

Abstract
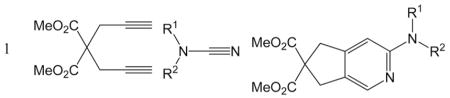
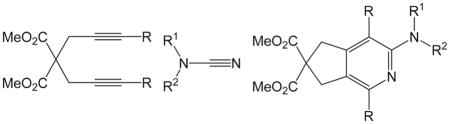
A variety of bicyclic N,N-disubstituted 2-aminopyridines have been prepared from diynes and cyanamides by nickel-catalyzed [2+2+2] cycloaddition reactions. The reactions proceeded at room temperature with low catalyst loading to afford 2-aminopyridines in good to excellent yields. The method is amenable to both internal and terminal diynes and proceeds in a regioselective manner. A number of cyanamides with diverse functional group tolerance were used. The intermolecular version employing 3-hexyne and N-cyanopyrrolidine also afforded the desired N,N-disubstituted 2-aminopyridine in good yield.
Keywords: Nickel, Cycloaddition, Cyanamides, Alkynes, Carbenes
Introduction
2-Aminopyridines are attractive synthetic targets due to their use in many fields of chemistry as organometallic ligands,[1–4] chromophores,[5–7] and intermediates in biologically active molecules.[8–14] A variety of methods for the synthesis of 2-aminopyridines exist, namely the amination of pyridines from sodium amide (the Chichibabin reaction),[15,16] Buchwald–Hartwig amination reactions,[17–20] the substitution of halopyridines,[21–24] multi-component condensations,[25,26] hetero-Diels–Alder reactions,[27] and Ullman-type aminations.[28,29] To complement existing methods, an easily envisioned route is the [2+2+2] cycloaddition of alkynes and cyanamides.
The metal-catalyzed [2+2+2] cycloaddition of alkynes and nitriles using a variety transition metals has a rich and extensive history and continues to be a highly studied field.[30,31] Although studies of cycloadditions of alkynes and nitriles are expansive, independent studies of analogous cyanamides are few. The seminal work of Bönnemann et al. demonstrated the cycloaddition of acetylene and cyanamide using a unique (η6-boranato)cobalt catalyst at high temperature and pressure.[32] Heller et al. accomplished a [CpCo(cod)]-catalyzed cycloaddition of dimethylcyanamide and acetylene as a single example, which afforded a moderate 46% yield.[33] Maryanoff and co-workers performed a more focused study demonstrating the effectiveness of the cycloaddition of diynes with cyanamides using [CpCo-(CO)2] as a catalyst.[34–36] More recently, Tanaka et al. developed a cationic [Rh(cod)]/BINAP cycloaddition catalyst that facilitated the [2+2+2] cycloaddition of a malonate-derived diyne and N-cyanomorpholine as a single example in 47% yield.[37] Heller and co-workers used a chiral cobalt catalyst to synthesize a variety of chiral 1-aryl-5,6,7,8-tetra-hydroquinolines from aryl-substituted diynes and nitriles. This system was also amenable to the cycloaddition of aryl-substituted diynes and piperidine-1-carbonitrile as a single example in an excellent yield.[38] Owing to our recent success in the mild and efficient nickel-catalyzed [2+2+2] cycloaddition of alkynes and nitriles to generate pyridines,[39] we believed that the use of cyanamides as coupling partners would be a practical extension of this methodology. Herein we report the nickel-catalyzed [2+2+2] cycloaddition of alkynes and cyanamides at room temperature with low catalyst loadings.
Results and Discussion
Reaction Optimization
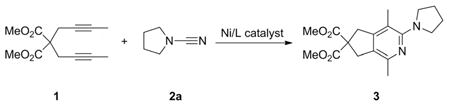
Diyne 1 and N-cyanopyrrolidine (2a) were chosen as model substrates due to our familiarity and simplicity of 1 as well as the simplicity, ease of handling, and commercial availability of 2a [Equation (1)]. Initial investigations focused on catalyst screening with [Ni(cod)2] (cod = 1,5-cyclooctadiene) as an Ni0 source in combination with a variety of ligands: Phosphanes, phosphites, amines, and N-heterocyclic carbenes (NHCs; Table 1). The resultant catalysts were then added to a 1:1 solution of diyne 1 and 2a in toluene. The reactions were stirred at room temperature for 3 h and analyzed by gas chromatography (GC). The product 3 was detected with most of the ligands tested (Entries 1–5), which is significantly different to the results of Ni-catalyzed cycloaddition reactions of simple nitriles for which only select NHC ligands (i.e., no phosphanes) afforded appreciable amounts of pyridine products.[39] The highest yields were obtained when either IMes or SIPr was employed as the ligand. The effectiveness of the [Ni(cod)2]/IMes catalyst is particularly surprising as this catalyst system typically produces significant amounts of a dimerized diyne as a side-product.[35–39] Brief optimization showed that increasing the concentration of the cyanamide retarded the reaction and that a 1:1 diyne/cyanamide ratio was ideal. In addition, catalyst loading could be reduced to 5 mol-% [Ni(cod)2] and 10 mol-% IMes or SIPr without loss of yield.
Table 1.
Ligand screening for the [2+2+2] cycloaddition reaction between 1 and 2a.[a]
| Entry | Ligand (mol-%) | Conv. [%][b] | Yield [%][b] |
|---|---|---|---|
| 1 | IPr (20)[c] | 83 | 78 |
| 2 | SIPr (20)[c] | 100 | 98 |
| 3 | IMes (20)[c] | 100 | 98 |
| 4 | P(tBu)3 (20) | 91 | 45 |
| 5 | P(Cy)3 (20) | 100 | 81 |
| 6 | PPh3 (20) | 80 | 25 |
| 7 | DPPE (10)[c] | 47 | 17 |
| 8 | BINAP (10)[c] | 13 | 6 |
| 9 | DPPF (10)[c] | 11 | 6 |
| 10 | P(OPh)3 (20) | 6 | 0 |
| 11 | P(OiPr)3 (20) | 86 | 11 |
| 12 | TMEDA (20)[c] | 65 | 0 |
| 13 | [Ni(cod)2] (10) | 91 | 0 |
Reaction conditions: 0.1 M diyne, 0.1 M cyanamide, 10 mol-% [Ni(cod)2], PhMe, room temp., 3 h.
Determined by GC relative to naphthalene as an internal standard.
SIPr = 1,3-bis(2,6-di-isopropylphenyl)-4,5-dihydroimidazolin-2-ylidine, IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, IMes = 1,3-bis(1,3,5-trimethylphenyl)imidazol-2-ylidene, DPPE = 1,2-bis(diphenylphosphanyl)ethane, BINAP = rac-(1,1′-binaphthalene-2,2′-diyl)bis(di-phenylphosphane), DPPF = 1,1′-bis(diphenylphosphanyl)ferrocene, TMEDA = N,N,N′,N′-tetramethylethylenediamine.
 |
(1) |
Upon addition of a solution of [Ni(cod)2]/IMes to the reaction mixture, an instant and dramatic color change occurred, possibly an indication of a rapid reaction. As such, the reaction times of the [Ni(cod)2]/IMes and [Ni(cod)2]/SIPr systems were explored (Table 2). Two reaction mixtures, both consisting of equimolar amounts of diyne 1 and 2a in toluene, were treated with either [Ni(cod)2]/IMes or [Ni(cod)2]/SIPr. Although the reactions with both SIPr and IMes as ligands each generated excellent yields of 2-aminopyridine 3, we found that reactions run with IMes produced 3 in only 15 min, whereas the analogous reaction run with SIPr required 60 min to achieve the same yield (Entry 3 vs. 4).
Table 2.
Comparison of the reaction times for the reaction of 1 and 2a.[a]
| Entry | Ligand | Time [min] | Conv. [%][b] | Yield [%][b] |
|---|---|---|---|---|
| 1 | SIPr | 15 | 30 | 29 |
| 2 | SIPr | 30 | 75 | 72 |
| 3 | SIPr | 60 | 100 | 98 |
| 4 | IMes | 15 | 98 | 98 |
Reaction conditions: 0.1 M diyne, 0.1 M cyanamide, 5 mol-% [Ni(cod)2], 10 mol-% ligand, PhMe, room temp.
Determined by GC relative to naphthalene as an internal standard.
A variety of solvents were screened for the [Ni(cod)2]/IMes-catalyzed cycloaddition of diyne 1 and cyanamide 2a (Table 3). Excellent yields were obtained in pentane, 1,4-dioxane, and toluene. Toluene was chosen for use in further cycloaddition reactions, because the yields were higher in toluene than in dioxane and also because many substrates are only moderately soluble in pentane.
Table 3.
Solvent screening for cycloaddition of 1 and 2a.[a]
| Entry | Solvent | Conv. [%][b] | Yield [%][b] |
|---|---|---|---|
| 1 | THF | 95 | 67 |
| 2 | Et2O | 100 | 86 |
| 3 | benzene | 33 | 4 |
| 4 | pentane | 100 | 98 |
| 5 | 1,4-dioxane | 100 | 95 |
| 6 | toluene | 100 | 98 |
Reaction conditions: 0.1 M diyne, 0.1 M cyanamide, 5 mol-%[Ni(cod)2], 10 mol-% IMes, room temp., 30 min.
Determined by GC relative to naphthalene as an internal standard.
Substrate Scope
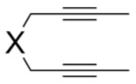
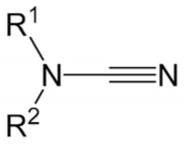
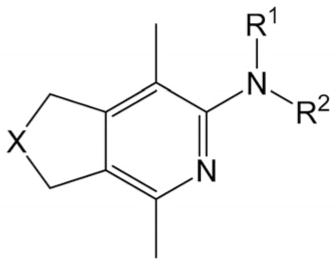
With the optimized conditions in hand, we examined the reactions of a range of diyne and cyanamide substrates with varying electronic and steric properties (Table 4). In addition to the model cyanamide 2a, other dialkylcyanamides readily underwent cyclization, affording excellent yields. The yields appear to decrease with increasing steric bulk: Me (4) > Et (5) > Pr (6; Entries 2–4). This is further highlighted by the complete inactivity of diisopropylcyanamide (2e, see below) towards cycloaddition. The heterocyclic cyanamide N-cyanomorpholine (Entry 5) was an excellent substrate, affording product 7 in 97% yield. In addition to alkyl substituents, a variety of amine-protected cyanamides were evaluated, these include methyl PMB cyanamide (PMB = p-methoxybenzyl; 2g), dibenzylcyanamide (2h), as well as carbonyl-protected cyanamides such as N-(tert-butoxycarbonyl)-N-butylcyanamide (2i) and N-butyl-N-cyanoacetamide (2j; Entries 6–9). Notable unreactive protected cyanamides are N-tosylated butylcyanamide (2k) and diallylcyanamide (2l; Figure 1). Note, N-tosylamines have been, for the most part, troublesome in many of our previous cycloaddition reactions.[39–43] It is unclear in this case whether the steric bulk or the reactivity of the N-tosyl moiety deactivates the catalyst. Diallylcyanamide was also employed but was also found to deactivate the catalyst. Free amine is incompatible with our catalyst system as seen by the lack of reaction of free cyanamide and N-butylcyanamide. Cyanamides containing pendant functional groups [methyl 2-(N-butylcyanamido)acetate (2m), N-butyl-N-(3-chloropropyl)cyanamide (2n), and N-butyl-N-(3-chloroethyl)cyanamide (2o)] were also evaluated and gave mixed results. Both chlorinated cyanamides 2n and 2o readily deactivated the catalyst as no conversion was observed in these reactions (Figure 1). Cyanamide 2m afforded aminopyridine 12 in 81% yield as a light-sensitive aminopyridine that required purification by chromatography to be performed in a darkened room (Entry 10). Arylcyanamides are also good substrates with cyanamide 2p affording aminopyridine 13 in excellent yield (Entry 11). Various diynes were subjected to the reaction conditions with model cyanamide 2a and afforded excellent yields. For example, internal N-protected amines (14) and ethers (16) as well as linear diyne 18 all reacted well under our conditions (Entries 12–14, respectively). Notably, diyne 20 readily reacted to afford an aminopyridine appended to a seven-membered ring in 76% yield despite the lack of Thorpe–Ingold assistance in the substrate (Entry 15).
Table 4.
Cycloaddition of cyanamides and internal diynes.[a]
| Entry | Diyne | Cyanamide | Yield[b] |
|---|---|---|---|
|
|
|
|
| 1 | X = C(CO2Me)2 1 |
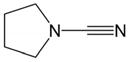 2a |
3, 93% |
| 2 | 1 | R1 = R2 = Me 2b |
4, 99% |
| 3 | 1 | R1 = R2 = Et 2c |
5, 93% |
| 4 | 1 | R1 = R2 = nPr 2d |
6, 89% |
| 5 | 1 |
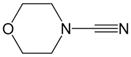 2f |
7, 97% |
| 6 | 1 | R1 = Me, R2 = PMB 2g |
8, 87% |
| 7 | 1 | R1 = R2 = Bn 2h |
9, 88% |
| 8 | 1 |
2i R1 = nBu R2 = Boc |
10, 82% |
| 9 | 1 | R1 = nBu, R2 = Ac 2j |
11, 87% |
| 10 | 1 | R1 = nBu, R2 = CH2CO2Me 2m |
12, 81% |
| 11 | 1 | R1 = Me, R2 = Ph 2p |
13, 90% |
| 12 | X = NTs 14 |
2a | 15, 84% |
| 13 | X = O 16 |
2a | 17, 94% |
| 14 |
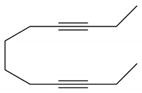 18 |
2a |
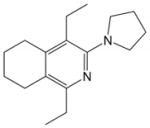 19, 92 |
| 15 |
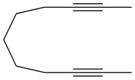 20 |
2a |
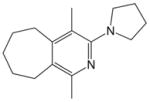 21, 76% |
Reaction conditions: 0.1 M diyne, 0.1 M cyanamide, 5 mol-% [Ni(cod)2], 10 mol-% IMes, PhMe, room temp., 30 min.
Isolated yield, average of at least two runs.
No reaction.
Figure 1.
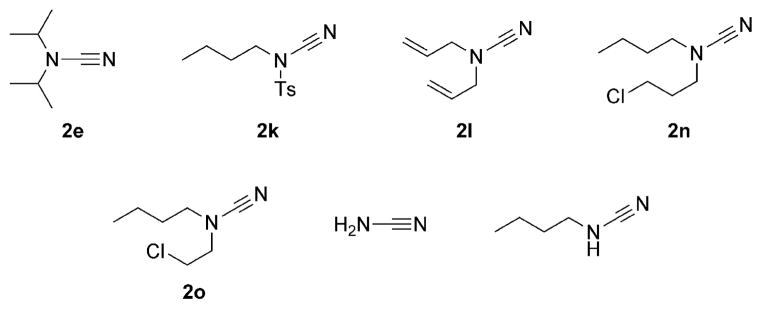
Examples of cyanamides that are incompatible with the [Ni(cod)2]/NHC-catalyzed cycloaddition.
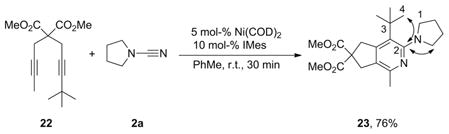
The regioselectivity of the reaction was investigated by treating diyne 22 and cyanamide 2a under the optimized reaction conditions; aminopyridine 23 was obtained as a single regioisomer in 76% yield [Equation (2)].[44] The structure was assigned through complementary HMQC and HMBC NMR experiments, with the regiochemistry being determined by exclusive correlations between C-2 and protons located on C-4 and C-1.
 |
(2) |
We also investigated the synthesis of N,N-disubstituted 2-aminopyridines by the cycloaddition of an untethered alkyne [Equation (3)]. Subjecting 3-hexyne (2 equiv.) and N-cyanopyrrolidine (2a) to the optimized conditions described above (5 mol-% of catalyst, room temp.) afforded 2-aminopyridine 24 in 86% yield.
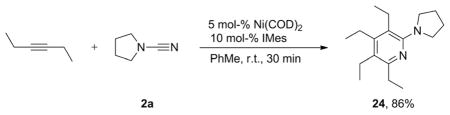 |
(3) |
Terminal diynes, a challenging substrate for the parent Ni-catalyzed nitrile cycloaddition reaction,[39] were also evaluated as potential substrates (Table 5). Initially, the [Ni(cod)2]/IMes-catalyzed reaction between diyne 25 and cyanamide 2a only yielded trace amounts of the desired product in an otherwise unidentifiable reaction mixture. However, when IMes was substituted for SIPr, the reaction was effective and generated aminopyridine 26 in 80% yield. Extended reaction times were required for the [Ni(cod)2]/SIPr-catalyzed reactions. Interestingly, unlike internal diynes, the cycloaddition of terminal diynes and cyanamides using [Ni(cod)2]/SIPr did not occur in 1,4-dioxane. The use of dioxane did lead to full conversion; however, no identifiable products were formed nor were any byproducts isolated from the complex reaction mixture. Thus, cycloaddition reactions using [Ni(cod)2]/SIPr were carried out exclusively in toluene.
Table 5.
Cycloaddition reactions of the terminal malonate diyne.[a]
| Entry | Diyne | Cyanamide | Yield[b] |
|---|---|---|---|
| |||
| 1 | 25 | 2a | 26, 80% |
| 2 | 25 | 2f | 27, 86% |
| 3 | 25 | 2i | 28, 82% |
| 4 | 25 | 2j | 29, 93% |
| 5 | 25 | 2g | 30, n.r.[c] |
| 6 | 25 | 2m | 31, n.r[c] |
| 7 | 25 | 2p | 32, 83% |
Reaction conditions: 0.1 M diyne, 0.1 M cyanamide, PhMe, 5 mol-% [Ni(cod)2], 10 mol-% SIPr, room temp., 60 min.
Isolated yield, average of at least two runs.
No reaction.
Under these revised conditions, terminal diynes were amenable to cycloaddition with most of the previously tested cyanamides (Table 5). Alkyl substituents work well with cyanamides 2a and 2f affording products 26 and 27 in 80 and 86% yields, respectively (Entries 1 and 2). Terminal diynes were also compatible with N-Boc- (2i) and N-acyl-protected (2j) cyanamides, with yields comparable to internal diynes (Entries 3 and 4). However, no reaction was observed with diyne 25 and either PMB-protected cyanamide 2g or ester-functionalized cyanamide 2m (Entries 5 and 6). In addition to the alkylcyanamides, phenyl-substituted cyanamides (2p) are excellent substrates affording 32 in good yield (Entry 7).
In Situ Catalyst Generation
Previously, we developed a method that generates the active Ni0/NHC catalyst in situ from air-stable, readily available precursors.[45] The method employed [Ni(acac)2] as a nickel source, an appropriate NHC·HCl or HBF4 salt, and nBuLi as a simultaneous reductant and base. We found this in situ method was also effective for the [2+2+2] cycloaddition reactions of diynes and cyanamides. When diyne 1 and cyanamide 2a were treated with a stirred solution of [Ni(acac)2], IMes·HCl, and nBuLi, aminopyridine 3 was obtained in 80% yield. Furthermore, a variety of aminopyridines were obtained in this fashion with yields comparable to the yields obtained in the initial substrate scope experiments (Table 6, Entries 1–5). In situ cycloaddition using terminal diyne 25 and cyanamide 2a was also successful when IMes·HCl was replaced by SIPr·HBF4. Further cyanamides were tested with diyne 25 with mixed results. The reactions with both cyanamides 2a and 2p went to completion and gave good yields, whereas the Boc (2i) and acyl (2j) cyanamides gave modest yields of 50 and 30% with 20 and 32% recovered starting material, respectively (Entries 6–9).
Table 6.
Yields from in situ generated Ni–carbene catalyst (rsm = recovered starting material).[a]
| Entry | Diyne | Cyanamide | Yield[a] |
|---|---|---|---|
| |||
| 1 | R = CH3 1 |
2a | 3[b], 80% |
| 2 | 1 | 2i | 10[b], 70% |
| 3 | 1 | 2j | 11[b], 79% |
| 4 | 1 | 2m | 12[b], 78% |
| 5 | 1 | 2p | 13[b], 85% |
| 6 | R = H 25 |
2a | 26[c], 75% |
| 7 | 25 | 2i | 28[c], 50% (20% rsm) |
| 8 | 25 | 2j | 29[c], 30% (32% rsm) |
| 9 | 25 | 2p | 30[c], 80% |
Isolated yields (average of at least two runs).
Reaction conditions: 0.1 M diyne, 0.1 M cyanamide, PhMe, 5 mol-% [Ni(acac)2], 10 mol-% IMes·HCl, 25 mol-% nBuLi, room temp., 60 min.
Reaction conditions: 0.1 M diyne, 0.1 M cyanamide, PhMe, 5 mol-% [Ni(acac)2], 10 mol-% SIPr·HBF4, 25 mol-% nBuLi, room temp., 60 min.
Conclusions
We have demonstrated that diynes undergo [2+2+2] cycloaddition reactions with cyanamides in the presence of an Ni–carbene catalyst to generate N,N-disubstituted 2-amino-pyridines. The method is effective for the cycloaddition of internal as well as terminal diynes with a variety of cyanamides, affording good to excellent yields. N,N-Disubstituted 2-aminopyridines were also obtained when using an in situ generated Ni–carbene catalyst prepared from air-stable, commercially available sources.
Experimental Section
General
Ligands 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidine (IPr), 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazolin-2-ylidine (SIPr), and 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidine (IMes) were prepared according to literature procedures.[46,47] The diynes dimethyl 2,2-bis(but-2-ynyl)malonate (1),[48] dimethyl 2,2-bis(prop-2-yn-1-yl)malonate (25),[48] N,N-bis(but-2-ynyl)-p-toluene-sulfonamide (14),[49] 1-(but-2-ynyloxy)but-2-yne (16),[50] and di-methyl 2-(but-2-ynyl)-2-(4,4-dimethylpent-2-ynyl)malonate (22)[44] were also prepared according to literature procedures. N-Butylcyanamide was also prepared by literature procedures.[51] The diynes 3,9-dodecadiyne (18) and 2,9-undecadiyne (20) were purchased from GFS and Lancaster chemical companies, respectively, and distilled from CaH2 before use. Bis(1,5-cyclooctadiene)nickel(0), [Ni(cod)2], was purchased from the Strem chemical company and used without further purification. All other reagents were purchased from commercial sources and used without further purification. All liquid reagents were degassed prior to use by the freeze-pump-thaw method. All reactions were preformed in a nitrogen-filled glove box or under nitrogen using standard Schlenk techniques unless otherwise noted.
N,N-Dipropylcyanamide (2d)
Dipropylamine (3 mL, 21.9 mmol, 1.0 equiv.) was added dropwise to a stirred solution of Et2O/THF (1:1) (50 mL) and BrCN (1.4 g, 13.3 mmol, 0.6 equiv.) over 10 min. The reaction mixture was stirred at ambient temperature for 3 h, after which time hexane (10 mL) was added followed by an additional 10 min of stirring. The mixture was then filtered through a pad of Celite and washed with H2O (3 × 50 mL) and brine (2 × 50 mL). The organic phase was then dried with anhydrous Na2SO4 and concentrated to yield 1.35 g of 2d as a colorless oil; yield 98%. 1H NMR (CDCl3): δ = 0.89 (t, J = 7.5 Hz, 3 H), 1.59 (dt, J1 = 5, J2 = 10 Hz, 2 H), 2.87 (t, J = 5 Hz, 2 H) ppm. 13C NMR (CDCl3): δ = 10.92, 20.9, 53.1, 117.8 ppm. IR (neat): ν̃ = 2968, 2878, 2208, 1462, 1090 cm−1. HRMS (ESI): calcd. for C7H15N2 [M + H]+ 127.1230; found 127.1230.
N-(tert-Butoxycarbonyl)-N-butylcyanamide (2i)
N-Butylcyanamide (740 mg, 7.6 mmol, 1 equiv.) in Et2O (5 mL) was added to a suspension of NaH (217 mg, 9.1 mmol, 1.2 equiv.) in THF (75 mL) at 0 °C. The mixture was warmed to room temperature, after which Boc2O (1.9 mL, 8.3 mmol, 1.1 equiv.) was added. The reaction mixture was stirred for 1 h, quenched with H2O (1 mL), and Et2O (10 mL) was added. The crude reaction mixture was poured into brine/water (1:1), extracted with Et2O (3 × 15 mL), and concentrated. The residue was purified by column chromatography using CH2Cl2 to afford 1.41 g of the title compound as a colorless oil; yield 95%. 1H NMR (CDCl3): δ = 0.94 (t, J = 7.5 Hz, 3 H), 1.38 (m, 2 H), 1.51 (s, 9 H), 1.66 (dt, 2 H), 3.46 (t, 2 H) ppm. 13C NMR (CDCl3): δ = 13.7, 19.5, 27.9, 29.9, 47.6, 85.4, 109.8, 151.2 ppm. IR (neat): ν̃ = 2966, 2361, 2241, 1748, 1461, 1152 cm−1. HRMS (ESI): calcd. for C10H19N2O2 [M + H]+ 199.1441; found 199.1441.
N-Butyl-N-cyanoacetamide (2j)
N-Butylcyanamide (770 mg, 7.9 mmol, 1 equiv.) in Et2O (5 mL) was added to a suspension of NaH (230 mg, 9.4 mmol, 1.2 equiv.) in THF (25 mL) at 0 °C. The mixture was warmed to room temperature, after which acetyl chloride (0.617 mL, 8.6 mmol, 1.1 equiv.) was added. The reaction mixture was stirred for 1 h, quenched with H2O (1 mL), and Et2O was added (10 mL). The crude reaction mixture was poured into brine/water (1:1), extracted with Et2O (3 × 15 mL), and concentrated. The residue was purified by column chromatography using CH2Cl2 to afford 1.07 g of the title compound as a colorless volatile oil; yield 98%. 1H NMR (CDCl3): δ = 0.93 (t, J = 7.5 Hz, 3 H), 1.35 (m, 2 H), 1.64 (dt, 2 H), 2.38 (s, 3 H), 3.54 (t, 2 H) ppm. 13C NMR (CDCl3): δ = 13.6, 19.6, 22.3, 29.7, 45.9, 111.1, 169.4 ppm. IR (neat): ν̃ = 2963, 2875, 2361, 2233, 1730, 1373, 1244 cm−1. HRMS (ESI): calcd. for C7H13N2O [M + H]+ 141.1022; found 141.1023.
N-Butyl-N-cyano-4-methylbenzenesulfonamide (2k)
N-Butylcyanamide (1.21 g, 12.3 mmol, 1 equiv.) in Et2O (5 mL) was added to a suspension of NaH (356 mg, 14.8 mmol, 1.2 equiv.) in THF (50 mL) at 0 °C. The mixture was warmed to room temperature, after which p-toluene sulfonyl chloride (2.9 g, 14.8 mmol, 1.2 equiv.) in THF (5 mL) was added. The reaction mixture was stirred for 4 h, quenched with H2O (1 mL), and Et2O (10 mL) was added. The crude reaction mixture was poured into brine/water (1:1), extracted with Et2O (3 × 15 mL), and concentrated. The residue was purified by column chromatography using CH2Cl2 to afford 1.3 g of the title compound as a viscous colorless oil; yield 97%. 1H NMR (CDCl3): δ = 0.85 (t, J = 7.5 Hz, 3 H), 1.29 (m, 2 H), 1.59 (m, 2 H), 2.44 (s, 3 H), 3.33 (m, 2 H), 7.38 (d, 2 H), 7.78 (d, 2 H) ppm. 13C NMR (CDCl3): δ = 13.3, 19.1, 21.7, 29.6, 49.8, 108.5, 127.7, 130.4, 133.5, 146.5 ppm. IR (neat): ν̃ = 2963, 2875, 2230, 1380, 1173 cm−1. HRMS (ESI): calcd. for C12H17N2O2S [M + H]+ 253.1005; found 253.1006.
Methyl 2-(N-Butylcyanamido)acetate (2m)
N-Butylcyanamide (1.01 g, 10.3 mmol, 1 equiv.) in Et2O (5 mL) was added to a suspension of NaH (295 mg, 12.3 mmol, 1.2 equiv.) in THF (50 mL) at 0 °C. The mixture was warmed to room temperature, after which methyl 2-bromoacetate (1.0 mL, 11 mmol, 1.1 equiv.) was added. The reaction mixture was stirred for 3 h, quenched with H2O (1 mL), and Et2O (10 mL) was added. The crude reaction mixture was poured into brine/water (1:1), extracted with Et2O (3 × 15 mL), and concentrated. The residue was purified by column chromatography using CH2Cl2 to afford 1.53 g of the title compound as a pale-yellow oil; yield 88%. 1H NMR (C6D6): δ = 0.59 (t, J = 7.5 Hz, 3 H), 0.98 (m, 2 H), 1.21 (dt, 2 H), 2.52 (t, 2 H), 3.19 (s, 2 H), 3.23 (s, 3 H) ppm. 13C NMR (C6D6): δ = 13.9, 20.1, 30.1, 52.1, 52.2, 52.6, 117.0, 168.9 ppm. IR (neat): ν̃ = 2960, 2875, 2360, 2216, 1752, 1217 cm−1. HRMS (ESI): calcd. for C8H15N2O2 [M + H]+ 171.1128; found 171.1128.
N-Butyl-N-(3-chloropropyl)cyanamide (2n)
N-Butylcyanamide (805 mg, 8.2 mmol, 1 equiv.) in Et2O (5 mL) was added to a suspension of NaH (250 mg, 9.9 mmol, 1.2 equiv.) in THF (50 mL) at 0 °C. The mixture was warmed to room temperature, after which 1-bromo-3-chloropropane (2.45 mL, 24.7 mmol, 3 equiv.) was added. The mixture was heated to reflux and stirred overnight. The reaction mixture was then quenched with H2O (1 mL), and Et2O (10 mL) was added. The crude reaction mixture was poured into brine/water (1:1), extracted with Et2O (3 × 15 mL), and concentrated. The residue was purified by column chromatography using CH2Cl2 to afford 1.3 g of the title compound as a colorless oil; yield 91%. 1H NMR (C6D6): δ = 0.59 (t, J = 7.5 Hz, 3 H), 0.93 (m, 2 H), 1.15 (dt, 2 H), 1.48 (m, 2 H), 2.30 (t, J = 7.5 Hz, 2 H), 2.46 (t, J = 7.5 Hz, 2 H), 3.04 (t, J = 7.5 Hz, 2 H) ppm. 13C NMR (CDCl3): δ = 13.99, 20.1, 30.1, 30.8, 41.9, 48.7, 51.9, 117.0 ppm. IR (neat): ν̃ = 2961, 2874, 2361, 2208, 1459 cm−1. HRMS (ESI): calcd. for C8H16ClN2 [M + H]+ 175.0997; found 175.0997.
N-Butyl-N-(2-chloroethyl)cyanamide (2o)
N-Butylcyanamide (1.1 g, 11.2 mmol, 1.0 equiv.) in THF (5 mL) was added dropwise to a stirring suspension of NaH (400 mg, 16.8 mmol, 1.5 equiv.) in THF/DCE (1,2-dichloroethane) (1:1) (20 mL). The reaction mixture was stirred at ambient temperature for 15 min, then heated at reflux for 8 h. The mixture was cooled to room temperature, quenched with MeOH (5 mL), and then poured into H2O (100 mL). The suspension was extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were dried with anhydrous Na2SO4, concentrated, and purified by column chromatography eluting with CH2Cl2 to afford 1.38 g of 2o as a pale-yellow oil; yield 77%. 1H NMR (C6D6): δ = 0.68 (t, J = 7.5 Hz, 3 H), 0.97 (m, 2 H), 1.18 (dt, 2 H), 2.28 (t, J = 7.5 Hz, 2 H), 2.42 (t, J = 7.5 Hz, 3 H), 2.92 (t, J = 5 Hz, 2 H) ppm. 13C NMR (CDCl3): δ = 13.6, 19.5, 29.8, 40.9, 51.7, 53.1, 116.8 ppm. IR (neat): ν̃ = 2962, 2874, 2210, 1461, 1106 cm−1. HRMS (ESI): calcd. for C7H13N2 [M + H]+ 161.0840; found 161.0841.
N-Methyl-N-phenylcyanamide (2p)
N-Methylaniline (1.7 mL, 15.6 mmol, 1 equiv.) was added to a solution of cyanogen bromide (1 g, 9.4 mmol, 0.6 equiv.) in Et2O/THF (1:1) (25 mL) at room temperature. The reaction mixture was then stirred overnight, after which time hexane (5 mL) was added, and the mixture was stirred for an additional 10 min. It was then filtered through a pad of Celite, and the filtrate was washed with H2O (4 × 25 mL) and brine (2 × 25 mL). The organic phase was dried with anhydrous Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography using ethyl acetate/hexane (1:1) to afford 2.19 g of the title compound as a colorless solid: yield 82%. M.p. 28–30 °C. 1H NMR (C6D6): δ = 2.21 (s, 3 H), 6.96 (m, 2 H), 6.76 (m, 1 H), 6.7 (m, 2 H) ppm. 13C NMR (C6D6): δ = 36.2, 115.1, 123.3, 129.9, 141.4 ppm. IR (neat): ν̃ = 2361, 2223, 1600, 1114 cm−1. HRMS (ESI): calcd. for C8H9N2 [M + H]+ 133.0760; found 133.0760.
General Co-Cycloaddition Procedure for Terminally Substituted Diynes (A)
In a nitrogen-filled glove-box, [Ni(cod)2] (1 equiv.) and IMes (2 equiv.) were diluted to 0.036 M in toluene or dioxane and stirred for at least 4 h. A solution of diyne (100 mg) and cyanamide (1 equiv.) in toluene or dioxane was placed in an oven-dried vial equipped with a stirring bar. The [Ni(cod)2/IMes solution was added to this stirred solution with a total reaction concentration of 0.1 M in the diyne. The reaction mixture was stirred at ambient temperature for 30 min, taken out of the glove-box, concentrated, and then purified by flash column chromatography to afford the aminopyridine product.
General Co-Cycloaddition Procedure for Terminal Diynes (B)
In a nitrogen-filled glove box, [Ni(cod)2] (1 equiv.) and SIPr (2 equiv.) were diluted to 0.036 M in toluene and stirred for at least 4 h. A solution of diyne (100 mg) and cyanamide (1 equiv.) in toluene was placed in an oven-dried vial equipped with a stirring bar. The [Ni(cod)2]/SIPr solution was added to this stirred solution with a total reaction concentration of 0.1 M in the diyne. The reaction mixture was stirred at ambient temperature for 1 h, taken out of the glove box, concentrated, and then purified by flash column chromatography to afford the aminopyridine product.
In Situ Cycloaddition Procedure
In a nitrogen-filled glove box, [Ni-(acac)2] (5.3 mg, 0.02 mmol) and either IMes·HCl (14.1 mg, 0.04 mmol) or SIPr·HBF4 (19.8 mg, 0.04 mmol) were suspended in pentane (1.5 mL). To this was added a 2.5 M solution of nBuLi in hexanes (45 mL, 0.103 mmol). The resultant suspension was stirred at room temperature for 10 min. Concurrently, the appropriate diyne (0.41 mmol) and cyanamide (0.41 mmol) were placed in an oven-dried vial equipped with a stirring bar and dissolved in toluene (3 mL). The catalyst solution was then added to the toluene solution and the reaction mixture was stirred at room temperature for 1 h. The reaction was quenched by the addition of MeOH (5 drops). The reaction solution was then concentrated and purified by column chromatography.
Dimethyl 1,4-Dimethyl-3-(pyrrolidin-1-yl)-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (3)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2a (42.7 mL, 0.42 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 130.7 mg of the title compound as a white solid; yield 93%. M.p. 85–87 °C. 1H NMR (CDCl3): δ = 1.885 (m, 4 H), 2.162 (s, 3 H), 2.30 (s, 3 H), 3.440 (m, 4 H), 3.475 (s, 2 H), 3.483 (s, 2 H), 3.763 (s, 6 H) ppm. 13C NMR (CDCl3): δ = 15.9, 21.9, 25.7, 38.7, 40.2, 50.3, 53.2, 60.0, 113.5, 124.7, 147.5, 150.3, 159.1, 172.4 ppm. IR (neat): ν̃ = 2955, 2870, 2211, 1738, 1600, 1430 cm−1. HRMS (ESI): calcd. for C18H25N2O4 [M + H]+ 333.1809; found 333.1808.
Dimethyl 3-(Dimethylamino)-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (4)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2b (34.2 mL, 0.42 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 129.5 mg of the title compound as a white solid; yield 99%. M.p. 63–64 °C. 1H NMR (CDCl3): δ = 2.16 (s, 3 H), 2.34 (s, 3 H), 2.30 (s, 3 H), 2.78 (s, 6 H), 3.49 (s, 2 H), 3.50 (s, 2 H), 3.77 (s, 6 H) ppm. 13C NMR (CDCl3): δ = 15.0, 21.8, 38.8, 40.1, 42.5, 53.3, 59.9, 117.2, 127.3, 147.9, 150.4, 172.3 ppm. IR (neat): ν̃ = 2953, 2360, 1735, 1588, 1442, 1277 cm−1. HRMS (ESI): calcd. for C16H23N2O4 [M + H]+ 307.1652; found 307.1652.
Dimethyl 3-(Diethylamino)-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (5)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2c (42.7 mL, 0.42 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 135.8 mg of the title compound as a colorless oil; yield 93%. 1H NMR (CDCl3): δ = 1.01 (t, J = 8 Hz, 6 H), 2.11 (s, 3 H), 2.30 (s, 3 H), 3.09 (m, 4 H), 3.48 (s, 2 H), 3.49 (s, 2 H), 3.74 (s, 6 H) ppm. 13C NMR (CDCl3): δ = 13.4, 14.6, 21.7, 38.8, 40.1, 45.7, 53.0, 59.7, 119.3, 127.3, 147.8, 149.9, 160.3, 172.2 ppm. IR (neat): ν̃ = 2967, 2361, 1738, 1588, 1433, 1264 cm−1. HRMS (ESI): calcd. for C18H27N2O4 [M + H]+ 335.1965; found 335.1968.
Dimethyl 3-(Dipropylamino)-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (6)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2d (53.5 mg, 0.42 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 136.4 mg of the title compound as a colorless oil; yield 89%. 1H NMR (CDCl3): δ = 0.839 (t, J = 8 Hz, 6 H), 1.24 (m, 4 H), 2.13 (s, 3 H), 2.32 (s, 3 H), 3.02 (m, 4 H), 3.49 (s, 2 H), 3.50 (s, 2 H), 3.77 (s, 6 H) ppm. 13C NMR (CDCl3): δ = 11.9, 14.7, 21.4, 21.9, 38.9, 40.2, 53.3, 53.9, 59.8, 119.2, 127.3, 147.9, 150.1, 160.8, 172.4 ppm. IR (neat): ν̃ = 2956, 2872, 1739, 1589, 1433, 1267 cm−1. HRMS (ESI): calcd. for C20H31N2O4 [M + H]+ 363.2278; found 363.2279.
Dimethyl 1,4-Dimethyl-3-morpholino-5H-cyclopenta[c]pyridine-6,6-(7H)-dicarboxylate (7)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2f (42.8 mL, 0.42 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 142.9 mg of the title compound as a white solid; yield 97%. M.p. 82–83 °C. 1H NMR (CDCl3): δ = 2.16 (s, 3 H), 2.34 (s, 3 H), 3.08 (m, 4 H), 3.49 (s, 2 H), 3.51 (s, 2 H), 3.77 (s, 6 H), 3.84 (m, 4 H) ppm. 13C NMR (CDCl3): δ = 14.5, 21.8, 38.8, 40.1, 50.8, 53.3, 59.9, 67.5, 117.9, 128.4, 148.6, 150.5, 160.3, 172.2 ppm. IR (neat): ν̃ = 2955, 2848, 2361, 1737, 1588, 1431, 1259 cm−1. HRMS (ESI): calcd. for C18H25N2O5 [M + H]+ 349.1758; found 349.1757.
Dimethyl 3-[(4-Methoxybenzyl)(methyl)amino]-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (8)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2g (74.6 mg, 0.42 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 151.9 mg of the title compound as a viscous colorless oil; yield 87%. 1H NMR (CDCl3): δ = 2.26 (s, 3 H), 2.41 (s, 3 H), 2.71 (s, 3 H), 3.57 (s, 2 H), 3.58 (s, 2 H), 3.82 (s, 6 H), 3.84 (s, 3 H), 6.92 (d, J = 8 Hz, 2 H), 7.37 (d, J = 8 Hz, 2 H) ppm. 13C NMR (CDCl3): δ = 14.7, 21.7, 38.7, 39.6, 40.0, 53.1, 55.3, 57.7, 59.8, 113.7, 117.8, 127.6, 129.3, 131.9, 148.0, 150.3, 172.1 ppm. IR (neat): ν̃ = 2953, 2838, 1737, 1588, 1512, 1436, 1246 cm−1. HRMS (ESI): calcd. for C23H29N2O5 [M + H]+ 413.2071; found 413.2069.
Dimethyl 3-(Dibenzylamino)-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (9)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2h (94.1 mg, 0.42 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 170.8 mg of the title compound as a viscous colorless oil; yield 88%. 1H NMR (CDCl3): δ = 2.30 (s, 3 H), 2.36 (s, 3 H), 3.55 (s, 2 H), 3.55 (s, 2 H), 3.82 (s, 6 H), 4.31 (s, 3 H), 7.24 (m, 2 H), 7.32 (m, 4 H), 7.38 (m, 4 H) ppm. 13C NMR (CDCl3): δ = 14.6, 21.7, 38.8, 40.1, 53.2, 55.4, 59.8, 118.9, 126.8, 128.2, 128.3, 128.6, 139.9, 148.2, 150.5, 159.9, 172.2 ppm. IR (neat): ν̃ = 3029, 2952, 2360, 1737, 1591, 1434, 1267 cm−1. HRMS (ESI): calcd. for C28H31N2O4 [M + H]+ 459.2278; found 459.2282.
Dimethyl 3-[(tert-Butoxycarbonyl)(butyl)amino]-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (10)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2i (87.2 mg, 0.44 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 149.7 mg of the title compound as a colorless solid; yield 82%. M.p. 64–66 °C. 1H NMR (C6D5CD3, 60 °C): δ = 0.85 (t, J = 8 Hz, 3 H), 1.28 (m, 2 H), 1.38 (s, 9 H), 2.11 (s, 3 H), 2.21 (s, 3 H), 3.37 (s, 6 H), 3.45 (s, 2 H), 3.51 (s, 2 H), 3.88 (br. s, 2 H) ppm. 13C NMR (C6D5CD3, 60 °C): δ = 14.1, 14.8, 20.4, 21.5, 30.4, 38.6, 40.0, 51.4, 52.0, 52.7, 53.1, 59.8, 116.6, 127.2, 147.4, 150.6, 159.1, 172.1, 172.7 ppm. IR (neat): ν̃ = 2955, 2361, 1738, 1595, 1434, 1271 cm−1. HRMS (ESI): calcd. for C23H35N2O6 [M + H]+ 435.2490; found 435.2497.
Dimethyl 3-(N-Butylacetamido)-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (11)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2j (63.1 mg, 0.45 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using 5 % MeOH/CH2CH2 to afford 137.5 mg of the title compound as a colorless yellow viscous oil; yield 87%. 1H NMR (C6D5CD3, 60 °C): δ = 0.81 (m, 3 H), 2.11 (br. s, 2 H), 1.59 (br. s, 2 H), 1.66 (s, 3 H), 1.87 (s, 3 H), 2.20 (s, 3 H), 3.35 (s, 6 H), 3.44 (s, 4 H), 4.03 (br. s, 1 H) ppm. 13C NMR (C6D5CD3, 60 °C): δ = 13.4, 13.9, 20.5, 21.4, 22.0, 30.4, 38.9, 39.9, 47.4, 52.7, 59.7, 123.9, 133.8, 137.4, 150.9, 151.5, 153.5, 168.9, 171.3 ppm. IR (neat): ν̃ = 2955, 2361, 1738, 1595, 1434, 1271 cm−1. HRMS (ESI): calcd. for C20H29N2O5 [M + H]+ 377.2071; found 377.2072.
Dimethyl 3-[(Butyl)(2-methoxy-2-oxoethyl)amino]-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (12)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2m (73 mg, 0.43 mmol) in toluene (3.6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 138.3 mg of the title compound as a viscous colorless oil; yield 81%. (Note: The title compound is light-sensitive, and chromatography was performed in a dimly lit room. The green degradation products were not isolated.) 1H NMR (CDCl3): δ = 0.87 (t, J = 7.5 Hz, 3 H), 1.27 (dt, J1 = 5, J2 = 10 Hz, 3 H), 1.55 (m, 2 H), 2.15 (s, 3 H), 2.23 (s, 3 H), 3.15 (m, 2 H), 3.45 (s, 2 H), 3.46 (s, 2 H), 3.65 (s, 3 H), 3.73 (s, 6 H), 3.91 (s, 2 H) ppm. 13C NMR (CDCl3): δ = 14.1, 14.8, 20.4, 21.5, 30.4, 38.6, 40.0, 51.4, 52.0, 52.7, 53.1, 59.8, 116.6, 127.2, 147.4, 150.6, 159.1, 172.1, 172.7 ppm. IR (neat): ν̃ = 2955, 2361, 1738, 1595, 1434, 1271 cm−1. HRMS (ESI): calcd. for C21H31N2O6 [M + H]+ 407.2177; found 407.2186.
Dimethyl 1,4-Dimethyl-3-[(methyl)(phenyl)amino]-5H-cyclopenta-[c]pyridine-6,6(7H)-dicarboxylate (13)
General Procedure A was used with diyne 1 (100 mg, 0.42 mmol) and cyanamide 2p (55.5 mg, 0.42 mmol) in toluene (3.0 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 139 mg of the title compound as a colorless waxy oil; yield 90%. 1H NMR (C6D5CD3): δ = 1.77 (s, 3 H), 2.29 (s, 3 H), 3.33 (s, 3 H), 3.34 (s, 6 H), 3.46 (s, 2 H), 3.53 (s, 2 H), 6.68 (d, J = 10 Hz, 2 H), 6.74 (t, J = 7.5 Hz, 1 H), 7.03 (t, J = 7.5 Hz, 2 H) ppm. 13C NMR (CDCl3): δ = 14.5, 21.6, 38.9, 39.8, 39.8, 39.9, 52.4, 59.9, 117.9, 119.9, 120.9, 129.2, 129.9, 137.4, 149.8, 149.9, 150.7, 157.1, 171.6 ppm. IR (neat): ν̃ = 2956, 1737, 1592, 1436, 1265 cm−1. HRMS (ESI): calcd. for C21H25N2O4 [M + H]+ 369.1809; found 369.1810.
4,7-Dimethyl-6-(pyrrolidin-1-yl)-2-tosyl-2,3-dihydro-1H-pyrrolo-[3,4-c]pyridine (15)
General Procedure A was used with diyne 14 (100 mg, 0.36 mmol) and cyanamide 2a (36.6 mL, 0.36 mmol) in toluene (3.0 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (2:1) to afford 113.3 mg of the title compound as a white solid; yield 84%. M.p. 110–112 °C (dec.). 1H NMR (CDCl3): δ = 1.88 (m, 4 H), 2.09 (s, 3 H), 2.23 (s, 3 H), 2.42 (s, 3 H), 3.44 (m, 4 H), 4.50 (s, 4 H), 7.33 (d, J = 9 Hz, 2 H), 7.79 (d, J = 9 Hz, 2 H) ppm. 13C NMR (CDCl3): δ = 15.9, 21.7, 21.8, 25.8, 50.3, 52.6, 53.6, 111.4, 120.8, 127.7, 130.1, 134.1, 143.9, 146.5, 146.7, 159.2 ppm. IR (neat): ν̃ = 2963, 2875, 2230, 1380, 1173 cm−1. HRMS (ESI): calcd. for C20H26N3O2S [M + H]+ 372.1740; found 372.1749.
4,7-Dimethyl-6-(pyrrolidin-1-yl)-1,3-dihydrofuro[3,4-c]pyridine (17)
General Procedure A was used with diyne 16 (103.4 mg, 0.85 mmol) and cyanamide 2a (85.3 mL, 0.85 mmol) in toluene (7.2 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (2:1) to afford 174.3 mg of the title compound as a colorless oil; yield 94%. 1H NMR (CDCl3): δ = 1.91 (m, 4 H), 2.13 (s, 1 H), 2.27 (s, 3 H), 3.49 (m, 4 H), 4.99(s, 2 H), 5.02 (s, 2 H) ppm. 13C NMR (CDCl3): δ = 16.1, 21.9, 25.7, 50.2, 72.8, 73.2, 110.3, 123.7, 145.1, 149.9, 159.1 ppm. IR (neat): ν̃ = 2960, 2865, 2361, 1591, 1430.5, 1345, 1060 cm−1. HRMS (ESI): calcd. for C13H19N2O [M + H]+ 219.1492; found 219.1493.
1,4-Diethyl-3-(pyrrolidin-1-yl)-5,6,7,8-tetrahydroisoquinoline (19)
General Procedure A was used with diyne 18 (107 mg, 0.66 mmol) and cyanamide 2a (66.5 mL, 0.66 mmol) in toluene (5.2 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 156.6 mg of the title compound as a colorless viscous oil; yield 92%. 1H NMR (CDCl3): δ = 1.21 (t, J = 7.5 Hz, 3 H), 1.31 (t, J = 7.5 Hz, 3 H), 1.81 (m, 4 H), 1.95 (m, 4 H), 2.69 (m, 8 H), 3.47 (m, 4 H) ppm. 13C NMR (CDCl3): δ = 12.3, 13.8, 20.5, 23.0, 23.1, 25.6, 25.8, 26.9, 27.6, 51.1, 122.2, 123.1, 145.5, 155.4, 157.2 ppm. IR (neat): ν̃ = 2931, 2866, 2361, 1739, 1563, 1416 cm−1. HRMS (ESI): calcd. for C17H27N2 [M + H]+ 259.2169; found 259. 2170.
1,4-Dimethyl-3-(pyrrolidin-1-yl)-6,7,8,9-tetrahydro-5H-cyclohepta-[c]pyridine (21)
General Procedure A was used with diyne 20 (104 mg, 0.70 mmol) and cyanamide 2a (71 mL, 0.70 mmol) in toluene (6 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 130.3 mg of the title compound as a colorless viscous oil; yield 76%. 1H NMR (CDCl3): δ = 1.59 (m, 4 H), 1.81 (m, 2 H), 1.89 (m, 4 H), 2.17 (s, 3 H), 2.42 (s, 3 H), 2.75 (m, 4 H), 3.38 (m, 4 H) ppm. 13C NMR (CDCl3): δ = 15.7, 23.1, 25.5, 26.8, 27.6, 29.2, 30.1, 32.2, 50.4, 116.1, 128.5, 148.9, 152.3, 158.2 ppm. IR (neat): ν̃ = 2920, 2853, 1570, 1420 cm−1. HRMS (ESI): calcd. for C16H25N2 [M + H]+ 245.2012; found 245.2014.
Dimethyl 4-tert-Butyl-1-methyl-3-(pyrrolidin-1-yl)-5H-cyclopenta-[c]pyridine-6,6(7H)-dicarboxylate (23)
General Procedure A was used with diyne 22 (70 mg, 0.30 mmol) and cyanamide 2a (31.0 mL, 0.30 mmol) in toluene (3.0 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 85.4 mg of the title compound as a colorless viscous oil; yield 76%. 1H NMR (CDCl3): δ = 1.34 (s, 9 H), 1.88 (m, 4 H), 2.18 (s, 3 H), 3.42 (s, 2 H), 3.48 (m, 4 H), 3.68 (s, 2 H), 3.75 (s, 6 H) ppm. 13C NMR (CDCl3): δ = 15.7, 25.7, 29.34, 29.55, 38.4, 39.2, 40.5, 50.2, 53.2, 60.5, 112.6, 122.3, 151.5, 157.2, 157.8, 172.3 ppm. IR (neat): ν̃ = 2959, 1739, 1704, 1612, 1569, 1480, 1389, 1275, 1141 cm−1. HRMS (ESI): calcd. for C21H31N2O4 [M + H]+ 375.2278; found 375.2284.
2,3,4,5-Tetraethyl-6-(pyrrolidin-1-yl)pyridine (24)
General Procedure A was used with 3-hexyne (75 mL, 0.66 mmol) and cyanamide 2a (33.3 mL, 0.33 mmol) in toluene (7.0 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 74 mg of the title compound as a waxy colorless oil; yield 86%. 1H NMR (CDCl3): δ = 1.17 (m, 9 H), 1.31 (t, J = 7.5 Hz, 3 H), 1.91 (m, 4 H), 2.67 (m, 8 H), 3.45 (m, 4 H) ppm. 13C NMR (CDCl3): δ = 13.5, 14.7, 15.58, 15.61, 20.9, 21.1, 22.0, 25.7, 27.9, 50.8, 122.8, 126.5, 150.3, 155.3, 157.5 ppm. IR (neat): ν̃ = 2966, 2871, 1562, 1416 cm−1. HRMS (ESI): calcd. for C17H29N2 [M + H]+ 261.2325; found 261.2328.
Dimethyl 3-(Pyrrolidin-1-yl)-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (26)
General Procedure B was used with diyne 25 (100 mg, 0.48 mmol) and cyanamide 2a (48.4 mL, 0.48 mmol) in toluene (4.1 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (3:1) to afford 116.8 mg of the title compound as a light-yellow solid; yield 80%. M.p. 98–99 °C. 1H NMR (CDCl3): δ = 1.95 (m, 4 H), 3.38 (m, 4 H), 3.45 (s, 4 H), 3.71 (s, 6 H), 6.20 (s, 1 H), 7.92 (s, 1 H) ppm. 13C NMR (CDCl3): δ = 25.6, 37.5, 40.5, 46.9, 53.1, 60.7, 101.8, 123.2, 143.1, 151.0, 157.0, 171.9 ppm. The melting point and spectra match known values.[32]
Dimethyl 3-Morpholino-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (27)
General Procedure B was used with diyne 25 (100 mg, 0.48 mmol) and cyanamide 2f (48.5 mL, 0.48 mmol) in toluene (4.1 mL). The remaining residue was purified by flash column chromatography using dichloromethane to afford 132.2 mg of the title compound as a light-yellow wax; yield 86%. 1H NMR (CDCl3): δ = 3.43 (m, 4 H), 3.49 (s, 4 H), 3.74 (s, 6 H), 3.79 (m, 4 H), 6.51 (s, 1 H), 8.01 (s, 1 H) ppm. 13C NMR (CDCl3): δ = 37.6, 40.6, 46.3, 53.2, 60.7, 66.9, 102.8, 126.32, 143.1, 151.7, 159.5, 171.8 ppm. The spectra match known values.[32]
Dimethyl 3-[(tert-Butoxycarbonyl)(butyl)amino]-5H-cyclopenta-[c]pyridine-6,6(7H)-dicarboxylate (28)
General Procedure B was used with diyne 25 (100 mg, 0.48 mmol) and cyanamide 2i (96 mg, 0.48 mmol) in toluene (4.1 mL). The remaining residue was purified by flash column chromatography using dichloromethane to afford 160.7 mg of the title compound as a light-yellow viscous oil; yield 82 %. 1H NMR (CDCl3): δ = 0.86 (t, J = 7.5 Hz, 3 H), 1.27 (dt, J1 = 5, J2 = 10 Hz, 3 H), 1.48 (s, 9 H), 3.55 (s, 2 H), 3.56 (s, 2 H), 3.74 (s, 6 H), 3.86 (m, 2 H), 7.42 (s, 1 H), 8.17 (s, 1 H) ppm. 13C NMR (CDCl3): δ = 13.9, 20.2, 28.5, 31.2, 37.8, 40.4, 47.1, 53.3, 60.6, 80.8, 116.1, 132.3, 143.0, 150.9, 153.8, 154.6, 171.6 ppm. IR (neat): ν̃ = 2959, 1739, 1704, 1390, 1276, 1142 cm−1. HRMS (ESI): calcd. for C21H31N2O6[M + H]+ 407.2177; found 407.2189.
Dimethyl 3-(N-Butylacetamido)-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (29)
General Procedure B was used with diyne 25 (100 mg, 0.48 mmol) and cyanamide 2j (67 mg, 0.48 mmol) in toluene (4.1 mL). The remaining residue was purified by flash column chromatography using dichloromethane to afford 155.5 mg of the title compound as a light-yellow viscous oil; yield 93%. 1H NMR (CDCl3): δ = 0.84 (t, J = 7.5 Hz, 3 H), 1.27 (dt, J1 = 5, J2 = 10 Hz, 2 H), 1.47 (m, 2 H), 1.93 (br. s, 3 H), 3.61 (s, 4 H), 3.76 (br. s, 8 H), 7.04 (s, 1 H), 8.30 (s, 1 H) ppm. 13C NMR (CDCl3): δ = 13.9, 20.2, 23.2, 30.5, 37.9, 40.4, 48.4, 53.4, 60.4, 117.7, 128.7, 135.1, 144.7, 152.5, 154.7, 170.2, 171.4 ppm. IR (neat): ν̃ = 2955, 2361, 1738, 1595, 1434, 1271 cm−1. HRMS (ESI): calcd. for C18H25N2O5 [M + H]+ 349.1758; found 349.1760.
Dimethyl 3-[(Methyl)(phenyl)amino]-5H-cyclopenta[c]pyridine-6,6-(7H)-dicarboxylate (32)
General Procedure B was used with diyne 25 (100 mg, 0.48 mmol) and cyanamide 2p (63.5 mg, 0.48 mmol) in toluene (4.1 mL). The remaining residue was purified by flash column chromatography using hexanes/ethyl acetate (1:2) to afford 135.5 mg of the title compound as a light-yellow viscous oil; yield 83%. 1H NMR (CDCl3): δ = 3.36 (s, 3 H), 3.43 (s, 3 H), 3.48 (s, 3 H), 3.71 (s, 6 H), 6.41 (s, 1 H), 7.19 (t, J = 7.5 Hz, 1 H), 7.21 (d, J = 10 Hz, 2 H), 7.37 (t, J = 7.5 Hz, 2 H), 8.04 (s, 1 H) ppm. 13C NMR (CDCl3): δ = 37.5, 38.9, 40.4, 53.2, 60.6, 104.8, 125.3, 125.7, 126.3, 129.8, 142.8, 147.2, 150.8, 158.5, 171.9 ppm. IR (neat): ν̃ = 3005, 2953, 2370, 1736, 1617, 1491, 1393, 1268 cm−1. HRMS (ESI): calcd. for C19H21N2O4 [M + H]+ 341.1496; found 341.1496.
Supplementary Material
Acknowledgments
The authors thank the Ryan Looper group for the generous donation of N-(4-methoxybenzyl)-N-methylcyanamide and general advice. We gratefully acknowledge the National Science Foundation (NSF) and the National Institute of General Medical Studies (NIGMS) (5RO1GM076125) for support of this research.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.201100428.
Supporting Information (see footnote on the first page of this article): 1H and 13C NMR spectra for all compounds, as well as (1H,13C) HMBC and (1H,13C) HMQC spectra for compound 23. HRMS and IR data available upon request from the authors.
References
- 1.Horie H, Koyama I, Kurahashi T, Matsubara S. Chem Commun. 2011;47:2658–2660. doi: 10.1039/c0cc04061d. [DOI] [PubMed] [Google Scholar]
- 2.Daka P, Xu Z, Alexa A, Wang H. Chem Commun. 2011;47:224–226. doi: 10.1039/c0cc00917b. [DOI] [PubMed] [Google Scholar]
- 3.Di Nicola C, Effendy, Marchetti F, Nervi C, Pettinari C, Robinson WT, Sobolev AN, White AH. Dalton Trans. 2010;39:908–922. doi: 10.1039/b913173f. [DOI] [PubMed] [Google Scholar]
- 4.Yip JHK, Suwarno, Vittal JJ. Inorg Chem. 2000;39:3537–3543. doi: 10.1021/ic9913482. [DOI] [PubMed] [Google Scholar]
- 5.Mutai T, Cheon J-D, Tsuchiya G, Araki K. J Chem Soc Perkin Trans. 2002;2:862–865. [Google Scholar]
- 6.Tardioli S, Gooijer C, van der Zwan G. J Phys Chem B. 2009;113:6949–6957. doi: 10.1021/jp9005907. [DOI] [PubMed] [Google Scholar]
- 7.Sathyamoorthi G, Soong ML, Ross TW, Boyer JH. Heteroat Chem. 1993;4:603. [Google Scholar]
- 8.Tursky M, Lorentz-Petersen LLR, Olsen LB, Madsen R. Org Biomol Chem. 2010;8:5576–5582. doi: 10.1039/c0ob00106f. [DOI] [PubMed] [Google Scholar]
- 9.Ueno M, Nobana T, Togo H. J Org Chem. 2003;68:6424–6426. doi: 10.1021/jo030045t. [DOI] [PubMed] [Google Scholar]
- 10.Kamal A, Reddy JS, Ramaiah MJ, Dastagiri D, Bharathi EV, Sagar MVP, Pushpavalli SNCVL, Ray P, Pal-Bhadra M. Med Chem Commun. 2010;1:355–360. [Google Scholar]
- 11.Nam TG, Nara SJ, Zagol-Ikapitte I, Cooper T, Valgimigli L, Oates JA, Porter NA, Boutaud O, Pratt DA. Org Biomol Chem. 2009;7:5103–5112. doi: 10.1039/b912528k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlucci G, Colanzi A, Mazzeo P, Quaglia MG. Int J Pharm. 1989;53:257–259. [Google Scholar]
- 13.Acker R-D, Hamprecht G. Preparation of 2-aminopyridine derivatives. 4395555. US Patent. 1981
- 14.Ji H, Delker SL, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. J Med Chem. 2010;53:7804–7824. doi: 10.1021/jm100947x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chichibabin AE, Zeide OA. J Russ Phys Chem Soc. 1914;46:1216–1236. [Google Scholar]
- 16.Chichibabin AE, Zeide OA. Ber Dtsch Chem Ges. 1923;56B:1879–1885. [Google Scholar]
- 17.Wagaw S, Buchwald SL. J Org Chem. 1996;61:7240. doi: 10.1021/jo9612739. [DOI] [PubMed] [Google Scholar]
- 18.Patriciu OI, Fînaru AL, Massip S, Léger JM, Jarry C, Guillaumet G. Eur J Org Chem. 2009;22:3753–3764. doi: 10.1021/ol902369v. [DOI] [PubMed] [Google Scholar]
- 19.Shen Q, Hartwig JF. Org Lett. 2008;10:4109–4112. doi: 10.1021/ol801615u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorimer AV, O’Connor PD, Brimble MA. Synthesis. 2008;17:2764–2770. [Google Scholar]
- 21.Matsumoto K, Fukuyama K, Iida H, Toda M, Lown JW. Heterocycles. 1995;41:237. [Google Scholar]
- 22.Perron-Sierra F, Saint Dizier D, Bertrand M, Genton A, Tucker GC, Casaram P. Bioorg Med Chem Lett. 2002;12:3291. doi: 10.1016/s0960-894x(02)00696-0. [DOI] [PubMed] [Google Scholar]
- 23.Gupton JT, Idoux JP, Baker G, Colon C, Crews AD, Jurss CD, Rampi RC. J Org Chem. 1983;48:2933. [Google Scholar]
- 24.Thomas S, Roberts S, Pasumansky L, Gamsey S, Singaram B. Org Lett. 2003;5:3867–3870. doi: 10.1021/ol035430j. [DOI] [PubMed] [Google Scholar]
- 25.Teague SJ. J Org Chem. 2008;73:9765–9766. doi: 10.1021/jo801303v. [DOI] [PubMed] [Google Scholar]
- 26.Ranu BC, Jana R, Sowmiah S. J Org Chem. 2007;72:3152–3154. doi: 10.1021/jo070015g. [DOI] [PubMed] [Google Scholar]
- 27.Sainte F, Serckx-Poncin B, Hesbain-Frisque AM, Ghosez L. J Am Chem Soc. 1982;104:1428–1430. [Google Scholar]
- 28.Liu ZJ, Vors JP, Gesing ERF, Bolm C. Adv Synth Catal. 2010;352:3158–3162. [Google Scholar]
- 29.Shafir A, Buchwald SL. J Am Chem Soc. 2006;128:8742–8743. doi: 10.1021/ja063063b. [DOI] [PubMed] [Google Scholar]
- 30.For reviews of transition-metal-catalyzed [2+2+2] cycloadditions, see: Varela JA, Saá C. Synlett. 2008:2571–2578.Heller B, Hapke M. Chem Soc Rev. 2007;36:1085–1094. doi: 10.1039/b607877j.Hua R, Abrenica MVA, Wang P. Curr Org Chem. 2011;15:712–729.Varela JA, Saá C. Chem Rev. 2003;103:3787–3801. doi: 10.1021/cr030677f.Chopade PR, Louie J. Adv Synth Catal. 2006;348:2307–2327.Tanaka K. Synlett. 2007:1977–1993.
- 31.For recent examples of transition-metal-catalyzed [2+2+2] cycloadditions of alkynes and nitriles, see: Iwayama T, Sato Y. Chem Commun. 2009:5245–5247. doi: 10.1039/b912022j.Garcia L, Pla-Quintana A, Roglans A, Parella T. Eur J Org Chem. 2010:3407–3415.Garcia P, Moulin S, Miclo Y, Leboeuf D, Gandon V, Aubert C, Malacria M. Chem Eur J. 2009;15:2129–2139. doi: 10.1002/chem.200802301.Kadlcikova A, Hrdina R, Valterova I, Kotora M. Adv Synth Catal. 2009;351:1279–1283.Young DD, Teske JA, Deiters A. Synthesis. 2009:3785–3790.Komine Y, Kamisawa A, Tanaka K. Org Lett. 2009;11:2361–2364. doi: 10.1021/ol900802d.Garcia P, Evanno Y, George P, Sevrin M, Ricci G, Malacria M, Aubert C, Gandon V. Org Lett. 2011;13:2030–2033. doi: 10.1021/ol200417p.Miclo Y, Garcia P, Evanno Y, George P, Sevrin M, Malacria M, Gandon V, Aubert C. Synlett. 2010:2314–2318.Geny A, Agenet N, Iannazzo L, Malacria M, Aubert C, Gandon V. Angew Chem. 2009;121:1842. doi: 10.1002/anie.200806001.Angew Chem Int Ed. 2009;48:1810–1813. doi: 10.1002/anie.200806001.Hsieh JC, Cheng CH. Chem Commun. 2008:2992–2994. doi: 10.1039/b801870g.Watanabe JI, Sugiyama YK, Nomura A, Azumatei S, Goswami A, Saino N, Okamoto S. Macromolecules. 2010;43:2213–2218.
- 32.Bönnemann H, Brijoux W, Brinkmann R, Meurers W. Helv Chim Acta. 1984;67:1616–1624. [Google Scholar]
- 33.Heller B, Reihsig J, Schulz W, Oehme G. Appl Organomet Chem. 1993;7:641–646. [Google Scholar]
- 34.Boñaga LVR, Zhang HC, Maryanoff BE. Chem Commun. 2004:2394–2395. doi: 10.1039/b410012c. [DOI] [PubMed] [Google Scholar]
- 35.Boñaga LVR, Zhang HC, Moretto AF, Ye H, Gauthier DA, Li J, Leo GC, Maryanoff BE. J Am Chem Soc. 2005;127:3473–3485. doi: 10.1021/ja045001w. [DOI] [PubMed] [Google Scholar]
- 36.Zhang HC, Boñaga LVR, Ye H, Derian CK, Damiano BP, Maryanoff BE. Bioorg Med Chem Lett. 2007;17:2863–2868. doi: 10.1016/j.bmcl.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka K, Suzuki N, Nishida G. Eur J Org Chem. 2006:3917–3922. [Google Scholar]
- 38.Hapko M, Kral K, Fischer C, Spannenberg A, Gutnov A, Redkin D, Heller B. J Org Chem. 2010;75:3993–4003. doi: 10.1021/jo100122d. [DOI] [PubMed] [Google Scholar]
- 39.McCormick MM, Duong HA, Zuo G, Louie J. J Am Chem Soc. 2005;127:5030–5031. doi: 10.1021/ja0508931. [DOI] [PubMed] [Google Scholar]
- 40.Louie J, Gibby JE, Farnworth MV, Tekavec TN. J Am Chem Soc. 2002;124:15188–15189. doi: 10.1021/ja027438e. [DOI] [PubMed] [Google Scholar]
- 41.Duong HA, Cross MJ, Louie J. J Am Chem Soc. 2004;126:11438–11439. doi: 10.1021/ja046477i. [DOI] [PubMed] [Google Scholar]
- 42.Tekavec TN, Louie J. Org Lett. 2005;7:4037–4039. doi: 10.1021/ol0515558. [DOI] [PubMed] [Google Scholar]
- 43.Tekavec TN, Louie J. J Org Chem. 2008;73:2641–2648. doi: 10.1021/jo702508w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tekavec TN, Arif AM, Louie J. Tetrahedron. 2004;60:7431–7437. [Google Scholar]
- 45.Tekavec TN, Zuo G, Simon K, Louie J. J Org Chem. 2006;71:5834–5836. doi: 10.1021/jo0608669. [DOI] [PubMed] [Google Scholar]
- 46.Böhm VPW, Weskammp T, Gstottmayr CWK, Herrmann WA. Angew Chem. 2000;112:1672. [Google Scholar]; Angew Chem Int Ed. 2000;39:1602–1604. doi: 10.1002/(sici)1521-3773(20000502)39:9<1602::aid-anie1602>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 47.Arduengo AJ, III, Krafczyk R, Schmutzler R, Craig HA, Goerlich JR, Marshall WJ, Unverzagt M. Tetrahedron. 1999;55:14523–14534. [Google Scholar]
- 48.Atkinson RS, Grimshire MJ. J Chem Soc Perkin Trans 1. 1986:1215–1217. [Google Scholar]
- 49.Nishida M, Shiga H, Mori M. J Org Chem. 1998;63:8606–8608. [Google Scholar]
- 50.Nugent WA, Thorn DL, Harlow RL. J Am Chem Soc. 1987;109:2788–2796. [Google Scholar]
- 51.Ross WJ, Harrison RG, Jolley MRJ, Neville MC, Todd A, Verge JP, Dawson W, Sweatman WJF. J Med Chem. 1979;22:412–417. doi: 10.1021/jm00190a011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.