells.35 Both the GM-CSF-α receptor and M-CSF receptor are required for macrophage and granulocyte development from myeloid progenitors, and their expressions are also activated by direct binding of PU.1 to the promoter loci.36,37 Because Mysm1 is found to mediate PU.1 recruitment to the Flt3 promoter, we speculated that Mysm1 may also involve in recruiting PU.1 to the GM-CSF-α receptor and M-CSF receptor promoter loci. To test this hypothesis, we first examined binding of PU.1 and Mysm1 at the promoter regions of GM-CSF-α receptor and M-CSF receptor by ChIP analysis in WT Lin− cells. The results showed high enrichment of both GM-CSF-α receptor and M-CSF receptor promoters from the anti-PU.1 pull-down samples, whereas no signal was found from anti-Mysm1 pull-down samples (Figure 7D). To further confirm that Mysm1 does not affect PU.1 recruitment to the GM-CSF-α receptor and M-CSFR promoters, chromatin from Mysm1−/− BM Lin− cells was pulled down by anti-PU.1 antibody or IgG control and precipitation of GM-CSF-α receptor and M−CSF receptor promoters was surveyed by site-specific primers. The result showed high associations of PU.1 at GM-CSF-α receptor and M-CSF receptor promoters in the absence of Mysm1 (Figure 7E). In addition, we observed mRNA expression levels of the GM-CSF-α receptor and the M-CSF receptor was intact in Mysm1−/− Lin− cells (Figure 7F). Collectively, the results indicate that PU.1 recruitment to the GM-CSF-α receptor and M-CSF receptor loci is Mysm1-independent. This data may provide a molecular basis for the selective regulatory mechanism of Mysm1 for DC lineage specification from CMP.
Discussion
Hematopoietic lineage commitment requires both temporal and spatial regulation of gene expression.38 One of the mechanisms to regulate gene expression is epigenetic control via histone modifications. In this study, we have demonstrated for the first time the essential role of histone H2A deubiquitinase Mysm1 in DC development by de-repressing Flt3 transcription.
We found that Mysm1 deletion caused defective DC development in a cell-intrinsic manner. We found further that Mysm1−/− BM was unresponsive to Flt3L stimulation, whereas GM-CSF stimulation caused normal DC development in vitro. This data strongly supports the role of Mysm1 in steady-state DC development, because Flt3L is required for steady-state DC development. From further phenotypical analysis, we found a significant reduction in all DC progenitors, from early progenitors including HSCs, LSKs, CLPs, and CMPs, to DC-committed precursors including MDPs, CDPs, and pre-cDCs. Furthermore, we found that the DC progenitors Lin−, LSKs, CMPs, and Lin−CD115+ from Mysm1−/− were unable to generate DCs upon Flt3L stimulation. Therefore, DC differentiation is defective in Mysm1−/− from early stage progenitors due to reduced numbers and defective function.
Flt3, the receptor of Flt3L, is required for DC development, as demonstrated in a KO study where the deletion of Flt3 results in a dramatic reduction of cDCs, pDCs, and interstitial dermal DCs.9 Furthermore, as DC development proceeds along a successive line of Flt3+ progenitors, expression of Flt3 predefines the DC lineage potential of hematopoietic progenitors.12,13,39 However, Mysm1−/− mice showed decreased Flt3-expressing CMPs in BM and decreased Flt3-expressing pre-cDCs in spleen. Thus, we speculated that Flt3 expression may be altered, which is responsible for defective DC development in Mysm1−/− mice. We confirmed that Flt3 expression was significantly reduced in Mysm1−/− progenitors including Lin−, LSKs, and CMPs, and DC-committed progenitors in both protein and mRNA levels. Additionally, our ChIP data revealed a direct association of Mysm1 at the Flt3 promoter region, indicating an essential role of Mysm1 in Flt3 expression and a close correlation between defective Flt3 expression and DC development in Mysm1−/− mice.
Our finding that Mysm1 is required for DC lineage, but not granulocyte and macrophage lineage differentiation begs a critical question: how is Mysm1 able to selectively drive the DC lineage program from CMP over other lineages? First, we found that Mysm1 is selectively expressed in the CMP subset. CMPs are a heterogeneous population that gives rise to multiple myeloid lineages.40 Approximately 40% of CMPs express Flt3, which can differentiate into DCs upon Flt3L stimulation. The remaining 60%, Flt3− CMPs, have less DC potential and favor to GMP and megakaryocyte and erythrocyte progenitor lineages. We found that Mysm1 is highly expressed in Flt3+ CMPs compared with Flt3− CMPs, implying that Mysm1 promotes Flt3 expression for DC differentiation.
The selective role of Mysm1 in DC differentiation is further supported by PU.1 dependency on Mysm1 for recruitment to the Flt3 loci. Pu.1 is a critical transcription factor in hematopoietic lineage determination and modulates expression of nearly 3000 genes expressed in hematopoietic cells including cell-surface proteins (CD11b) cytokines, and their receptors (granulocyte-CSF, GM-CSF, M-CSF, and Flt3).11,36,41 In our study, we showed that Mysm1 mediates PU.1 recruitment to the Flt3 locus for its transcription, but recruitment of PU.1 to the GM-CSF-α receptor and M-CSF receptor loci is Mysm1-indepenent. Moreover, we observed that the reduction of GMP was not as severe as the reduction of DC-committed precursors (CDPs, pre-cDCs) in Mysm1−/− mice, presumably because Flt3 expression is dispensable for GMP development. Thus, this study indicates that, in addition to its differential expression in subsets of CMPs, a lineage-specific defect of DC differentiation in Mysm1−/− is attained by Mysm1-dependent or Mysm1-independent recruitment of PU.1 to different gene loci.
This unexpected finding certainly raises additional questions about how Mysm1 regulates histone modifications and recruitment of transcription factors in a gene-specific manner. One possible scenario is that Mysm1 may find its binding target in a DNA sequence-specific manner because Mysm1 contains a DNA-binding motif in its SWI3, RSC8 and MOIRA domain. The consensus sequence and detailed mechanisms need to be investigated in the future. Besides its distinct roles in hematopoietic cell differentiation, Mysm1 was found to play important roles in HSC renewal and maintenance.16,17 Further studies are needed to investigate how Mysm1 plays such diverse roles.
Acknowledgments
This work was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (AI084811, AI068472), and National Cancer Institute (CA090427, CA116677), the Leukemia & Lymphoma Society SCOR Award, and the Norris Comprehensive Cancer Center/University of Southern California.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: H.W. and S.-Y.C. designed experiments; H.W., V.N., P.Y., S.S., L.J., X.F.H., and S.-Y.C. performed experiments and analyzed data; H.W. wrote the manuscript; and S.-Y.C. supervised research.
Conflict of interest disclosure: The authors declare no competing financial interests.
Correspondence: Si-Yi Chen, Norris Research Tower 7506, 1450 Biggy St, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033; e-mail: siyichen@usc.edu.
References
- 1.Schmid MA, Kingston D, Boddupalli S, Manz MG. Instructive cytokine signals in dendritic cell lineage commitment. Immunol Rev. 2010;234(1):32–44. doi: 10.1111/j.0105-2896.2009.00877.x. [DOI] [PubMed] [Google Scholar]
- 2.Ebner S, Ratzinger G, Krösbacher B, et al. Production of IL-12 by human monocyte-derived dendritic cells is optimal when the stimulus is given at the onset of maturation, and is further enhanced by IL-4. J Immunol. 2001;166(1):633–641. doi: 10.4049/jimmunol.166.1.633. [DOI] [PubMed] [Google Scholar]
- 3.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179(4):1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol. 2007;179(11):7577–7584. doi: 10.4049/jimmunol.179.11.7577. [DOI] [PubMed] [Google Scholar]
- 6.Vremec D, Lieschke GJ, Dunn AR, Robb L, Metcalf D, Shortman K. The influence of granulocyte/macrophage colony-stimulating factor on dendritic cell levels in mouse lymphoid organs. Eur J Immunol. 1997;27(1):40–44. doi: 10.1002/eji.1830270107. [DOI] [PubMed] [Google Scholar]
- 7.Brasel K, De Smedt T, Smith JL, Maliszewski CR. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood. 2000;96(9):3029–3039. [PubMed] [Google Scholar]
- 8.Gilliet M, Boonstra A, Paturel C, et al. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3-ligand and granulocyte/macrophage colony-stimulating factor. J Exp Med. 2002;195(7):953–958. doi: 10.1084/jem.20020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenna HJ. Role of hematopoietic growth factors/flt3 ligand in expansion and regulation of dendritic cells. Curr Opin Hematol. 2001;8(3):149–154. doi: 10.1097/00062752-200105000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Waskow C, Liu K, Darrasse-Jèze G, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. 2008;9(6):676–683. doi: 10.1038/ni.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kingston D, Schmid MA, Onai N, Obata-−Onai A, Baumjohann D, Manz MG. The concerted action of GM-CSF and Flt3-ligand on in vivo dendritic cell homeostasis. Blood. 2009;114(4):835–843. doi: 10.1182/blood-2009-02-206318. [DOI] [PubMed] [Google Scholar]
- 12.Karsunky H, Merad M, Cozzio A, Weissman IL, Manz MG. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med. 2003;198(2):305–313. doi: 10.1084/jem.20030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Amico A, Wu L. The early progenitors of mouse dendritic cells and plasmacytoid predendritic cells are within the bone marrow hemopoietic precursors expressing Flt3. J Exp Med. 2003;198(2):293–303. doi: 10.1084/jem.20030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu P, Zhou W, Wang J, et al. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol Cell. 2007;27(4):609–621. doi: 10.1016/j.molcel.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang XX, Nguyen Q, Chou Y, et al. Control of B cell development by the histone H2A deubiquitinase MYSM1. Immunity. 2011;35(6):883–896. doi: 10.1016/j.immuni.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Nandakumar V, Jiang XX, et al. The control of hematopoietic stem cell maintenance, self-−renewal, and differentiation by Mysm1-mediated epigenetic regulation. Blood. 2013;122(16):2812–2822. doi: 10.1182/blood-2013-03-489641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nijnik A, Clare S, Hale C, et al. Sanger Institute Microarray Facility; Sanger Mouse Genetics Project. The critical role of histone H2A-deubiquitinase Mysm1 in hematopoiesis and lymphocyte differentiation. Blood. 2012;119(6):1370–1379. doi: 10.1182/blood-2011-05-352666. [DOI] [PubMed] [Google Scholar]
- 18.Song XT, Evel-Kabler K, Shen L, Rollins L, Huang XF, Chen SY. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008;14(3):258–265. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharabi AB, Aldrich M, Sosic D, et al. Twist-−2 controls myeloid lineage development and function. PLoS Biol. 2008;6(12):e316. doi: 10.1371/journal.pbio.0060316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou W, Zhu P, Wang J, et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008;29(1):69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen L, Evel-−Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. 2004;22(12):1546–1553. doi: 10.1038/nbt1035. [DOI] [PubMed] [Google Scholar]
- 22.Carotta S, Dakic A, D’Amico A, et al. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity. 2010;32(5):628–641. doi: 10.1016/j.immuni.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-−producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19(1):59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 24.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev. 2010;234(1):45–54. doi: 10.1111/j.0105-2896.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 25.Liu K, Victora GD, Schwickert TA, et al. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324(5925):392–397. doi: 10.1126/science.1170540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satpathy AT, Murphy KM, Kc W. Transcription factor networks in dendritic cell development. Semin Immunol. 2011;23(5):388–397. doi: 10.1016/j.smim.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 28.Gelato KA, Fischle W. Role of histone modifications in defining chromatin structure and function. Biol Chem. 2008;389(4):353–363. doi: 10.1515/BC.2008.048. [DOI] [PubMed] [Google Scholar]
- 29.Atanassov BS, Koutelou E, Dent SY. The role of deubiquitinating enzymes in chromatin regulation. FEBS Lett. 2011;585(13):2016–2023. doi: 10.1016/j.febslet.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 31.Nutt SL, Metcalf D, D’Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med. 2005;201(2):221–231. doi: 10.1084/jem.20041535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Back J, Allman D, Chan S, Kastner P. Visualizing PU.1 activity during hematopoiesis. Exp Hematol. 2005;33(4):395–402. doi: 10.1016/j.exphem.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 33.Guerriero A, Langmuir PB, Spain LM, Scott EWPU. PU.1 is required for myeloid-derived but not lymphoid-derived dendritic cells. Blood. 2000;95(3):879–885. [PubMed] [Google Scholar]
- 34.Anderson KL, Perkin H, Surh CD, Venturini S, Maki RA, Torbett BE. Transcription factor PU.1 is necessary for development of thymic and myeloid progenitor-derived dendritic cells. J Immunol. 2000;164(4):1855–1861. doi: 10.4049/jimmunol.164.4.1855. [DOI] [PubMed] [Google Scholar]
- 35.Kastner P, Chan S. PU.1: a crucial and versatile player in hematopoiesis and leukemia. Int J Biochem Cell Biol. 2008;40(1):22–27. doi: 10.1016/j.biocel.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 36.Hohaus S, Petrovick MS, Voso MT, Sun Z, Zhang DE, Tenen DGPU. PU.1 (Spi-−1) and C/EBP alpha regulate expression of the granulocyte-macrophage colony-stimulating factor receptor alpha gene. Mol Cell Biol. 1995;15(10):5830–5845. doi: 10.1128/mcb.15.10.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang DE, Hetherington CJ, Chen HM, Tenen DG. The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol. 1994;14(1):373–381. doi: 10.1128/mcb.14.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zon LI. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature. 2008;453(7193):306–313. doi: 10.1038/nature07038. [DOI] [PubMed] [Google Scholar]
- 39.Mende I, Karsunky H, Weissman IL, Engleman EG, Merad M. Flk2+ myeloid progenitors are the main source of Langerhans cells. Blood. 2006;107(4):1383–1390. doi: 10.1182/blood-2005-05-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11(11):750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 41.Dakic A, Metcalf D, Di Rago L, Mifsud S, Wu L, Nutt SLPU. PU.1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J Exp Med. 2005;201(9):1487–1502. doi: 10.1084/jem.20050075. [DOI] [PMC free article] [PubMed] [Google Scholar]