

Abstract
Over the past 15 years protein kinases have become the pharmaceutical industry’s most important class of drug target in the field of cancer. Some 20 drugs that target kinases have been approved for clinical use over the past decade, and hundreds more are undergoing clinical trials. However, the recent approval of the first protein kinase inhibitors for the treatment of inflammatory diseases, coupled with an enhanced understanding of the signaling networks that control the immune system, suggests that there will be a surge of interest in this area over the next 10 years. In this connection, we discuss opportunities for targeting protein kinases in the MyD88 signaling network for the development of drugs to treat chronic inflammatory and autoimmune diseases. Activating mutations in protein kinases underlie many other diseases and conditions, and we also discuss why the protein kinases SPAK/OSR1 and LRRK2 have recently become interesting targets for the treatment of hypertension and Parkinson’s disease, respectively, and the progress that has been made in developing LRRK2 inhibitors. Finally we suggest that more focus on the identification of inhibitors of kinase activation, rather than kinase activity, may pay dividends in identifying exquisitely specific inhibitors of signal transduction cascades, and we also highlight “pseudo-kinases” as an attractive and unexplored area for drug development that merits much more attention in the years to come.

One focus of this special issue of ACS Chemical Biology is on “enzymes that are difficult to target”. Twenty years ago protein kinases would have been included in this category and there were two major reasons for this state of affairs. First, the similarities between their ATP-binding pockets made it seem a near impossible task to develop compounds that would inhibit just one of the 500 plus protein kinases encoded by the human genome, without inhibiting a number of the others. Second, making compounds with the requisite potency to compete with the millimolar ATP concentrations present in cells seemed an almost equally insurmountable mountain to climb. Indeed, developing potent and specific protein kinase inhibitors remains a challenge, but in a number of cases, these problems have been solved or found to less problematic than was once feared. For example, the identification of compounds that not only interact with the ATP-binding pocket but also target hydrophobic pockets in the vicinity of the ATP binding site and that are unique to particular protein kinases has allowed a number of potent and relatively specific kinase inhibitors to be developed. On the other hand, in the field of oncology, lack of specificity has not proved to be the barrier to clinical approval it was once thought to be, since many kinase inhibitors have turned out to be tolerated rather well. Moreover, the simultaneous inhibition of several kinases can be advantageous in preventing drug resistance or by enabling the same drug to be used for the treatment of several cancers. For example, Imatinib (also known as Gleevec) is used to treat chronic myelogenous leukemia (CML) because it inhibits the oncogenic BCR-Abl tyrosine kinase fusion protein, gastrointestinal tumors (GIST) because it inhibits the c-Kit receptor tyrosine kinase, and myelo-proliferative diseases because it inhibits the PDGF receptor. For these reasons, oncology has been the therapeutic area on which the great majority of kinase drug discovery programs have been focused. The great majority of small molecule inhibitors of protein kinases that have been approved or are nearing approval for clinical use and most of the 150 plus kinase inhibitors undergoing clinical trials target protein tyrosine kinases and are used to treat one or more cancers. They include several drugs that have reached “blockbuster” status with sales exceeding US$1 billion per annum. Global revenues for kinase inhibitors were US$29 billion in 2011 and are projected to reach US$40 billion by 2015. Imatinib has transformed CML and GIST from rapidly fatal diseases into manageable conditions, so much so that CML is no longer a rare leukemia and the number of patients requiring this drug increases year by year. From our discussions with pharmaceutical companies it would appear that something like 50–70% of current cancer drug discovery programmes are focused on protein kinase inhibitors.
Although oncology will undoubtedly remain a major focus of kinase drug discovery for many years to come, the number of kinase inhibitors undergoing clinical trials for the treatment of other diseases is increasing. For example, the Janus Kinase (JAK) inhibitors Tofacitinib and Ruxolitinib were recently approved for the treatment of rheumatoid arthritis and myelofibrosis, respectively (Table 1). These are the first drugs to be approved for the treatment of inflammatory diseases that were developed by targeting a specific protein kinase, and we predict that this will lead to a surge of interest in the development of protein kinase inhibitors for the treatment of diseases of the immune system, similar to the impact that Imatinib had on the development of kinase inhibitors for the treatment of cancer after its spectacular efficacy for the treatment of CML was revealed 15 years ago. In the following sections, we discuss a number of protein kinases that are being targeted for the development of drugs to treat diseases other than cancer. We focus our discussion on serine/threonine-specific protein kinases, because although they are far more numerous than protein tyrosine kinases, only a relatively small proportion of these enzymes have been explored as potential drug targets so far, which suggests that kinase drug discovery may still be in its infancy.
Table 1. Small Molecule Inhibitors of Protein Kinases Approved for Clinical Use or in Advanced Clinical Trials.
| Name | Structure | Reported target | Company | Approved for clinical use |
|---|---|---|---|---|
| Eril |
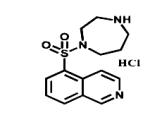
|
ROCK | Eisai | 1995 cerebral vasospasm (Japan) |
| Rapamune |
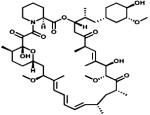
|
mTOR | Wyeth Pfizer | 2000 kidney transplantation |
| Temsirolimus |
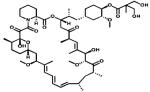
|
mTOR | Wyeth Pfizer | 2007 advanced renal cell carcinoma |
| Everolimus |
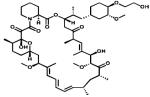
|
mTOR | Novartis | 2009 several cancers |
| Imatinib |
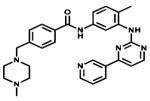
|
Bcr-Abl c-KIT PDGFR | Novartis | 2001 chronic myelogenous leukaemia |
| Gefitinib |
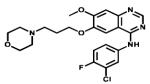
|
EGFR | Astra Zeneca | 2005 lung cancer |
| Erlotinib |
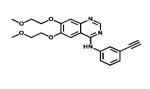
|
ErbB1 | Genetech Roche | 2005 lung, pancreatic and others cancers |
| Sorafenib |
|
Multiple Tyrosine kinases targeted | Onyx Bayer | 2005 renal cancer |
| Dasatinib |
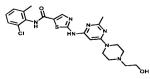
|
Multiple Tyrosine-kinases targeted | Bristol Myers Squibb | 2006 chronic myeloenous leukaemia, All |
| Sunitinib |
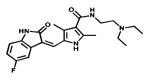
|
Multiple Tyrosine-kinases targeted | SUGEN Pfizer | 2006 renal cancer and GIST |
| Nilotinib |
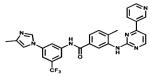
|
Bcr-Abl | Novartis | 2007 chronic myelogenous leukaemia |
| Lapatinib |
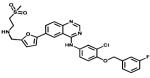
|
Her2 EGFR | GlaxoSmith Kline | 2009 renal cancer |
| Pazopanib |
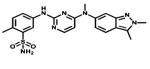
|
VEGFR2 PDGFR c-KIT | GlaxoSmith Kline | 2009 renal cancer |
| Ruxolitinib |
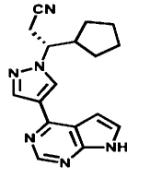
|
JAKs | Incyte | 2011 myelofibrosis |
| Crizotinib |
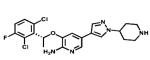
|
ALK/Met | Pfizer | 2011 NSCLC with Alk mutation |
| Vemurafenib |
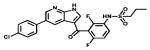
|
BRAF | Roche | 2011 melanoma |
| Vadetinib |
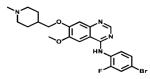
|
Multiple Tyrosine kinases targeted | Caprelsa IPR Pharms | 2012 thyroid cancer |
| Axitinib |
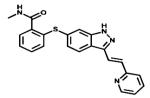
|
VEGFRs PDGFRB c-KIT | Pfizer | 2012 renal cell carcinoma |
| Bosutinib |
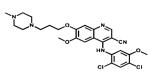
|
BcrAbi SRC | Pfizer | 2012 chronic myelogenous leukemia |
| Tivozanib |
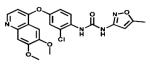
|
VGFRs | AVEO Pharms | 2012 kidney cancer |
| Tofacitinib |
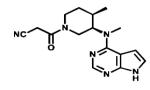
|
JAKs | Pfizer | 2012 rheumatoid arthritis |
| Regorafenib |
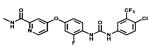
|
Multiple Tyrosine kinases targeted | Stivarga Bayer | 2012 thyroid cancer |
| Lenvatinib |
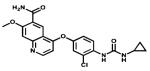
|
VEGFR2/VEGFR2 | Eisai | 2012 thyroid cancer (Japan) |
| Toceranib |
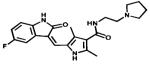
|
Multiple Tyrosine kinases targeted | Pfizer | 2009 canine mastocytoma |
| Masivet Kinavet |
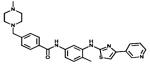
|
c-KIT PDGFR | AB Science | 2010 canine mastocytoma |
| Cabozantinib |
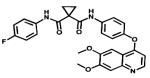
|
VEGFRs KIT / Axl | Cometriq Exelixis | 2012 canine thyroid cancer |
| Afatinib |
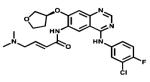
|
Her2 EGFR | Boehringer Ingelheim | Not yet NSCLC |
| Dabrafenib |
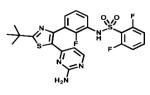
|
BRaf | GlaxoSmith Kline | Not yet metastatic melanoma |
| Trametinib |
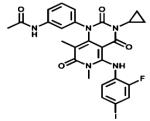
|
MEK1/2 | GlaxoSmith Kline | Not yet metastatic melanoma |
TARGETING SERINE/THREONINE PROTEIN KINASES FOR THE TREATMENT OF DISEASE
Hypertension
About 25% of adults in the developed world have high blood pressure, a largely asymptomatic condition that is a major risk factor for stroke, congestive heart failure, and kidney disease, with an estimated US$130 billion being spent globally on the treatment of this condition in 2010.1 There was much excitement in 2001 when autosomal dominant inherited mutations that enhanced the expression of a poorly characterized protein kinase WNK1 [with-no-K(Lys)] or missense mutations in the gene encoding the related WNK4 were found to underlie an inherited hypertension called Gordon’s syndrome.2 These findings stimulated research into understanding how WNKs regulate blood pressure, which culminated in the finding that they phosphorylate and activate two related protein kinases termed STE20/SPS1-related proline/alanine-rich kinase (SPAK) and oxidative stress-responsive kinase 1 (OSR1).3,4 More recently, the catalytic activities of the phosphorylated forms of SPAK and OSR1 were shown to be enhanced a further 100-fold through binding to another protein termed MO25.5 Once activated, SPAK and OSR1 phosphorylate and stimulate the activities of key ion co-transporters, including the Na+/Cl− co-transporter (NCC) and the Na+/K+/2Cl− co-transporter-2 (NKCC2) that play critical roles in reabsorbing salt from the urine back into the blood6 (Figure 1). Remarkably, these are the same ion co-transporters that are the targets for thiazide-diuretic (NCC) and loop-diuretic (NKCC2) drugs that are commonly deployed to lower blood pressure.6 The critical roles that SPAK and OSR1 play in controlling blood pressure was subsequently confirmed by demonstrating that SPAK- and OSR1-deficient mice display reduced phosphorylation of NCC and NKCC2 and markedly reduced blood pressure.7-9 Moreover, a significant number of people in the Chinese population with an inherited low blood pressure condition, termed Gittleman’s syndrome, possess a mutation (Thr60Met) in the key residue of NCC whose phosphorylation by SPAK/OSR1 stimulates NCC-mediated salt reabsorption.10,11 A genome-wide association study of systolic and diastolic blood pressure has also reported strong association with common variants of SPAK, further emphasizing the importance of this enzyme in hypertension.12
Figure 1.

WNK signaling network that activates the protein kinases SPAK and OSR1 and the NCC and NKCC2 ion co-transporters that control blood pressure. Hyperosmotic and hypotonic low chloride conditions induce the autoactivation of WNK isoforms, which then activate the SPAK and OSR1 protein kinases by phosphorylating their activation loops. The interaction of SPAK and OSR1 with the MO25 component is also needed to generate maximal activity. SPAK and OSR1 phosphorylate and activate the NCC and NKCC2 ion cotransporters that play a vital role in regulating salt reabsorption in the kidney and are the targets of the widely used thiazide and loop diuretic drugs. It should be noted that in some studies where overexpression systems were employed, WNK4 was reported to negatively regulate NCC. Whether this inhibitory effect is of physiological significance or mediated through SPAK or OSR1 requires further investigation.
Although hypertension can be moderated by lifestyle changes, for most patients drugs are required to lower blood pressure significantly. Many patients on current therapies nevertheless have poor control of blood pressure and/or suffer from side effects, and there is therefore a need for improved therapies. Targeting the WNK pathway represents a new opportunity to develop improved treatments that lower blood pressure, and one potential advantage of targeting the WNK isoforms themselves is the unusual position of the catalytic lysine residue on WNK isoforms compared with other kinases, a feature that might be exploited to develop WNK-specific ATP-competitive inhibitors. However, it is unclear which combination of the four mammalian WNK isoforms would need to be inhibited to reduce the activity of NCC and NKCC2 in the kidney. Moreover, some reports suggest that WNK4 may function as a negative regulator of the WNK1 signaling pathway, and inhibiting this isoform might therefore stimulate kidney salt reabsorption and enhance blood pressure.4
For the reasons discussed above, SPAK and OSR1 are arguably simpler enzymes to target than WNK isoforms, and all the evidence points to SPAK and OSR1 inhibitors having a major impact in reducing the activity of both NCC and NKCC2, thereby suppressing renal salt reabsorption and reducing blood pressure. SPAK and OSR1 are likely to function redundantly in the regulation of NCC and NKCC2, so that a dual inhibitor of both isoforms might be more efficient at lowering blood pressure. The kinase domains of SPAK and OSR1 are 89% identical, which suggests that drugs inhibiting both isoforms could be developed. A potential advantage of a dual SPAK/OSR1 inhibitor over current therapies that target either NCC or NKCC2 is that it would coordinately reduce the activities of both NCC and NKCC2, as well as other uncharacterized substrates of these kinases. SPAK and OSR1 also possess a specific substrate docking CCT domain that mediates interaction with the RFxV motifs present in WNKs, NCC, and NKCC2 (where x is any amino acid residue),4 and the three-dimensional structure of this domain complexed to the RFxV motif has been solved at high resolution.13 These studies raise the possibility of developing a drug that binds to the CCT domain pocket and that would prevent the activation of SPAK/OSR1 as well as the phosphorylation of NCC/NKCC2. The CCT domain is unique to SPAK and OSR1 and would not be expected to inhibit any other kinase, which is an important consideration in treating a chronic condition, such as hypertension, where treatment may need to be sustained for decades.
Parkinson’s Disease
Autosomal dominant missense mutations within the leucine rich repeat kinase 2 (LRRK2) are thought to be responsible for ~1% of all cases of Parkinson’s disease.14 Patients with LRRK2 mutations develop disease symptoms that are largely indistinguishable from idiopathic Parkinson’s both in age of onset and disease course. LRRK2 is a large (2527-residue) multidomain protein that possesses a GTPase domain as well as the kinase domain (Figure 2). Although over 50 mutations have been reported, by far the most common replaces the conserved glycine residue Gly2019 in the Mg2+-binding motif of the kinase domain with a Ser residue, increasing kinase activity around 3-fold.15,16 These findings suggests that hyper-activation of LRRK2 is a cause of Parkinson’s disease and that LRRK2 inhibitors may have therapeutic potential for the prevention and/or treatment of Parkinson’s. To date, several relatively specific LRRK2 inhibitors have been reported, including compounds that do not cross the blood-brain barrier (LRRK2-IN117 or GSK2578215A18) as well as brain penetrant inhibitors (HG-10-102-0119 or GENE-791520) (Figure 2).
Figure 2.

Domain structure of LRRK2 and some reported inhibitors of this protein kinase. LRRK2 is a large 2527-residue protein kinase that, in addition to a protein kinase domain, contains an active GTPase domain. The figure shows the structures of the specific LRRK2 inhibitors that are being used to dissect the cellular functions of LRRK2. To date no direct physiological substrates of LRRK2 have been identified. However, LRRK2 induces its own phosphorylation at Ser910 and Ser935 by an indirect mechanism that remains to be identified, and this allows it to bind to 14-3-3 isoforms.58 Monitoring the state of phosphorylation of LRRK2 at Ser910 and Ser935 with phospho-specific antibodies is therefore widely employed to assess the in vivo efficacy of LRRK2 inhibitors that are being elaborated for the treatment of Parkinson’s disease.
Although there is much excitement in the field about the therapeutic potential of LRRK2 inhibitors, several key challenges remain. First, despite intensive research, little is known about how LRRK2 is regulated, what its substrates might be, and how phosphorylation of these substrates might be linked to Parkinson’s disease. Second, several missense mutations in LRRK2 that cause Parkinsonism lie outside the catalytic domain (Figure 3), and at least in biochemical assays, these mutations do not affect catalytic activity.21 Thus alterations in its intrinsic kinase activity may not be the only way in which the huge multidomain kinase LRRK2 impacts on Parkinson’s disease.
Figure 3.

Simplified outline of the MyD88-signaling pathway by which TLR agonists induce the production of inflammatory mediators. Reasons why the protein kinases highlighted in red may be particularly attractive drug targets are discussed in the text. The activation of TLRs in myeloid cells recruits MyD88 and protein kinases of the IRAK family to the receptor, which induce the E3 ligase TRAF6 to produce Lys63-linked polyubiquitin (K63-pUb) chains and the E3 ligase LUBAC to produce linear pUb chains. The binding of K63-pUb chains to the TAB2 and TAB3 components of TAK1 kinase complex and K63-pUb and/or linear-pUb chains to the NEMO component of the canonical IKK complex is thought to induce conformational changes that activate these protein kinases. The IKKs phosphorylate the inhibitory IκBα component of the transcription factor NFκB and the inhibitory NFκB1/p105 component of the protein kinase Tpl2, triggering the proteasomal degradation of these inhibitors and the activation of NFκB and Tpl2. TAK1 not only initiates the activation of the IKK complex but also activates the pathways that switch on the mitogen-activated protein kinases (MAPKs) termed p38 MAP kinase and c-Jun N-terminal kinases (JNKs), while Tpl2 switches on the signaling pathway that leads to the activation of extracellular signalregulated protein kinases 1 and 2 (ERK1, ERK2). The p38 MAP kinases, JNKs, and ERK1/2, collectively termed MAPKs in the figure, phosphorylate many proteins, which regulate the transcription, translation, processing, and secretion of inflammatory mediators, including the pro-inflammatory cytokines TNFα, IL-6, and IL-12. The protein kinase MK2, which is activated by p38α MAP kinase, stimulates post-transcriptional events required for the production of pro-inflammatory cytokines, such as TNFα and IL-6. The p38α MAP kinase and ERK1/2 also activate the protein kinases MSK1 and MSK2, which phosphorylate the transcription factor CREB, stimulating the transcription of genes encoding anti-inflammatory cytokines, such as IL-10 and IL-1ra.59 CREB transcriptional activity is greatly enhanced by the coactivator CRTC3 in macrophages. The phosphorylation of CRTC3, which is catalyzed by the SIK subfamily of protein kinases, prevents CRTC3 from activating CREB, restricting the production of IL-10, which can be overcome by inhibition of the SIKs.25 Following its secretion, IL-10 activates the IL-10 receptor by autocrine and paracrine mechanisms switching on members of the JAK family of protein kinases that phosphorylate and activate the transcription factor STAT3. These lead to the synthesis of proteins that suppress the production of pro-inflammatory cytokines and drive the conversion of classically activated M1 macrophages to regulatory M2b macrophages.
Finally, recent analysis of LRRK2 knockout22 and catalytically inactive knock-in mice23 has revealed that these animals develop moderate kidney pathology as they age, raising concern that long-term treatment of patients with potent LRRK2 inhibitors that ablate activity might induce kidney toxicity. One way to circumvent this problem would be to develop compounds that selectivity target the LRRK2[G2019S] mutant over the wild type enzyme, and indeed, several of the reported inhibitors have ~10-fold greater potency toward this variant.17,19 The three-dimensional structure of a Dictyostelium discoideum orthologue of LRRK2 has recently been determined and has suggested that the G2019S mutation may induce the formation of an additional hydrogen bond between the Ser2019 residue and an arginine residue in the αC-helix, thereby stabilizing the conformation of the active kinase domain. This structural feature could provide opportunities to elaborate compounds that bind selectively to this form of the enzyme.24
Inflammatory and Autoimmune Diseases
Adalimumab (Humira) and other biological drugs that neutralize tumour necrosis factor α (TNFα) have had a major impact on the treatment of several inflammatory diseases, including rheumatoid and psoriatic arthritis, ankylosing spondalitis, and Crohn’s and ulcerative colitis. Indeed, with sales expected to approach US$9 billion this year, Adalumimab will be the world’s best selling drug in 2012. However, like all drugs that are antibodies, Adalumimab is expensive and has to be injected repeatedly, and only about half of rheumatoid arthritis patients are good responders. There is therefore undiminished interest in the development of orally active drugs that suppress the production and secretion of TNFα and other pro-inflammatory cytokines by the innate immune system.
TNFα is mainly produced by myeloid cells via the MyD88 signaling network, which is activated in response to ligands that activate Toll-like receptors (TLRs). Many serine/threonine-specific protein kinases participate in this pathway to control the transcription, translation, processing, and secretion of TNFα and other pro-inflammatory cytokines (Figure 3), raising the question as to which protein kinase(s) should be targeted to develop improved drugs. This is a complex issue, because the MyD88 signaling network plays numerous roles in addition to the production of TNFα, IL-6, and IL-12 (Figure 3). For example, it is required for the production of type 1 interferons by plasmacytoid dendritic cells in response to viral nucleic acids that activate TLR7, 8, and 9 and also mediates signaling by interleukins (IL) 1, 18, and 33 that play other roles in innate immunity. Moreover, the MyD88 signaling pathway is required not just for the production of pro-inflammatory cytokines but also the production of anti-inflammatory molecules, such as IL-1 receptor antagonist (IL-1ra) and IL-10. These molecules keep the innate immune system in check by restricting the strength of IL-1 signaling (IL-1ra) and by preventing the overproduction of TNFα and other pro-inflammatory cytokines (IL-10) (Figure 3). Moreover, IL-10 also promotes the conversion of “classically activated” or M1 macrophages to the “regulatory” or M2b macrophages25 that are important for the resolution of inflammation once it has done its job.
For these reasons, the recent finding that the SIK family of protein kinases suppress the transcription of the genes encoding IL-10 and IL-1ra is of great interest.25 The SIKs suppress IL-10 gene transcription by phosphorylating CRTC3 (CREB-regulated transcriptional co-activator 3), an essential coactivator of the transcriptional factor CREB (Figure 3). This causes CRTC3 to dissociate from CREB and exit the nucleus, where it binds to 14-3-3 proteins. Conversely, SIK inhibitors induce the dephosphorylation of CRTC3 leading to its nuclear entry where it activates CREB and stimulates IL-10 gene transcription. The IL-10 that is secreted activates the IL-10 receptor and hence the JAK-STAT3 signaling pathway, which stimulates the production of proteins needed to suppress the production of pro-inflammatory cytokines (Figure 3) and/or drive the conversion of macrophages from the M1 to the M2b phenotype.25 The development of potent, selective, and drug-like inhibitors of the SIKs and the effects that they have in animal models of chronic inflammatory and autoimmune diseases is awaited with interest.
IRAK4 lies at the head of the MyD88 signaling network, and this pathway cannot be activated in the absence of this protein. Consequently, young children with inherited IRAK4 deficiency are extremely susceptible to infection by pyogenic bacteria, such as Streptococcus pneumoniae, and require antibiotics from birth to keep them alive.26 One might therefore have anticipated that inhibitors of IRAK4 catalytic activity would suppress the production of both pro-inflammatory and anti-inflammatory molecules, but interestingly, this is not the case. We recently found that pharmacological inhibition of IRAK4 prevents the production of pro-inflammatory cytokines in mouse macrophages but not the production of IL-10.27 A likely explanation for this finding is that the kinase activities of IRAK4 and IRAK1 function redundantly in this system, so that IRAK4 only becomes rate-limiting after IRAK1 has been degraded, which occurs 2-3 h after the MyD88 signaling network has been activated by TLR ligands.27 At this time IL-10 and other anti-inflammatory molecules have already been produced, but pro-inflammatory cytokine production has barely started. Hence IRAK4 inhibitors inhibit the production of proinflammatory cytokines but not IL-10. A number of potent and relatively selective inhibitors of IRAK4 have been identified,28 but these projects still appear to be at the preclinical stage of development. Remarkably, invasive bacterial infection no longer occurs after children with IRAK4 deficiency reach the age of about 14, indicating that IRAK4 is vital only for childhood immunity.26 Thus IRAK4 inhibitors may alleviate chronic inflammatory diseases in adults without increasing their susceptibility to infection significantly.
Plasmacytoid dendritic cells (pDCs) are unique in secreting copious amounts of type 1 interferons when the MyD88-dependent signaling network is activated by viral nucleic acids that engage TLR7, 8, and 9, and this is thought to be a major route by which interferons are produced during viral infection. However, the activation of pDCs by interferogenic immune complexes may also lead to the overproduction of interferon α, which is associated with predisposition to lupus.29 Therefore drugs that suppress type 1 interferon production by pDCs may have efficacy for the treatment of this autoimmune disease. Interestingly, IRAK1 catalytic activity is required for the production of type 1 interferons by mouse pDCs,30,31 but not for the production of pro-inflammatory cytokines by myeloid cells. The development of specific IRAK1 inhibitors therefore merits consideration as a potential therapy for the prevention and/or treatment of lupus.
The protein kinase activity of Tpl2 (Figure 3) is not required for TNFα gene transcription or the translation of its mRNA into protein but is rate-limiting for the processing of pre-TNFα to the mature secreted form of this cytokine.32 Moreover, Tpl2-deficient mice are protected against TNFα-induced inflammatory bowel disease, suggesting that Tpl2 is an interesting target for the development of anti-inflammatory drugs.33 Potent and relatively selective inhibitors of Tpl234,35 have been described and shown to suppress TNFα secretion, but drugs that target this protein kinase have yet to enter clinical trials.
In the mid 1990s a novel class of cytokine synthesis anti-inflammatory drugs (CSAIDs) that suppressed the LPS-stimulated production of TNFα by human monocytes were found to be potent and relatively selective inhibitors of the protein kinase, now called p38α MAP kinase.36,37 These findings stimulated many companies to develop inhibitors of this protein kinase, many of which entered clinical trials for the treatment of rheumatoid arthritis and other inflammatory diseases. However, none of these compounds advanced to late stage clinical trials due to lack of efficacy and/or unacceptable side effects. As our knowledge of the MyD88 signaling network has improved in recent years, the potential disadvantages of targeting p38α MAP kinase have become increasingly apparent. They include its involvement in the production of IL-10 (Figure 3) and other anti-inflammatory molecules, as well as pro-inflammatory cytokines, and its participation in feedback control loops that suppress the activation of other protein kinases that are positive regulators of the MyD88 signaling network.38 Moreover, and in contrast to IRAK4 or Tpl2, p38α MAP kinase is activated by many physiological stimuli and not just by inflammatory stimuli. Thus the inhibition of p38α MAP kinase may have many unforeseen consequences.
For these reasons there has been increasing interest in developing drugs that inhibit MAP kinase-activated protein kinase-2 (MK2), a protein kinase that is activated by p38α MAP kinase (Figure 3). Macrophages from MK2-deficient mice produce far less TNFα than macrophages from wild type mice, and MK2-deficient mice are protected in animal models of septic shock39 and collagen-induced arthritis.40 Drugs that inhibit MK2 and the closed related MK3 may therefore retain the efficacy of p38 MAP kinase inhibitors in suppressing TNFα production, without suppressing the production of anti-inflammatory molecules and be free of other potential problems associated with inhibition of p38α MAP kinase. However, it has proved challenging to develop sufficiently potent, drug-like inhibitors of MK2. Nevertheless, progress is now being reported, and MK2 inhibitors capable of suppressing TNFα production in LPS-stimulated peripheral blood mononuclear cells have been described.41-43 Information on the efficacy of MK2 inhibitors in the clinic is awaited with interest.
In summary, there is no shortage of serine/threonine protein kinases that operate in the MyD88 signaling network and that appear be attractive drug targets. In retrospect, progress in developing kinase inhibitors for the treatment of inflammatory and autoimmune disease has been hampered by the huge amount of effort devoted to the development of p38α MAP kinase inhibitors, which seems misdirected now that the full complexity of the MyD88 signaling network is being revealed. Targeting the MyD88 signaling network and other signaling pathways of the immune system to develop drugs to treat inflammatory and autoimmune diseases could be about to undergo a renaissance.
NOVEL APPROACHES FOR THE DEVELOPMENT OF KINASE INHIBITORS
Identifying Inhibitors of Kinase Activation
Many protein kinases are components of so-called “cascade systems” in which several protein kinases activate one another sequentially (e.g., Figures 1 and 3). Kinase cascades are important in amplifying and diversifying the effects of a signal and in permitting interactions between different signaling pathways to create networks. The existence of kinase cascades creates the opportunity to identify and develop compounds that are not kinase inhibitors, but by binding to kinases prevent them from being activated by the next “upstream” kinase in the cascade. Indeed PD98059, the first inhibitor of the Ras-Raf-MEK-ERK kinase cascade to be developed (Figure 4), acts in this way by binding to the protein kinase MEK1 and preventing it from being activated by the kinase Raf.44 More recently, the compound Akti1/2, was found to bind to the protein kinase Akt and prevent it from being activated by the “upstream” kinase PDK1.45 An improved version of this compound, termed MK220646 (Figure 4), is being evaluated in at least 47 clinical trials (http://www.clinicaltrials.gov/ct2/results?term=mk2206&Search=Search). These compounds appear to interact with a region of Akt located between the pleckstrin homology (PH) domain and the catalytic domain and prevent the conformational change that accompanies the binding of phosphatidylinositol 3,4,5-trisphosphate to the PH domain and which permits PDK1 to phosphorylate and activate Akt.47,48 The crystal structure of full length Akt bound to Akti1/2 has revealed the intricate web of interactions between the PH and kinase domains, as well as the critical amino residues that mediate binding of the inhibitor to Akt1.49 This study also demonstrated the critical role played by Trp80 in the PH domain, a surface-exposed residue that forms a ring-stacking interaction with Akti1/2.49 These findings explain previous work demonstrating that mutation of Trp80 has no effect on kinase activity but renders Akt1 resistant to Akti1/2.47
Figure 4.

Structures of compounds that prevent the activation of one kinase by another. The compound PD98059 prevents the activation of MEK1 by Raf, while Akti1/2 and MK2206 prevent the activation of Akt1 by PDK1. There are 47 clinical trials of MK2206 in progress for the treatment of different cancers.
Both PD98059 and Akti1/2/MK2206 display exquisite specificity, making one wonder why more inhibitors of this type have not been developed, since they have the additional advantage of not competing with ATP for binding to the ATP-binding site. To systematically identify inhibitors of kinase activation, it is necessary to set up screens in which two or more kinases activate one another sequentially and then “deconvolute” the screen to identify which compounds are direct inhibitors of the kinase components in the screen or which prevent the activation of one kinase by another. Perhaps pharmaceutical and biotechnology companies have been reluctant to conduct the secondary assays that are needed to deconvolute screens in which an entire kinase cascade has been targeted, explaining why such screens have rarely been performed. However they were set up and run successfully many years ago.50,51 We suggest that, now that the “low hanging fruit” on the kinome has been picked, it may be timely to set up such screens to identify compounds that inhibit the activation of kinases that are attractive drug targets, but where sufficiently potent or specific inhibitors of kinase activity have proved impossible to develop.
Targeting Pseudo-kinases
The human kinome encodes about 50 proteins that possess a domain that resembles the canonical kinase domain, but which lacks amino acid residues that are required for catalysis by most protein kinases.52,53 A number of these “pseudo-kinases” have been studied and do indeed appear to be devoid of catalytic activity. They include the STRAD pseudo-kinases that regulate the LKB1 tumor suppressor kinase, the KSR pseudo-kinases that control Raf signaling, ILK (integrin-like kinase) that controls integrin signaling, Erb3 that modulates EGF signaling, the N-terminal kinase domain of the JAK tyrosine kinases, and IRAK2 (Figure 3), which plays an important role in controlling innate immunity.52 It is becoming clear that, despite lacking catalytic activity, many pseudo-kinases possess an intact ATP-binding site and retain the ability to bind to ATP.52 For example, the interaction of ATP with the pseudo-kinases STRADα and STRADβ,54 in conjunction with binding to the same MO25 scaffolding component discussed in the section Hypertension, induces STRAD to adopt a conformation resembling an active protein kinase that is capable of binding to LKB1 and stabilizing it in a conformation required for optimal catalytic activity.55,56 It is therefore likely that many pseudo-kinases, although lacking intrinsic catalytic activity, are able to interconvert between inactive non ATP-binding and active ATP-binding conformations and that this regulates their ability to bind to other target proteins to regulate biological responses.
Another striking example of a mutation in a pseudo-kinase linked to human disease is the V617F mutation in the JAK2 pseudo-kinase, which causes the C-terminal tyrosine kinase domain to become constitutively active, and this mutation is responsible for >95% of cases of polycythemia vera and ~50% of cases of essential thrombocythemia and primary myelofibrosis.57 It would be interesting to identify compounds that bind to the pseudo-kinase domain of the JAK2[V617F] mutant and suppress the inappropriate “activity” of this mutant protein. It would also be interesting to explore whether compounds could be developed that interact with the KSR and Erb3 pseudo-kinases, trapping them in their “inactive” conformations, and to investigate whether they could be deployed to inhibit Raf and the EGF receptor in cancers whose growth is driven by the inappropriate activation of these kinases. A further advantage of targeting pseudo-kinases is that the structures of their ATP binding sites are likely to be quite different from those of catalytically active protein kinases, suggesting that rather selective molecules could be developed.
There seems no a priori reason why it should not be possible to develop drugs that target the ATP-binding sites of pseudo-kinases and inhibit their functions. Indeed we have recently identified compounds that target the ATP-binding site of the IRAK2 pseudo-kinase and that mimic the effects of replacing IRAK2 with a functionally inactive mutant27 by suppressing the secretion of TNFα and IL-6, but not the secretion of IL-10 in mouse macrophages (S. Nanda, P. Cohen, and T. Haystead; unpublished work). The targeting of pseudo-kinases is a fascinating but virtually unexplored area of drug discovery.
ACKNOWLEDGMENTS
We thank N. Shpiro for collation of all the chemical structures displayed in the figures and Table 1. Research in the authors’ laboratories is supported by the UK Medical Research Council (D.A. and P.C.), The M. J. Fox Foundation (D.A.), AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Janssen Pharmaceutica, Merck-Serono and Pfizer (D.A. and P.C.).
KEYWORDS
- Cytokines
protein molecules secreted by cells that are used extensively in intercellular communication
- Hypertension
a chronic medical condition in which the blood pressure in the artieries is elevated
- Inflammation
biological response of vascular tissues to harmful stimuli, such as pathogens
- Kinome
the set of protein kinases encoded by the human genome
- Parkinson’s disease
a degenerative disorder of the central nervous system
- Protein kinase
enzyme that catalyses the transfer of the γ-phosphoryl group of ATP to an amino acid residue in a protein
- Protein kinase cascade
signal transduction network in which different protein kinases activate or inhibit one another to amplify or diversify a response
- Pseudo-kinases
proteins that possess a recognizable kinase domain, but lack amino acids considered to be essential for catalysis
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, Lloyd-Jones DM, Nelson SA, Nichol G, Orenstein D, Wilson PW, Woo YJ. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123:933–944. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- (2).Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- (3).Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 2005;391:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Richardson C, Alessi DR. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J. Cell Sci. 2008;121:3293–3304. doi: 10.1242/jcs.029223. [DOI] [PubMed] [Google Scholar]
- (5).Filippi BM, de Los Heros P, Mehellou Y, Navratilova I, Gourlay R, Deak M, Plater L, Toth R, Zeqiraj E, Alessi DR. MO25 is a master regulator of SPAK/OSR1 and MST3/MST4/YSK1 protein kinases. EMBO J. 2011;30:1730–1741. doi: 10.1038/emboj.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- (7).Rafiqi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanovic S, Jovanovic A, O’Shaughnessy KM, Alessi DR. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol. Med. 2010;2:63–75. doi: 10.1002/emmm.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chiga M, Rafiqi FH, Alessi DR, Sohara E, Ohta A, Rai T, Sasaki S, Uchida S. Phenotypes of pseudohypoaldosteronism type II caused by the WNK4 D561A missense mutation are dependent on the WNK-OSR1/SPAK kinase cascade. J. Cell Sci. 2011;124:1391–1395. doi: 10.1242/jcs.084111. [DOI] [PubMed] [Google Scholar]
- (9).Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 2010;21:1868–1877. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl-cotransporter by the WNK-regulated kinases SPAK and OSR1. J. Cell Sci. 2008;121:675–684. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- (11).Shao L, Lang Y, Wang Y, Gao Y, Zhang W, Niu H, Liu S, Chen N. High-frequency variant p.T60M in NaCl cotransporter and blood pressure variability in Han Chinese. Am. J. Nephrol. 2012;35:515–519. doi: 10.1159/000339165. [DOI] [PubMed] [Google Scholar]
- (12).Wang Y, O’Connell JR, McArdle PF, Wade JB, Dorff SE, Shah SJ, Shi X, Pan L, Rampersaud E, Shen H, Kim JD, Subramanya AR, Steinle NI, Parsa A, Ober CC, Welling PA, Chakravarti A, Weder AB, Cooper RS, Mitchell BD, Shuldiner AR, Chang YP. From the Cover: Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc. Natl. Acad. Sci. U.S.A. 2009;106:226–231. doi: 10.1073/pnas.0808358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Villa F, Goebel J, Rafiqi FH, Deak M, Thastrup J, Alessi DR, van Aalten DM. Structural insights into the recognition of substrates and activators by the OSR1 kinase. EMBO Rep. 2007;8:839–845. doi: 10.1038/sj.embor.7401048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68:201–206. doi: 10.1016/j.neuron.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Greggio E, Cookson MR. Leucine Rich Repeat Kinase 2 mutations and Parkinson’s disease: Three questions. ASN Neuro. 2009;1 doi: 10.1042/AN20090007. No. e00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR, Gray NS. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011;7:203–205. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Reith AD, Bamborough P, Jandu K, Andreotti D, Mensah L, Dossang P, Choi HG, Deng X, Zhang J, Alessi DR, Gray NS. GSK2578215A; a potent and highly selective 2-arylmethyloxy-5-substitutent-N-arylbenzamide LRRK2 kinase inhibitor. Bioorg. Med. Chem. Lett. 2012;22:5625–5629. doi: 10.1016/j.bmcl.2012.06.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Choi HG, Zhang J, Deng X, Hatcher JM, Patricelli MP, Zhao Z, Alessi DR, Gray NS. Brain penetrant LRRK2 inhibitor. ACS Med. Chem. Lett. 2012;3:658–662. doi: 10.1021/ml300123a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Estrada AA, Liu X, Baker-Glenn C, Beresford A, Burdick DJ, Chambers M, Chan BK, Chen H, Ding X, Dipasquale AG, Dominguez SL, Dotson J, Drummond J, Flagella M, Flynn S, Fuji R, Gill A, Gunzner-Toste J, Harris SF, Heffron TP, Kleinheinz T, Lee DW, Le Pichon CE, Lyssikatos JP, Medhurst AD, Moffat JG, Mukund S, Nash K, Scearce-Levie K, Sheng Z, Shore DG, Tran T, Trivedi N, Wang S, Zhang S, Zhang X, Zhao G, Zhu H, Sweeney ZK. Discovery of highly potent, selective, and brain-penetrable Leucine-Rich Repeat Kinase 2 (LRRK2) small molecule inhibitors. J. Med. Chem. 2012;55:9416–9433. doi: 10.1021/jm301020q. [DOI] [PubMed] [Google Scholar]
- (21).Nichols RJ, Dzamko N, Morrice NA, Campbell DG, Deak M, Ordureau A, Macartney T, Tong Y, Shen J, Prescott AR, Alessi DR. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 2010;430:393–404. doi: 10.1042/BJ20100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of {α}-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. U.S.A. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, Schnell CR, Mueller M, Kinzel B, Grevot A, Bolognani F, Stirn M, Kuhn RR, Kaupmann K, van der Putten PH, Rovelli G, Shimshek DR. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet. 2011;20:4209–4223. doi: 10.1093/hmg/ddr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gilsbach BK, Ho FY, Vetter IR, van Haastert PJ, Wittinghofer A, Kortholt A. Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc. Natl. Acad. Sci. U.S.A. 2012;109:10322–10327. doi: 10.1073/pnas.1203223109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Clark K, Mackenzie KF, Petkevicius K, Kristariyanto Y, Zhang J, Choi HG, Peggie M, Plater L, Pedrioli PG, McIver E, Gray NS, Arthur JS, Cohen P. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl. Acad. Sci. U.S.A. 2012;109:16986–16991. doi: 10.1073/pnas.1215450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ku CL, von Bernuth H, Picard C, Zhang SY, Chang HH, Yang K, Chrabieh M, Issekutz AC, Cunningham CK, Gallin J, Holland SM, Roifman C, Ehl S, Smart J, Tang M, Barrat FJ, Levy O, McDonald D, Day-Good NK, Miller R, Takada H, Hara T, Al-Hajjar S, Al-Ghonaium A, Speert D, Sanlaville D, Li X, Geissmann F, Vivier E, Marodi L, Garty BZ, Chapel H, Rodriguez-Gallego C, Bossuyt X, Abel L, Puel A, Casanova JL. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J. Exp. Med. 2007;204:2407–2422. doi: 10.1084/jem.20070628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pauls E, Nanda SK, Smith H, Toth R, Shpiro N, Arthur JSC, Cohen P. A reinvestigation of the roles of the IRAK family in regulating cytokine production in mouse macrophages. Submitted for publication. [Google Scholar]
- (28).Wang Z, Wesche H, Stevens T, Walker N, Yeh WC. IRAK-4 inhibitors for inflammation. Curr. Top. Med. Chem. 2009;9:724–737. doi: 10.2174/156802609789044407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ronnblom L, Alm GV, Eloranta ML. Type I interferon and lupus. Curr. Opin. Rheumatol. 2009;21:471–477. doi: 10.1097/BOR.0b013e32832e089e. [DOI] [PubMed] [Google Scholar]
- (30).Pauls E, Enesa K, Shpiro N, Toth R, Arthur JSC, Cohen P. The role of the IRAK family in the regulation of interferon production by plasmacytoid dendritic cells. Submitted for publication. [Google Scholar]
- (31).Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{α} induction. J. Exp. Med. 2005;201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Rousseau S, Papoutsopoulou M, Symons A, Cook D, Lucocq JM, Prescott AR, O’Garra A, Ley SC, Cohen P. TPL2-mediated activation of ERK1 and ERK2 regulates the processing of pre-TNFα in LPS-stimulated macrophages. J. Cell Sci. 2008;121:149–154. doi: 10.1242/jcs.018671. [DOI] [PubMed] [Google Scholar]
- (33).Kontoyiannis D, Boulougouris G, Manoloukos M, Armaka M, Apostolaki M, Pizarro T, Kotlyarov A, Forster I, Flavell R, Gaestel M, Tsichlis P, Cominelli F, Kollias G. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel disease. J. Exp. Med. 2002;196:1563–1574. doi: 10.1084/jem.20020281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).George D, Salmeron A. Cot/Tpl-2 protein kinase as a target for the treatment of inflammatory disease. Curr. Top. Med. Chem. 2009;9:611–622. doi: 10.2174/156802609789007345. [DOI] [PubMed] [Google Scholar]
- (35).Hu Y, Cole D, Denny RA, Anderson DR, Ipek M, Ni Y, Wang X, Thaisrivongs S, Chamberlain T, Hall JP, Liu J, Luong M, Lin LL, Telliez JB, Gopalsamy A. Discovery of indazoles as inhibitors of Tpl2 kinase. Bioorg. Med. Chem. Lett. 2011;21:4758–4761. doi: 10.1016/j.bmcl.2011.06.065. [DOI] [PubMed] [Google Scholar]
- (36).Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- (37).Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- (38).Cohen P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 2009;21:317–324. doi: 10.1016/j.ceb.2009.01.015. [DOI] [PubMed] [Google Scholar]
- (39).Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel M, Telliez JB. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol. Cell. Biol. 2007;27:170–181. doi: 10.1128/MCB.01456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hegen M, Gaestel M, Nickerson-Nutter CL, Lin LL, Telliez JB. MAPKAP kinase 2-deficient mice are resistant to collagen-induced arthritis. J. Immunol. 2006;177:1913–1917. doi: 10.4049/jimmunol.177.3.1913. [DOI] [PubMed] [Google Scholar]
- (41).Anderson DR, Meyers MJ, Vernier WF, Mahoney MW, Kurumbail RG, Caspers N, Poda GI, Schindler JF, Reitz DB, Mourey RJ. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2) J. Med. Chem. 2007;50:2647–2654. doi: 10.1021/jm0611004. [DOI] [PubMed] [Google Scholar]
- (42).Mourey RJ, Burnette BL, Brustkern SJ, Daniels JS, Hirsch JL, Hood WF, Meyers MJ, Mnich SJ, Pierce BS, Saabye MJ, Schindler JF, South SA, Webb EG, Zhang J, Anderson DR. A benzothiophene inhibitor of mitogen-activated protein kinase-activated protein kinase 2 inhibits tumor necrosis factor α production and has oral anti-inflammatory efficacy in acute and chronic models of inflammation. J. Pharmacol. Exp. Ther. 2010;333:797–807. doi: 10.1124/jpet.110.166173. [DOI] [PubMed] [Google Scholar]
- (43).Oubrie A, Kaptein A, de Zwart E, Hoogenboom N, Goorden R, van de Kar B, van Hoek M, de Kimpe V, van der Heijden R, Borsboom J, Kazemier B, de Roos J, Scheffers M, Lommerse J, Schultz-Fademrecht C, Barf T. Novel ATP competitive MK2 inhibitors with potent biochemical and cell-based activity throughout the series. Bioorg. Med. Chem. Lett. 2012;22:613–618. doi: 10.1016/j.bmcl.2011.10.071. [DOI] [PubMed] [Google Scholar]
- (44).Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- (45).Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE, Kahana JA, Kral AM, Leander K, Lee LL, Malinowski J, McAvoy EM, Nahas DD, Robinson RG, Huber HE. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 2005;385:399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- (47).Green CJ, Goransson O, Kular GS, Leslie NR, Gray A, Alessi DR, Sakamoto K, Hundal HS. Use of Akt inhibitor and a drug-resistant mutant validates a critical role for protein kinase B/Akt in the insulin-dependent regulation of glucose and system A amino acid uptake. J. Biol. Chem. 2008;283:27653–27667. doi: 10.1074/jbc.M802623200. [DOI] [PubMed] [Google Scholar]
- (48).Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:e17. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wu WI, Voegtli WC, Sturgis HL, Dizon FP, Vigers GP, Brandhuber BJ. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One. 2010;5:e12913. doi: 10.1371/journal.pone.0012913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Alessi DR, Cohen P, Ashworth A, Cowley S, Leevers SJ, Marshall CJ. Assay and expression of mitogen-activated protein kinase, MAP kinase kinase, and Raf. Methods Enzymol. 1995;255:279–290. doi: 10.1016/s0076-6879(95)55031-3. [DOI] [PubMed] [Google Scholar]
- (51).Hall-Jackson CA, Eyers PA, Cohen P, Goedert M, Boyle FT, Hewitt N, Plant H, Hedge P. Paradoxical activation of Raf by a novel Raf inhibitor. Chem. Biol. 1999;6:559–568. doi: 10.1016/s1074-5521(99)80088-x. [DOI] [PubMed] [Google Scholar]
- (52).Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16:443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- (53).Zeqiraj E, van Aalten DM. Pseudokinases-remnants of evolution or key allosteric regulators? Curr. Opin. Struct. Biol. 2010;20:772–781. doi: 10.1016/j.sbi.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- (55).Zeqiraj E, Filippi BM, Deak M, Alessi DR, van Aalten DM. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science. 2009;326:1707–1711. doi: 10.1126/science.1178377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zeqiraj E, Filippi BM, Goldie S, Navratilova I, Boudeau J, Deak M, Alessi DR, van Aalten DM. ATP and MO25α regulate the conformational state of the STRADα pseudokinase and activation of the LKB1 tumour suppressor. PLoS Biol. 2009;7:e1000126. doi: 10.1371/journal.pbio.1000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Passamonti F, Rumi E. Clinical relevance of JAK2 (V617F) mutant allele burden. Haematologica. 2009;94:7–10. doi: 10.3324/haematol.2008.001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR, Nichols RJ. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem. J. 2010;430:405–413. doi: 10.1042/BJ20100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ananieva O, Darragh J, Johansen C, Carr JM, McIlrath J, Park JM, Wingate A, Monk CE, Toth R, Santos SG, Iversen L, Arthur JS. The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat. Immunol. 2008;9:1028–1036. doi: 10.1038/ni.1644. [DOI] [PubMed] [Google Scholar]